Combining NMR Spectroscopy and Chemometrics to Monitor Structural Features of Crude Hep-arin

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Protocol for Sample Preparation
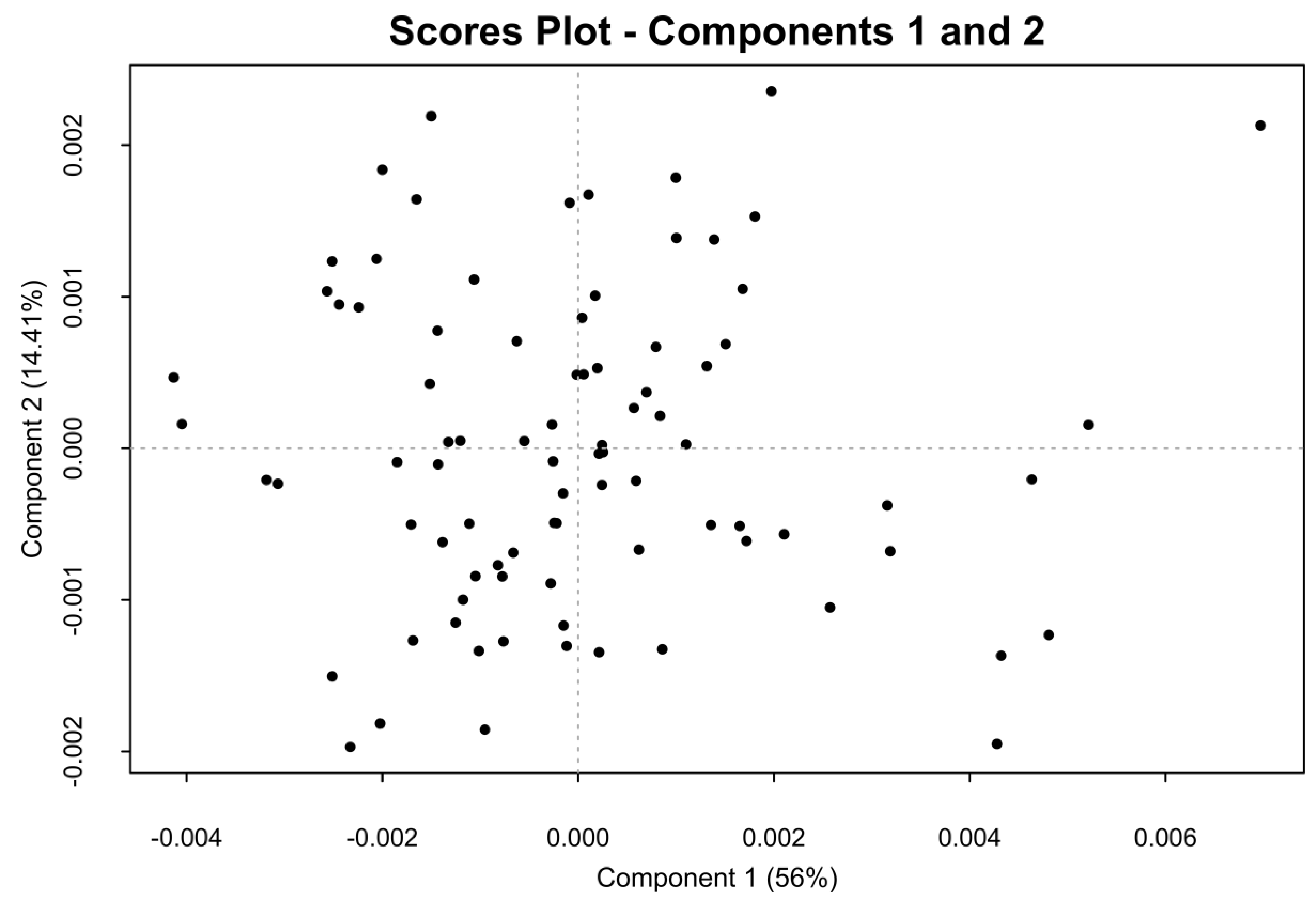
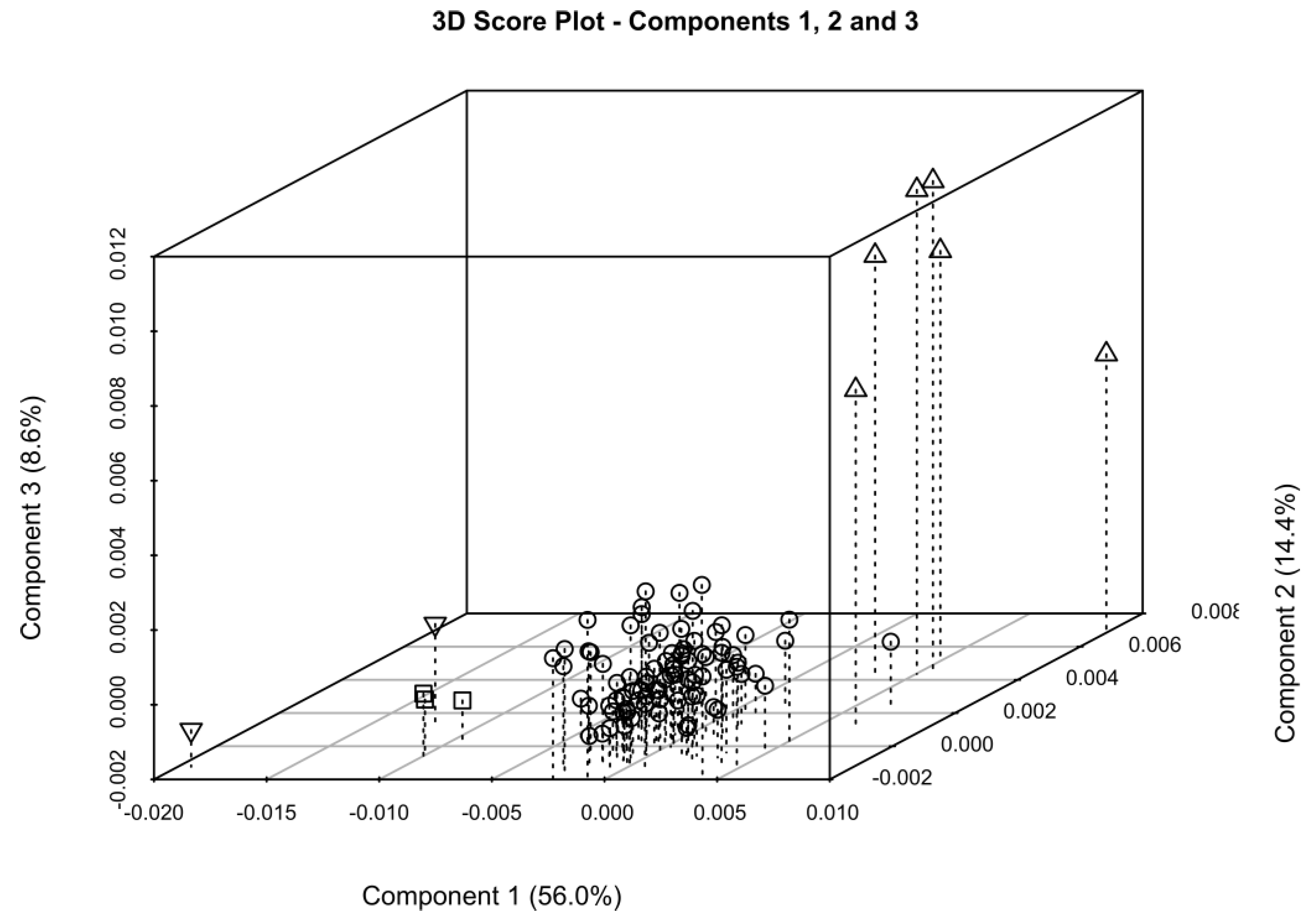
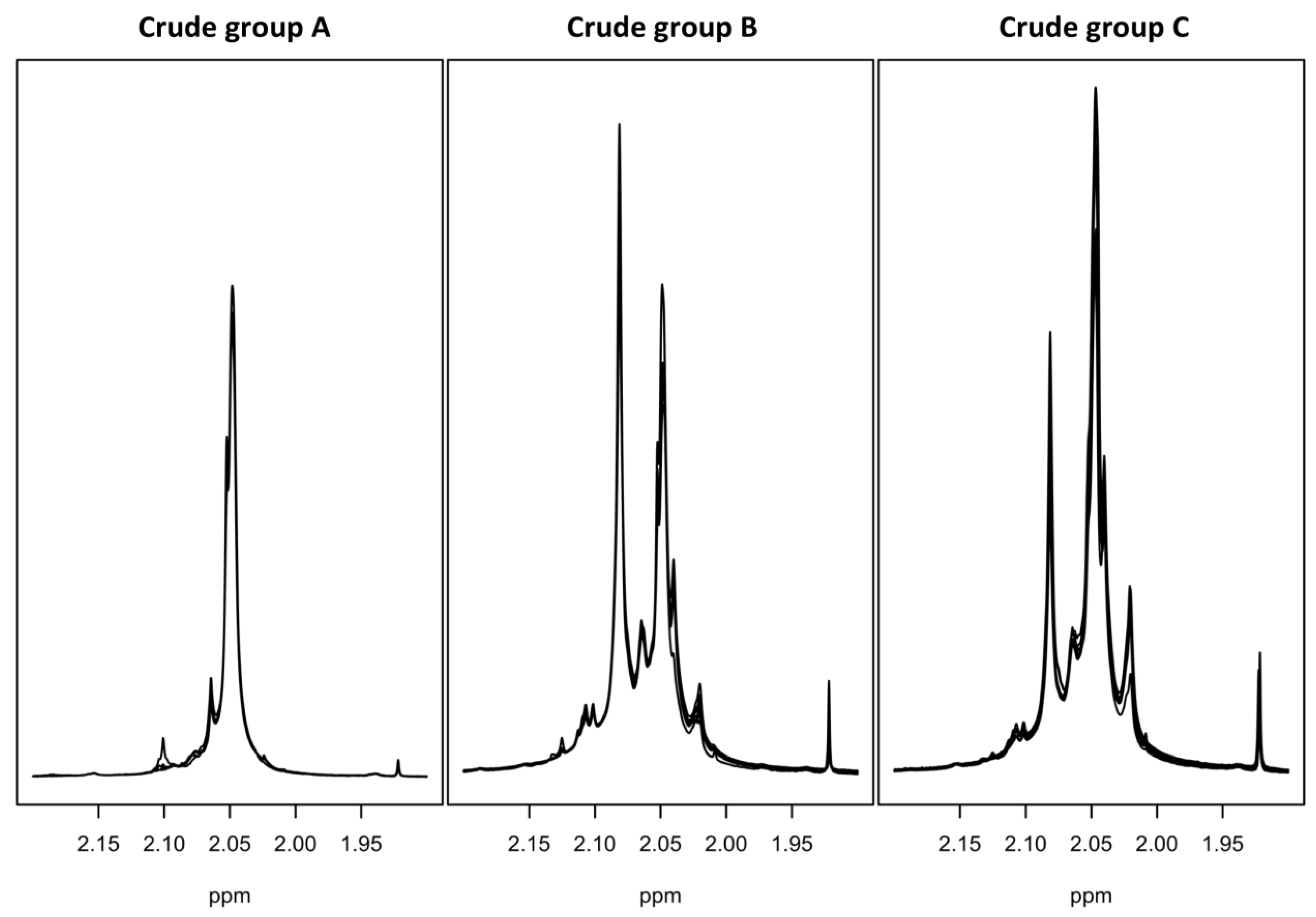
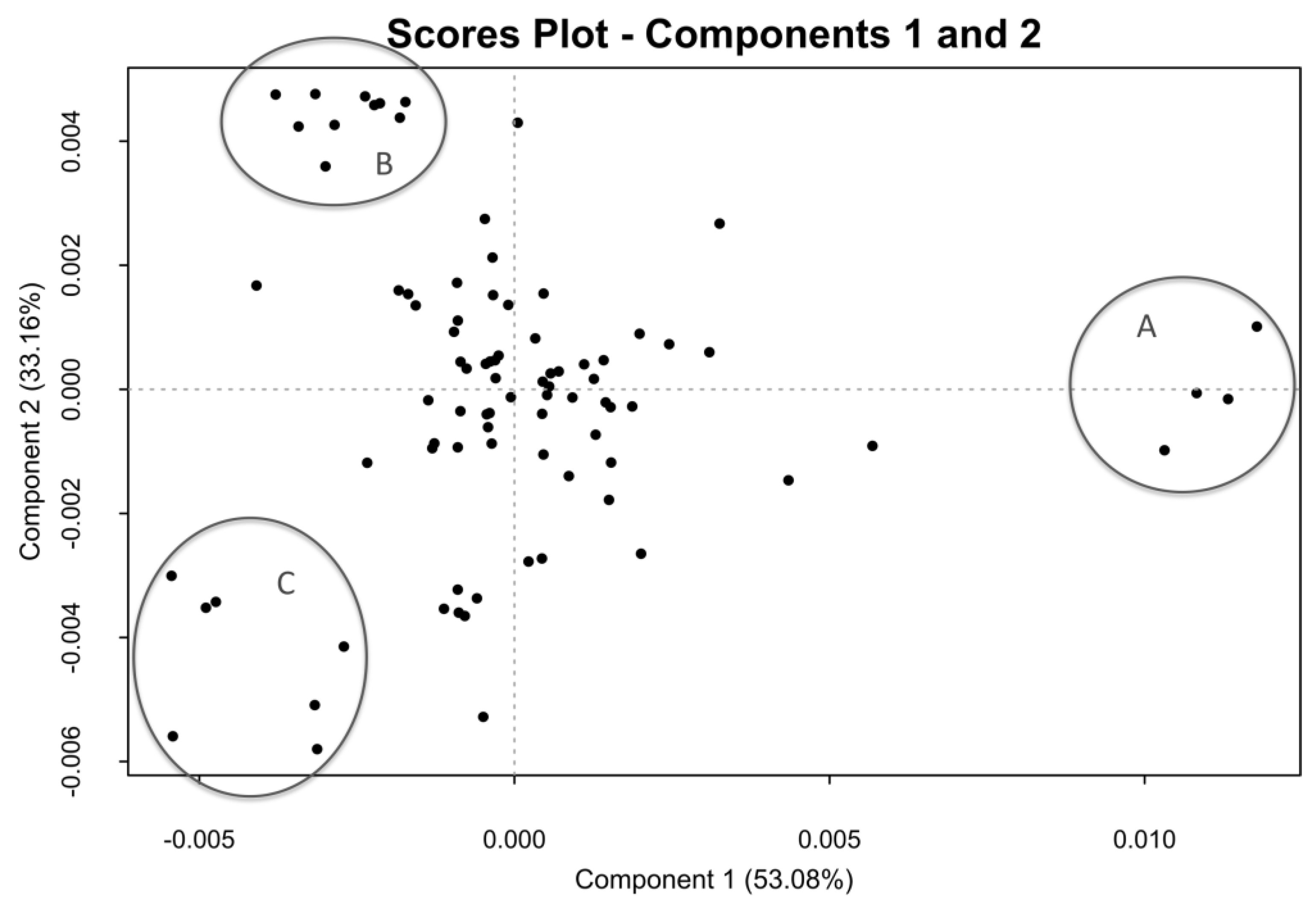
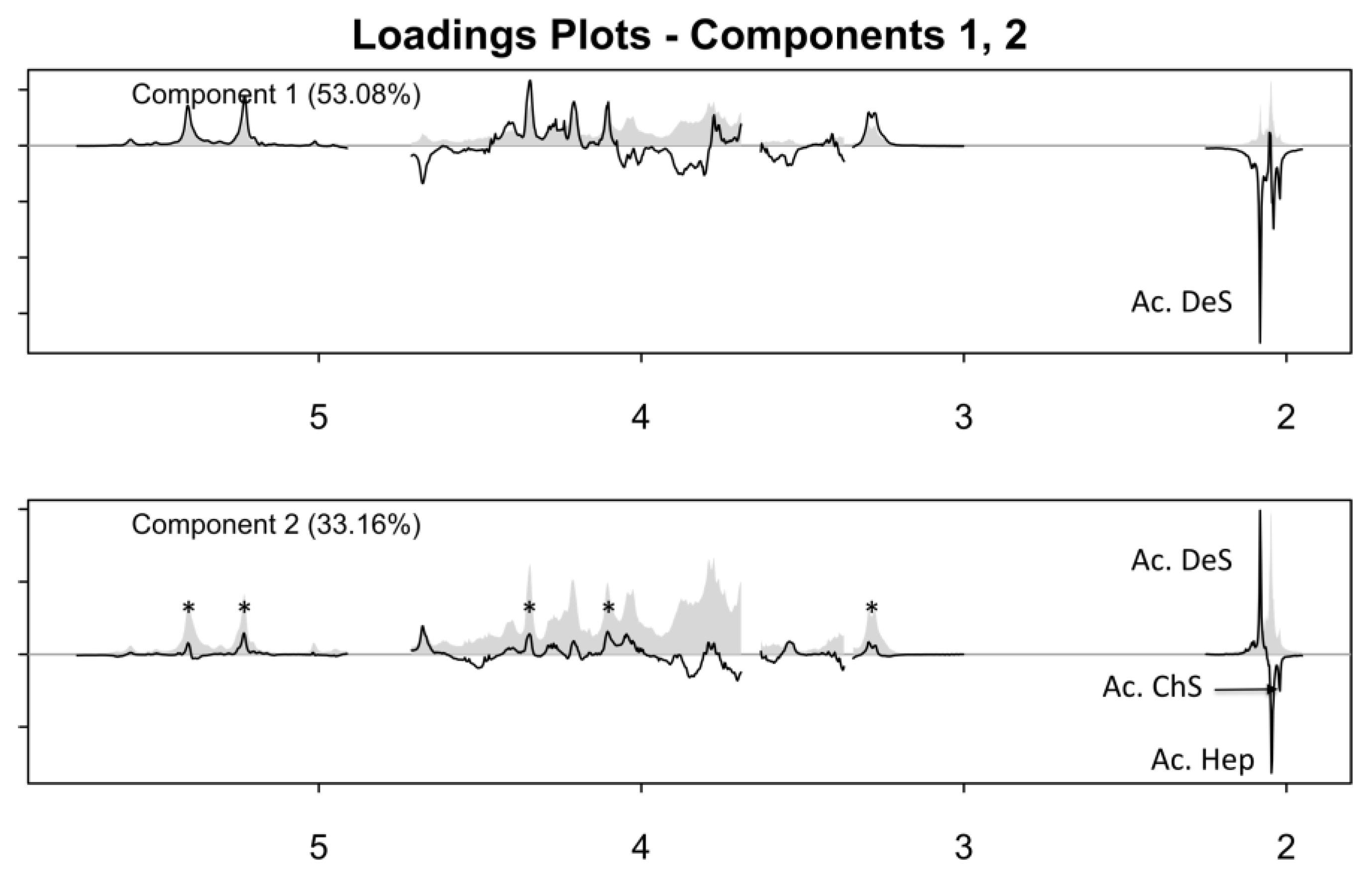
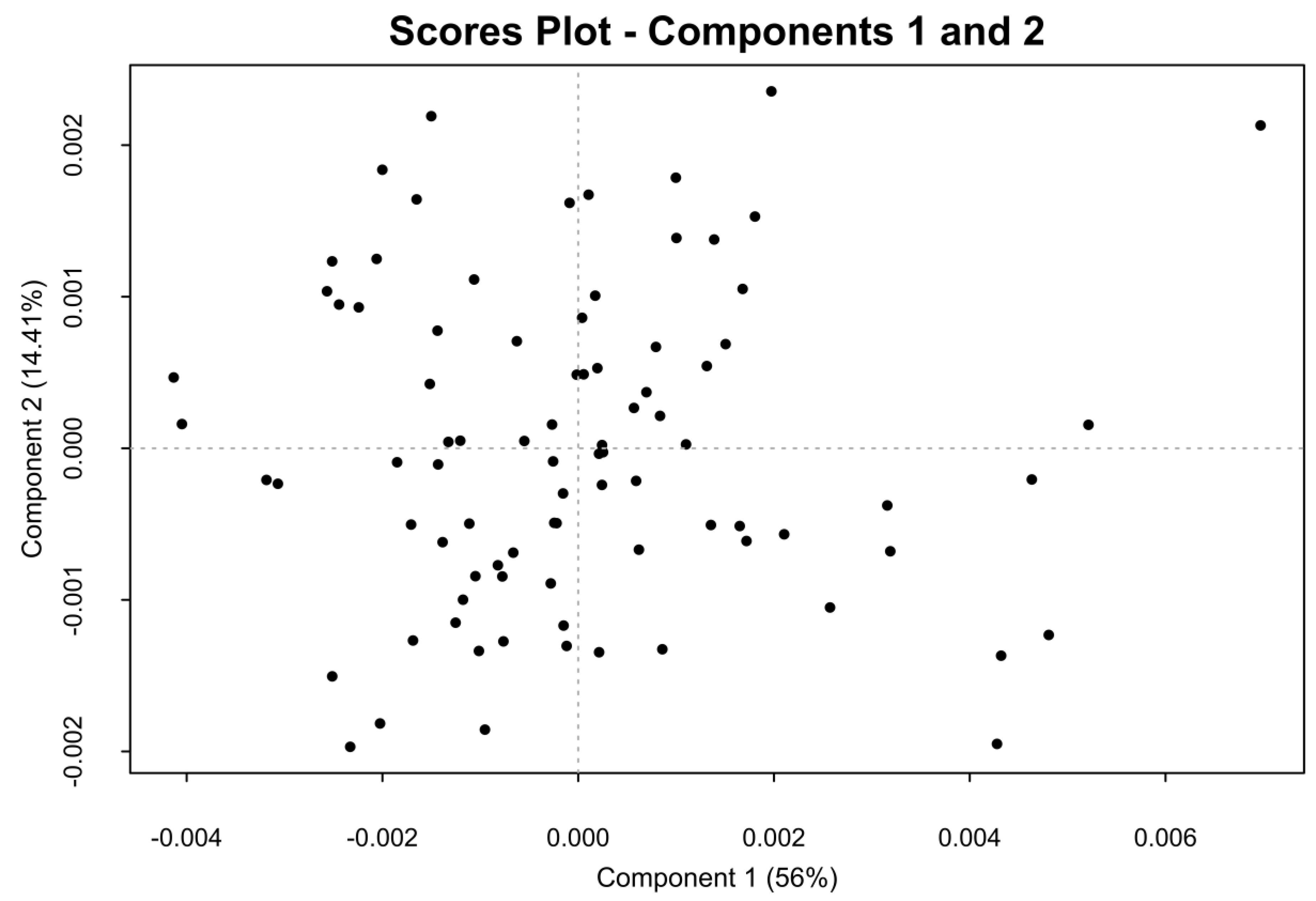
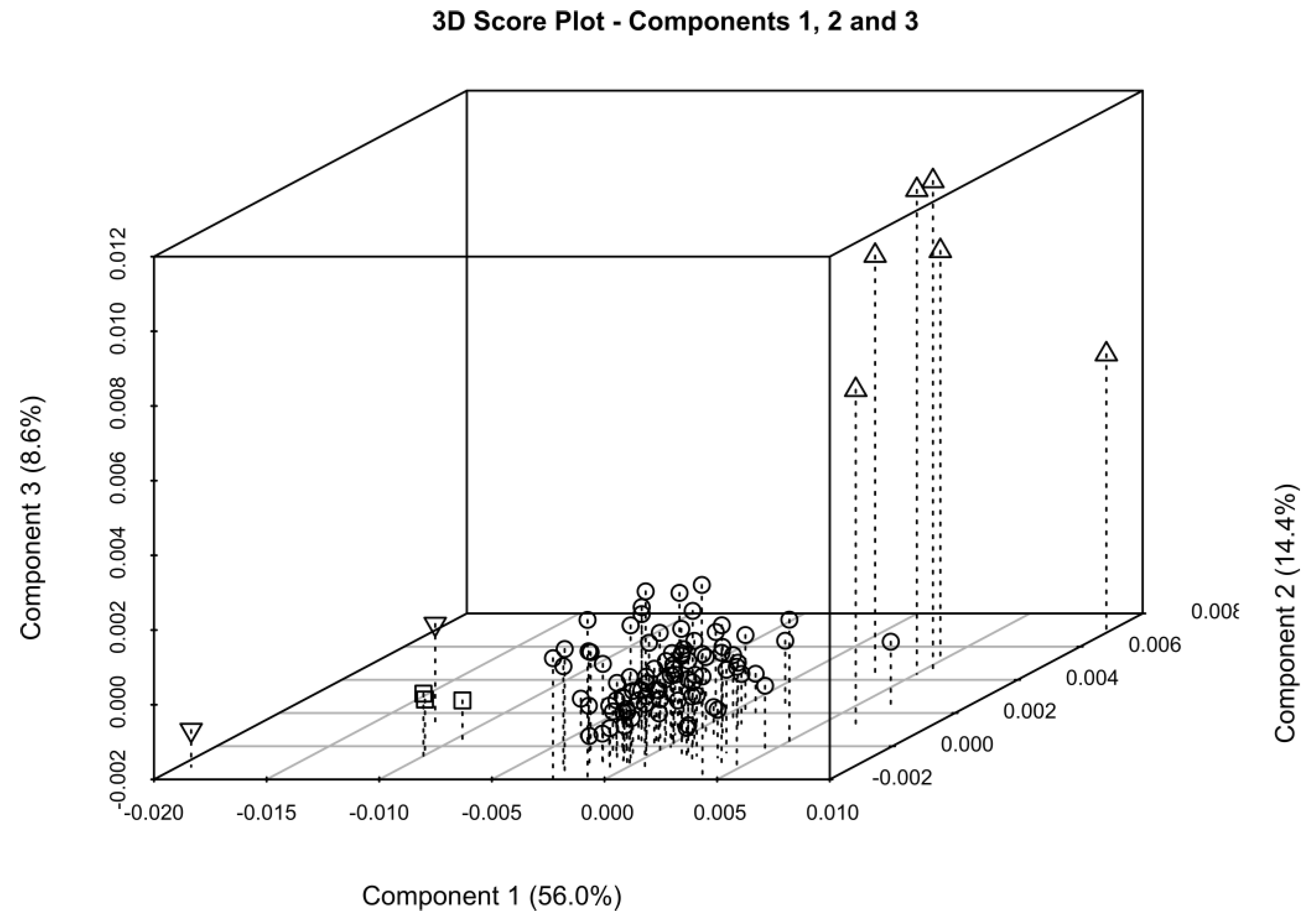
2.2. One-Dimensional (1D) Spectra Library and PCA
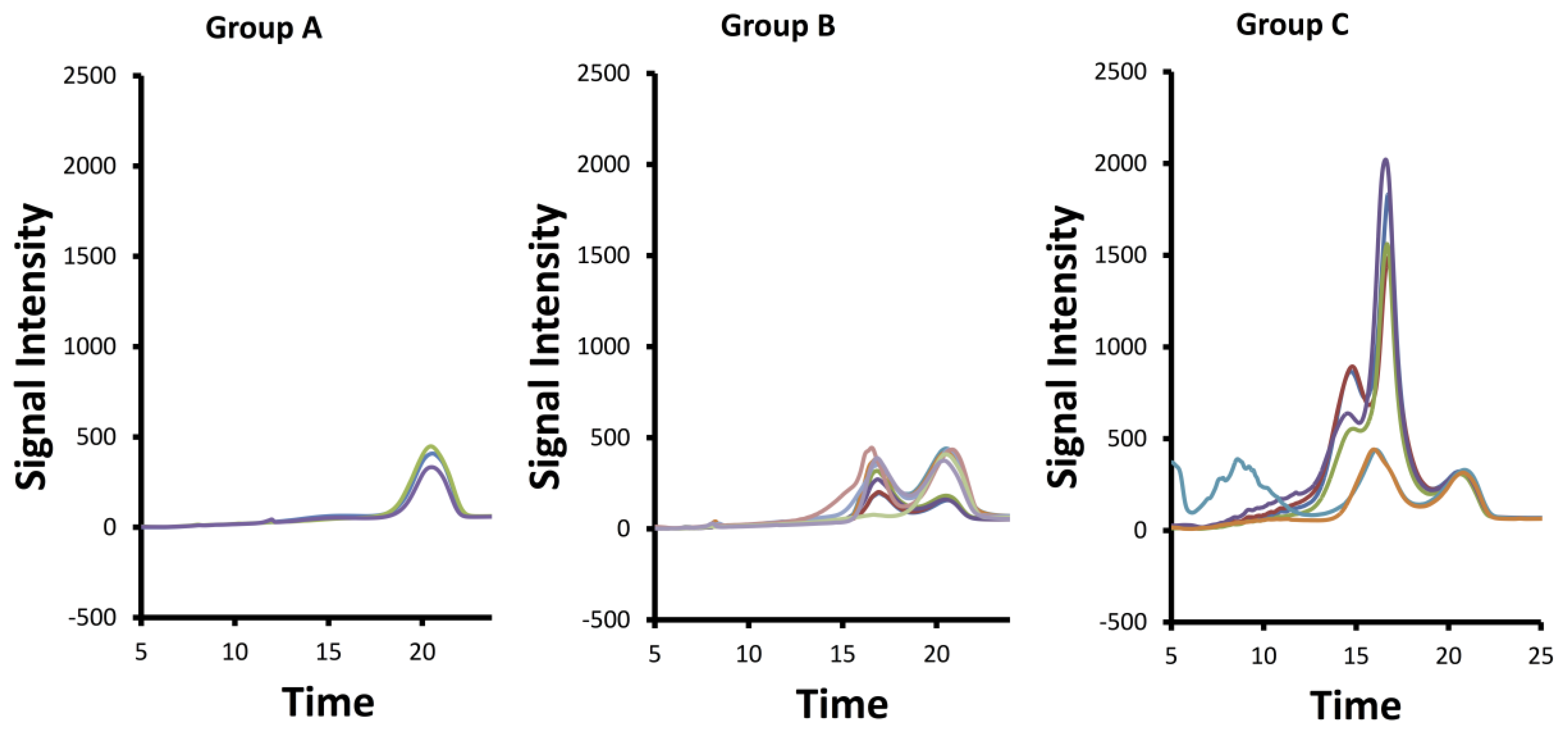
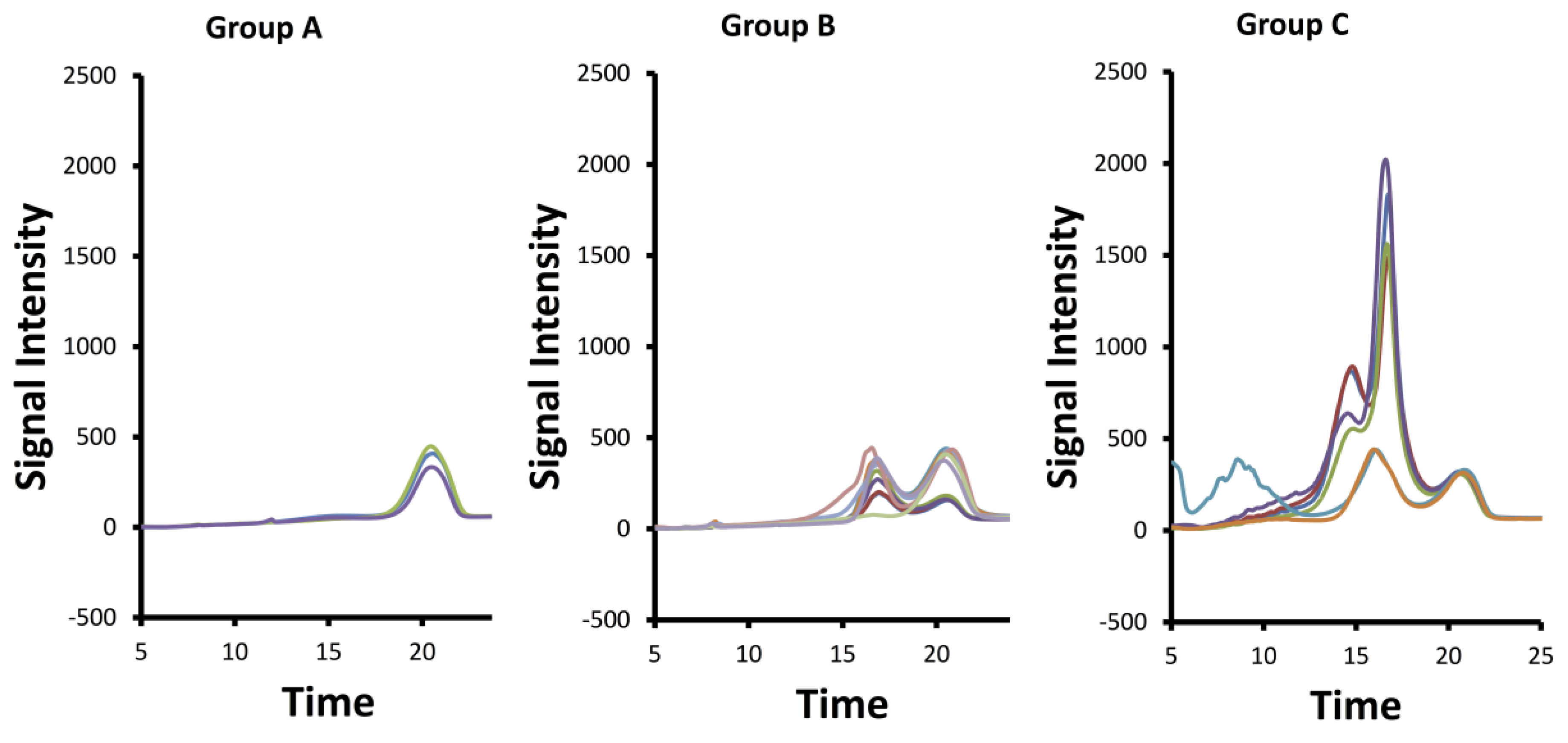
2.3. SAX-HPLC Analysis
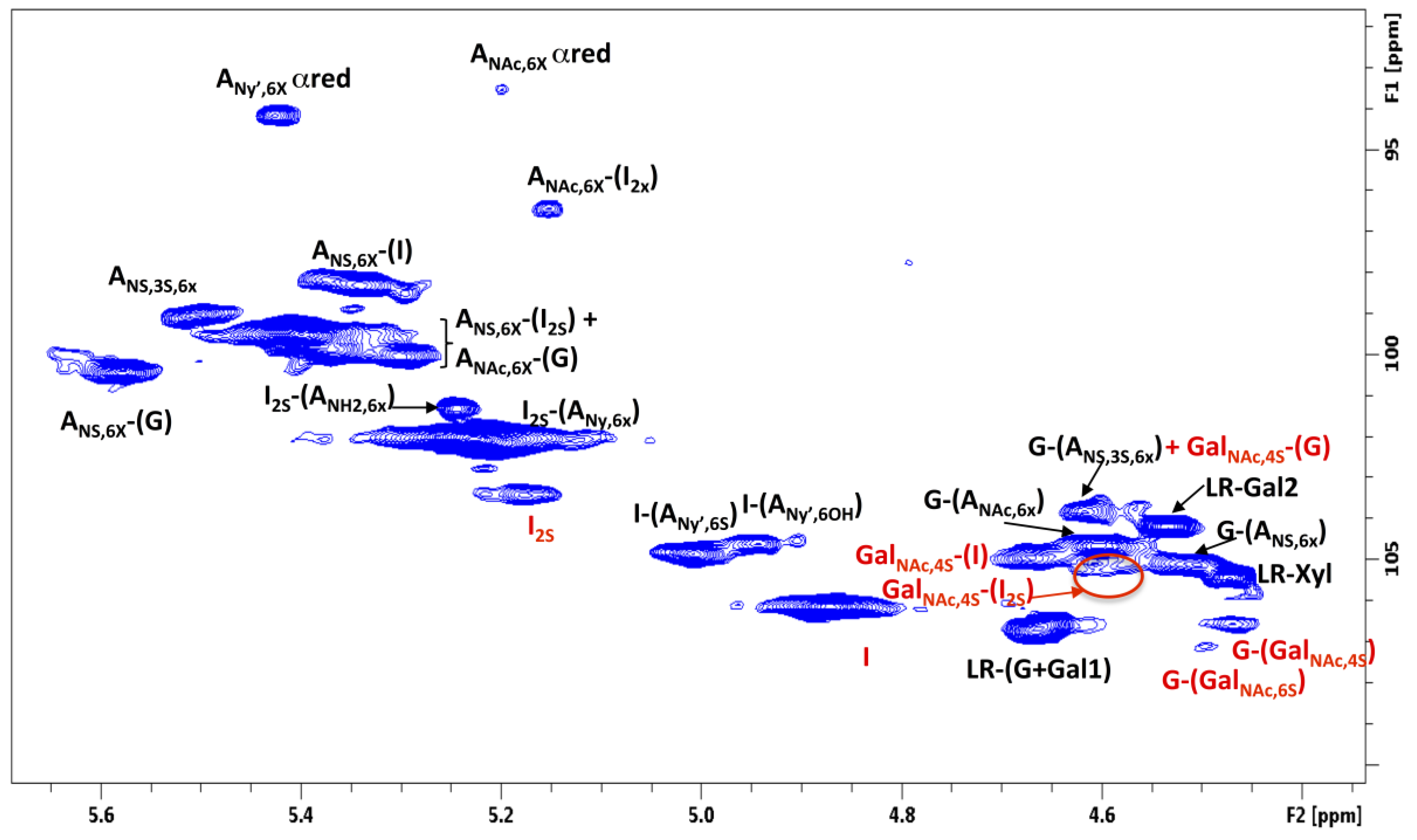
2.4. HSQC Analysis
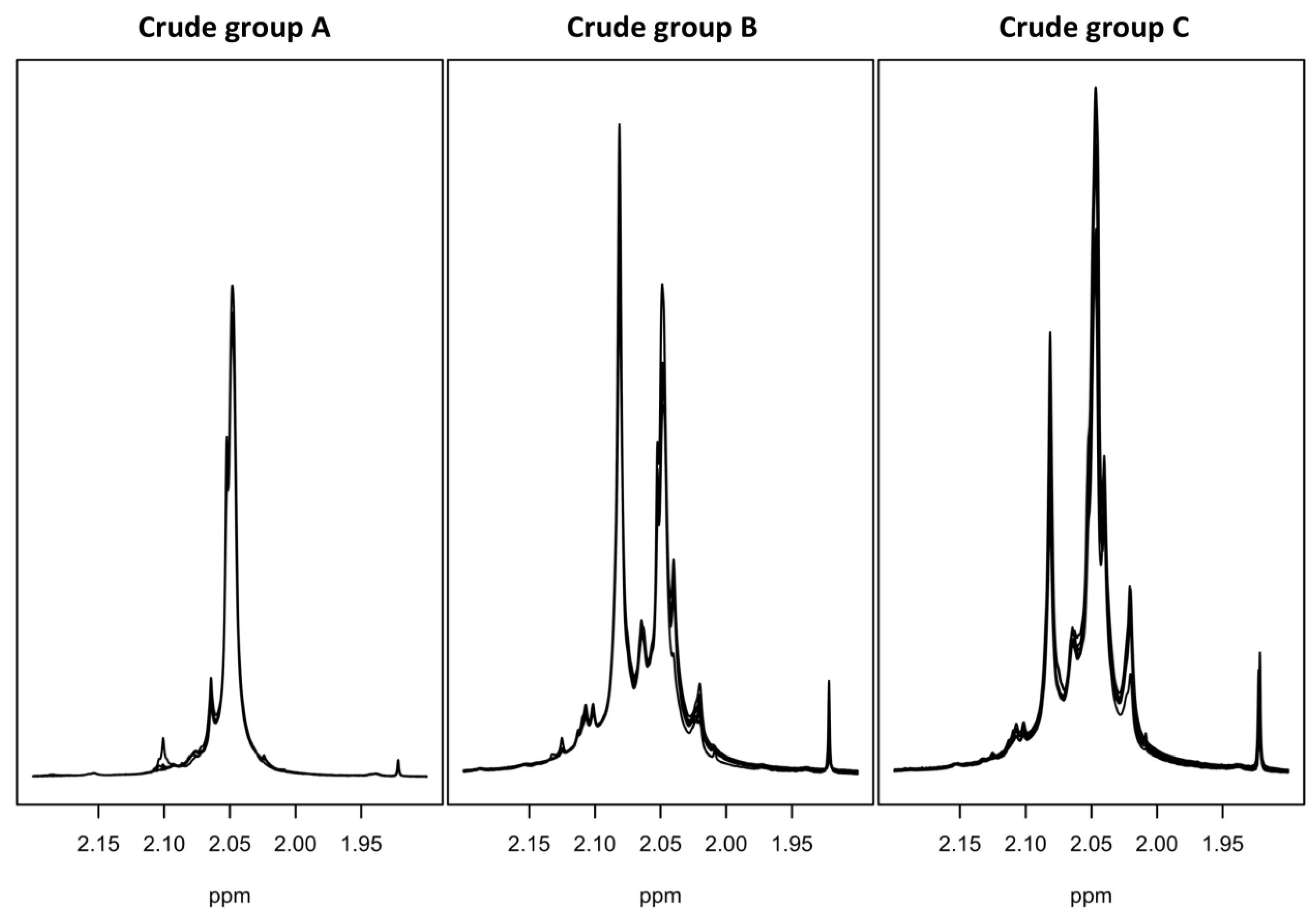
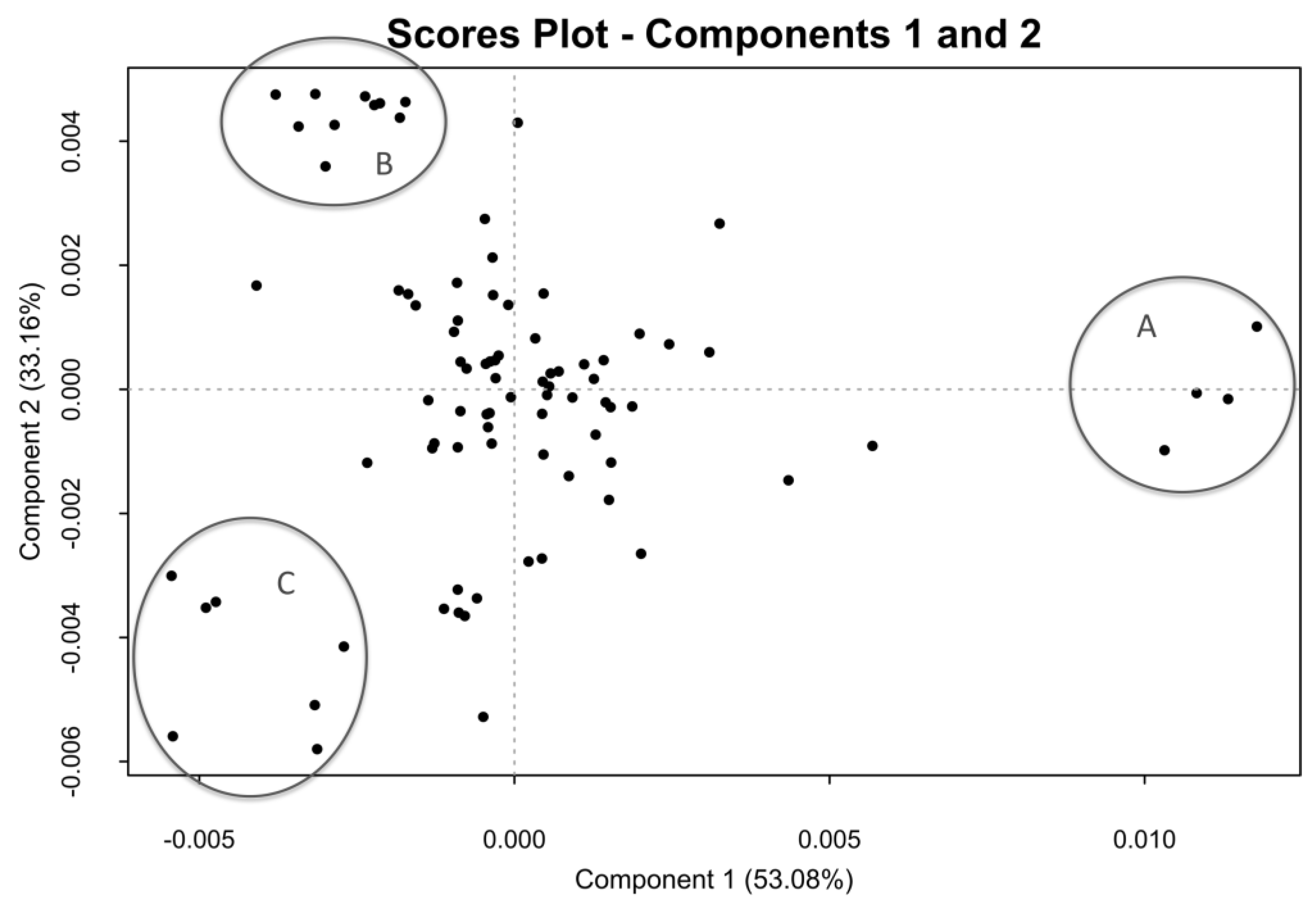
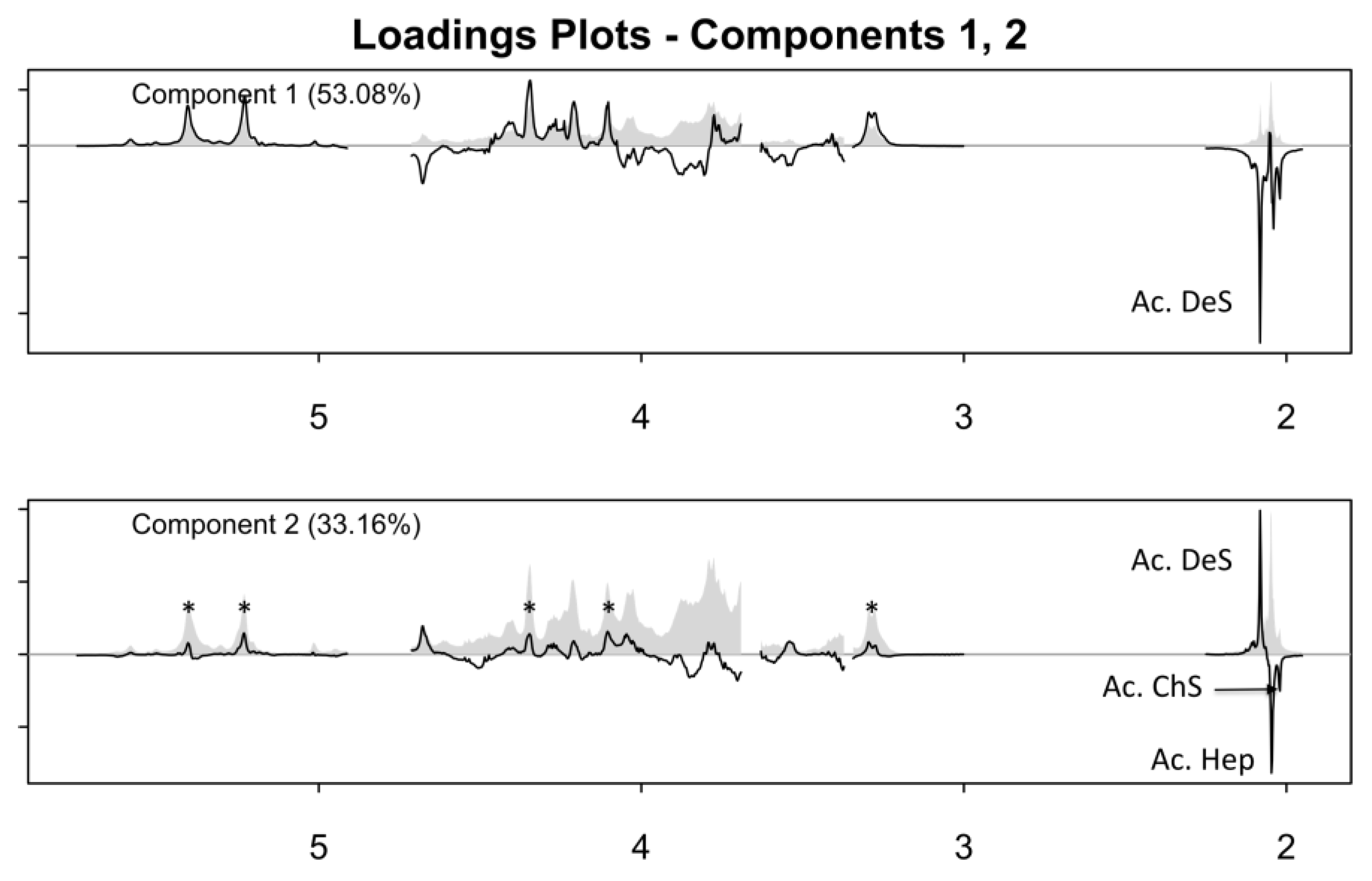
2.5. Crude of Different Origin
3. Discussion
4. Methods
4.1. Reagents and Starting Material
4.2. Strong Anion Exchange HPLC:
4.3. Sample Preparation
4.4. 1H-NMR
4.5. HSQC-NMR
4.6. PCA
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lever, R.; Mulloy, B.; Page, C.P. (Eds.) Heparin—A Century of Progress, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Casu, B. Structure and active domains of heparin. In Chemistry and Biology of Heparin and Heparan Sulphate, 1st ed.; Garg, H.G., Linhardt, R.J., Hales, C.A., Eds.; Elsevier Ltd: Oxford, UK, 2005; pp. 1–28. [Google Scholar]
