Carotenoid-Chlorophyll Interactions in a Photosynthetic Antenna Protein: A Supramolecular QM/MM Approach



Abstract
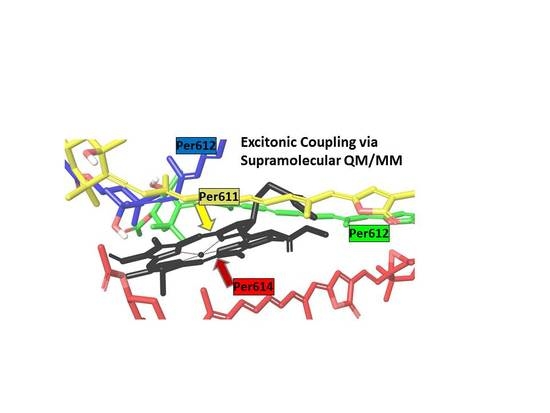
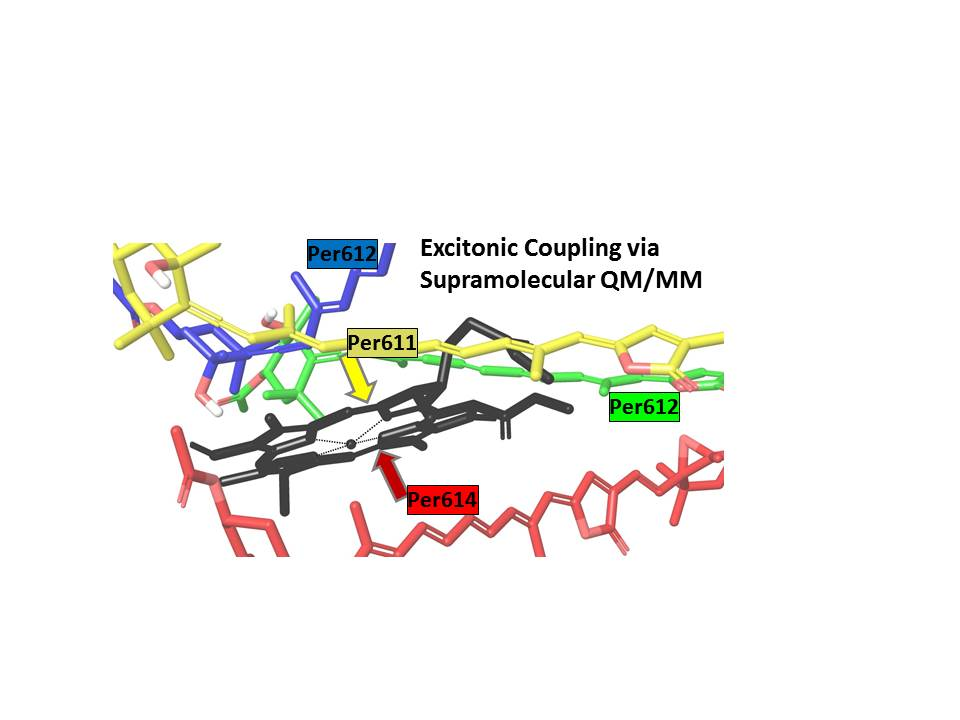
1. Introduction
2. Materials and Methods
2.1. Model Preparation
2.2. Structure Optimizations and QM/MM Simulations
2.3. Excited States and Electronic Coupling Analysis
2.4. Natural Transition Orbital (NTO) Analysis
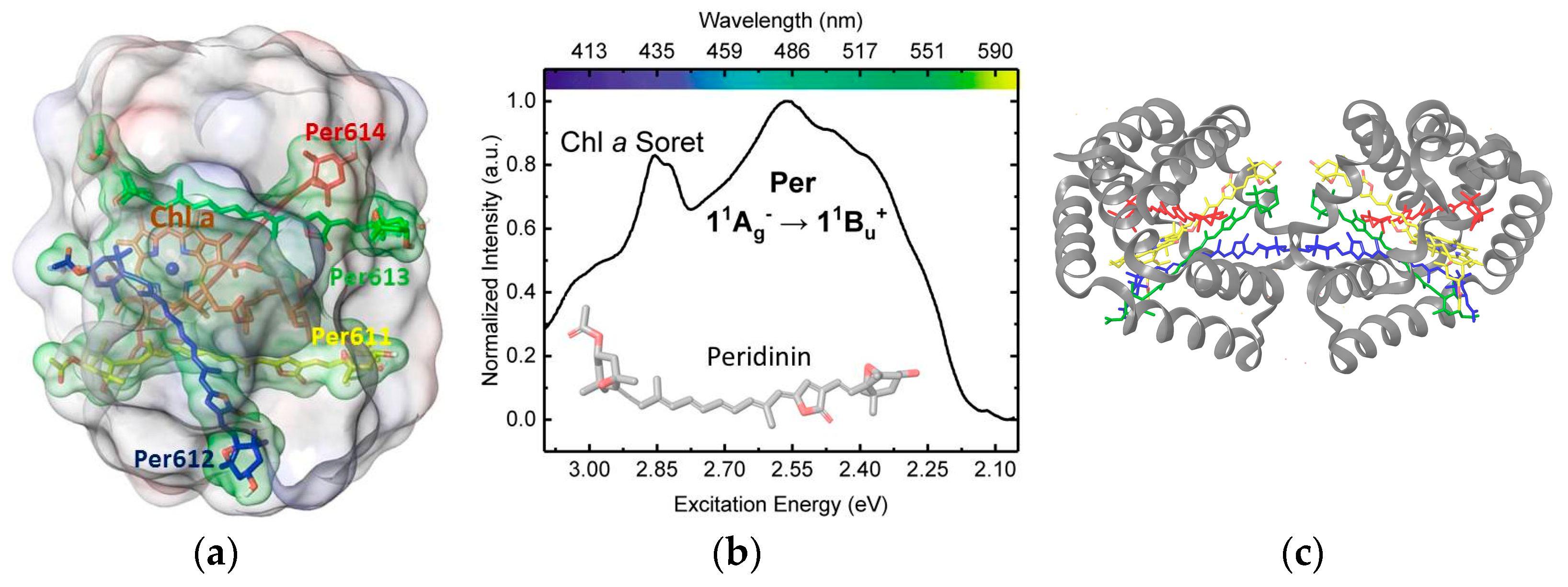
3. Results
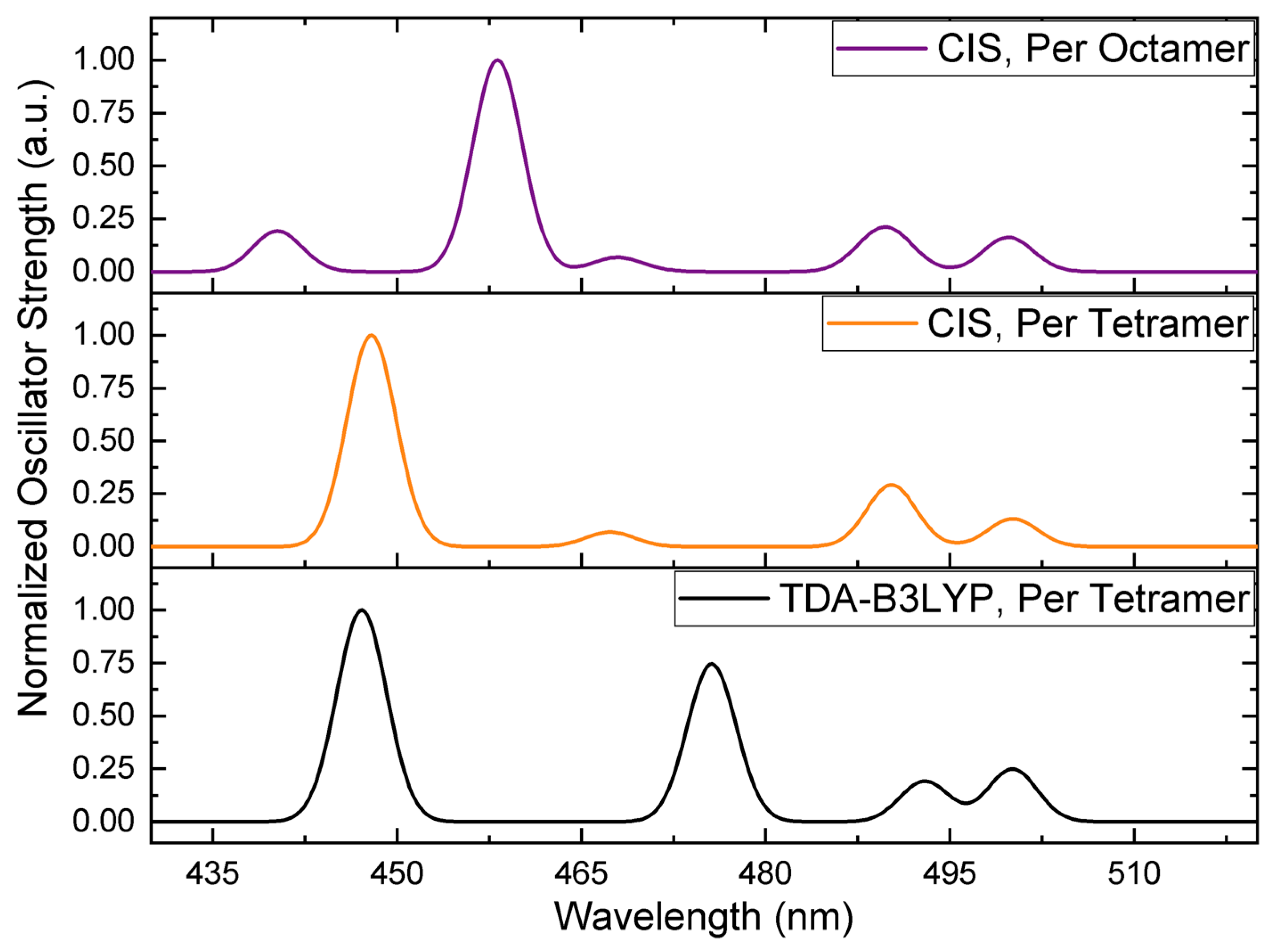
3.1. On the Accuracy of Excitonic Coupling Models
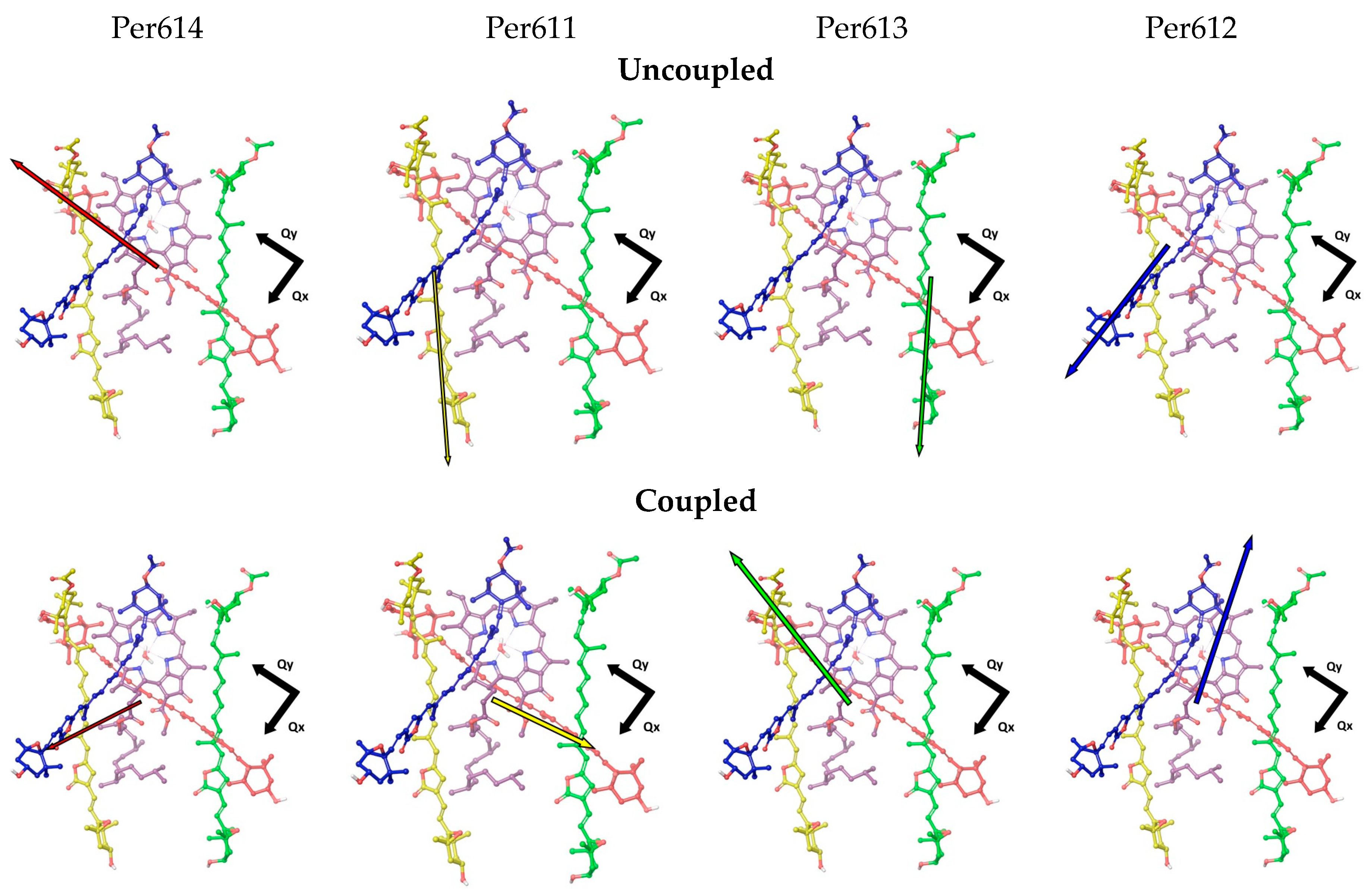
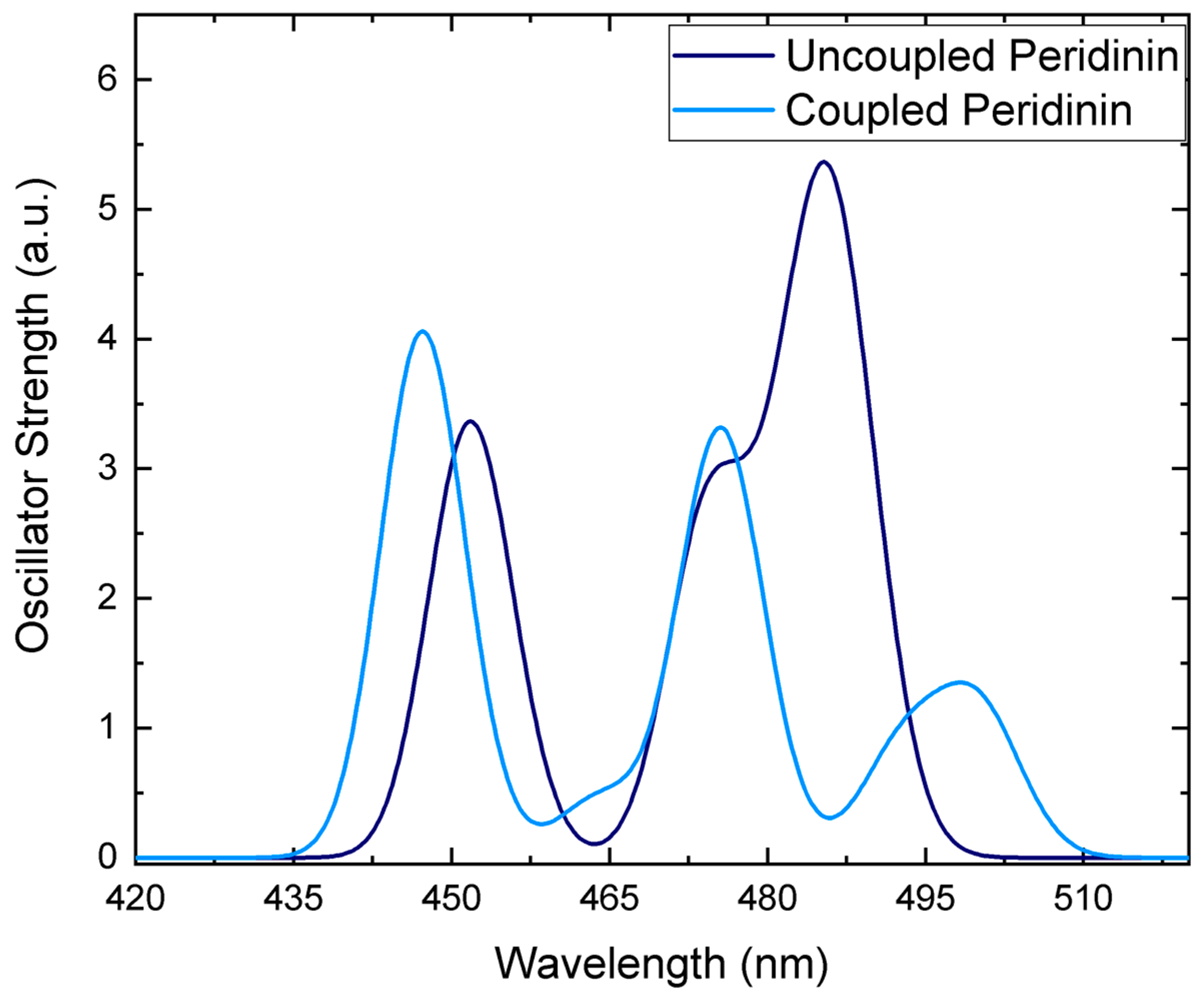
3.2. Environment Effect on Chromophore Coupling
3.3. Relating Coupling Constants to Experiment
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Scholes, G.D.; Fleming, G.R.; Olaya-Castro, A.; Van Grondelle, R. Lessons from nature about solar light harvesting. Nat. Chem. 2011, 3, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, T.; Ostroumov, E.E.; Anna, J.M.; van Grondelle, R.; Scholes, G.D. Light absorption and energy transfer in the antenna complexes of photosynthetic organisms. Chem. Rev. 2016, 117, 249–293. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.; Niedzwiedzki, D.M.; Birge, R.R.; Hiller, R.G.; Polívka, T.; Hofmann, E.; Frank, H.A. Identification of a single peridinin sensing Chl-a excitation in reconstituted PCP by crystallography and spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 20764–20769. [Google Scholar] [CrossRef] [PubMed]
