
Synthesis and Evaluation of Novel Ligustrazine Derivatives as Multi-Targeted Inhibitors for the Treatment of Alzheimer’s Disease

 ,
,
Abstract
1. Introduction
2. Results and Discussion
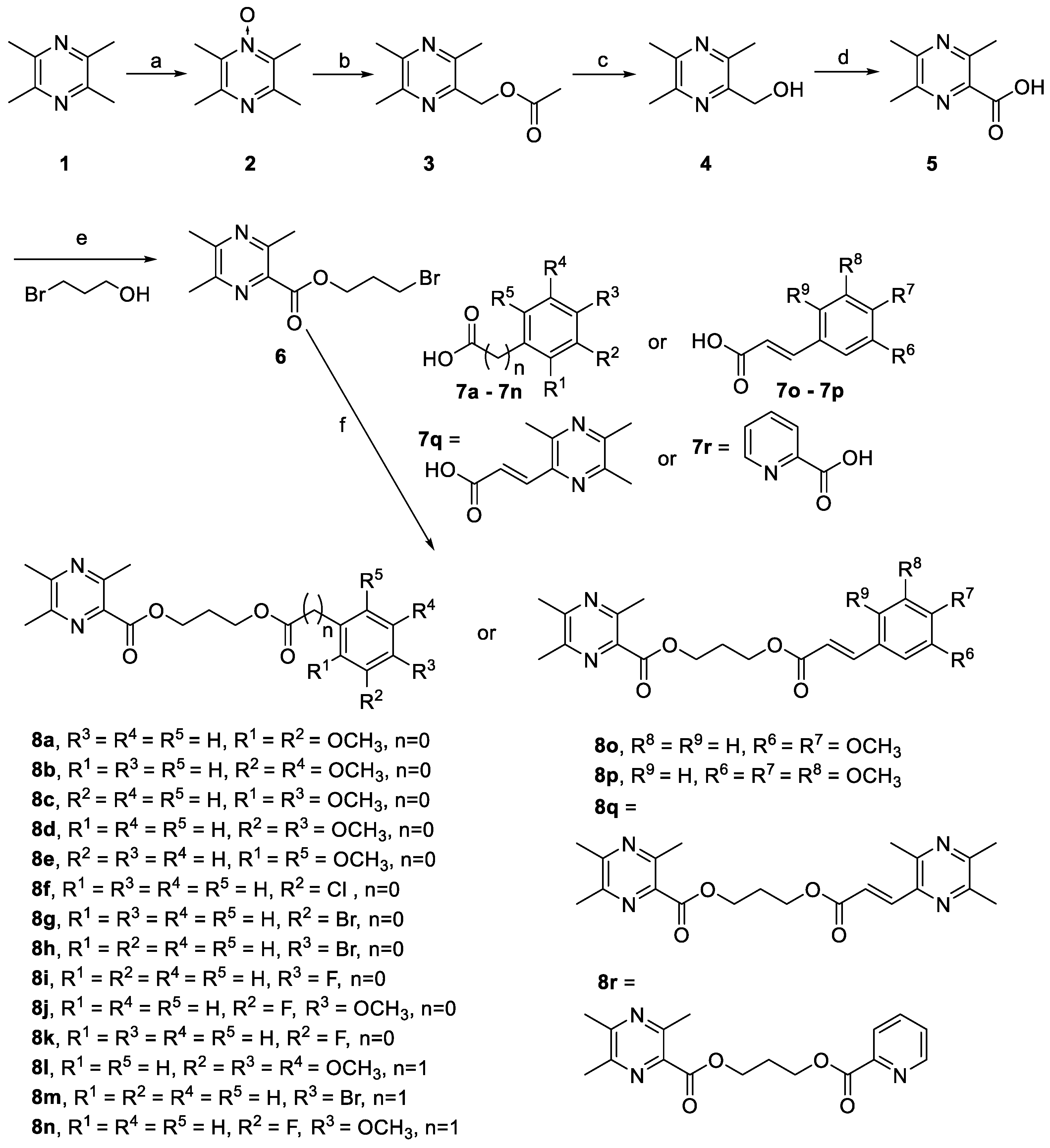
2.1. Chemistry
2.2. In Vitro Inhibition Studies on AChE, BuChE and Aβ (1-42) Self-Induced Aggregation.
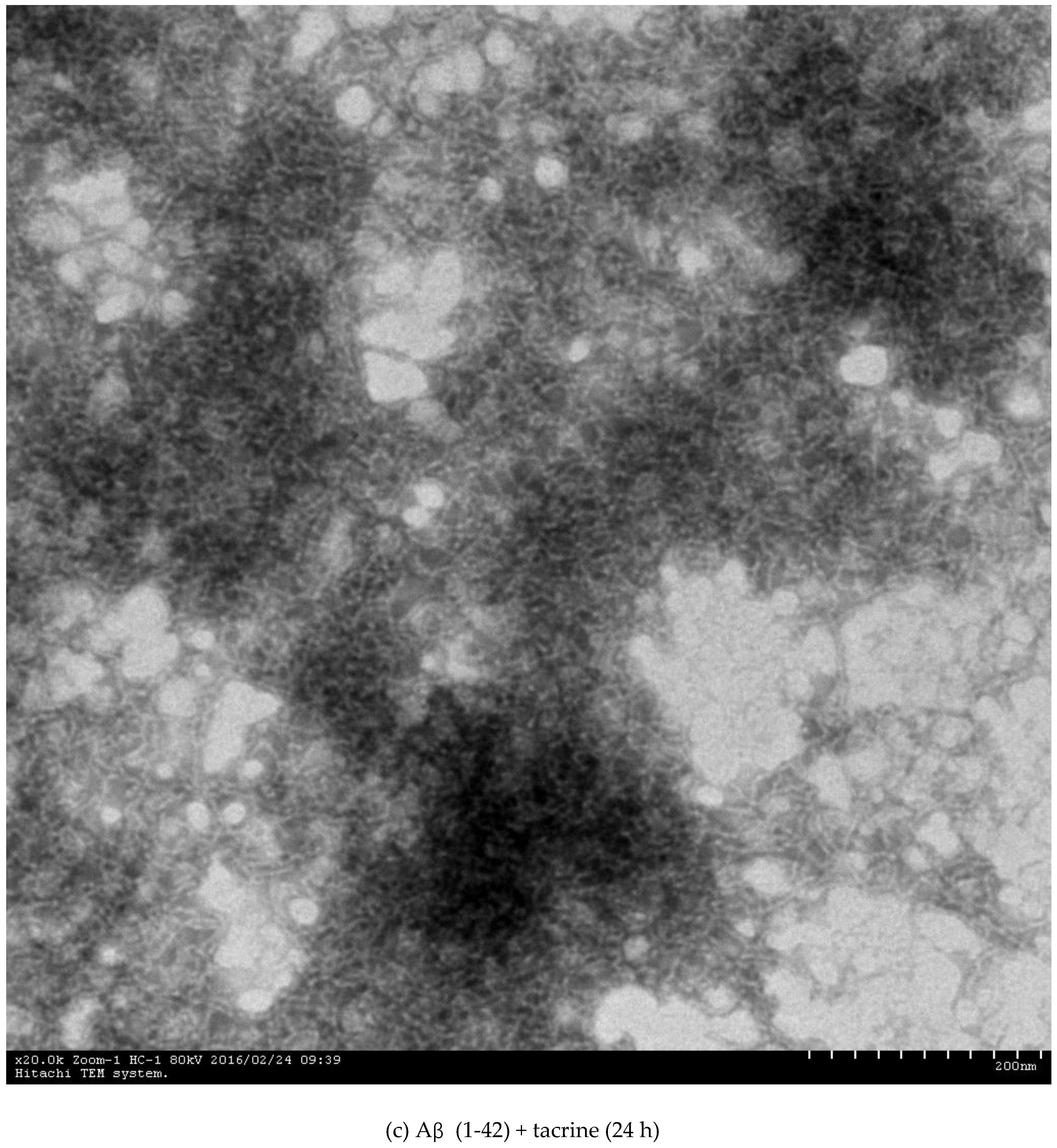
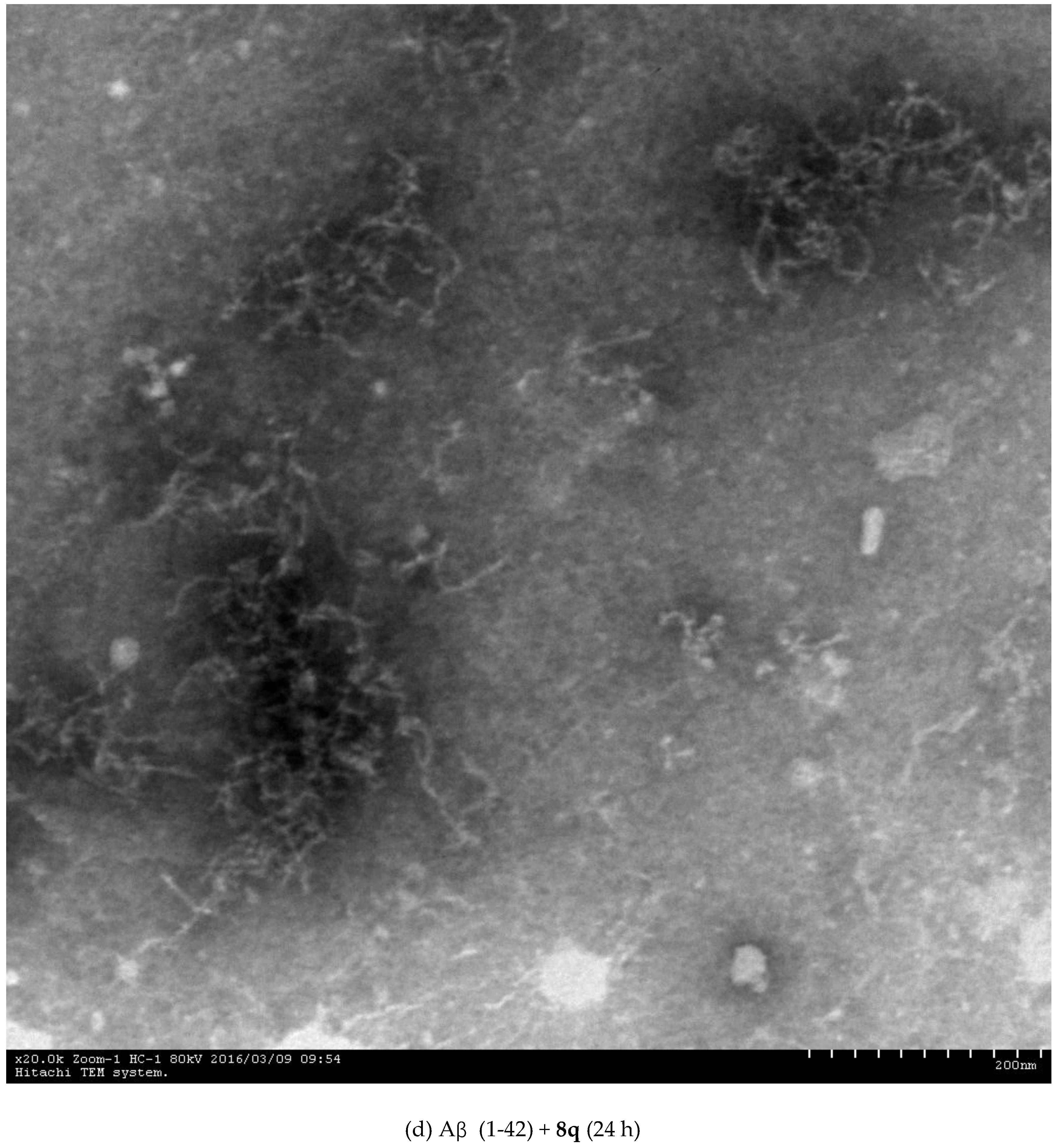
2.3. Inhibition of Aβ (1-42) Fibril Formation Monitored by Transmission Electron Microscopy (TEM)
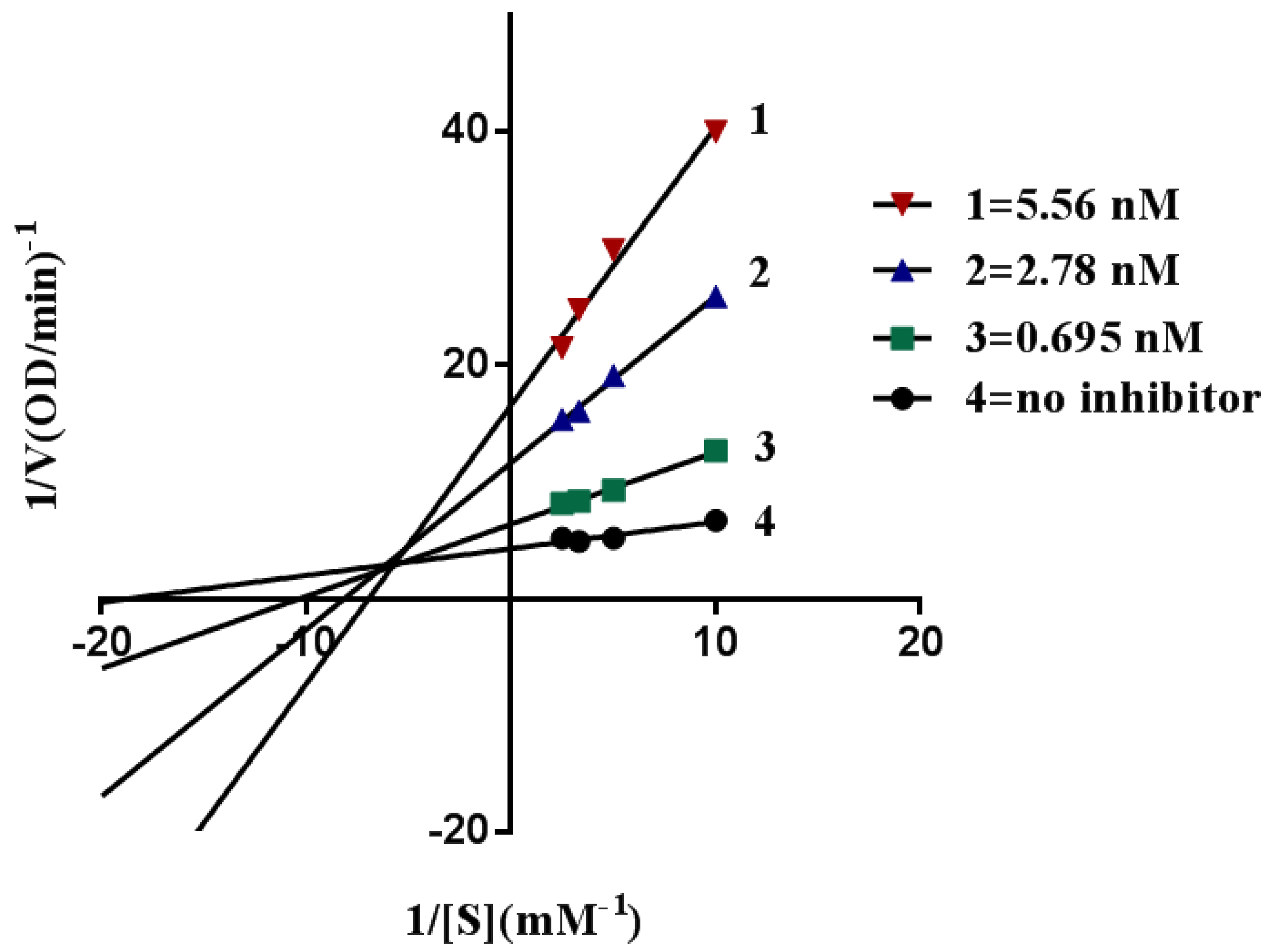
2.4. Kinetic Characterization of AChE Inhibition
2.5. Molecular Modeling Study of AChE
2.6. Cytotoxicity Assay for 8q and 8r
3. Materials and Methods
3.1. Chemistry
3.2. Synthesis of Intermediate 6
3.3. General Procedures for the Preparation of 8a–r
3.4. Biological Activity
3.4.1. Inhibitory Activities of AChE and BuChE
3.4.2. Inhibition of Self-Mediated Aβ (1-42) Aggregation
3.4.3. Transmission Electron Microscopy (TEM) Assay
3.4.4. Kinetic Study of AChE Inhibition
3.4.5. In Vitro Cytotoxicity of Tacrine and Ligustrazine Hybrids 8q, 8r in HepG2 Cells
3.4.6. In Vitro Cytotoxicity of Tacrine and Compounds 8q, 8r in SH-SY5Y Cells
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mazon, J.N.; Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Fox, M. ‘Evolutionary medicine’ perspectives on Alzheimer’s Disease: Review and new directions. Ageing Res. Rev. 2018, 47, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s, A. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar]
