Metal-Mediated Addition of N-Nucleophiles to Isocyanides: Mechanistic Aspects



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Details
3. Results and Discussion
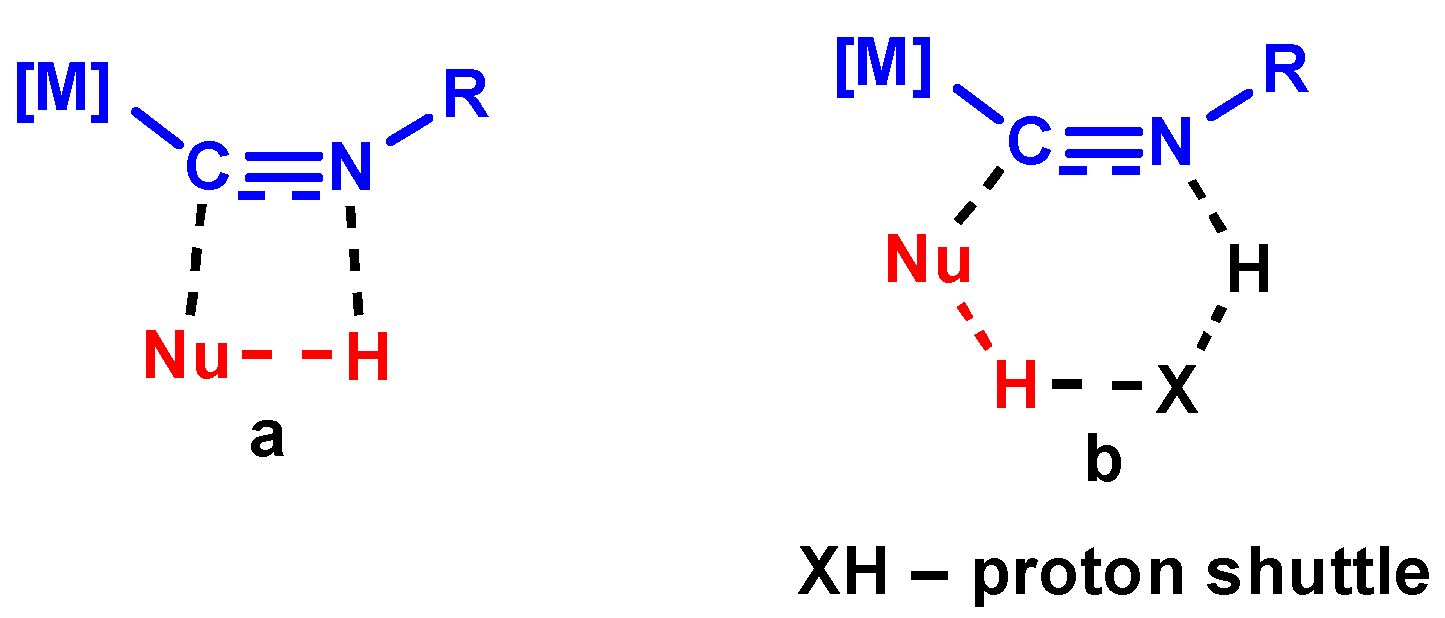
3.1. Mechanisms of NA to Isocyanides
3.2. Reaction of the sp3-N Nucleophiles. Comparison with the Experimental Kinetic Data
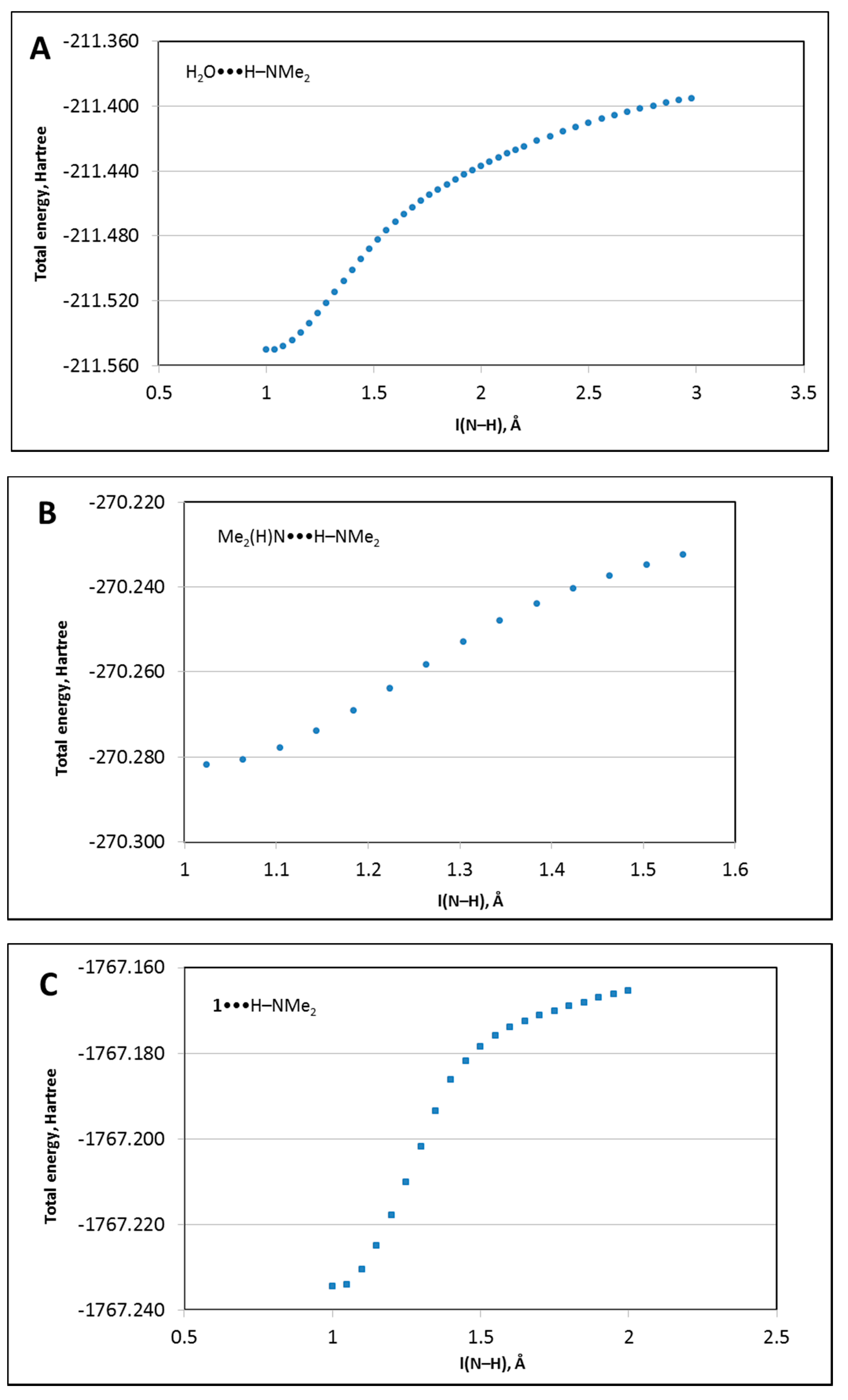
3.2.1. Concerted Mechanism
3.2.2. Dissociative Mechanism
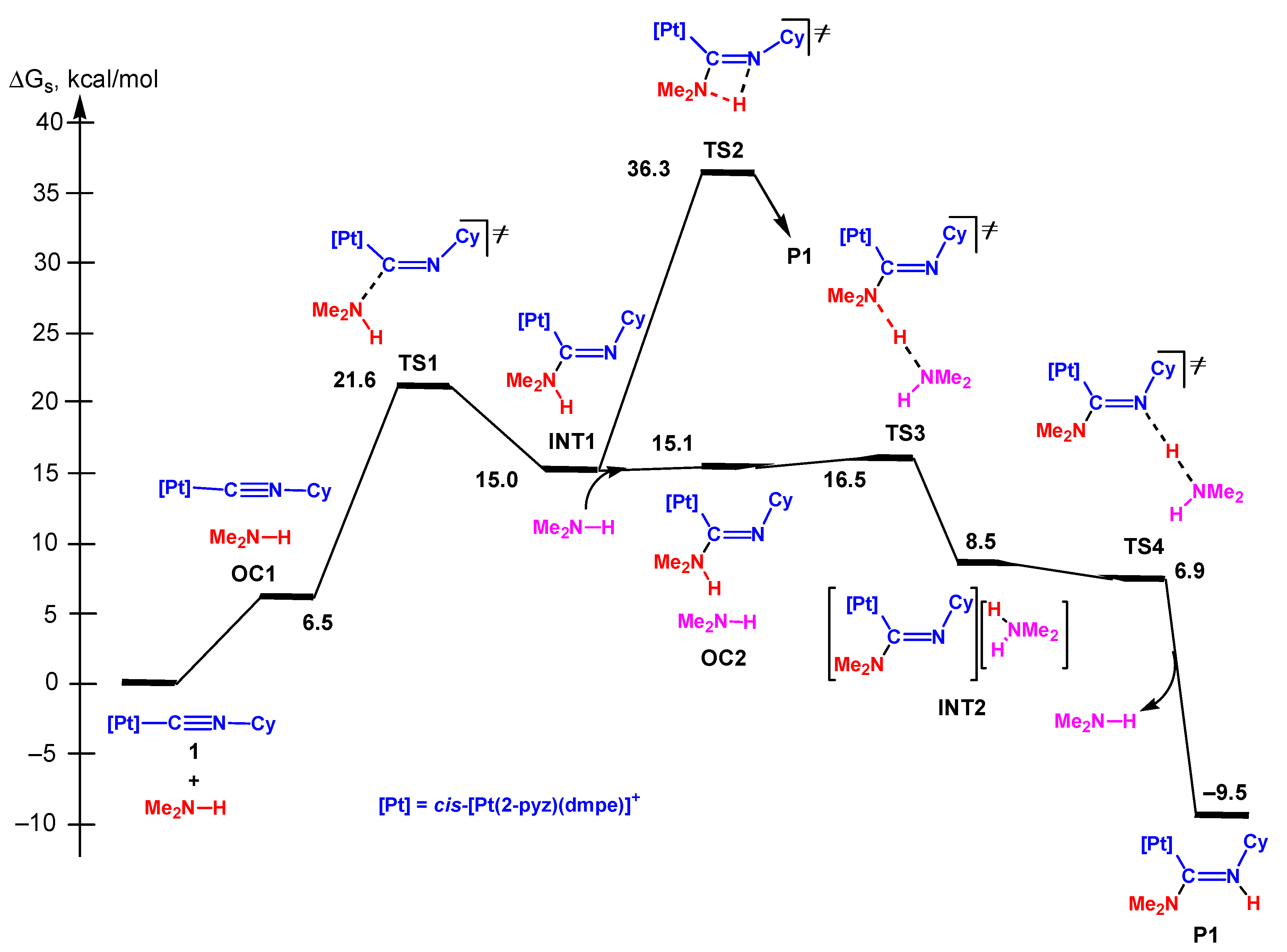
3.2.3. Associative Mechanism
3.3. Reactions of the sp2-N and sp2/sp3-N Nucleophiles
3.3.1. Concerted and Dissociative Mechanisms
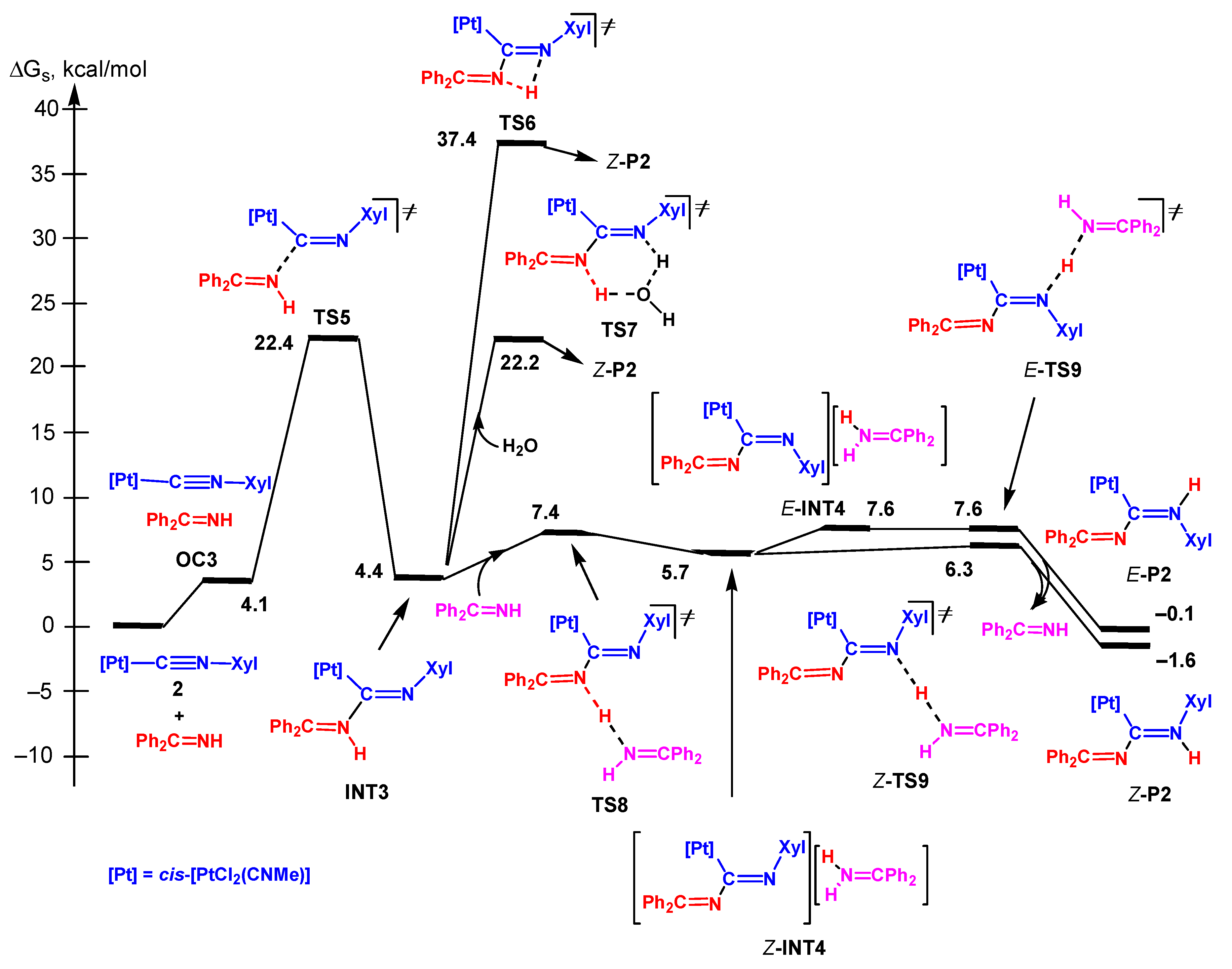
3.3.2 Associative Mechanism
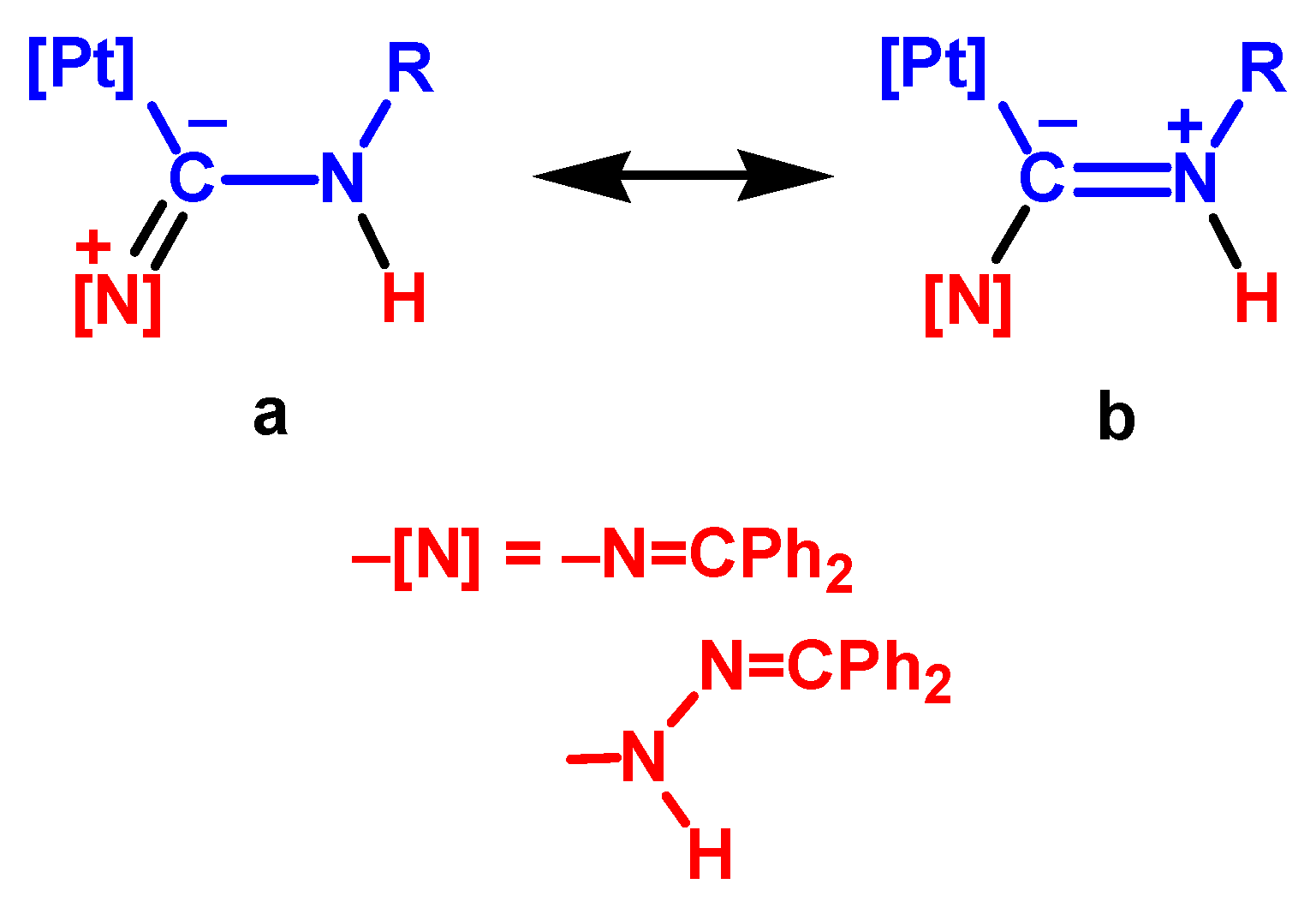
3.3.3. Nucleophilic Addition by the Imino N Atom of Hydrazone
3.3.4. Mechanism Involving the Dipole Tautomeric form of Hydrazone
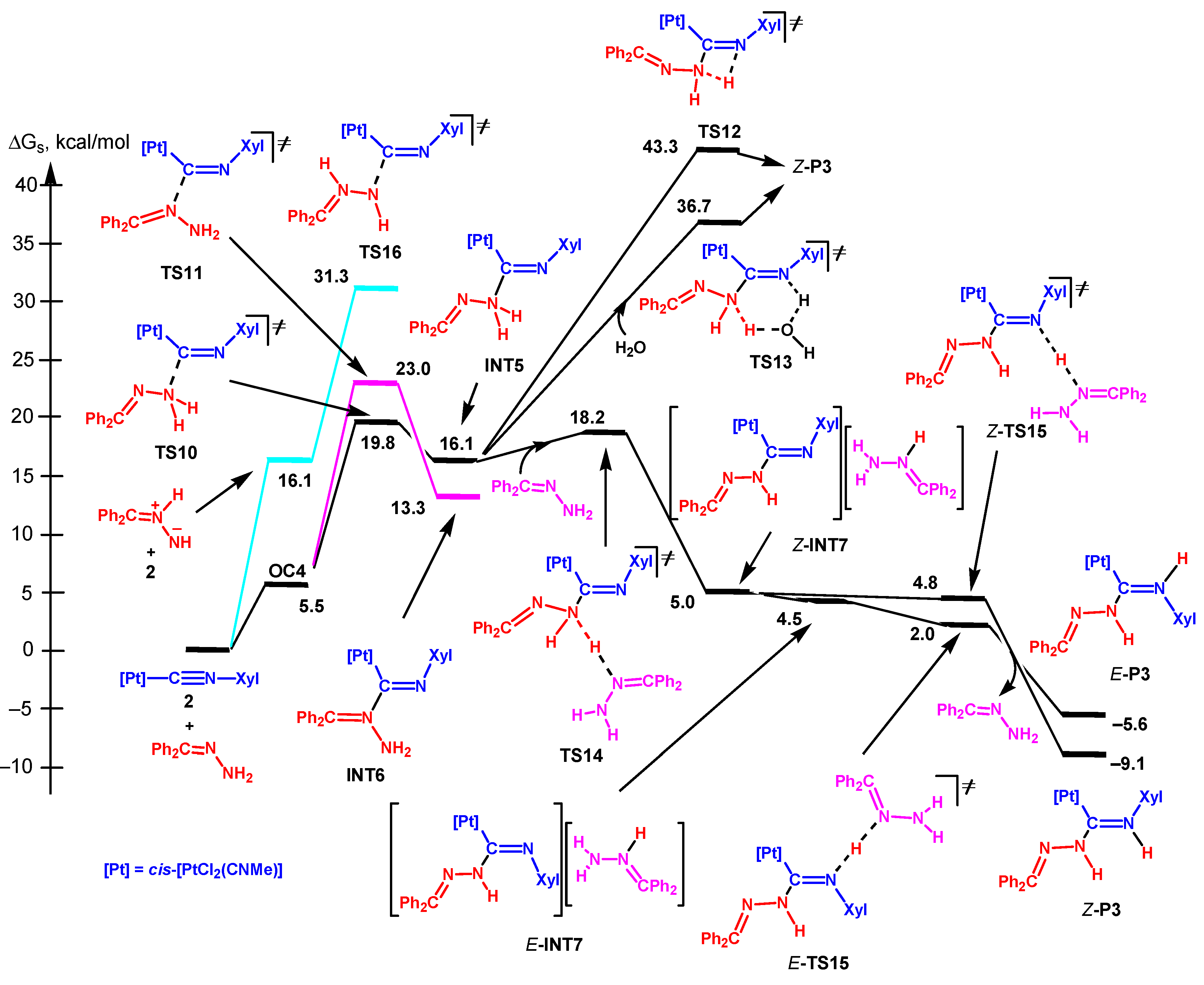
3.3.5. Analysis of the Energy Profiles
4. Final Remarks
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Yu. Metal-mediated and metal-catalyzed reactions of isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef] [PubMed]
- Gulevich, A.V.; Zhdanko, A.G.; Orru, R.V.A.; Nenajdenko, V.G. Isocyanoacetate derivatives: Synthesis, reactivity, and application. Chem. Rev. 2010, 110, 5235–5331. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef] [PubMed]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.P. Recent developments in the isonitrile-based multicomponent synthesis of heterocycles. Eur. J. Org. Chem. 2003, 7, 1133–1144. [Google Scholar] [CrossRef]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef] [PubMed]
- Hulme, C.; Lee, Y.-S. Emerging approaches for the syntheses of bicyclic imidazo[1,2-x]-heterocycles. Mol. Divers. 2008, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- El Kaim, L.; Grimaud, L. Beyond the Ugi reaction: Less conventional interactions between isocyanides and iminium species. Tetrahedron 2009, 65, 2153–2171. [Google Scholar] [CrossRef]
- Sadjadi, S.; Heravi, M.M. Recent application of isocyanides in synthesis of heterocycles. Tetrahedron 2011, 67, 2707–2752. [Google Scholar] [CrossRef]
- Heravi, M.M.; Moghimi, S. Catalytic multicomponent reactions based on isocyanides. J. Iran. Chem. Soc. 2011, 8, 306–373. [Google Scholar] [CrossRef]