- Croce, R.; Van Amerongen, H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 2014, 10, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Bautista, J.A.; Hiller, R.G.; Sharples, F.P.; Gosztola, D.; Wasielewski, M.; Frank, H.A. Singlet and triplet energy transfer in the peridinin–chlorophyll a–protein from amphidinium carterae. J. Phys. Chem. A 1999, 103, 2267–2273. [Google Scholar] [CrossRef]
- Hofmann, E.; Wrench, P.M.; Sharples, F.P.; Hiller, R.G.; Welte, W.; Diederichs, K. Structural basis of light harvesting by carotenoids: Peridinin-chlorophyll-protein from. Science 1996, 272, 1788–1791. [Google Scholar] [CrossRef] [PubMed]
- Guberman-Pfeffer, M.J.; Greco, J.A.; Birge, R.R.; Frank, H.A.; Gascón, J.A. Light harvesting by equally contributing mechanisms in a photosynthetic antenna protein. J. Phys. Chem. Lett. 2018, 9, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.S.; Kohler, B.E. A low-lying weak transition in the polyene α, ω-diphenyloctatetraene. Chem. Phys. Lett. 1972, 14, 299–304. [Google Scholar] [CrossRef]
- Fiedor, L.; Fiedor, J.; Pilch, M. Effects of molecular symmetry on the electronic transitions in carotenoids. J. Phys. Chem. Lett. 2016, 7, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Song, P.-S.; Koka, P.; Prezelin, B.B.; Haxo, F.T. Molecular topology of the photosynthetic light-harvesting pigment complex, peridinin-chlorophyll a-protein, from marine dinoflagellates. Biochemistry 1976, 15, 4422–4427. [Google Scholar] [CrossRef] [PubMed]
- Pilch, M.; Pawlikowski, M. Circular dichroism (CD) study of peridinin–chlorophyll a protein (PCP) complexes from marine dinoflagellate algae The tetramer approach. J. Chem. Soc. Faraday Trans. 1998, 94, 227–232. [Google Scholar] [CrossRef]
- Carbonera, D.; Giacometti, G.; Segre, U.; Hofmann, E.; Hiller, R.G. Structure-based calculations of the optical spectra of the light-harvesting peridinin−chlorophyll−protein complexes from Amphidinium carterae and Heterocapsa pygmaea. J. Phys. Chem. B 1999, 103, 6349–6356. [Google Scholar] [CrossRef]
- Bricker, W.P.; Lo, C.S. Efficient pathways of excitation energy transfer from delocalized S 2 excitons in the peridinin–chlorophyll a–protein complex. J. Phys. Chem. B 2015, 119, 5755–5764. [Google Scholar] [CrossRef] [PubMed]
- Meneghin, E.; Volpato, A.; Cupellini, L.; Bolzonello, L.; Jurinovich, S.; Mascoli, V.; Carbonera, D.; Mennucci, B.; Collini, E. Coherence in carotenoid-to-chlorophyll energy transfer. Nat. Commun. 2018, 9, 3160. [Google Scholar] [CrossRef] [PubMed]
