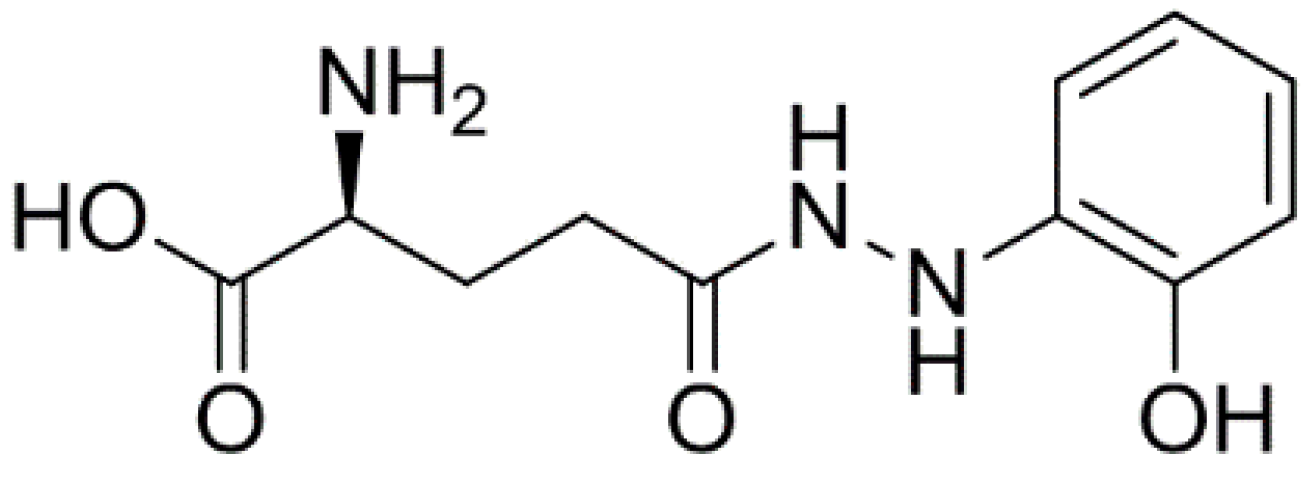
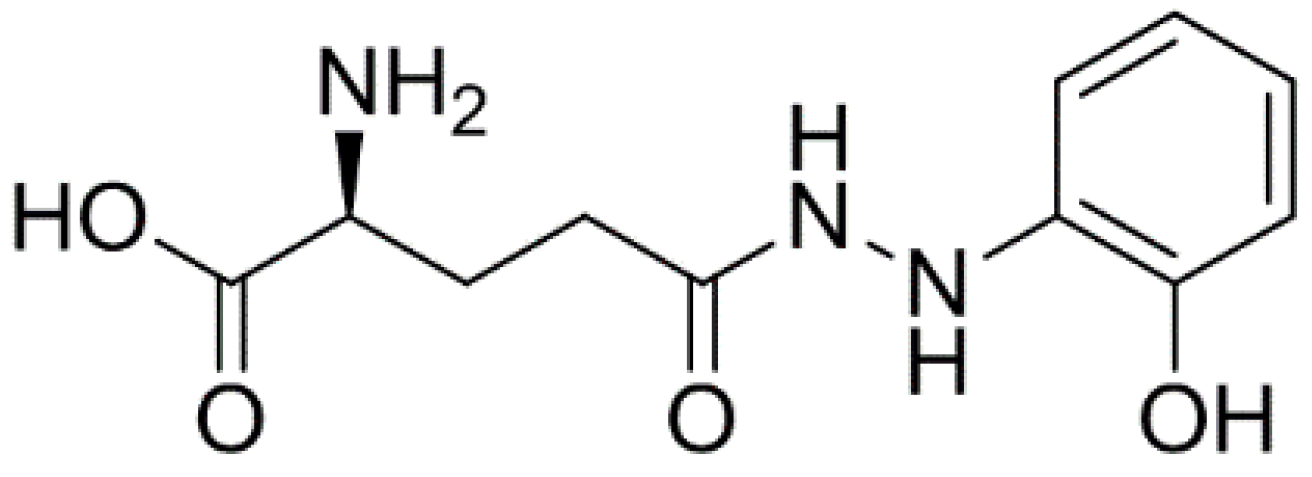
Anticancer Activity of Ramalin, a Secondary Metabolite from the Antarctic Lichen Ramalina terebrata, against Colorectal Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
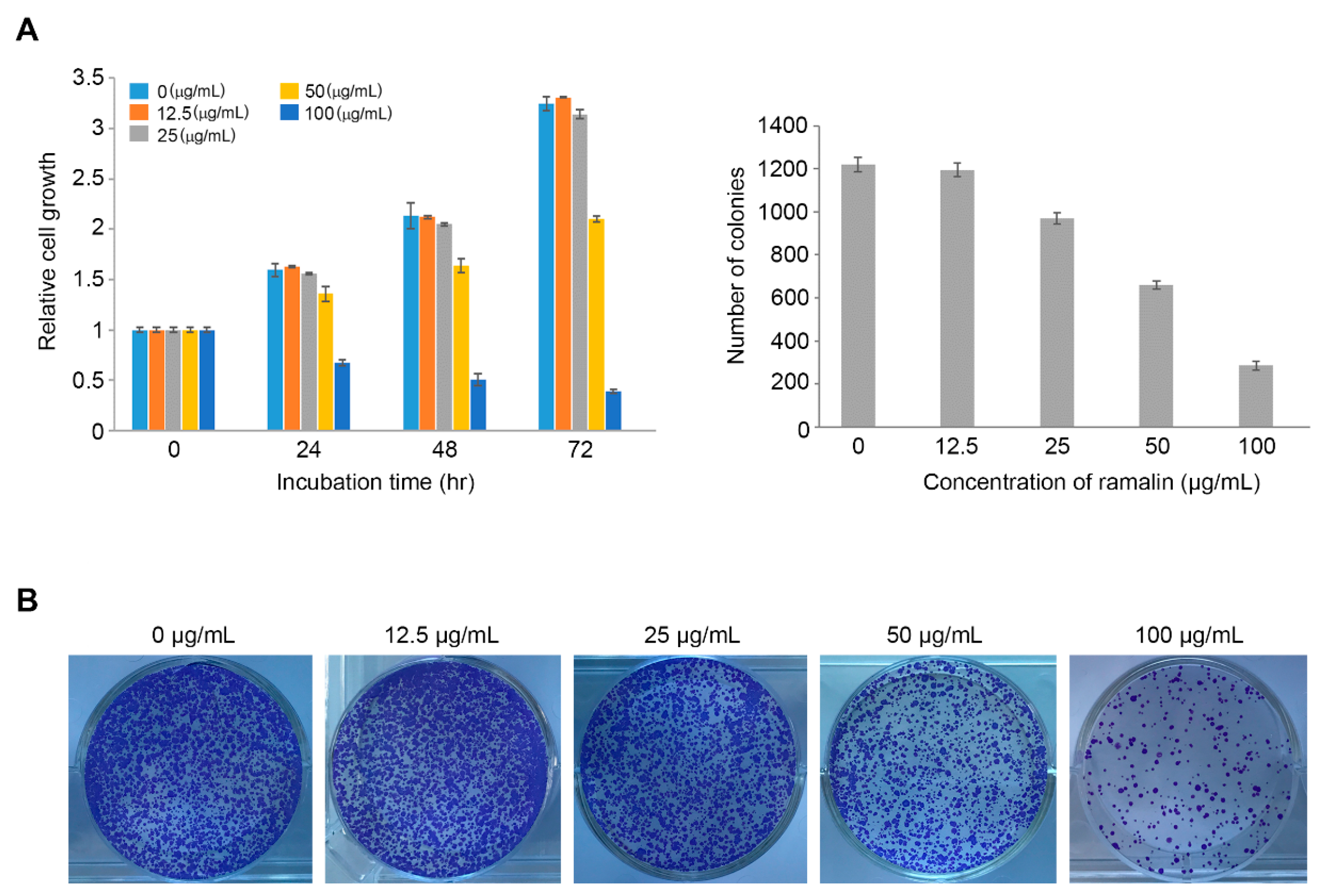
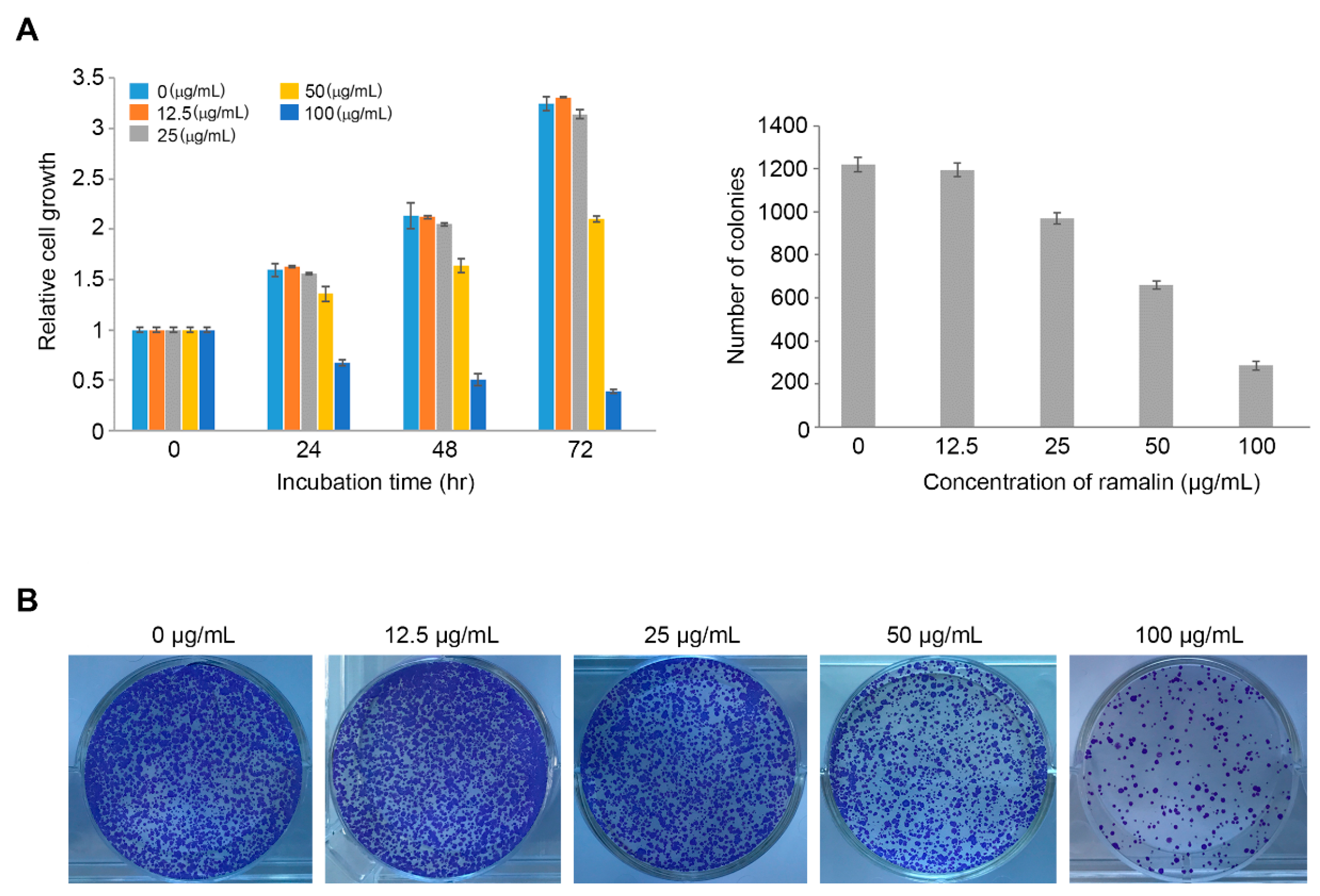
2.1. Antiproliferative Activity of Ramalin
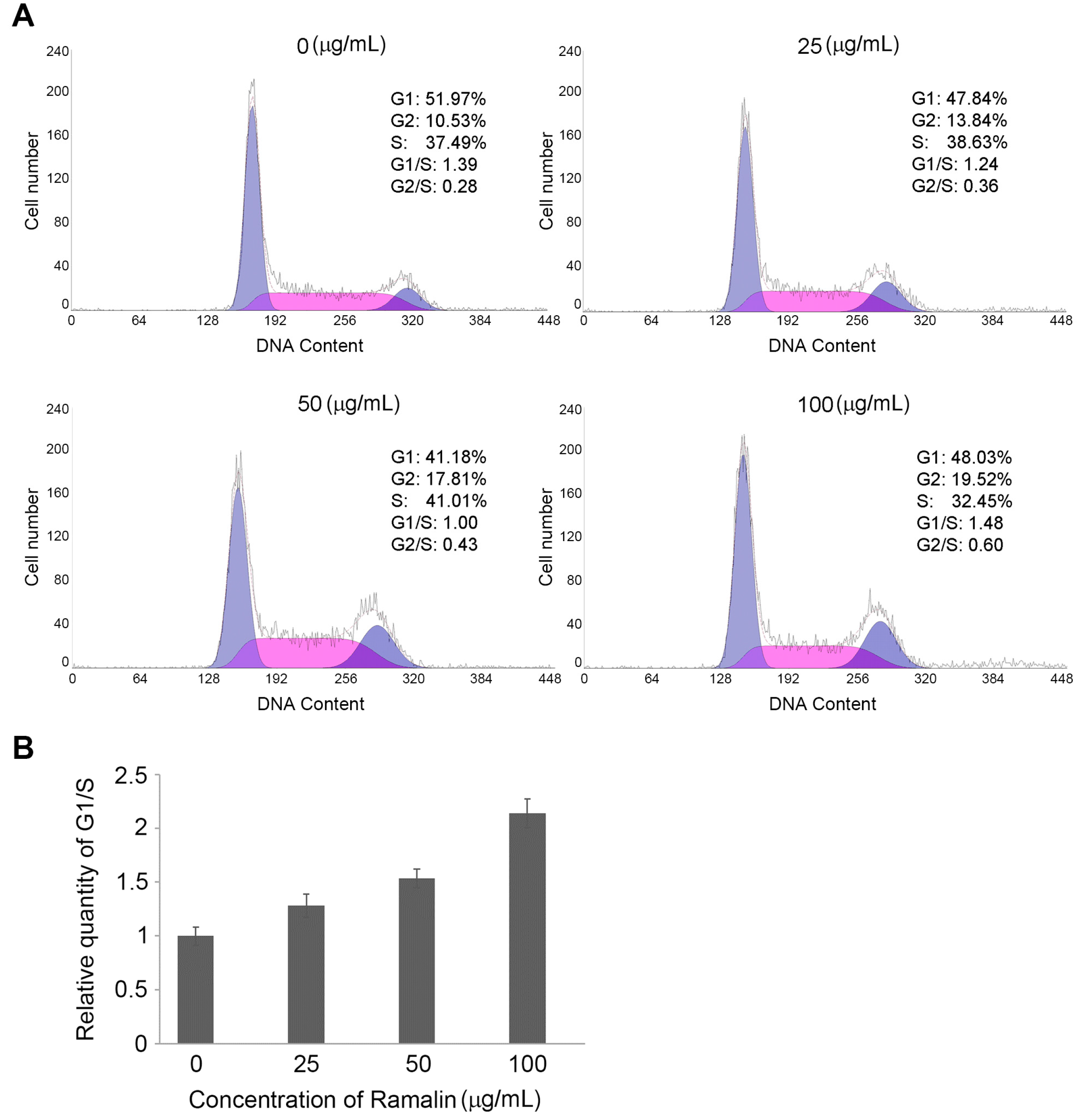
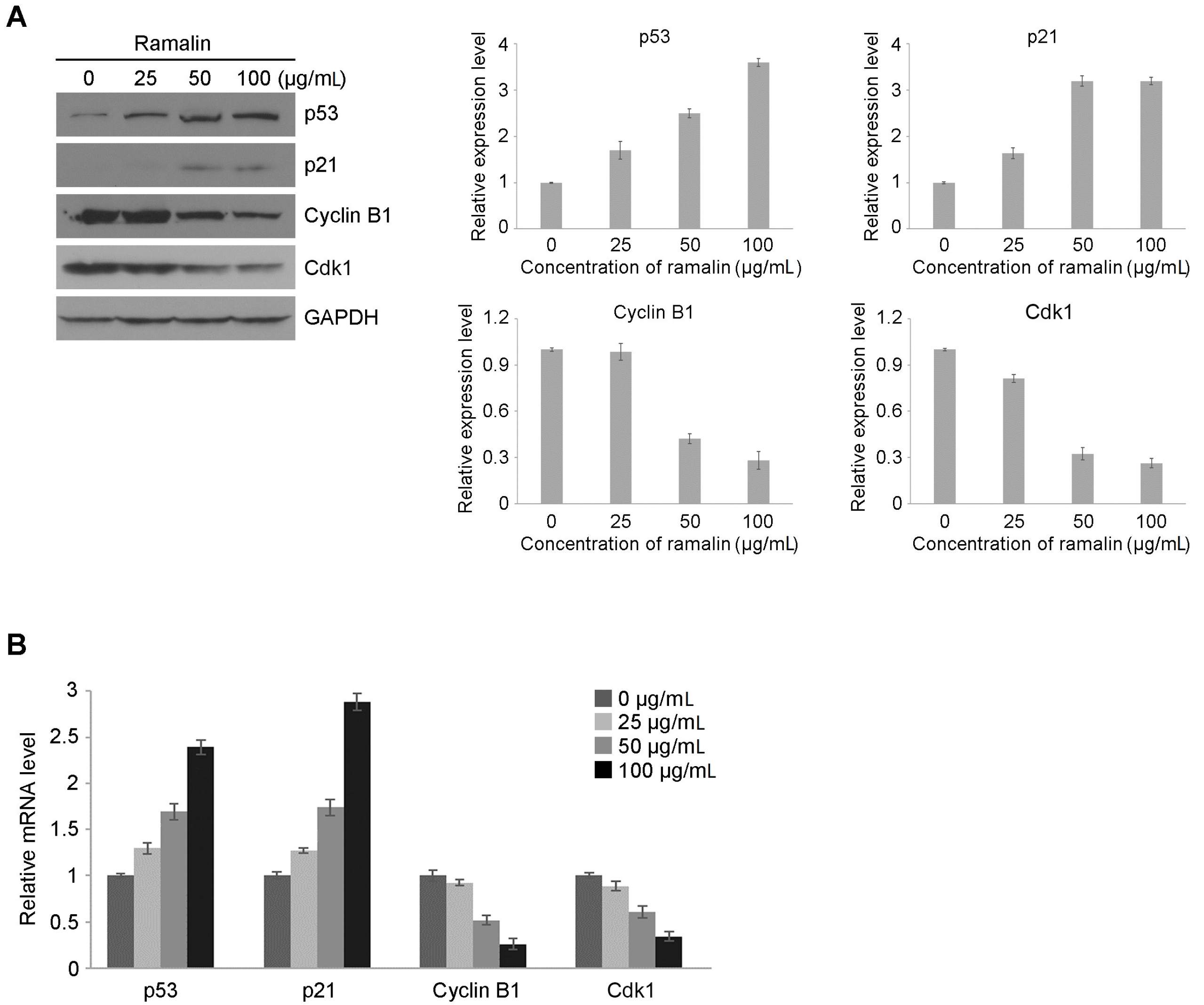
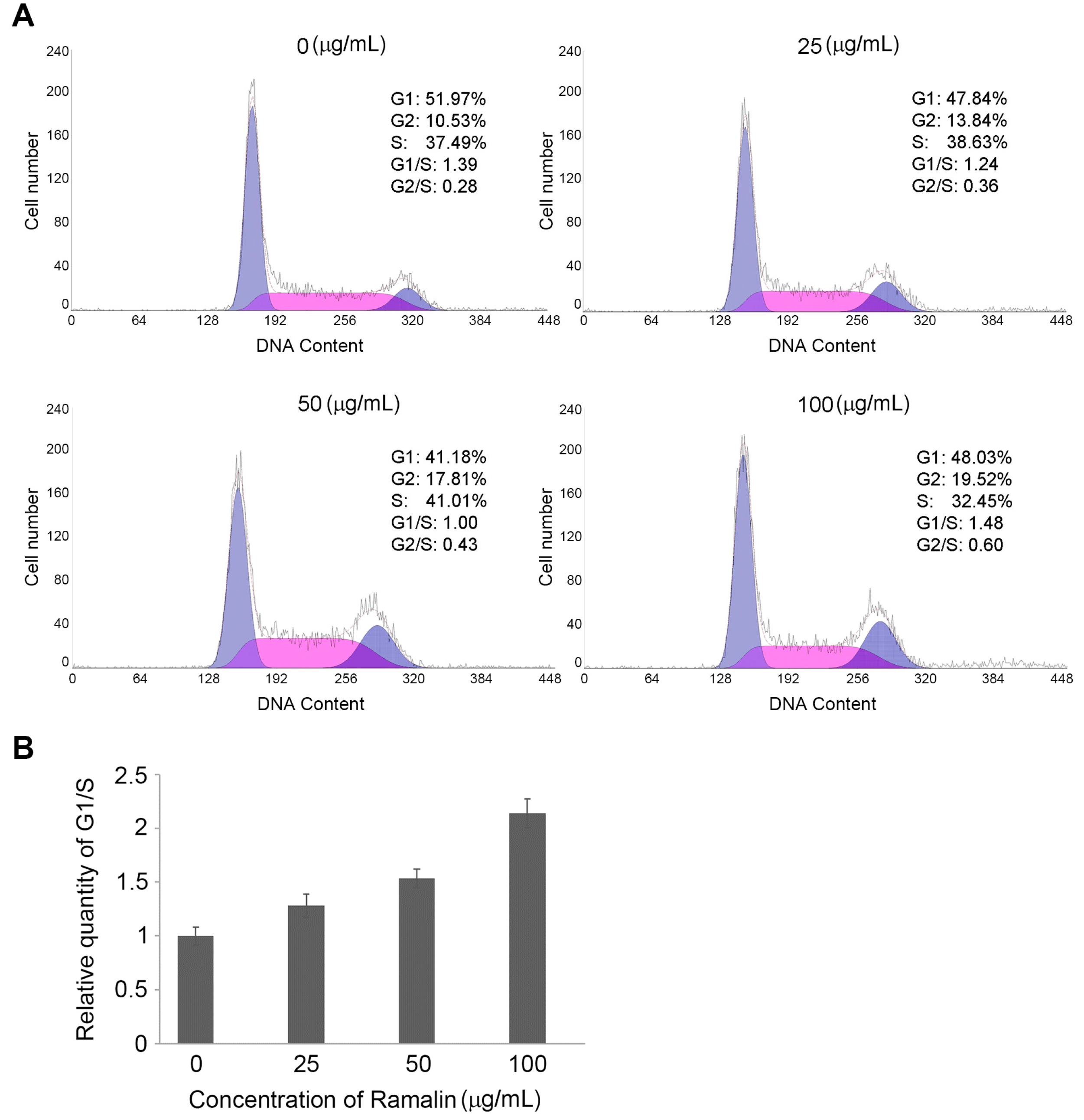
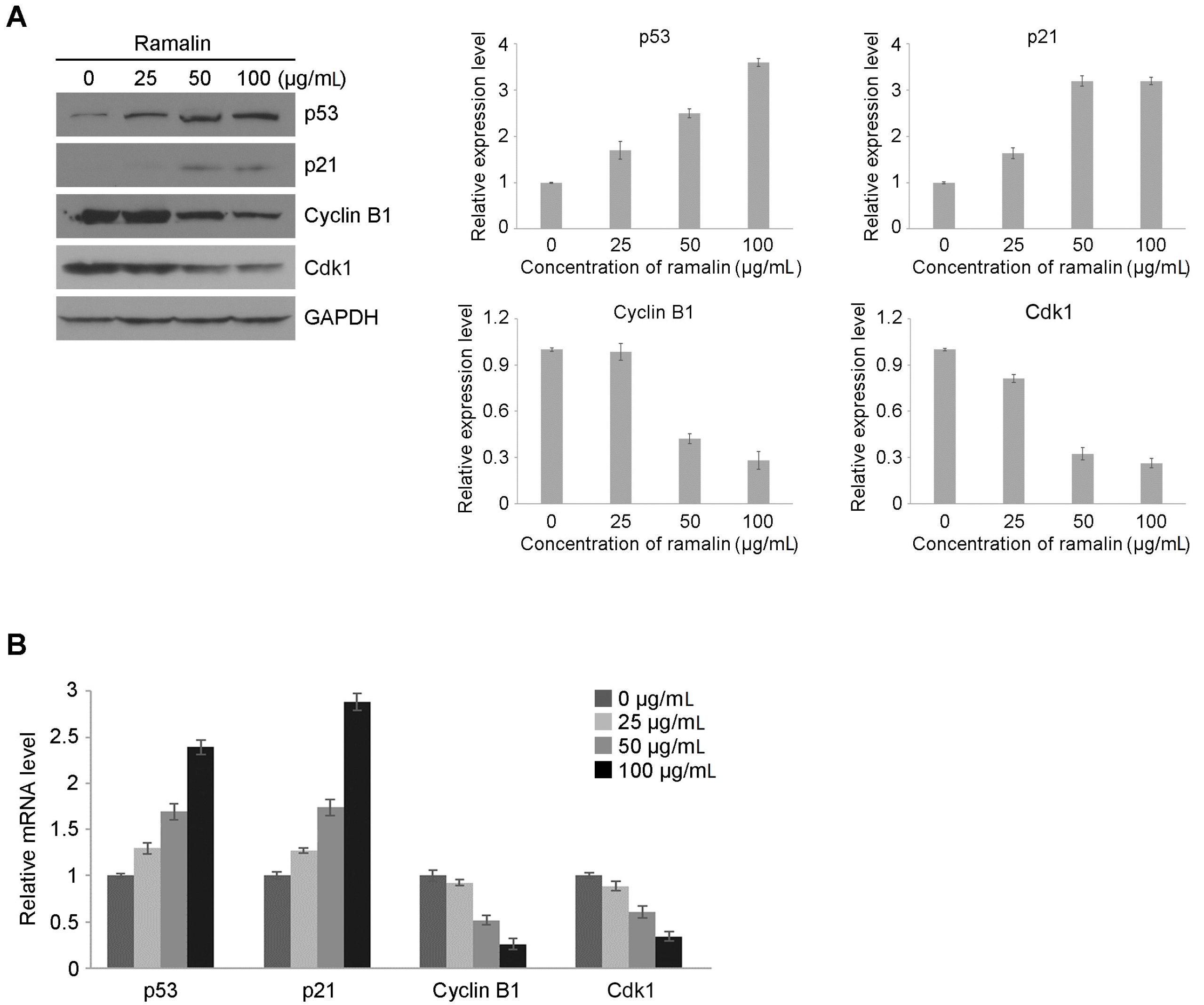
2.2. Ramalin-Induced Cell Cycle Arrest through the Modulation of Cell Cycle Marker Genes
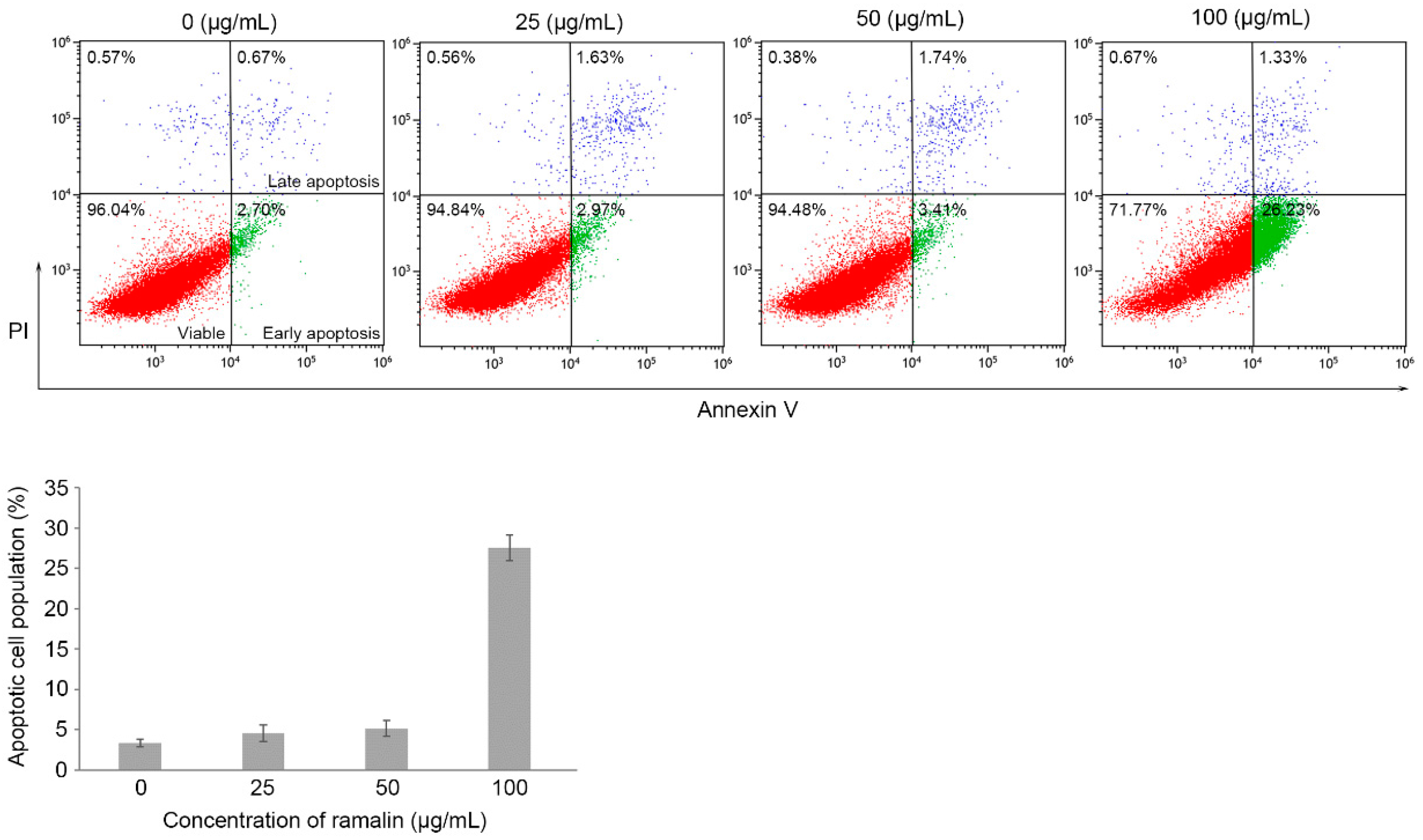
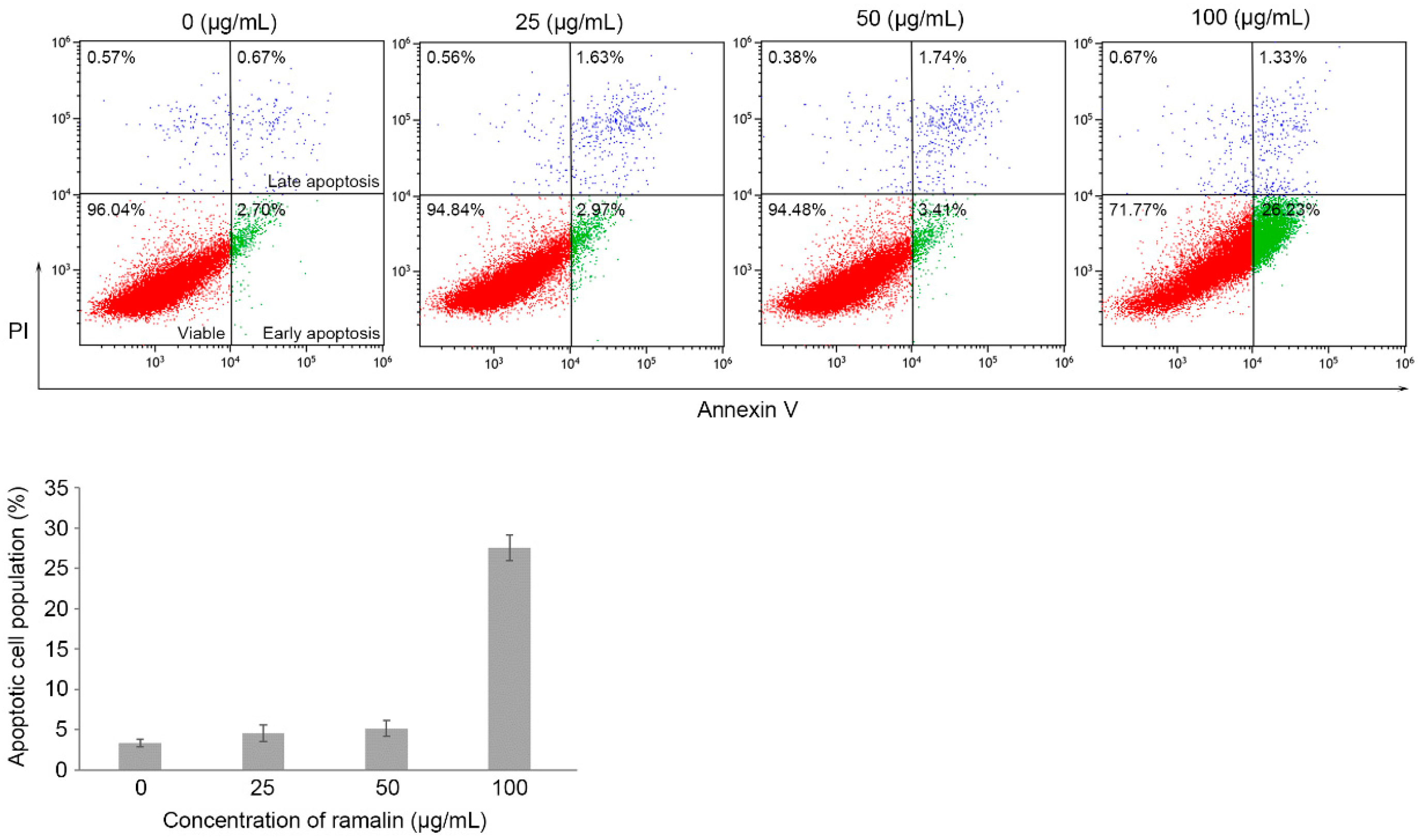
2.3. Ramalin-Mediated Induction of Apoptosis in HCT116 Cells
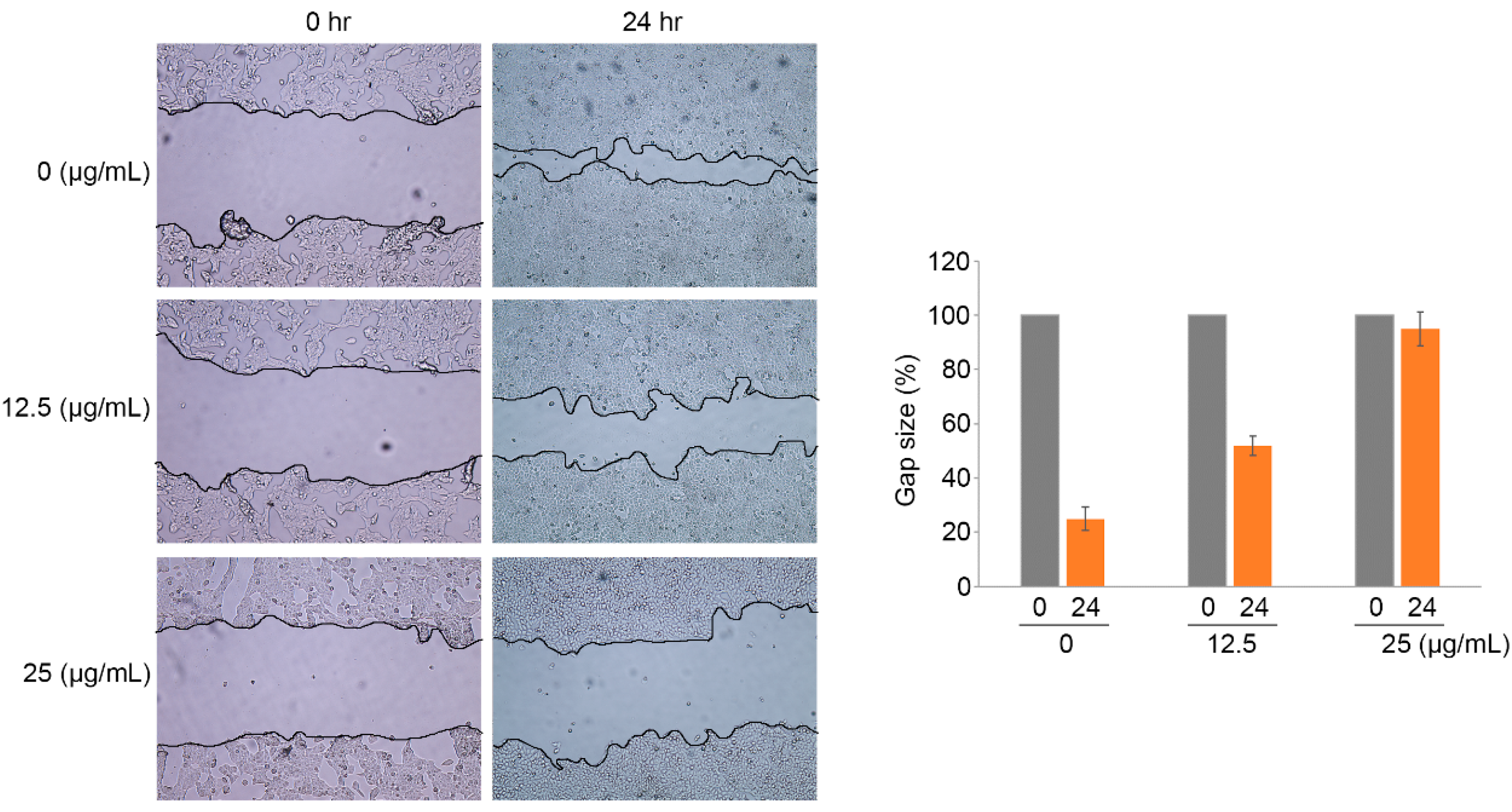
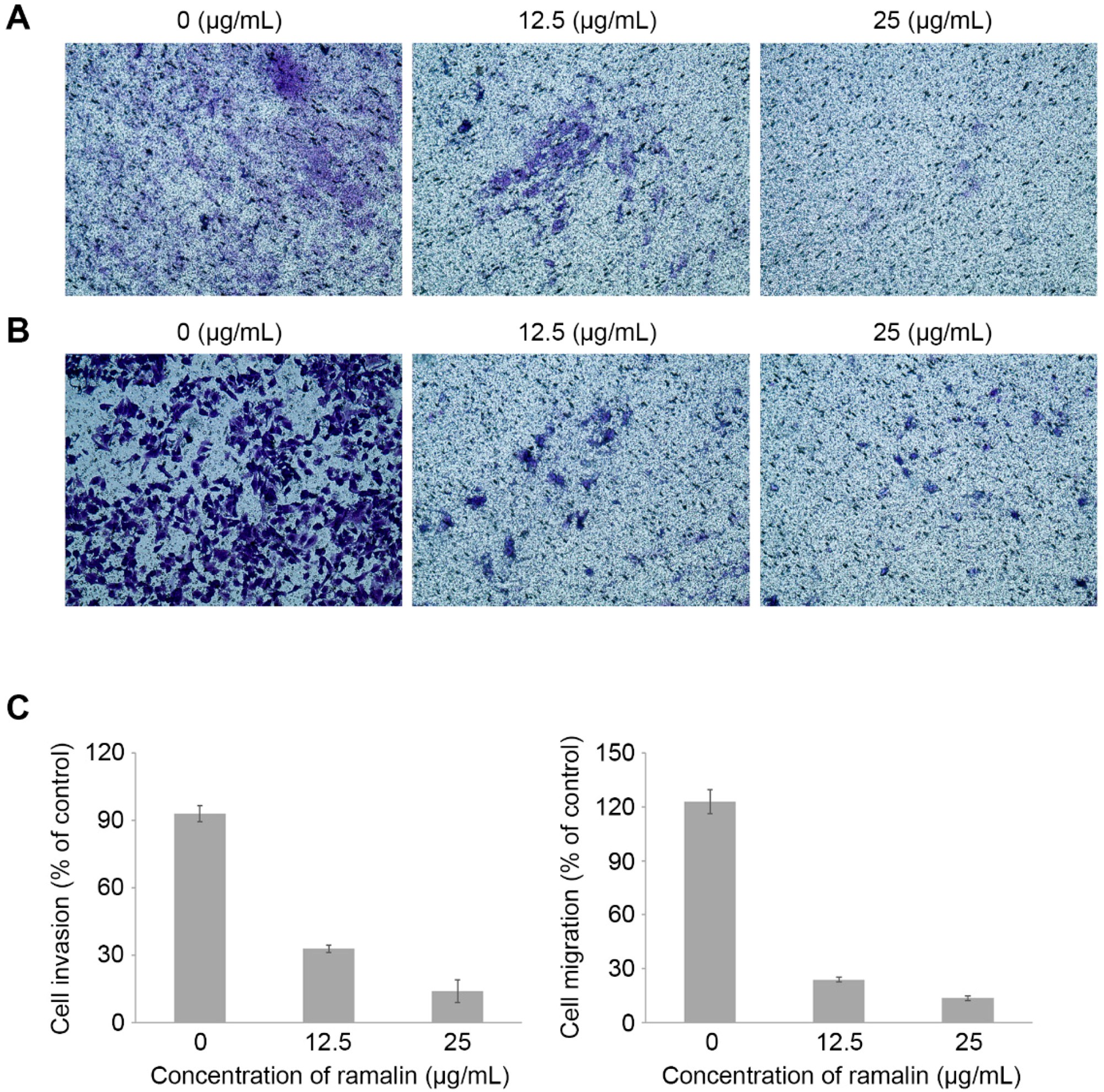