- van Berkel, S.S.; Bogels, B.G.M.; Wijdeven, M.A.; Westermann, B.; Rutjes, F.P.J.T. Recent advances in asymmetric isocyanide-based multicomponent reactions. Eur. J. Org. Chem. 2012, 19, 3543–3559. [Google Scholar] [CrossRef]
- Ramon, R.; Kielland, N.; Lavilla, R. Recent Progress in Nonclassical Isocyanide-Based MCRs; Wiley-VCH Verlag GmbH & Co., KGaA.: New York, NY, USA, 2012; p. 299. [Google Scholar]
- Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 2013, 42, 4867–4880. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Ding, Q.; Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef] [PubMed]
- Suginome, M.; Ito, Y. Transition metal-mediated polymerization of isocyanides. Adv. Polym. Sci. 2004, 171, 77–136. [Google Scholar] [CrossRef]
- Gómez, M. (Alkyl)(monocyclopentadienyl)niobium and -tantalum complexes in insertion processes. Eur. J. Inorg. Chem. 2003, 20, 3681–3697. [Google Scholar] [CrossRef]
- Antiñolo, A.; Garcıá-Yuste, S.; Otero, A.; Villaseñor, E. On the insertion processes of unsaturated molecules into the Nb–X σ-bond of “Cp′2NbX” moieties (Cp′ = η5-C5H4SiMe3; X = H, C, P). J. Organomet. Chem. 2007, 692, 4436–4447. [Google Scholar] [CrossRef]
- Lauzon, J.M.P.; Schafer, L.L. Tantallaaziridines: From synthesis to catalytic applications. Dalton Trans. 2012, 41, 11539–11550. [Google Scholar] [CrossRef] [PubMed]
- Boyarskiy, V.P.; Luzyanin, K.V.; Kukushkin, V.Yu. Acyclic diaminocarbenes (ADCs) as a promising alternative to N-heterocyclic carbenes (NHCs) in transition metal catalyzed organic transformations. Coord. Chem. Rev. 2012, 256, 2029–2056. [Google Scholar] [CrossRef]
- Ito, H.; Kato, T.; Sawamura, M. Design and synthesis of isocyanide ligands for catalysis: Application to Rh-catalyzed hydrosilylation of ketones. Chem. Asian J. 2007, 2, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, X.; Shao, J.; Yang, G.; Wu, Y.; Zhang, Z. Hydration of alkynes at room temperature catalyzed by gold(I) isocyanide compounds. Green Chem. 2015, 17, 532–537. [Google Scholar] [CrossRef]
- Liu, M.; Reiser, O. A copper(I) isonitrile complex as a heterogeneous catalyst for azide–alkyne cycloaddition in water. Org. Lett. 2011, 13, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Mohan, R.; Singh, J.K.; Samantaray, M.K.; Shaikh, M.M.; Panda, D.; Ghosh, P. Anticancer and antimicrobial metallopharmaceutical agents based on palladium, gold, and silver N-heterocyclic carbene complexes. J. Am. Chem. Soc. 2007, 129, 15042–15053. [Google Scholar] [CrossRef] [PubMed]
