Representation of the QM Subsystem for Long-Range Electrostatic Interaction in Non-Periodic Ab Initio QM/MM Calculations



Abstract

1. Introduction
2. Theory
2.1. Mechanical Embedding ai-QM/MM Calculations
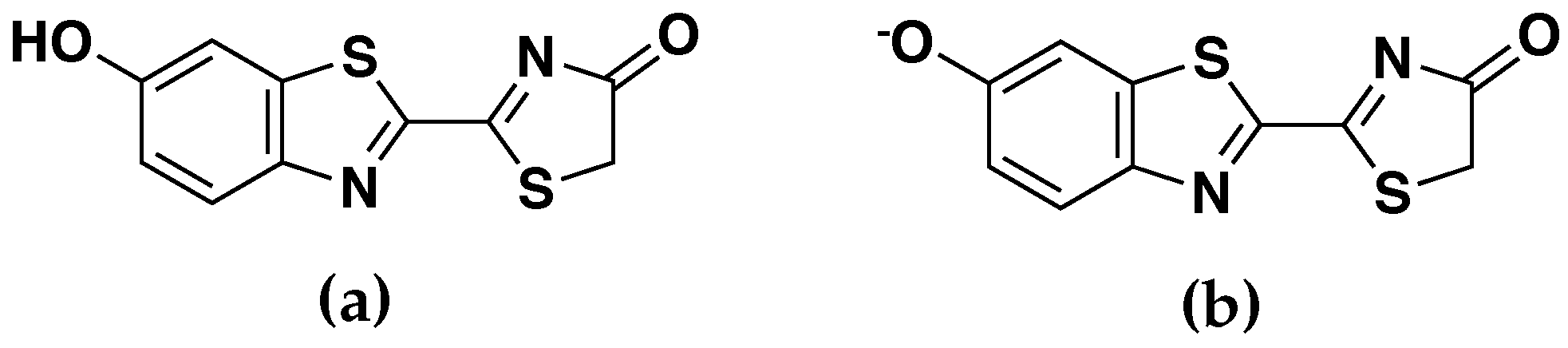
2.2. Electrostatic Embedding ai-QM/MM Calculations
2.3. Multipolar Representation with Mulliken Charges and Dipoles
2.4. Multipolar Representation with ESP Charges and Dipoles
2.5. A Hybrid Scheme with Switching Functions
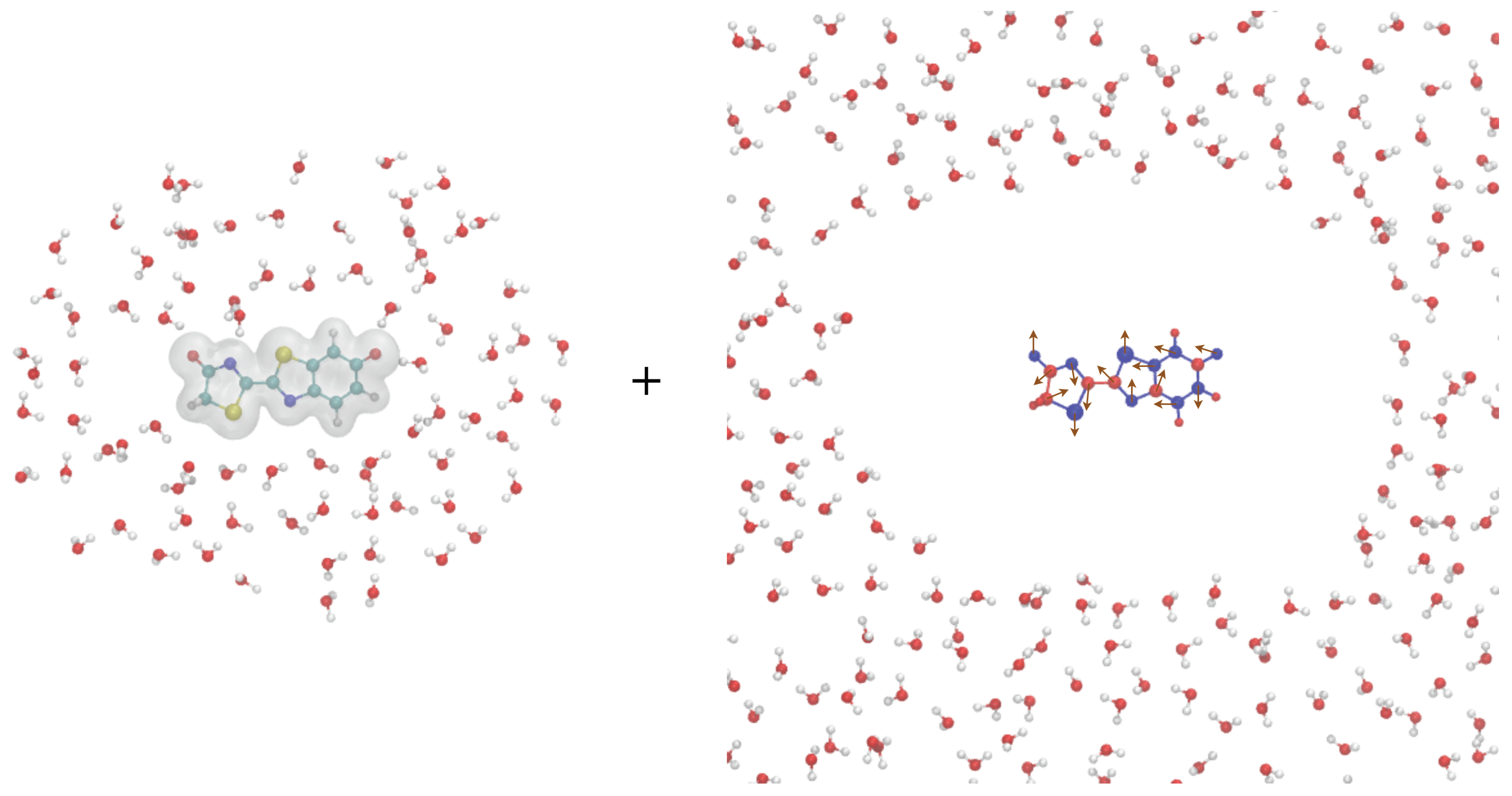
3. Computational Details
4. Results and Discussion
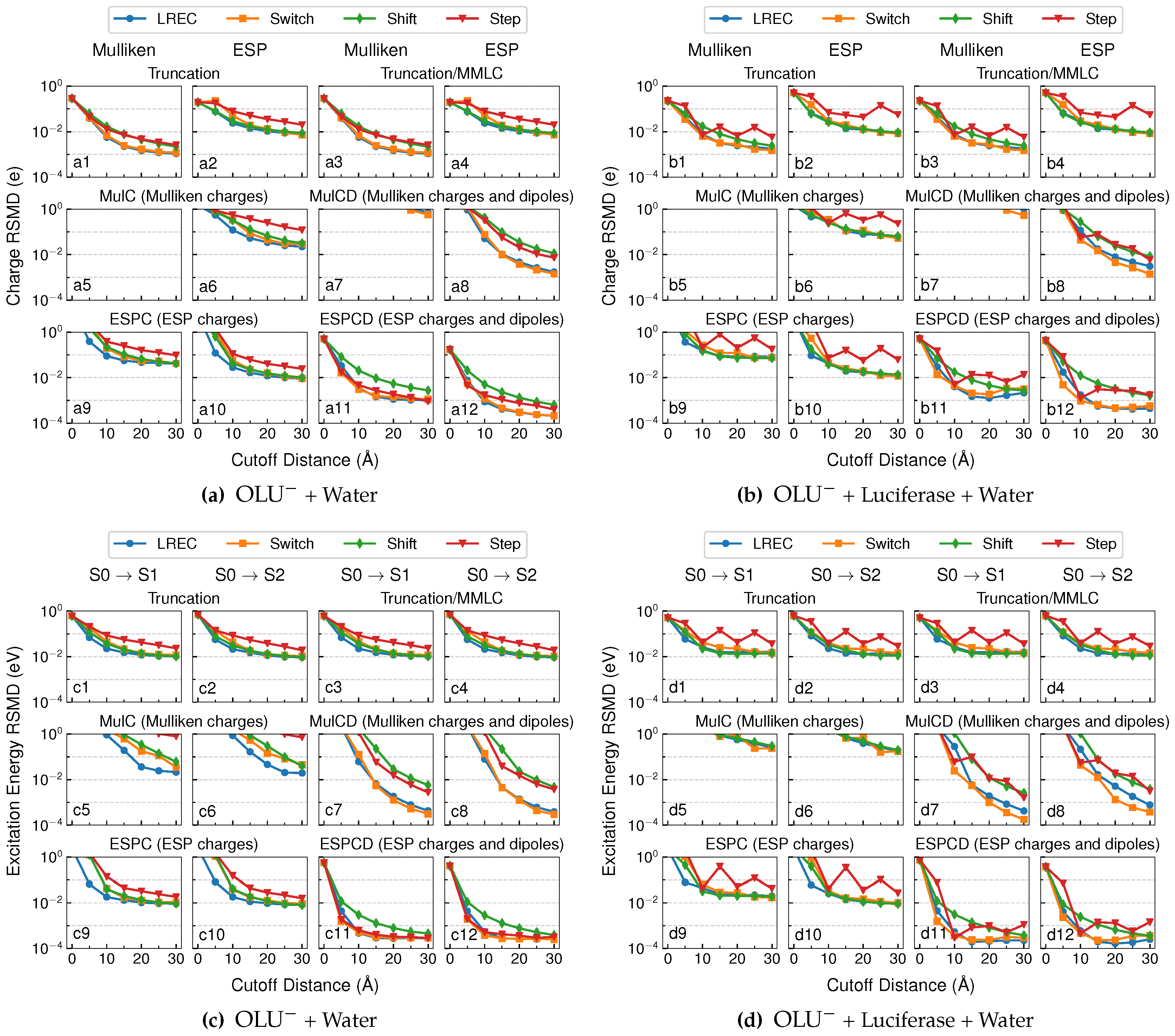
4.1. Electrostatic and Polarization Energies
4.1.1. Truncation and Truncation/MMLC Models
4.1.2. MulC and MulCD Models
4.1.3. ESPC and ESPCD Models
4.2. Population Analysis and Excitation Energies
4.3. Computational Timings
5. Conclusions
- The long-range QM/MM electrostatic interactions can be rather significant. At cutoff distances of 30 Å, their average contribution was found to be still around 10 kcal/mol with the anionic QM subsystem and around 1 kcal/mol with the neutral QM subsystem.
- In truncated QM/MM calculations, where only MM charges within a cutoff distance are kept, like Fang et al. [25], we found it to be beneficial to use LREC/Switch functions to scale down MM charge values to compensate for neglecting all far-field MM charges.
- The MulC model should generally be avoided because it can cause extremely large errors in QM/MM electrostatic and polarization energies. While the MulCD model offered an improvement upon the MulC model, its use is still not recommended because large distance cutoffs are needed and because the Mulliken charges on QM atoms obtained with this model still looked erroneous.
- While the ESPC model performed significantly better than MulC, it polarized the QM region only as well as the truncation model (as indicated by atomic charge populations, and vertical excitation energies) when LREC or Switch functions were used. Its only significant advantage over the truncation model occurred with the computation of QM/MM electrostatics energy with an anionic QM subsystem (the oxyluciferin anion).