- Robertson, D.S. Proposed biochemistry of Parkinson’s and Alzheimer′s diseases. Med. Hypotheses 2017, 109, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Soler-Lopez, M.; Badiola, N.; Zanzoni, A.; Aloy, P. Towards Alzheimer’s root cause: ECSIT as an integrating hub between oxidative stress, inflammation and mitochondrial dysfunction. Hypothetical role of the adapter protein ECSIT in familial and sporadic Alzheimer′s disease pathogenesis. Bioessays 2012, 34, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Osmana, W.; Tina, G.; Raoa, P.P. Selective inhibition of human acetylcholinesterase by xanthine derivatives: In vitro inhibition and molecular modeling investigations. Bioorg. Med. Chem. Lett. 2013, 23, 4336–4341. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, D.; Fallarero, A.; Brunhofer, G.; Mayer, C.; Prakash, O.; Mohan, C.G.; Vuorela, P.; Erker, T. The exploration of thienothiazines as selective butyrylcholinesterase inhibitors. Eur. J. Pharm. Sci. 2012, 47, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Cash, M.K.; Reid, G.A.; Martin, E.; Mitnitski, A.; Geula, C. Butyrylcholinesterase is associated with β-amyloid plaques in the transgenic APP SWE/PSEN1dE9 mouse model of alzheimer disease. J. Neuropathol. Exp. Neurol. 2012, 71, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; He, G. Toward a Rational Design to Regulate β-Amyloid Fibrillation for Alzheimer’s Disease Treatment. ACS Chem. Neurosci. 2018, 9, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Turner, A.J. AChE and the amyloid precursor protein (APP)–Cross-talk in Alzheimer’s disease. Chem. Biol. Interact. 