
The Internal Relation between Quantum Chemical Descriptors and Empirical Constants of Polychlorinated Compounds

Abstract
1. Introduction
2. Results and Discussion
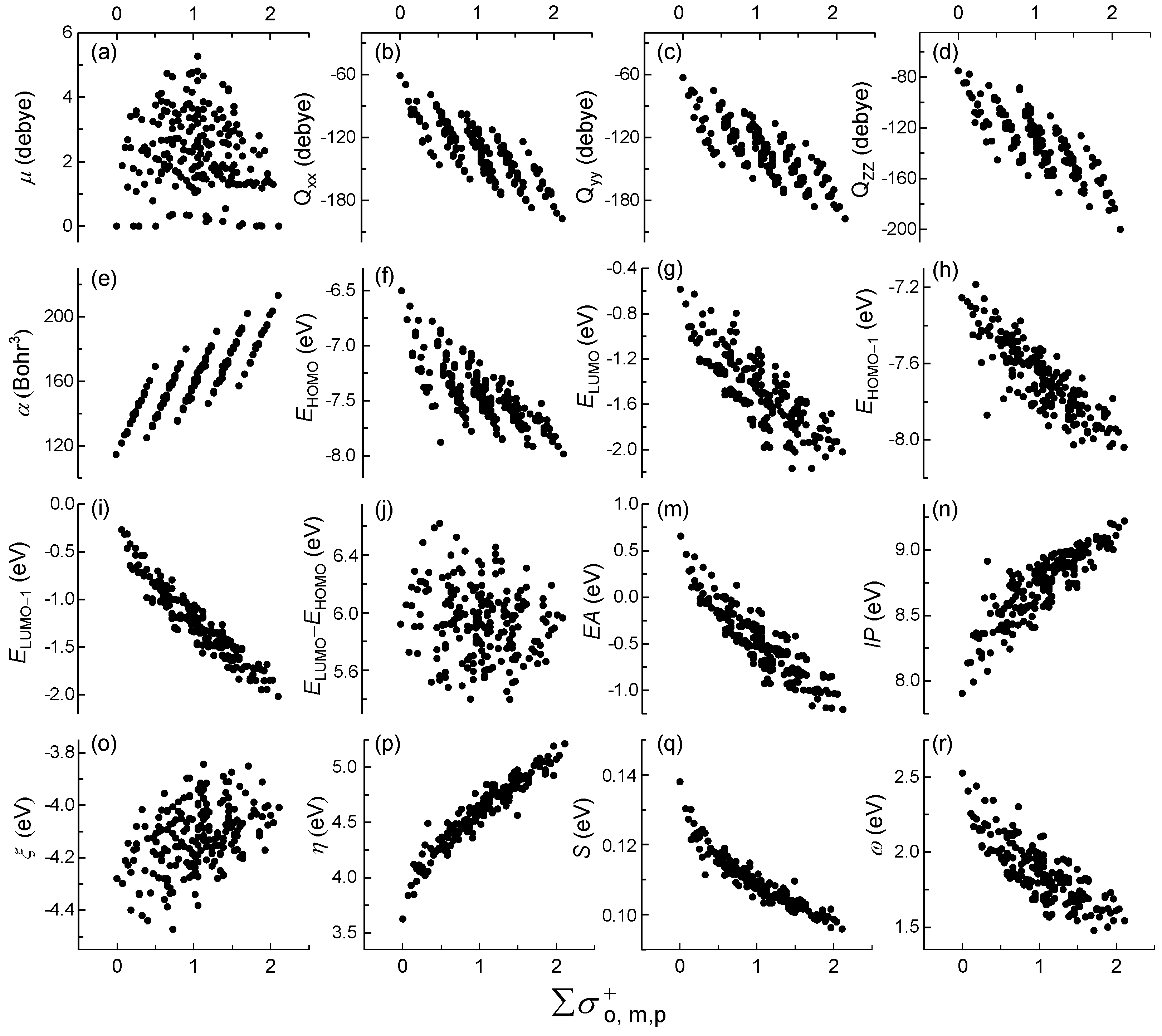
2.1. Reveal Relationships between Quantum Descriptors and Hammett Constants
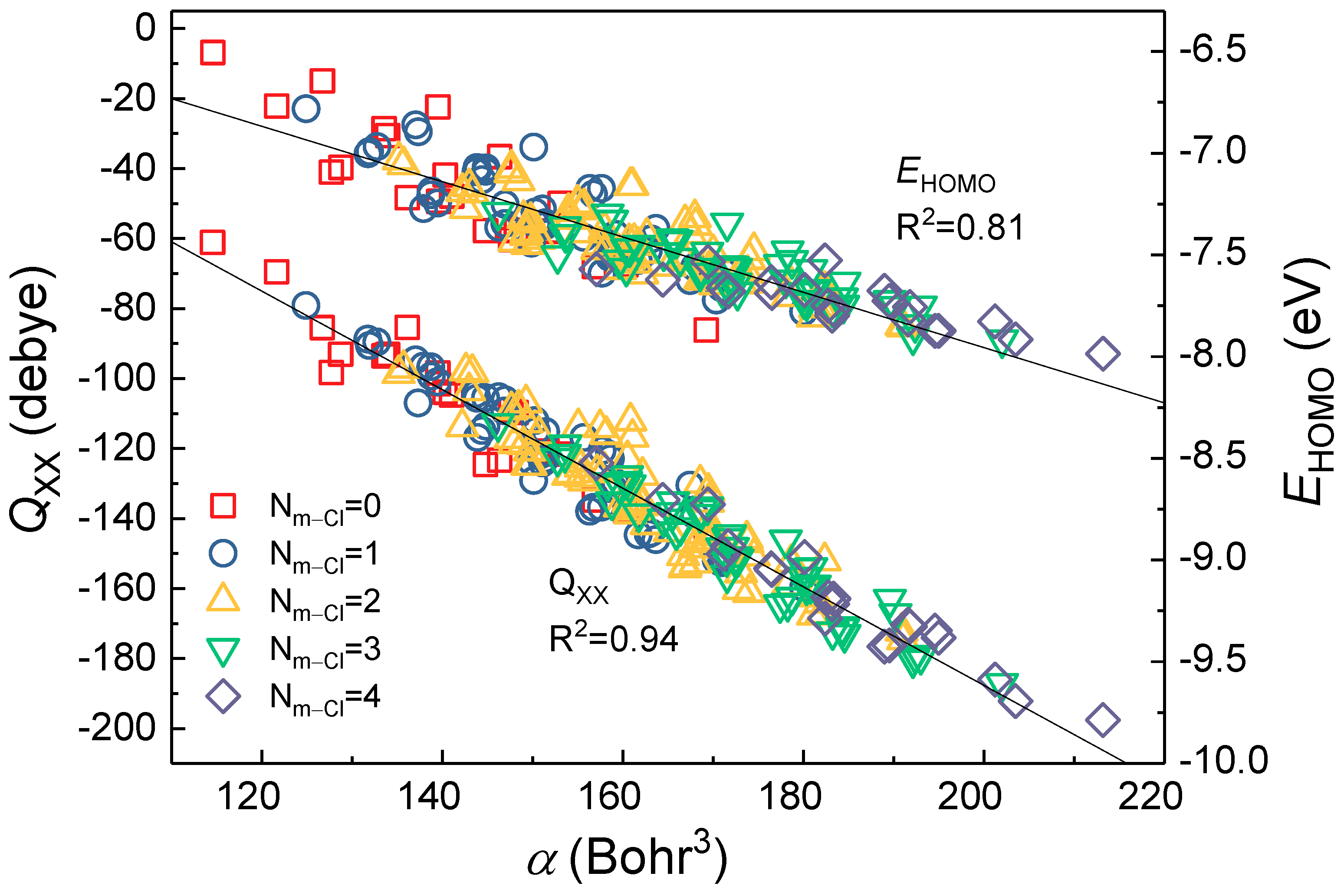
2.2. Mechanistic Interpretation of Internal Relationship
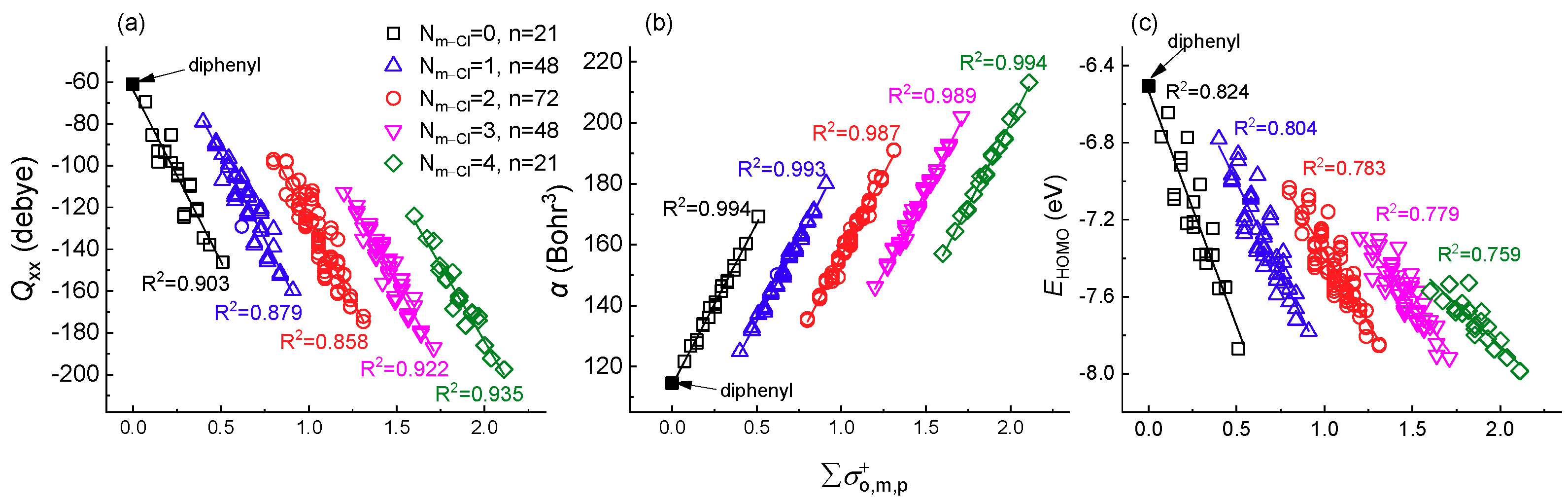
2.3. Application of Meta-Substituent Grouping
2.3.1. Application in Similar Compounds
2.3.2. Application in Prediction Model
3. Methods
3.1. Data Collection
3.2. Quantum Chemical Descriptors Calculation
3.3. Model Development
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1044. [Google Scholar] [CrossRef] [PubMed]
- Carbó, R. Molecular Similarity and Reactivity: From Quantum Chemical to Phenomenological Approaches; Springer Science & Business Media: Berlin, Germany, 1995; Volume 14. [Google Scholar]
- Primas, H. Chemistry, Quantum Mechanics and Reductionism: Perspectives in Theoretical Chemistry; Springer Science & Business Media: Berlin, Germany, 2013; Volume 24. [Google Scholar]
- Luo, S.; Wei, Z.; Spinney, R.; Villamena, F.A.; Dionysiou, D.D.; Chen, D.; Tang, C.-J.; Chai, L.; Xiao, R. Quantitative structure–activity relationships for reactivities of sulfate and hydroxyl radicals with aromatic contaminants through single–electron transfer pathway. J. Hazard. Mater. 2018, 344, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.T.; Walker, J.D.; Jaworska, J.S.; Comber, M.H.; Watts, C.D.; Worth, A.P. Use of QSARs in international decision-making frameworks to predict ecologic effects and environmental fate of chemical substances. Environ. Health Perspect. 2003, 111, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Von Gunten, U. Quantitative structure–activity relationships (QSARs) for the transformation of organic micropollutants during oxidative water treatment. Water Res. 2012, 46, 6177–6195. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. ATLA-Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar]