- The ESPCD model yielded the best performances. At a 10 Å cutoff distance, it reproduces QM/MM electrostatics energy within 0.1 kcal/mol, polarization energy within 10−3 kcal/mol and TDDT vertical excitation energy within 10−3 eV from the reference values. Therefore, ESPCD with LREC/Switch smoothing functions and a 10 Å cutoff would be our recommended combination for a hybrid representation for the QM subsystem in the treatment of short-range and long-range QM/MM electrostatics.
- Besides avoiding discontinuity at the cutoff distance, a LREC/Switch smoothing function between the near- and far-field interactions can also lead to better results in most cases, and thus should be applied in general.
- Only the oxyluciferin systems were studied using very short MD simulations. Testing on longer simulations and more systems, including enzymatic reactions, needs to be carried out for more general conclusions.
- Our ESPCD scheme should be readily extendable to ai-QM/MM PBC calculations, where both ESP charges and dipoles are used to represent the QM subsystem in the long-range electrostatics calculations. However, this needs to be thoroughly tested.
- We used the standard ESP grid (discretized four layers of vdW surfaces) in the computation of ESP charges and dipoles. Other grids should be explored.
- Only energy values were reported. The corresponding analytical gradient has yet to be coded.
- For the MM region, the TIP3P water model and C36 protein force field were employed. Therefore, it would be interesting to see if other force fields might offer a physically better description for the QM/MM electrostatics/polarization interactions.
- Our focus was placed on QM/MM electrostatics/polarization energies. Therefore, we have not considered QM/MM vdW and charge-transfer interactions, which can also significantly affect the simulation results.
- We studied how to more quickly converge QM/MM electrostatic energy with the number of MM charges included in the short-range evaluation. Therefore, we have not addressed the equally important issue of how to achieve quicker convergence of QM/MM results with the size of the QM region.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- York, D.M.; Darden, T.A.; Pedersen, L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: A comparison of the Ewald and truncated list methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- York, D.M.; Wlodawer, A.; Pedersen, L.G.; Darden, T.A. Atomic-level accuracy in simulations of large protein crystals. Proc. Natl. Acad. Sci. USA 1994, 91, 8715–8718. [Google Scholar] [CrossRef] [PubMed]
- York, D.M.; Yang, W.; Lee, H.; Darden, T.; Pedersen, L.G. Toward the accurate modeling of DNA: The importance of long-range electrostatics. J. Am. Chem. Soc. 1995, 117, 5001–5002. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Cisneros, G.A.; Karttunen, M.; Ren, P.; Sagui, C. Classical electrostatics for biomolecular simulations. Chem. Rev. 2014, 114, 779. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xia, X. A priori evaluation of aqueous polarization effects. Science 1992, 258, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; VandeVondele, J.; Rothlisberger, U. A Hamiltonian electrostatic coupling scheme for hybrid Car-Parrinello molecular dynamics simulations. J. Chem. Phys. 2002, 116, 6941–6947. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM studies of enzymes. Curr. Op. Chem. Biol. 2007, 11, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Truhlar, D. QM/MM: What have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef]