2018, 259B, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Gupta, S.K.; Ganeshpurkar, A.; Gutti, G.; Krishnamurthy, S.; Modi, G.; Singh, S.K. Development of Piperazinediones as dual inhibitor for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Khanam, H.; Ali, A.; Asif, M. Neurodegenerative diseases linked to misfolded proteins and their therapeutic approaches: A review. Eur. J. Med. Chem. 2016, 124, 1121–1141. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.K.; Dandapat, J.; Dash, U.C.; Kanhar, S. Features and outcomes of drugs for combination therapy as multi-targets strategy to combat Alzheimer’s disease. J. Ethnopharmacol. 2018, 215, 42–73. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Van der Schyf, C.J. Rationally designed multi-targeted agents against neurodegenerative diseases. Curr. Med. Chem. 2013, 20, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Leon, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Dias, K.S.; Viegas, J.C. Multi-Target Directed Drugs: A Modern Approach for Design of New Drugs for the treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.U. Polypharmacology-foe or friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wu, F.; Lin, X.; Shen, L.; Feng, Y. Developments in drug delivery of bioactive alkaloids derived from traditional Chinese medicine. Drug Deliv. 2018, 25, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wang, P.; Xu, X.; Chu, F.; Lin, J.; Zhang, Y.; Lei, H. An overview on structural modifications of ligustrazine and biological evaluation of its synthetic derivatives. Res. Chem. Intermed. 2015, 41, 1385–1411. [Google Scholar] [CrossRef]