- Papp, T.; Kollar, L.; Kegl, T. Employment of quantum chemical descriptors for Hammett constants: Revision Suggested for the acetoxy substituent. Chem. Phys. Lett. 2013, 588, 51–56. [Google Scholar] [CrossRef]
- Cocchi, M.; Menziani, M.C.; De Benedetti, P.G.; Cruciani, G. Theoretical versus empirical molecular descriptors in monosubstituted benzenes: A chemometric study. Chemom. Intell. Lab. 1992, 14, 209–224. [Google Scholar] [CrossRef]
- Nakano, T.; Kaminuma, T.; Sato, T.; Fukuzawa, K.; Akiyama, Y.; Uebayasi, M.; Kitaura, K. Fragment molecular orbital method: Use of approximate electrostatic potential. Chem. Phys. Lett. 2002, 351, 475–480. [Google Scholar] [CrossRef]
- Shi, J.; Qu, R.; Feng, M.; Wang, X.; Wang, L.; Yang, S.; Wang, Z. Oxidative degradation of decabromodiphenyl ether (BDE 209) by potassium permanganate: Reaction pathways, kinetics, and mechanisms assisted by density functional theory calculations. Environ. Sci. Technol. 2015, 49, 4209–4217. [Google Scholar] [CrossRef] [PubMed]
- Labanowski, J.K.; Andzelm, J.W. Density Functional Methods in Chemistry; Springer: New York, NY, USA, 1991. [Google Scholar]
- Edwards, J.O.; Pearson, R.G. The factors determining nucleophilic reactivities. J. Am. Chem. Soc. 1962, 84, 16–24. [Google Scholar] [CrossRef]
- Yang, Z.; Luo, S.; Wei, Z.; Ye, T.; Spinney, R.; Chen, D.; Xiao, R. Rate constants of hydroxyl radical oxidation of polychlorinated biphenyls in the gas phase: A single−descriptor based QSAR and DFT study. Environ. Pollut. 2016, 211, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Kohanoff, J.; Gidopoulos, N. Density functional theory: Basics, new trends and applications. In Handbook of Molecular Physics and Quantum Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2003; Volume 2, pp. 532–568. [Google Scholar]