- Barnard, P.J.; Wedlock, L.E.; Baker, M.V.; Berners-Price, S.J.; Joyce, D.A.; Skelton, B.W.; Steer, J.H. Luminescence studies of the intracellular distribution of a dinuclear gold(I) N-heterocyclic carbene complex. Angew. Chem. Int. Ed. 2006, 45, 5966–5970. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P.; Navarro, O. C–C bond formation by cross-coupling. In Comprehensive Organometallic Chemistry III, 1st ed.; Canty, A., Ed.; Elsevier: Oxford, UK, 2007; Volume 11, pp. 1–38. [Google Scholar]
- Marion, N.; Nolan, S.P. Well-defined N-heterocyclic carbenes−palladium(II) precatalysts for cross-coupling reactions. Acc. Chem. Res. 2008, 41, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Tskhovrebov, A.G.; Luzyanin, K.V.; Kuznetsov, M.L.; Sorokoumov, V.N.; Balova, I.A.; Haukka, M.; Kukushkin, V.Yu. Substituent R-dependent regioselectivity switch in nucleophilic addition of N-phenylbenzamidine to PdII- and PtII-complexed isonitrile RN≡C giving aminocarbene-like species. Organometallics 2011, 30, 863–874. [Google Scholar] [CrossRef]
- Díez-González, S.; Marion, N.; Nolan, S.P. N-heterocyclic carbenes in late transition metal catalysis. Chem. Rev. 2009, 109, 3612–3676. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, L.M. Acyclic aminocarbenes in catalysis. ACS Catal. 2012, 2, 1802–1816. [Google Scholar] [CrossRef]
- Kremzow, D.; Seidel, G.; Lehmann, C.W.; Furstner, A. diaminocarbene- and Fischer-carbene complexes of palladium and nickel by oxidative insertion: Preparation, structure, and catalytic activity. Chem. Eur. J. 2005, 11, 1833–1853. [Google Scholar] [CrossRef] [PubMed]
- Dhudshia, B.; Thadani, A.N. Acyclic diaminocarbenes: Simple, versatile ligands for cross-coupling reactions. Chem. Commun. 2006, 6, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Luzyanin, K.V.; Tskhovrebov, A.G.; Carias, M.C.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L.; Kukushkin, V.Yu. Novel metal-mediated (M = Pd, Pt) coupling between isonitriles and benzophenone hydrazone as a route to aminocarbene complexes exhibiting high catalytic activity (M = Pd) in the Suzuki−Miyaura reaction. Organometallics 2009, 28, 6559–6566. [Google Scholar] [CrossRef]
- Slaughter, L. M. Covalent self-assembly of acyclic diaminocarbene ligands at metal centers. Commun. Inorg. Chem. 2008, 29, 46–72. [Google Scholar] [CrossRef]