- Keire, D.; Mulloy, B.; Chase, C.; Al-Hakim, A.; Cairatti, D.; Gray, E.; Hogwood, J.; Morris, T.; Mourão, P.; Da Luz Carvalho Soares, M.; et al. Diversifying the Global Heparin Supply Chain: Reintroduction of Bovine Heparin in the United States? Pharm. Technol. 2015, 11, 2–9. [Google Scholar]
- Guerrini, M.; Beccati, D.; Shriver, Z.; Naggi, A.; Viswanathan, K.; Bisio, A.; Capila, I.; Lansing, J.C.; Guglieri, S.; Fraser, B.; et al. Oversulfated chondroitinsulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol. 2008, 26, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhanga, Z.; Linhardt, R.J. Lessons learned from the contamination of heparin. Nat. Prod. Rep. 2008, 26, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Gunay, N.S. Production and chemical processing of low molecular weight heparins. Semin. Thromb. Hemost. 1999, 25, 5–16. [Google Scholar] [PubMed]
- U.S. Department of Health and Human Services. Food and Drug Administration. Guidance for Industry: Heparin for Drug and Medical Device Use: Monitoring Crude Heparin for Quality; FDA: Silver Spring, MD, USA, 2013. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291390.pdf (accessed on 24 April 2017).