- Luo, S.; Gao, L.; Wei, Z.; Spinney, R.; Dionysiou, D.D.; Hu, W.-P.; Chai, L.; Xiao, R. Kinetic and mechanistic aspects of hydroxyl radical‒mediated degradation of naproxen and reaction intermediates. Water Res. 2018, 137, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Wei, Z.; Dionysiou, D.D.; Spinney, R.; Hu, W.-P.; Chai, L.; Yang, Z.; Ye, T.; Xiao, R. Mechanistic insight into reactivity of sulfate radical with aromatic contaminants through single-electron transfer pathway. Chem. Eng. J. 2017, 327, 1056–1065. [Google Scholar] [CrossRef]
- Qu, R.; Liu, H.; Feng, M.; Yang, X.; Wang, Z. Investigation on intramolecular hydrogen bond and some thermodynamic properties of polyhydroxylated anthraquinones. J. Chem. Eng. Data 2012, 57, 2442–2455. [Google Scholar] [CrossRef]
- Luo, S.; Wei, Z.; Spinney, R.; Zhang, Z.; Dionysiou, D.D.; Gao, L.; Chai, L.; Wang, D.; Xiao, R. UV direct photolysis of sulfamethoxazole and ibuprofen: An experimental and modelling study. J. Hazard. Mater. 2018, 343, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Ye, T.; Wei, Z.; Luo, S.; Yang, Z.; Spinney, R. Quantitative structure-activity relationship (QSAR) for the oxidation of trace organic contaminants by sulfate radical. Environ. Sci. Technol. 2015, 49, 13394–13402. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Wei, Z.; Spinney, R.; Yang, Z.; Chai, L.; Xiao, R. A novel model to predict gas–phase hydroxyl radical oxidation kinetics of polychlorinated compounds. Chemosphere 2017, 172, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Wei, Z.; Spinney, R.; Tang, C.-J.; Luo, S.; Xiao, R.; Dionysiou, D.D. Chemical structure-based predictive model for the oxidation of trace organic contaminants by sulfate radical. Water Res. 2017, 116, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Gross, K.C.; Seybold, P.G.; Peralta-Inga, Z.; Murray, J.S.; Politzer, P. Comparison of quantum chemical parameters and Hammett constants in correlating pKa values of substituted anilines. J. Org. Chem. 2001, 66, 6919–6925. [Google Scholar] [CrossRef] [PubMed]
- Amat, L.; Carbó-Dorca, R.; Cooper, D.L.; Allan, N.L.; Ponec, R. Structure-property relationships and momentum space quantities: Hammett σ-Constants. Mol. Phys. 2003, 101, 3159–3162. [Google Scholar] [CrossRef]