- Moncada, A.I.; Manne, S.; Tanski, J.M.; Slaughter, L.M. Modular chelated palladium diaminocarbene complexes: Synthesis, characterization, and optimization of catalytic Suzuki−Miyaura cross-coupling activity by ligand modification. Organometallics 2006, 25, 491–505. [Google Scholar] [CrossRef]
- Bartolomé, C.; Ramiro, Z.; García-Cuadrado, D.; Pérez-Galán, P.; Raducan, M.; Bour, C.; Echavarren, A. M.; Espinet, P. Nitrogen acyclic gold(I) carbenes: Excellent and easily accessible catalysts in reactions of 1,6-enynes. Organometallics 2010, 29, 951–956. [Google Scholar] [CrossRef]
- Bartolomé, C.; García-Cuadrado, D.; Ramiro, Z.; Espinet, P. Synthesis and catalytic activity of gold chiral nitrogen acyclic aarbenes and gold hydrogen bonded heterocyclic carbenes in cyclopropanation of vinyl arenes and in intramolecular hydroalkoxylation of allenes. Inorg. Chem. 2010, 49, 9758–9764. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Hengst, T.; Lothschütz, C.; Rominger, F. New and easily accessible nitrogen acyclic gold(I) carbenes: Structure and application in the gold-catalyzed phenol synthesis as well as the hydration of alkynes. Adv. Synth. Catal. 2010, 352, 1315–1337. [Google Scholar] [CrossRef]
- Seo, H.; Snead, D.R.; Abboud, K.A.; Hong, S. Bulky acyclic aminooxycarbene ligands. Organometallics 2011, 30, 5725–5730. [Google Scholar] [CrossRef]
- Cavarzan, A.; Scarso, A.; Sgarbossa, P.; Strukul, G.; Reek, J.N.H. Supramolecular control on chemo- and regioselectivity via encapsulation of (NHC)-Au catalyst within a hexameric self-assembled host. J. Am. Chem. Soc. 2011, 133, 2848–2851. [Google Scholar] [CrossRef] [PubMed]
- Catalano, V.J.; Etogo, A.O. Preparation of Au(I), Ag(I), and Pd(II) N-heterocyclic carbene complexes utilizing a methylpyridyl-substituted NHC ligand. Formation of a luminescent coordination polymer. Inorg. Chem. 2007, 46, 5608–5615. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, C.; Carrasco-Rando, M.; Coco, S.; Cordovilla, C.; Martín-Alvarez, J.M.; Espinet, P. Luminescent gold(I) carbenes from 2-pyridylisocyanide complexes: Structural consequences of intramolecular versus intermolecular hydrogen-bonding interactions. Inorg. Chem. 2008, 47, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.-M.; Chan, M.C.-W.; Su, Z.-M.; Cheung, K.-K.; Liu, S.-T.; Che, C.-M. Spectroscopic and excited-state properties of luminescent rhenium(I) N-heterocyclic carbene complexes containing aromatic diimine ligands. Organometallics 1998, 17, 1622–1630. [Google Scholar] [CrossRef]