- Nam, K.; Gao, J.L.; York, D.M. An efficient linear-scaling Ewald method for long-range electrostatic interactions in combined QM/MM calculations. J. Chem. Theory Comput. 2005, 1, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.C.; Crowley, M.F.; Case, D.A. The implementation of a fast and accurate QM/MM potential method in Amber. J. Comput. Chem. 2008, 29, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-May, P.; Pu, J. Isotropic periodic sum treatment of long-range electrostatic interactions in combined quantum mechanical and molecular mechanical calculations. J. Chem. Theory Comput. 2014, 10, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-May, P.; Pu, J. Treating electrostatics with Wolf summation in combined quantum mechanical and molecular mechanical simulations. J. Chem. Phys. 2015, 143, 174111. [Google Scholar] [CrossRef] [PubMed]
- Holden, Z.C.; Richard, R.M.; Herbert, J.M. Periodic boundary conditions for QM/MM calculations: Ewald summation for extended Gaussian basis sets. J. Chem. Phys. 2013, 139, 244108. [Google Scholar] [CrossRef] [PubMed]
- Holden, Z.C.; Richard, R.M.; Herbert, J.M. Erratum: “Periodic boundary conditions for QM/MM calculations: Ewald summation for extended Gaussian basis sets” [J. Chem. Phys. 139, 244108 (2013)]. J. Chem. Phys. 2015, 142, 059901. [Google Scholar] [CrossRef] [PubMed]
- Vasilevskaya, T.; Thiel, W. Periodic boundary conditions in QM/MM calculations: Implementation and tests. J. Chem. Theory Comput. 2016, 12, 3561–3570. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.; Li, Y.; Zhang, Y. Born-Oppenheimer ab initio QM/MM molecular dynamics simulations of enzyme reactions. In Methods in Enzymology: Computational Approaches for Studying Enzyme Mechanism Part A; Voth, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 577, pp. 105–118. [Google Scholar]
- Melo, M.C.R.; Bernardi, R.C.; Rudack, T.; Scheurer, M.; Riplinger, C.; Phillips, J.C.; Maia, J.D.C.; Rocha, G.B.; Ribeiro, J.V.; Stone, J.E.; et al. NAMD goes quantum: An integrative suite for hybrid simulations. Nat. Methods 2018, 15, 351. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; De Vivo, L.; Ferré, N.; Ghigo, G.; Malmqvist, P.Å.; Neogrady, P.; Pedersen, T.B.; Pitonak, M.; Reiher, M.; Roos, B.O.; et al. MOLCAS: The next generation. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fdez Galván, I.; Ferré, N.; Frutos, L.M.; et al. MOLCAS 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [PubMed]
- Ferré, N.; Ángyán, J.G. Approximate electrostatic interaction operator for QM/MM calculations. Chem. Phys. Lett. 2002, 356, 331–339. [Google Scholar] [CrossRef]
- Dziedzic, J.; Mao, Y.; Shao, Y.; Ponder, J.; Head-Gordon, T.; Head-Gordon, M.; Skylaris, C.K. TINKTEP: A fully self-consistent, mutually polarizable QM/MM approach based on the AMOEBA force field. J. Chem. Phys. 2016, 145, 124106. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Duke, R.E.; Cisneros, G.A. A new smoothing function to introduce long-range electrostatic effects in QM/MM calculations. J. Chem. Phys. 2015, 143, 044103. [Google Scholar] [CrossRef] [PubMed]
- Kratz, E.G.; Duke, R.E.; Cisneros, G.A. Long-range electrostatic corrections in multipolar/polarizable QM/MM simulations. Theor. Chem. Acc. 2016, 135, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Benighaus, T.; Thiel, W. A general boundary potential for hybrid QM/MM simulations of solvated biomolecular systems. J. Chem. Theory Comput. 2009, 5, 3114–3128. [Google Scholar] [CrossRef] [PubMed]
- Benighaus, T.; Thiel, W. Long-range electrostatic effects in QM/MM studies of enzymatic reactions: Application of the solvated macromolecule boundary potential. J. Chem. Theory Comput. 2011, 7, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Im, W.; Bernèche, S.; Roux, B. Generalized solvent boundary potential for computer simulations. J. Chem. Phys. 2001, 114, 2924–2937. [Google Scholar] [CrossRef]