- Santiago, C.B.; Milo, A.; Sigman, M.S. Developing a modern approach to account for steric effects in Hammett-type correlations. J. Am. Chem. Soc. 2016, 138, 13424–13430. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995; Volume 557. [Google Scholar]
- Sudhakaran, S.; Amy, G.L. QSAR models for oxidation of organic micropollutants in water based on ozone and hydroxyl radical rate constants and their chemical classification. Water Res. 2013, 47, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.J.; Dixon, S.L.; Jurs, P.C. Computer-assisted study of the relationship between molecular structure and Henry’s law constant. Anal. Chem. 1992, 64, 1350–1355. [Google Scholar] [CrossRef]
- Kusic, H.; Rasulev, B.; Leszczynska, D.; Leszczynski, J.; Koprivanac, N. Prediction of rate constants for radical degradation of aromatic pollutants in water matrix: A QSAR study. Chemosphere 2009, 75, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Zetzsch, C. Predicting the rate of OH-addition to aromatics using σ+-electrophilic substituent constants for mono-and polysubstituted benzene. In Proceedings of the 15 th Informal Conference on Photochemistry, Stanford, CA, USA, 27 June–1 July 1982. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Zeng, X.-L.; Wang, H.-J.; Wang, Y. QSPR models of n-octanol/water partition coefficients and aqueous solubility of halogenated methyl-phenyl ethers by DFT method. Chemosphere 2012, 86, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.J. Additivity methods in molecular polarizability. J. Am. Chem. Soc. 1990, 112, 8533–8542. [Google Scholar] [CrossRef]
- Soteras, I.; Curutchet, C.; Bidon-Chanal, A.; Dehez, F.; Angyan, J.G.; Orozco, M.; Chipot, C.; Luque, F.J. Derivation of distributed models of atomic polarizability for molecular simulations. J. Chem. Theory Comput. 