- Lai, S.-W.; Chan, M.C.-W.; Cheung, K.-K.; Che, C.-M. Carbene and isocyanide ligation at luminescent cyclometalated 6-phenyl-2,2‘-bipyridyl platinum(II) complexes: Structural and spectroscopic studies. Organometallics 1999, 18, 3327–3336. [Google Scholar] [CrossRef]
- Tschugajeff, L.; Skanawy-Grigorjewa, M. Reaction of K2PtCl4 with isonitriles and hydrazine. J. Russ. Chem. Soc. 1915, 47, 776. [Google Scholar]
- Tschugajeff, L.; Grigorjewa, M.; Posnjak, A. Über die hydrazin-carbilamin-komplexe des platins. Z. Anorg. Allg. Chem. 1925, 148, 37–42. [Google Scholar] [CrossRef]
- Rouschias, G.; Shaw, B.L. The chemistry and structure of Chugaev’s salt and related compounds Containing a cyclic carbene ligand. J. Chem. Soc. A 1971, 2097–2104. [Google Scholar] [CrossRef]
- Burke, A.; Balch, A.L.; Enemark, J.H. Palladium and platinum complex resulting from the addition of hydrazine to coordinated isocyanide. J. Am. Chem. Soc. 1970, 92, 2555–2557. [Google Scholar] [CrossRef]
- Butler, W.M.; Enemark, J.H.; Parks, J.; Balch, A.L. Chelative addition of hydrazines to coordinated isocyanides. Structure of Chugaev’s red salt. Inorg. Chem. 1973, 12, 451–457. [Google Scholar] [CrossRef]
- Badley, M.; Chatt, J.; Richards, R.L. Isonitrile complexes of platinum(II) and their reactions with alcohols and amines to give carbene complexes. J. Chem. Soc. A 1971, 21–25. [Google Scholar] [CrossRef]
- Fehlhammer, W.P.; Bartel, K.; Metzner, R.; Beck, W. Reactions of cationic isocyanide platinum(II) complexes with water (from hexafluoroacetone-hydrate): Carboxamido and isocyanato complexes. Z. Anorg. Allg. Chem. 2008, 634, 1002–1004. [Google Scholar] [CrossRef]
- Crociani, B.; Boschi, T.; Belluco, U. Synthesis and reactivity of novel palladium(II)-isocyanide complexes. Inorg. Chem. 1970, 9, 2021–2025. [Google Scholar] [CrossRef]
- Michelin, R.A.; Zanotto, L.; Braga, D.; Sabatino, P.; Angelici, R.J. Transition-metal-promoted cyclization reactions of isocyanide ligands. Synthesis of cyclic diaminocarbenes from isocyanide complexes of palladium(II) and platinum(II) and X-ray structure of cis-Br2Pt[CN(C6H4-p-Me)CH2CH2N(H)](PPh3). Inorg. Chem. 1988, 27, 93–99. [Google Scholar] [CrossRef]