- Sommers, C.D.; Mans, D.J.; Mecker, L.C.; Keire, D.A. Sensitive detection of oversulfated chondroitin sulfate in heparin sodium or crude heparin with a colorimetric microplate based assay. Anal. Chem. 2011, 83, 3422–3430. [Google Scholar] [CrossRef] [PubMed]
- Keire, D.A.; Trehy, M.L.; Reepmeyer, J.C.; Kolinski, R.E.; Ye, W.; Dunn, J.; Westenberger, B.J.; Buhse, L.F. Analysis of crude heparin by 1H-NMR, capillary electrophoresis, and strong-anion-exchange-HPLC for contamination by over sulfated chondroitin sulfate. J. Pharm. Biomed. Anal. 2010, 11, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Mauri, L.; Boccardi, G.; Torri, G.; Karfunkle, M.; Macchi, E.; Muzi, L.; Keire, D.; Guerrini, M. Qualification of HSQC methods for quantitative composition of heparin and low molecular weight heparins. J. Pharm. Biomed. Anal. 2017, 136, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Casu, B.; Hamer, G.K.; Perlin, A.S. Studies on the conformation of heparin by 1H and 13C NMR spectroscopy. Macromolecules 1979, 12, 1001–1007. [Google Scholar] [CrossRef]
- Nguyen, K.; Rabenstein, D.L. Determination of the primary structure and carboxyl pKAs of heparin-derived oligosaccharides by band-selective homonuclear-decoupled two-dimensional 1H-NMR. Anal. Bioanal. Chem. 2011, 399, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T. Principal Component Analysis, 2nd ed.; Bickel, P., Diggle, P., Fienberg, S., Krickeberg, K., Olkin, I., Wermuth, N., Zeger, S., Eds.