- Zhu, X.L.; Xiong, L.Z.; Wang, Q.; Liu, Z.G.; Ma, X.; Zhu, Z.H.; Hu, S.; Gong, G.; Chen, S.Y. Therapeutic time window and mechanism of tetramethylpyrazine on transient focal cerebral ischemia/reperfusion injury in rats. Neurosci. Lett. 2009, 449, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, W.; Zhao, R.; Xu, B.; Fang, X.; Yan, M.; Zhang, Y.; Wang, P.; Lei, H. Design, synthesis and evaluation of new ligustrazine derivatives as potential plasma-stable neuroprotective agents. MedChemComm 2017, 8, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qin, H.L.; Leng, J.; Ameeduzzafar, Z.; Amjad, M.W.; Raja, M.A.G.; Hussain, M.A.; Bukhari, S.N.A. Synthesis and biological evaluation of new tetramethylpyrazine-based chalcone derivatives as potential anti-Alzheimer agents. Chem. Biol. Drug. Des. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hong, G.; Li, X.; Zhang, Y.; Xu, Z.; Mao, L.; Feng, X.; Liu, T. Synthesis and activity towards Alzheimer’s disease in vitro: Tacrine, phenolic acid and ligustrazine hybrids. Eur. J. Med. Chem. 2018, 14, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Du, Q.Y.; Chen, L.D.; Wu, W.H.; Liao, S.Y.; Yu, L.H.; Liang, X.T. Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as multi-targeted compounds against Alzheimer’s disease. Eur. J. Med. Chem. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, L.D.; Liang, X.T.; Liu, W.X.; Wu, W.H. Synthesis and Biological Evaluation of Ligustrazine Derivatives. Chem. Nat. Comp. 2017, 53, 114–117. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres Jr., V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Huang, L.; Miao, H.; Sun, Y.; Meng, F.; Li, X. Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease. Eur. J. Med. Chem. 2014, 87, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional compounds for controlling metal-mediated aggregation of the aβ42 peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef] [PubMed]
- Nepovimova, E.; Uliassi, E.; Korabecny, J.; Peña-Altamira, L.E.; Samez, S.; Pesaresi, A.; Garcia, G.E.; Bartolini, M.; Andrisano, V.; Bergamini, C.; et al. Multitarget drug design strategy: Quinone-tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J. Med. Chem. 2014, 57, 8576–8589. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.P.; Sanson, B.; Nachon, F.; Gabellieri, E.; Fattorusso, C.; Campiani, G.; Weik, M. Conformational flexibility in the peripheral site of Torpedo californica acetylcholinesterase revealed by the complex structure with a bifunctional inhibitor. J. Am. Chem. Soc. 2006, 128, 4526–4527. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalk, I.; Ehretsabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary Ligand Binding to Aromatic Residues in the Active-Site Gorge of Acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, E.H.; Brumshtein, B.; Greenblatt, H.M.; Wong, D.M.; Shaya, D.; Williams, L.D.; Carlier, P.R.; Pang, Y.P.; Silman, I.; Sussman, J.L. Complexes of alkylene-linked tacrine dimers with Torpedo californica acetylcholinesterase: Binding of Bis5-tacrine produces a dramatic rearrangement in the active-site gorge. J. Med. Chem. 2006, 49, 5491–5500. [Google Scholar] [CrossRef] [PubMed]