2007, 3, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Kurup, A.; Hansch, C. On the role of polarizability in QSAR. Bioorg. Med. Chem. 2005, 13, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Ghanty, T.K.; Ghosh, S.K. A density functional approach to hardness, polarizability, and valency of molecules in chemical reactions. J. Phys. Chem. 1996, 100, 12295–12298. [Google Scholar] [CrossRef]
- Kim, K.S.; Tarakeshwar, P.; Lee, J.Y. Molecular clusters of π-systems: Theoretical studies of structures, spectra, and origin of interaction energies. Chem. Rev. 2000, 100, 4145–4186. [Google Scholar] [CrossRef] [PubMed]
- Mhin, B.J.; Lee, J.E.; Choi, W. Understanding the congener-specific toxicity in polychlorinated dibenzo-p-dioxins: Chlorination pattern and molecular quadrupole moment. J. Am. Chem. Soc. 2002, 124, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Jiang, X.; Yan, D.; Bian, Y. DFT study on the relationship between the structure and water solubility of dioxin compounds. Huan jing ke xue = Huanjing kexue 2007, 28, 2651–2656. [Google Scholar] [PubMed]
- Gu, C.; Jiang, X.; Yan, D.; Bian, Y.; Yu, G. Study on the relationship between dioxin structures and n-octanol/water partition coefficients using density functional theory (DFT). Acta Sci. Circumstantiae 2008, 2, 185–191. [Google Scholar]
- Yan, C.L.; Chen, J.W.; Huang, L.P.; Ding, G.H.; Huang, X.Y. Linear free energy relationships on rate constants for the gas-phase reactions of hydroxyl radicals with PAHs and PCDD/Fs. Chemosphere 2005, 61, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.N.; Locke, M.A. Relationships between molecular properties and log P and soil sorption (Koc) of substituted phenylureas: QSAR models. Chemosphere 1994, 28, 1929–1941. [Google Scholar] [CrossRef]
- Yang, G.Y.; Yu, J.; Wang, Z.Y.; Zeng, X.L.; Ju, X.H. QSPR Study on the Aqueous Solubility (− lgSw) and n-Octanol/Water Partition Coefficients (lgKow) of Polychlorinated Dibenzo-p-dioxins (PCDDs). QSAR Comb. Sci. 2007, 26, 352–357. [Google Scholar] [CrossRef]