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Keire, D.A.; Mans, D.J.; Ye, H.; Kolinski, R.E.; Buhse, L.F. Assay of possible economically motivated additives or native impurities levels in heparin by 1H-NMR, SAX-HPLC, and anticoagulation time approaches. J. Pharm. Biomed. Anal. 2010, 52, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Naggi, A.; Guglieri, S.; Santarsiero, R.; Torri, G. Complex glycosaminoglycans: Profiling substitution patterns by two dimensional NMR spectroscopy. Anal. Biochem. 2005, 337, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Guglieri, S.; Naggi, A.; Torri, G. LMWHs: Structural differentiation by bidimensional NMR spectroscopy. Semin. Thromb. Hemost. 2007, 33, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Keire, D.; Buhse, L.F.; al-Hakim, A. Characterization of currently marketed heparin products: Composition analysis by 2D-NMR. Anal. Methods 2013, 5, 2984–2994. [Google Scholar] [CrossRef]
- Mascellani, G.; Liverani, L.; Prete, A.; Bergonzini, G.; Bianchini, P.; Torri, G.; Guerrini, M.; Casu, B. Quantitation of dermatan sulfate active site for the heparin cofactor II by 1H Nuclear Magnetic Resonance spectroscopy. Anal. Biochem. 1994, 223, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Mucci, A.; Schenetti, L.; Volpi, N. 1H and 13C nuclear magnetic resonance identification and characterization of components of chondroitin sulfates of various origin. Carbohydr. Polym. 2000, 41, 37–45. [Google Scholar] [CrossRef]
- Mourier, P.; Anger, P.; Martinez, C.; Herman, F.; Viskov, C. Quantitative compositional analysis of heparin using exhaustive heparinase digestion and strong anion exchange chromatography. Anal. Chem. Res. 2015, 3, 46–53. [Google Scholar] [CrossRef]
- Casu, B.; Guerrini, M.; Naggi, A.; Torri, G.; De-Ambrosi, L.; Boveri, G.; Gonella, S. Differentiation of beef and pig mucosal heparins by NMR spectroscopy. Thromb. Haemost. 1995, 74, 1205. [Google Scholar] [PubMed]
- Watt, D.K.; Yorke, S.C.; Slimb, G.C. Comparison of ovine, bovine and porcine mucosal heparins and low molecular weight heparins by disaccharide analyses and 13C NMR. Carbohydr. Polym. 1997, 33, 5–11. [Google Scholar] [CrossRef]
- Tovar, A.M.F.; Teixeira, A.C.; Rembold, S.M.; Leite, M., Jr.; Lugon, J.R.; Mourão, P.A.S. Bovine and porcine heparins: Different drugs with similar effects on human haemodialysis. BMC Res. Notes 2013, 6, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Li, G.; Yang, B.; Onishi, A.; Li, L.; Sun, P.; Zhang, F.; Linhardt, R.J. Structural Characterization of Pharmaceutical Heparins Prepared from Different Animal