- Schaefer, P.; Riccardi, D.; Cui, Q. Reliable treatment of electrostatics in combined QM/MM simulation of macromolecules. J. Chem. Phys. 2005, 123, 014905. [Google Scholar] [CrossRef] [PubMed]
- Giese, T.J.; York, D.M. Ambient-potential composite Ewald method for ab initio quantum mechanical/molecular mechanical molecular dynamics simulation. J. Chem. Theory Comput. 2016, 12, 2611–2632. [Google Scholar] [CrossRef] [PubMed]
- Füsti-Molnár, L.; Pulay, P. The Fourier transform Coulomb method : Efficient and accurate calculation of the Coulomb operator in a Gaussian basis. J. Chem. Phys. 2002, 117, 7827. [Google Scholar] [CrossRef]
- Chang, C.M.; Shao, Y.; Kong, J. Ewald mesh method for quantum mechanical calculations. J. Chem. Phys. 2012, 136, 114112. [Google Scholar] [CrossRef] [PubMed]
- White, C.A.; Johnson, B.G.; Gill, P.M.W.; Head-Gordon, M. The continuous fast multipole method. Chem. Phys. Lett. 1994, 230, 8–16. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. An approach to computing electrostatic charges for molecules. J. Comput. Chem. 1984, 5, 129–145. [Google Scholar] [CrossRef]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nakatsu, T.; Ichiyama, S.; Hiratake, J.; Saldanha, A.; Kobashi, N.; Sakata, K.; Kato, H. Structural basis for the spectral difference in luciferase bioluminescence. Nature 2006, 440, 372–376. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain X1 and X2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Chem. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Casida, M.E.; Huix-Rotllant, M. Progress in time-dependent density-functional theory. Ann. Rev. Phys. Chem. 2012, 63, 287–323. [Google Scholar] [CrossRef] [PubMed]
- Slipchenko, L.V.; Gordon, M.S. Damping functions in the effective fragment potential method. Mol. Phys. 2009, 107, 999–1016. [Google Scholar] [CrossRef]
- Wang, B.; Truhlar, D.G. Including charge penetration effects in molecular modeling. J. Chem. Theory Comput. 2010, 6, 3330–3342. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Lu, Z.; Yang, W. Fitting molecular electrostatic potentials from quantum mechanical calculations. J. Chem. Theory Comput. 2007, 3, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Truhlar, D.G. Tuned and balanced redistributed charge scheme for combined quantum mechanical and molecular mechanical (QM/MM) methods and fragment methods: Tuning based on the CM5 charge model. J. Chem. Theory Comput. 2013, 9, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cutoff (Å) | # (Charges) | Time (s) | Cutoff (Å) | # (Charges) | Time (s) | ||
|---|---|---|---|---|---|---|---|
| Energy | Force | Energy | Force | ||||
| 5 | 144 | 0.1 | 0.4 | 25 | 9297 | 3.7 | 28.2 |
| 10 | 837 | 0.3 | 2.0 | 30 | 15,291 | 5.8 | 42.5 |
| 15 | 2418 | 0.9 | 5.8 | 60 | 107,535 | 39.4 | 262.7 |
| 20 | 5121 | 2.2 | 15.3 | Unit Cell | 166,114 | 58.2 | 403.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Rosta, E.; Shao, Y. Representation of the QM Subsystem for Long-Range Electrostatic Interaction in Non-Periodic Ab Initio QM/MM Calculations. Molecules 2018, 23, 2500. https://doi.org/10.3390/molecules23102500
Pan X, Rosta E, Shao Y. Representation of the QM Subsystem for Long-Range Electrostatic Interaction in Non-Periodic Ab Initio QM/MM Calculations. Molecules. 2018; 23(10):2500. https://doi.org/10.3390/molecules23102500
Chicago/Turabian StylePan, Xiaoliang, Edina Rosta, and Yihan Shao. 2018. "Representation of the QM Subsystem for Long-Range Electrostatic Interaction in Non-Periodic Ab Initio QM/MM Calculations" Molecules 23, no. 10: 2500. https://doi.org/10.3390/molecules23102500
APA StylePan, X., Rosta, E., & Shao, Y. (2018). Representation of the QM Subsystem for Long-Range Electrostatic Interaction in Non-Periodic Ab Initio QM/MM Calculations. Molecules, 23(10), 2500. https://doi.org/10.3390/molecules23102500