- Raves, M.L.; Harel, M.; Pang, Y.P.; Silman, I.; Kozikowski, A.P.; Sussman, J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (−)-huperzine A. Nat. Struct. Biol. 1997, 4, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Esquivias-Pérez, M.; Maalej, E.; Romero, A.; Chabchoub, F.; Samadi, A.; Marco-Contelles, J.; Oset-Gasque, M.J. Nontoxic and neuroprotective β-naphthotacrines for Alzheimer’s disease. Chem. Res. Toxicol. 2013, 26, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhao, L.; Zhu, B.; Li, Q.; Yew, D.T.; Yao, Z.; Xu, J. Protective effects of Ginkgo biloba extract (EGb761) and its constituents quercetin and ginkgolide B against β-amyloid peptide-induced toxicity in SH-SY5Y cells. Chem. Biol. Interact. 2009, 181, 115–123. [Google Scholar] [CrossRef] [PubMed]
- González-Muñoz, G.C.; Arce, M.P.; López, B.; Pérez, C.; Romero, A.; del Barrio, L.; Martìn-de-Saavedra, M.D.; Egea, J.; León, R.; Villarroya, M.; et al. N-acylaminophenothiazines: Neuroprotective agents displaying multifunctional activities for a potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 c for AChE (nM) | IC50 for BuChE (mM) | Selectivity Index f | IC50 for Aβ (1-42) Aggregation (µM) g |
|---|---|---|---|---|
| 8a | 167.9 ± 0.39 | 3.63 ± 0.07 | 2.16 × 104 | NA |
| 8b | 4.43 ± 0.35 | 5.64 ± 0.38 | 1.27 × 106 | 3.66 ± 0.104 |
| 8c | 387.9 ± 0.45 | 2.70 ± 0.03 | 5.16 × 103 | 22.54 ± 0.014 |
| 8d | 4.12 ± 0.24 | 4.29 ± 0.12 | 1.02 × 106 | 51.81 ± 0.047 |
| 8e | 143.8 ± 0.25 | 0.34 ± 0.03 | 2.36 × 103 | 55.86 ± 0.035 |
| 8f | 6.61 ± 0.13 | 3.27 ± 0.06 | 4.95 × 105 | 45.88 ± 0.024 |
| 8g | NA d | 21.13 ± 0.46 | 6.23 | NA |