Tissues. J. Pharm. Sci. 2013, 102, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Naggi, A.; Gardini, C.; Pedrinola, P.; Mauri, L.; Urso, E.; Alekseeva, A.; Casu, B.; Cassinelli, G.; Guerrini, M.; Iacomini, M.; et al. Structural peculiarity andantithrombin binding region profile of mucosal bovine and porcine heparins. J. Pharm. Bioanal. Anal. 2016, 118, 52–63. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Crude (mg/mL) | Stirring | Supernatant (mg) | Precipitate (mg) | Weight loss % (%w) | Precipitate (%w) |
|---|---|---|---|---|---|---|
| G9709 | 40.4 | No | 35.3 | 1.2 | 9.7 | 3.3 |
| G9709 | 40.8 | No | 35.4 | 1.0 | 10.8 | 2.7 |
| G9709 | 38.9 | No | 33.7 | 1.2 | 10.3 | 3.4 |
| G9709 | 39.0 | Yes | 34.0 | 1.1 | 10.0 | 3.1 |
| G9709 | 79.7 | No | 60.4 | 10.0 | 11.7 | 14.2 |
| G9709 | 81.4 | Yes | 62.4 | 9.4 | 11.8 | 13.1 |
| G9710 | 39.0 | No | 33.8 | 2.7 | 6.4 | 7.4 |
| G9710 | 39.3 | Yes | 33.2 | 2.1 | 10.2 | 5.9 |
| G9710 | 79.5 | No | 56.8 | 13.8 | 11.2 | 19.5 |
| G9710 | 80.5 | Yes | 59.3 | 13.0 | 10.2 | 18.0 |
| PMHC | Hep | DeS | ChS | ANH26X | ANAc6X | ANS3S6X | A6S | G2OH | G2S | I2OH | I2S | SDEG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| average | 89.3 | 8.6 | 2.4 | 1.8 | 16.3 | 4.5 | 73.3 | 18.0 | 0.01 | 10.5 | 71.5 | 2.31 |
| median | 89.2 | 8.7 | 2.1 | 1.9 | 16.3 | 4.4 | 73.7 | 17.9 | 0.00 | 10.6 | 71.4 | 2.32 |