- Huang, J.; Yu, G.; Zhang, Z.-L. Application of TLSER method in predicting the aqueous solubility and n-octanol/water partition coefficient of PCBs, PCDDs and PCDFs. J. Environ. Sci. 2004, 16, 21–29. [Google Scholar]
- Kim, M.; Li, L.Y.; Grace, J.R. Predictability of physicochemical properties of polychlorinated dibenzo-p-dioxins (PCDDs) based on single-molecular descriptor models. Environ. Pollut. 2016, 213, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Canonica, S.; Tratnyek, P.G. Quantitative structure-activity relationships for oxidation reactions of organic chemicals in water. Environ. Toxicol. Chem. 2003, 22, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView 5.0.8; Gaussian Inc.: Wallingford, CT, USA, 2008. [Google Scholar]
- Deppmeier, B.; Driessen, A.; Hehre, T.; Hehre, W.; Johnson, J.; Klunzinger, P.; Leonard, J.; Ohlinger, W.; Pham, I.; Pietro, W. Spartan’10. Wavefunction Inc 2011. [Google Scholar]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field. IV. Conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Frisc, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09 (Revision C. 01); Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Perdew, J.P.; Kieron, B.; Yue, W. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. [Google Scholar] [CrossRef]
- Easton, R.E.; Giesen, D.; Welch, A.; Cramer, C.; Truhlar, D. The MIDI! basis set for quantum mechanical calculations of molecular geometries and partial charges. Theor. Chim. Acta 1996, 93, 281–301. [Google Scholar] [CrossRef]
- Thompson, J.D.; Winget, P.; Truhlar, D.G. MIDIX basis set for the lithium atom: Accurate geometries and atomic partial charges for lithium compounds with minimal computational cost. PhysChemComm 2001, 4, 72–77. [Google Scholar] [CrossRef]
- Lynch, B.J.; Truhlar, D.G. Small basis sets for calculations of barrier heights, energies of reaction, electron affinities, geometries, and dipole moments. Theor. Chem. Acc. 2004, 111, 335–344. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- SPSS Statistics for Windows, V. 17.0; SPSS Inc: Chicago, IL, USA, 2008.