- Yu, I.; Wallis, C.J.; Patrick, B.O.; Diaconescu, P.L.; Mehrkhodavandi, P. Phosphine-tethered carbene ligands: Template synthesis and reactivity of cyclic and acyclic functionalized carbenes. Organometallics 2010, 29, 6065–6076. [Google Scholar] [CrossRef]
- Arias, J.; Bardají, M.; Espinet, P. Luminescence and mesogenic properties in crown-ether-isocyanide or carbene gold(I) complexes: Luminescence in solution, in the solid, in the mesophase, and in the isotropic liquid state. Inorg. Chem. 2008, 47, 3559–3567. [Google Scholar] [CrossRef] [PubMed]
- Crociani, B.; Uguagliati, P.; Belluco, U. Steric role of aromatic ring ortho-substituents in the mechanism of carbene formation from palladium(II) arylisocyanide complexes and anilines. J. Organomet. Chem. 1976, 117, 189–199. [Google Scholar] [CrossRef]
- Lazar, M.; Zhu, B.; Angelici, R.J. Non-nanogold catalysis of reactions of isocyanides, secondary amines, and oxygen to give ureas. J. Phys. Chem. C 2007, 111, 4074–4076. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Levi, C.; Bertolasi, V. Synthesis and mechanism of formation of novel NHC−NAC bis-carbene complexes of gold(I). Organometallics, 2011, 30, 875–883. [Google Scholar] [CrossRef]
- Ruiz, J.; García, L.; Mejuto, C.; Vivanco, M.; Díaz, M.R.; García-Granda, S. Strong electron-donating metalla-N-heterocyclic carbenes. Chem. Commun. 2014, 50, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Anding, B.J.; Ellern, A.; Woo, L.K. Comparative study of rhodium and iridium porphyrin diaminocarbene and N-heterocyclic carbene complexes. Organometallics 2014, 33, 2219–2229. [Google Scholar] [CrossRef]
- Vicente, J.; Chicote, M.T.; Huertas, S.; Jones, P.G. 1,1-Ethylenedithiolato complexes of palladium(II) and platinum(II) with isocyanide and carbene ligands. Inorg. Chem. 2003, 42, 4268–4274. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huynh, H.V. Mixed carbene–isocyanide Pd(II) complexes: Synthesis, structures and reactivity towards nucleophiles. Dalton Trans. 2009, 12, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Moncada, A.I.; Tanski, J.M.; Slaughter, L.M. Sterically Controlled formation of monodentate versus chelating carbene ligands from phenylhydrazine. J. Organomet. Chem. 2005, 690, 6247–6251. [Google Scholar] [CrossRef]