| st.dev. | 4.1 | 3.1 | 1.9 | 0.7 | 2.0 | 0.7 | 3.4 | 1.9 | 0.09 | 0.9 | 2.5 | 0.08 |
| min | 79.9 | 0.0 | 0.0 | 0.0 | 11.6 | 3.1 | 63.3 | 14.0 | 0.00 | 8.2 | 65.4 | 2.13 |
| max | 100.0 | 16.6 | 7.7 | 3.0 | 21.1 | 5.9 | 79.8 | 22.4 | 0.75 | 12.4 | 76.8 | 2.46 |
| BMHC | Hep | DeS | ChS | ANH26X | ANAc6X | ANS3S6X | A6S | G2OH | G2S | I2OH | I2S | SDEG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| average | 94.7 | 4.2 | 0.4 | 1.5 | 10.6 | 2.0 | 51.7 | 14.1 | 1.5 | 6.8 | 77.1 | 2.20 |
| median | 98.1 | 1.1 | 0.0 | 1.9 | 9.6 | 1.8 | 52.4 | 13.8 | 2.0 | 6.3 | 76.9 | 2.19 |
| st.dev. | 5.9 | 5.8 | 0.9 | 1.2 | 3.3 | 0.7 | 4.0 | 2.0 | 1.2 | 1.2 | 2.8 | 0.07 |
| min | 85.3 | 0.0 | 0.0 | 0.0 | 8.2 | 1.3 | 47.1 | 11.9 | 0.0 | 6.0 | 73.4 | 2.12 |
| max | 98.8 | 12.5 | 2.1 | 2.6 | 17.2 | 3.5 | 57.2 | 17.4 | 2.5 | 9.2 | 81.0 | 2.31 |
| BLHC | Hep | DeS | ChS | ANH26X | ANAc6X | ANS3S6X | A6S | G2OH | G2S | I2OH | I2S | SDEG |
| Sample 1 | 94.7 | 4.2 | 0.4 | 1.9 | 6.6 | 2.8 | 85.8 | 7.8 | 0.0 | 4.6 | 87.6 | 2.68 |
| Sample 2 | 98.1 | 1.1 | 0.0 | 0.0 | 1.6 | 2.5 | 90.2 | 2.3 | 0.5 | 2.8 | 93.4 | 2.85 |
| OMHC | Hep | DeS | ChS | ANH26X | ANAc6X | ANS3S6X | A6S | G2OH | G2S | I2OH | I2S | SDEG |
| Sample 1 | 98.9 | 1.1 | 0.0 | 1.5 | 8.8 | 5.0 | 80.5 | 10.2 | 0.0 | 7.0 | 82.8 | 2.58 |