| 8h | NA | 2.49 ± 0.29 | 2.62 | NA |
| 8i | NA | 112.74 ± 0.47 | 1.14 × 102 | 211.18 ± 0.039 |
| 8j | NA | 0.857 ± 0.002 | 2.97 | 7.12 ± 0.012 |
| 8k | NA | 5.49 ± 0.15 | 9.49 | 5.10 ± 0.040 |
| 8l | 303.4 ± 0.26 | 3.96 ± 0.26 | 1.31 × 104 | 77.45 ± 0.029 |
| 8m | NA | 373.52 ± 0.78 | 52.17 | 200.84 ± 0.016 |
| 8n | NA | 3.06 ± 0.34 | 20.67 | NA |
| 8o | 3.24 ± 0.10 | 3.97 ± 0.10 | 1.23 × 106 | 45.29 ± 0.033 |
| 8p | 93.51 ± 0.20 | 4.02 ± 0.03 | 4.30 × 104 | 71.28 ± 0.044 |
| 8q | 1.39 ± 0.33 | 4.04 ± 0.91 | 2.91 × 106 | 17.36 ± 0.027 |
| 8r | 0.25 ± 0.39 | 3.30 ± 0.06 | 1.32 × 107 | 49.14 ± 0.025 |
| Tacrine | 73.36 ± 0.22 | 14.45 ± 0.06 e | 0.20 | 12.21 ± 0.02 |
| Galanthamine | 23.74 ± 0.06 | 0.180 ± 0.004 | 7.58 × 103 | NA |
| Compounds | Control | 25 µM | 50 µM | 125 µM | 200 µM | 300 µM | 400 µM | 500 µM |
|---|---|---|---|---|---|---|---|---|
| 8q | 100 ± 0.82 | 96.81 ± 3.05 ns | 91.09 ± 0.99 ns | 81.19 ± 1.48 ** | 64.81 ± 0.54 ** | 66.75 ± 1.11 ** | 68.89 ± 0.51 ** | 70.10 ± 1.98 ** |
| 8r | 100 ± 0.86 | 102.83 ± 0.57 ns | 88.05 ± 1.12 ns | 81.72 ± 0.87 ** | 71.10 ± 1.06 ** | 71.10 ± 0.80 ** | 68.85 ± 1.11 ** | 68.31 ± 0.53 ** |
| Tacrine | 100 ± 2.87 | 99.78 ± 1.07 ns | 95.02 ± 2.30 ns | 83.72 ± 0.73 ** | 81.80 ± 1.86 ** | 74.32 ± 3.93 ** | 23.61 ± 0.05 ** | 19.92 ± 0.05 ** |
| Compounds | Control | 25 µM | 50 µM | 125 µM | 200µM | 300 µM | 400 µM | 500 µM |
|---|---|---|---|---|---|---|---|---|
| 8q | 100 ± 0.28 | 104.30 ± 0.29 ns | 96.08 ± 0.78 ns | 80.25 ± 1.53 ** | 72.17 ±0.54 ** | 58.92 ± 0.70 ** | 55.14 ± 0.50 ** | 55.28 ± 0.55 ** |