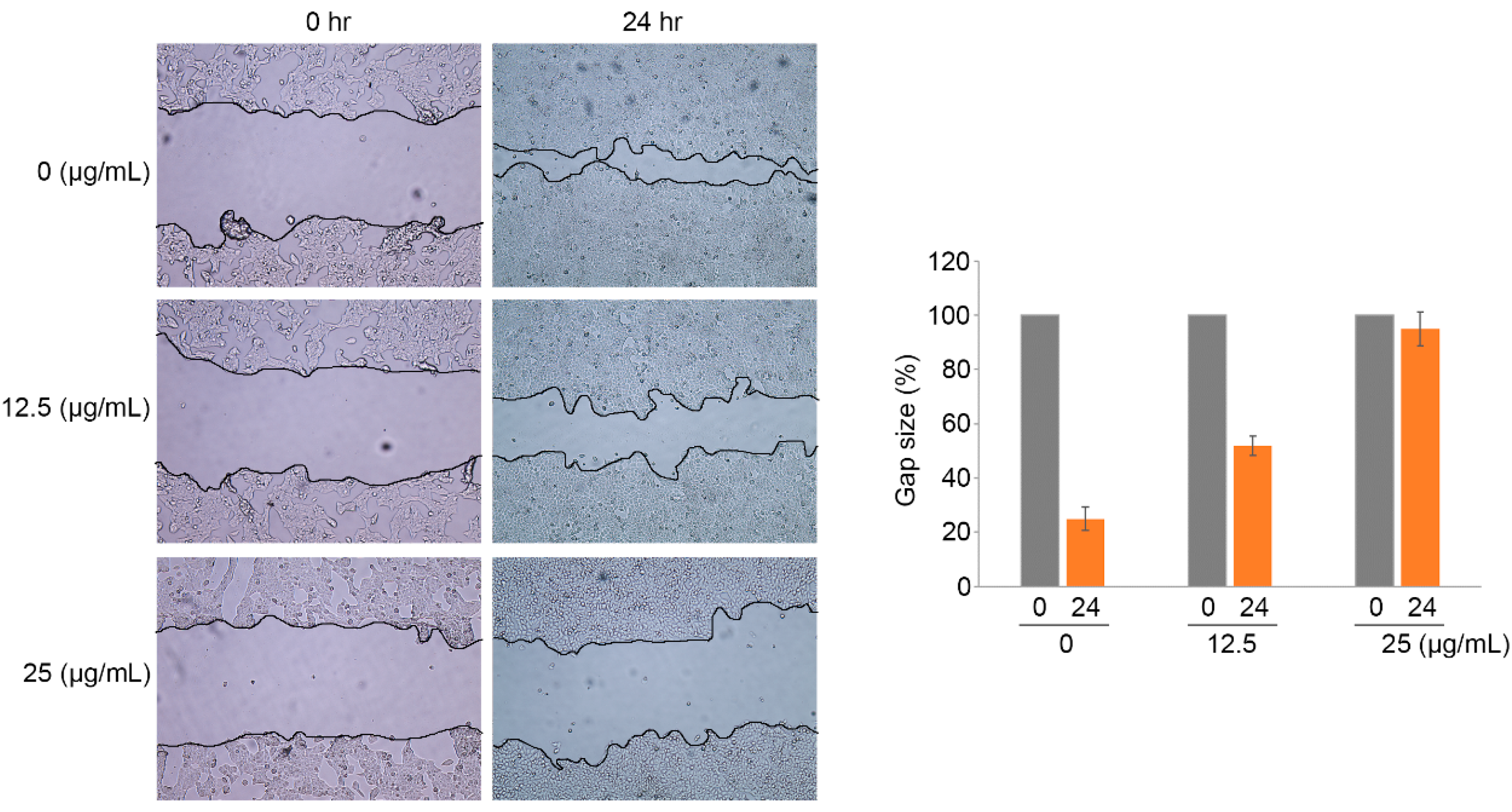
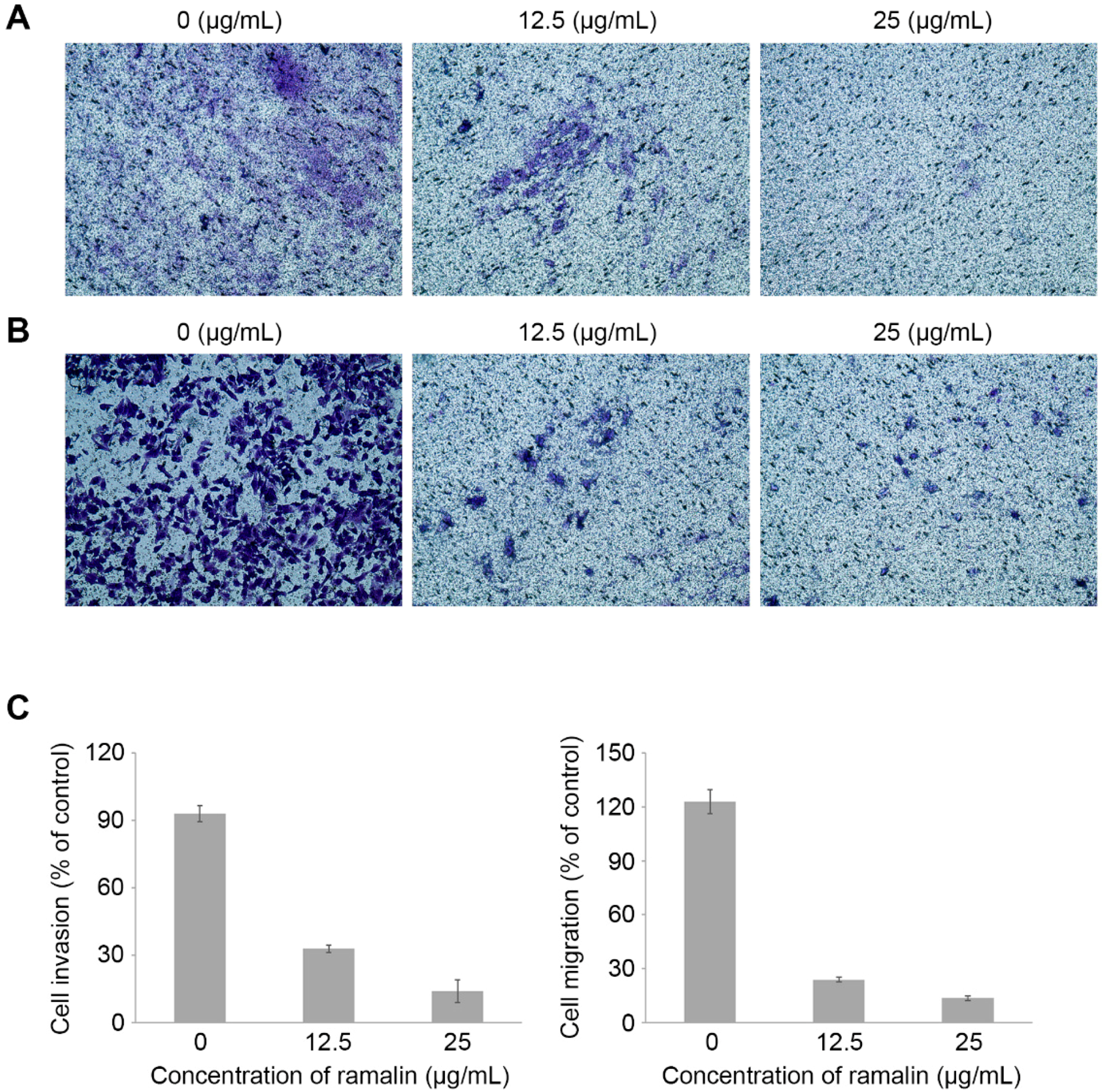
2.4. Ramalin-Mediated Inhibition of Cellular Migration and Invasion
3. Materials and Methods
3.1. Cell Culture and Cell Growth Inhibition Assay
3.2. Cell Cycle Analysis
3.3. RNA Extraction and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
3.4. Western Blot Analysis
3.5. Apoptosis Assay
3.6. Wound-Healing Assay
3.7. Colony-Forming Assay
3.8. Invasion and Migration Assays
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Riihiӓki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed]
- Colussi, D.; Brandi, G.; Bazzoli, F.; Ricciardiello, L. Molecular pathways involved in colorectal cancer: Implications for disease behavior and prevention. Int. J. Mol. Sci. 2013, 14, 16365–16385. [Google