- Roscioli, J.D.; Ghosh, S.; LaFountain, A.M.; Frank, H.A.; Beck, W.F. Quantum coherent excitation energy transfer by carotenoids in photosynthetic light harvesting. J. Phys. Chem. Lett. 2017, 8, 5141–5147. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Bishop, M.M.; Roscioli, J.D.; LaFountain, A.M.; Frank, H.A.; Beck, W.F. Excitation energy transfer by coherent and incoherent mechanisms in the peridinin–chlorophyll a protein. J. Phys. Chem. Lett. 2017, 8, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, R.P.; Shima, S.; Melkozernov, A.; Lin, S.; Blankenship, R.E.; Sharples, F.P.; Hiller, R.G.; Birge, R.R.; Frank, H.A. Spectroscopic properties of the main-form and high-salt peridinin− chlorophyll a proteins from Amphidinium carterae. Biochemistry 2004, 43, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Tannor, D.J.; Marten, B.; Murphy, R.; Friesner, R.A.; Sitkoff, D.; Nicholls, A.; Honig, B.; Ringnalda, M.; Goddard, W.A., III. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar] [CrossRef]
- Bricker, W.P.; Lo, C.S. Excitation energy transfer in the peridinin-chlorophyll a-protein complex modeled using configuration interaction. J. Phys. Chem. B 2014, 118, 9141–9154. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Curutchet, C.; Mennucci, B. Toward a molecular scale interpretation of excitation energy transfer in solvated bichromophoric systems. J. Am. Chem. Soc. 2005, 127, 16733–16744. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Curutchet, C.; Mennucci, B. Towards a molecular scale interpretation of excitation energy transfer in solvated bichromophoric systems. II. the through-bond contribution. J. Phys. Chem. B 2007, 111, 853–863. [Google Scholar] [PubMed]
- Murphy, R.B.; Philipp, D.M.; Friesner, R.A. A mixed quantum mechanics/molecular mechanics (QM/MM) method for large-scale modeling of chemistry in protein environments. J. Comp. Chem. 2000, 21, 1442–1457. [Google Scholar] [CrossRef]
- Andreussi, O.; Knecht, S.; Marian, C.M.; Kongsted, J.; Mennucci, B. Carotenoids and light-harvesting: From DFT/MRCI to the tamm–dancoff approximation. J. Chem. Theory Comput. 2015, 11, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Knecht, S.; Marian, C.M.; Kongsted, J.; Mennucci, B. On the photophysics of carotenoids: A multireference DFT study of peridinin. J. Phys. Chem. B 2013, 117, 13808–13815. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Damjanović, A.; Ritz, T.; Schulten, K. Excitation transfer in the peridinin-chlorophyll-protein of Amphidinium carterae. Biophys. J. 2000, 79, 1695–1705. [Google Scholar] [CrossRef]
- Mennucci, B.; Curutchet, C.; Cammi, R.; Scholes, G.D. How the molecular environment controls excitation energy transfer and light harvesting: A quantum mechanical model. AIP Conf. Proc. 2007, 963, 346. [Google Scholar] [CrossRef]