| 8r | 100 ± 1.75 | 94.51 ± 0.45 ns | 91.46 ± 0.29 ns | 83.23 ± 0.78 * | 83.26 ± 2.74 * | 68.43 ± 0.48 ** | 62.68 ± 0.56 ** | 66.38 ± 1.57 ** |
| Tacrine | 100 ± 1.86 | 87.89 ± 0.85 ns | 88.17 ± 1.55 ns | 75.42 ± 0.78 ** | 48.75 ± 0.43 ** | 43.39 ± 2.69 ** | 34.69 ± 2.86 ** | 27.93 ± 1.44 ** |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, W.; Liang, X.; Xie, G.; Chen, L.; Liu, W.; Luo, G.; Zhang, P.; Yu, L.; Zheng, X.; Ji, H.; et al. Synthesis and Evaluation of Novel Ligustrazine Derivatives as Multi-Targeted Inhibitors for the Treatment of Alzheimer’s Disease. Molecules 2018, 23, 2540. https://doi.org/10.3390/molecules23102540
Wu W, Liang X, Xie G, Chen L, Liu W, Luo G, Zhang P, Yu L, Zheng X, Ji H, et al. Synthesis and Evaluation of Novel Ligustrazine Derivatives as Multi-Targeted Inhibitors for the Treatment of Alzheimer’s Disease. Molecules. 2018; 23(10):2540. https://doi.org/10.3390/molecules23102540
Chicago/Turabian StyleWu, Wenhao, Xintong Liang, Guoquan Xie, Langdi Chen, Weixiong Liu, Guolin Luo, Peiquan Zhang, Lihong Yu, Xuehua Zheng, Hong Ji, and et al. 2018. "Synthesis and Evaluation of Novel Ligustrazine Derivatives as Multi-Targeted Inhibitors for the Treatment of Alzheimer’s Disease" Molecules 23, no. 10: 2540. https://doi.org/10.3390/molecules23102540
APA StyleWu, W., Liang, X., Xie, G., Chen, L., Liu, W., Luo, G., Zhang, P., Yu, L., Zheng, X., Ji, H., Zhang, C., & Yi, W. (2018). Synthesis and Evaluation of Novel Ligustrazine Derivatives as Multi-Targeted Inhibitors for the Treatment of Alzheimer’s Disease. Molecules, 23(10), 2540. https://doi.org/10.3390/molecules23102540