| Sample 2 | 97.7 | 2.3 | 0.0 | 1.8 | 8.8 | 4.7 | 82.2 | 10.5 | 0.0 | 7.2 | 82.3 | 2.59 |
| Sample 3 | 98.7 | 1.3 | 0.0 | 1.6 | 7.8 | 5.3 | 82.2 | 9.7 | 0.0 | 7.0 | 83.4 | 2.61 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauri, L.; Marinozzi, M.; Mazzini, G.; Kolinski, R.E.; Karfunkle, M.; Keire, D.A.; Guerrini, M. Combining NMR Spectroscopy and Chemometrics to Monitor Structural Features of Crude Hep-arin. Molecules 2017, 22, 1146. https://doi.org/10.3390/molecules22071146
Mauri L, Marinozzi M, Mazzini G, Kolinski RE, Karfunkle M, Keire DA, Guerrini M. Combining NMR Spectroscopy and Chemometrics to Monitor Structural Features of Crude Hep-arin. Molecules. 2017; 22(7):1146. https://doi.org/10.3390/molecules22071146
Chicago/Turabian StyleMauri, Lucio, Maria Marinozzi, Giulia Mazzini, Richard E. Kolinski, Michael Karfunkle, David A. Keire, and Marco Guerrini. 2017. "Combining NMR Spectroscopy and Chemometrics to Monitor Structural Features of Crude Hep-arin" Molecules 22, no. 7: 1146. https://doi.org/10.3390/molecules22071146
APA StyleMauri, L., Marinozzi, M., Mazzini, G., Kolinski, R. E., Karfunkle, M., Keire, D. A., & Guerrini, M. (2017). Combining NMR Spectroscopy and Chemometrics to Monitor Structural Features of Crude Hep-arin. Molecules, 22(7), 1146. https://doi.org/10.3390/molecules22071146