- Kleima, F.J.; Wendling, M.; Hofmann, E.; Peterman, E.J.; van Grondelle, R.; van Amerongen, H. Peridinin chlorophyll a protein: Relating structure and steady-state spectroscopy. Biochemistry 2000, 39, 5184–5195. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Coordinates of all the calculated structures are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excitonic State | Vacuum | Solvated PCP Complex | ||
|---|---|---|---|---|
| Supramolecular QM/MM | EET | Supramolecular QM/MM | EET | |
| State 1 | 476 | 478 | 502 | 513 |
| State 2 | 469 | 468 | 495 | 501 |
| State 3 | 456 | 458 | 476 | 482 |
| State 4 | 434 | 434 | 447 | 454 |
| Uncoupled Chromophores | Coupling Constants (cm−1) | Coupled Excitonic State | Coupling Constants (cm−1) |
|---|---|---|---|
| Per611-Qy | 298 | Per611 *-Qy | 290 |
| Per612-Qy | 151 | Per612 *-Qy | 48 |
| Per613-Qy | 129 | Per613 *-Qy | 121 |
| Per614-Qy | 343 | Per614 *-Qy | 274 |
| Per611-Qx | 67 | Per611 *-Qx | 113 |
| Per612-Qx | 27 | Per612 *-Qx | 40 |
| Per613-Qx | 62 | Per613 *-Qx | 89 |
| Per614-Qx | 102 | Per614 *-Qx | 113 |
| Coupled Fragments | Coupling Constants (cm−1) | ||
|---|---|---|---|
| Vacuum | Vacuum PCP | Solvated PCP | |
| Per611-Per612 | 651 | 609 | 620 |
| Per611-Per613 | 311 | 302 | 278 |
| Per611-Per614 | 379 | 372 | 367 |
| Per612-Per613 | 372 | 365 | 353 |
| Per612-Per614 | 203 | 200 | 189 |
| Per613-Per614 | 473 | 487 | 468 |