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| # | [21] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | R2 | A | B | R2 | A | B | R2 | ||
| Nm-Cl = 0 | 0~0.51 | −164.46 | −63.53 | 0.903 | 106.13 | 113.94 | 0.994 | −2.44 | −6.53 | 0.824 |
| Nm-Cl = 1 | 0.4~0.91 | −162.23 | −13.51 | 0.879 | 106.38 | 81.72 | 0.993 | −1.88 | −6.06 | 0.804 |
| Nm-Cl = 2 | 0.8~1.31 | −156.71 | 31.14 | 0.858 | 107.73 | 48.49 | 0.987 | −1.49 | −5.88 | 0.783 |
| Nm-Cl = 3 | 1.2~1.71 | −152.50 | 72.28 | 0.922 | 107.67 | 16.33 | 0.989 | −1.20 | −5.84 | 0.779 |
| Nm-Cl = 4 | 1.6~2.11 | −144.53 | 104.95 | 0.935 | 108.87 | −17.84 | 0.994 | −0.87 | −6.12 | 0.759 |
| Congeners | Model | Statistical Diagnostic | |
|---|---|---|---|
| PCBs | Nm-Cl = 0 | lnk = ‒5.73 × − 25.64 | = −0.03 |
| Nm-Cl = 1 | lnk = ‒5.74 × − 23.90 | Q2 = 0.825 | |
| Nm-Cl = 2 | lnk = ‒5.82 × − 22.11 | F = 113 | |
| Nm-Cl = 3 | lnk = ‒5.81 × − 20.37 | p < 0.01 | |
| Nm-Cl = 4 | lnk = ‒5.88 × − 18.53 | [21] | |
| PCDDs | Nm-Cl = 0 | logKOW = 4.60 × + 4.46 | = 0.45 |
| Nm-Cl = 1 | logKOW = 4.70 × + 2.97 | Q2 = 0.954 | |
| Nm-Cl = 2 | logKOW = 4.70 × + 1.48 | F = 314 | |
| Nm-Cl = 3 | logKOW = 4.69 × + 0.02 | p < 0.01 | |
| Nm-Cl = 4 | logKOW = 4.69 × 1.45 | [43] * | |
| PCDDs | Nm-Cl = 0 | −logSW = 9.52 × + 4.78 | = −0.92 |
| Nm-Cl = 1 | −logSW = 9.73 × + 1.69 | Q2 = 0.981 | |
| Nm-Cl = 2 | −logSW = 9.74 × − 1.38 | F = 659 | |
| Nm-Cl = 3 | −logSW = 9.72 × − 4.42 | p < 0.01 | |
| Nm-Cl = 4 | −logSW = 9.72 × − 7.46 | [45] * | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fei, J.; Mao, Q.; Peng, L.; Ye, T.; Yang, Y.; Luo, S. The Internal Relation between Quantum Chemical Descriptors and Empirical Constants of Polychlorinated Compounds. Molecules 2018, 23, 2935. https://doi.org/10.3390/molecules23112935
Fei J, Mao Q, Peng L, Ye T, Yang Y, Luo S. The Internal Relation between Quantum Chemical Descriptors and Empirical Constants of Polychlorinated Compounds. Molecules. 2018; 23(11):2935. https://doi.org/10.3390/molecules23112935
Chicago/Turabian StyleFei, Jiangchi, Qiming Mao, Lu Peng, Tiantian Ye, Yuan Yang, and Shuang Luo. 2018. "The Internal Relation between Quantum Chemical Descriptors and Empirical Constants of Polychlorinated Compounds" Molecules 23, no. 11: 2935. https://doi.org/10.3390/molecules23112935
APA StyleFei, J., Mao, Q., Peng, L., Ye, T., Yang, Y., & Luo, S. (2018). The Internal Relation between Quantum Chemical Descriptors and Empirical Constants of Polychlorinated Compounds. Molecules, 23(11), 2935. https://doi.org/10.3390/molecules23112935