- Martínez-Martínez, A.-J.; Chicote, M.-T.; Bautista, D.; Vicente, J. Synthesis of palladium(II), -(III), and -(IV) complexes with acyclic diaminocarbene ligands. Organometallics 2012, 31, 3711–3719. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Böhling, C.; Lothschütz, C.; Rominger, F. From isonitriles to carbenes: Synthesis of new NAC− and NHC−palladium(II) compounds and their catalytic activity. Organometallics 2011, 30, 2411–2417. [Google Scholar] [CrossRef]
- Heathcote, R.; Howell, J.A.S.; Jennings, N.; Cartlidge, D.; Cobden, L.; Coles, S.; Hursthouse, M. Gold(I)–isocyanide and gold(I)–carbene complexes as substrates for the laser decoration of gold onto ceramic surfaces. Dalton Trans. 2007, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, C.; Carrasco-Rando, M.; Coco, S.; Cordovilla, C.; Espinet, P.; Martín-Alvarez, J.M. Gold(I)–carbenes Derived from 4-Pyridylisocyanidecomplexes: Supramolecular macrocycles supported by hydrogen bonds, and luminescent behavior. Dalton Trans. 2007, 45, 5339–5345. [Google Scholar] [CrossRef]
- Handa, S.; Slaughter, L.M. Enantioselective alkynylbenzaldehyde cyclizations catalyzed by chiral gold(I) acyclic fiaminocarbene complexes containing weak Au–arene interactions. Angew. Chem. Int. Ed. 2012, 51, 2912–2915. [Google Scholar] [CrossRef] [PubMed]
- Crespo, O.; Gimeno, M.C.; Laguna, A.; Montanel-Pérez, S.; Villacampa, M.D. Facile synthesis of gold(III) aryl–carbene metallacycles. Organometallics 2012, 31, 5520–5526. [Google Scholar] [CrossRef]
- Bertani, R.; Mozzon, M.; Michelin, R.A. Reactions of aziridine, thiirane, and oxirane with isocyanide ligands in complexes of palladium(II) and platinum(II): Syntheses of neutral five-membered cyclic diamino-, aminothio-, and aminooxycarbene compounds. Inorg. Chem. 1988, 27, 2809–2815. [Google Scholar] [CrossRef]
- Uguagliati, P.; Crociani, B.; Belluco, U.; Calligaro, L. Solvent, ligand and temperature effects on the rates of reactions of cis-[PdCl2(CNR)(L)] with secondary amines. J. Organomet. Chem. 1976, 112, 111–121. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, E.; Rust, J.; Alcarazo, M. Synthesis and reactivity of metal complexes with acyclic (amino) (ylide) carbene ligands. Angew. Chem. Int. Ed. 2013, 52, 11392–11395. [Google Scholar] [CrossRef] [PubMed]