| Chromophore Pair | Coupling Constant | Chromophore Pair | Coupling Constant |
|---|---|---|---|
| Per611-Per612 | 619 | Per612-Per613′ | 262 |
| Per613-Per614 | 470 | Per613-Per612′ | 262 |
| Per612-Per613 | 377 | Per613-Per613′ | 255 |
| Per611-Per614 | 365 | Per612-Per614 | 194 |
| Per612-Per612′ | 351 | Per612-Qy | 151 |
| Per614-Qy | 343 | Per613-Qx | 129 |
| Per611-Per613 | 310 | Per614-Qx | 102 |
| Per611-Qy | 298 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guberman-Pfeffer, M.J.; Gascón, J.A. Carotenoid-Chlorophyll Interactions in a Photosynthetic Antenna Protein: A Supramolecular QM/MM Approach. Molecules 2018, 23, 2589. https://doi.org/10.3390/molecules23102589
Guberman-Pfeffer MJ, Gascón JA. Carotenoid-Chlorophyll Interactions in a Photosynthetic Antenna Protein: A Supramolecular QM/MM Approach. Molecules. 2018; 23(10):2589. https://doi.org/10.3390/molecules23102589
Chicago/Turabian StyleGuberman-Pfeffer, Matthew J., and José A. Gascón. 2018. "Carotenoid-Chlorophyll Interactions in a Photosynthetic Antenna Protein: A Supramolecular QM/MM Approach" Molecules 23, no. 10: 2589. https://doi.org/10.3390/molecules23102589
APA StyleGuberman-Pfeffer, M. J., & Gascón, J. A. (2018). Carotenoid-Chlorophyll Interactions in a Photosynthetic Antenna Protein: A Supramolecular QM/MM Approach. Molecules, 23(10), 2589. https://doi.org/10.3390/molecules23102589