Scholar] [CrossRef] [PubMed]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Delker, D.A.; McGettigan, B.M.; Kanth, P.; Pop, S.; Neklason, D.W.; Bronner, M.P.; Burt, R.W.; Hagedorn, C.H. RNA sequencing of sessile serrated colon polyps identifies differentially expressed genes and immunohistochemical markers. PLoS ONE 2014, 9, e88367. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, B.; Grieu, F.; Amanuel, B. Microsatellite instability in colorectal cancer. Asia Pac. J. Clin. Oncol. 2010, 6, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Mojarad, E.N.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol. Hepatol. Bed Bench 2013, 6, 120–128. [Google Scholar]
- Ng, J.M.K.; Yu, J. Promoter hypermethylation of tumour suppressor genes as potential biomarkers in colorectal cancer. Int. J. Mol. Sci. 2015, 16, 2472–2496. [Google Scholar] [CrossRef] [PubMed]
- Okayama, H.; Schetter, A.J.; Harris, C.C. MicroRNAs and inflammation in the pathogenesis and progression of colon cancer. Dig. Dis. 2012, 30, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nishihara, R.; VanderWeele, T.J.; Wang, M.; Nishi, A.; Lochhead, P.; Qian, Z.R.; Zhang, X.; Wu, K.; Nan, H.; et al. Review article: The role of molecular pathological epidemiology in the study of neoplastic and non-neoplastic diseases in the era of precision medicine. Epidemiology 2016, 27, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. Gut 2011, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Zambare, V.P.; Christopher, L.P. Biopharmaceutical potential of lichens. Pharm. Biol. 2012, 50, 778–798. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Moriano, C.; Gómez-Serranillos, M.P.; Crespo, A. Antioxidant potential of lichen species and their secondary metabolites. A systematic review. Pharm. Biol. 2016, 54, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, V.M.; Karunaratne, V. Potential of lichen compounds as antidiabetic agents with antioxidative properties: A review. Oxid. Med. Cell. Longev. 2017, 2079697. [Google Scholar] [CrossRef] [PubMed]
- Müller, K. Pharmaceutically relevant metabolites from lichens. Appl. Microbiol. Biotechnol. 2001, 56, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Paudel, B.; Bhattarai, H.D.; Koh, H.Y.; Lee, S.G.; Han, S.J.; Lee, H.K.; Oh, H.; Shin, H.W.; Yim, J.H. Ramalin, a novel nontoxic antioxidant compound from the Antarctic lichen Ramalina terebrata. Phytomedicine 2011, 18, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Jang, Y.J.; Yim, J.H.; Lee, H.K.; Pyo, S. Ramalin isolated from Ramalina terebrata attenuates atopic dermatitis-like skin lesions in Balb/c mice and cutaneous immune responses in keratinocytes and mast cells. Phytother. Res. 2016, 30, 1978–1987. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, C.G.; Yim, J.H.; Lee, H.K.; Pyo, S. Ramalin-mediated apoptosis is enhanced by autophagy inhibition in human breast cancer cells. Phytother. Res. 2016, 30, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S. Cell signaling and cancer. Cancer Cell 2003, 4, 167–174. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Labi, V.; Erlacher, M. How cell death shapes cancer. Cell Death Dis. 2015, 6, e1675. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and molecular targeting therapy in cancer. Biomed. Res. Int. 2014, 2014, 150845. [Google Scholar] [CrossRef] [PubMed]
- Baig, S.; Seevasant, I.; Mohamad, J.; Mukheem, A.; Huri, H.Z.; Kamarul, T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis. 2016, 7, e2058. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Xue, D. The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.S.; Yang, E.J.; Lee, S.G.; Youn, U.J.; Han, S.J.; Kim, I.C.; Kim, S. Bioactivities of ethanol extract from the Antarctic freshwater microalga, Chloromonas sp. Int. J. Med. Sci. 2017, 14, 560–569. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: The compound, ramalin, is available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suh, S.-S.; Kim, T.K.; Kim, J.E.; Hong, J.-M.; Nguyen, T.T.T.; Han, S.J.; Youn, U.J.; Yim, J.H.; Kim, I.-C. Anticancer Activity of Ramalin, a Secondary Metabolite from the Antarctic Lichen Ramalina terebrata, against Colorectal Cancer Cells. Molecules 2017, 22, 1361. https://doi.org/10.3390/molecules22081361
Suh S-S, Kim TK, Kim JE, Hong J-M, Nguyen TTT, Han SJ, Youn UJ, Yim JH, Kim I-C. Anticancer Activity of Ramalin, a Secondary Metabolite from the Antarctic Lichen Ramalina terebrata, against Colorectal Cancer Cells. Molecules. 2017; 22(8):1361. https://doi.org/10.3390/molecules22081361
Chicago/Turabian StyleSuh, Sung-Suk, Tai Kyoung Kim, Jung Eun Kim, Ju-Mi Hong, Trang Thu Thi Nguyen, Se Jong Han, Ui Joung Youn, Joung Han Yim, and Il-Chan Kim. 2017. "Anticancer Activity of Ramalin, a Secondary Metabolite from the Antarctic Lichen Ramalina terebrata, against Colorectal Cancer Cells" Molecules 22, no. 8: 1361. https://doi.org/10.3390/molecules22081361
APA StyleSuh, S.-S., Kim, T. K., Kim, J. E., Hong, J.-M., Nguyen, T. T. T., Han, S. J., Youn, U. J., Yim, J. H., & Kim, I.-C. (2017). Anticancer Activity of Ramalin, a Secondary Metabolite from the Antarctic Lichen Ramalina terebrata, against Colorectal Cancer Cells. Molecules, 22(8), 1361. https://doi.org/10.3390/molecules22081361