- Viguri, M.E.; Huertos, M.A.; Perez, J.; Riera, L. Imidazole–nitrile or imidazole–isonitrile CC coupling on rhenium tricarbonyl complexes. Chem. Eur. J. 2013, 19, 12974–12977. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, O.; Stasch, A.; Pape, T.; Hepp, A.; Edwards, P.G.; Newman, P.D.; Hahn, F.E. Metal template controlled formation of [11]ane-P2CNHC macrocycles. J. Am. Chem. Soc. 2009, 131, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E.; Tamm, M.; Lügger, T. Equilibrium between isocyanide and carbene complexes in coordination compounds of 2,6-dihydroxyphenyl isocyanide. Angew. Chem. Int. Ed. Engl. 1994, 33, 1356–1359. [Google Scholar] [CrossRef]
- Menezes, F.M.C.; Kuznetsov, M.L.; Pombeiro, A.J.L. Isocyanide complexes with platinum and palladium and their reactivity toward cycloadditions with nitrones to form aminooxycarbenes: A theoretical study. Organometallics 2009, 28, 6593–6602. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L. Theoretical study of Re(IV) and Ru(II) bis-isocyanide complexes and their reactivity in cycloaddition reactions with nitrones. Inorg. Chim. Acta 2012, 380, 78–89. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L.; Pombeiro, A. J. L. Theory of the formation and decomposition of N-heterocyclic aminooxycarbenes through metal-assisted [2+3]-dipolar cycloaddition/retro-cycloaddition. Chem. Eur. J. 2013, 19, 2874–2888. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, M.L.; Kukushkin, V.Yu. Diversity of reactivity modes upon interplay between Au(III)-bound isocyanides and cyclic nitrones: A theoretical consideration. Dalton Trans. 2017, 46, 786–802. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.A. N-Heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Spallek, M.J.; Riedel, D.; Rominger, F.A.; Hashmi, A.S.K.; Trapp, O. Six-membered, chiral NHCs derived from camphor: Structure–reactivity relationship in asymmetric oxindole synthesis. Organometallics 2012, 31, 1127–1132. [Google Scholar] [CrossRef]
- Ruiz, J.; Perandones, B.F.; García, G.; Mosquera, M.E.G. Synthesis of N-heterocyclic carbene complexes of manganese(I) by coupling isocyanide ligands with propargylamines and propargylic alcohols. Organometallics 2007, 26, 5687–5695. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Lothschütz, C.; Graf, K.; Häffner, T.; Schuster, A.; Rominger, F. A short way to switchable carbenes. Adv. Synth. Catal. 2011, 353, 1407–1412. [Google Scholar] [CrossRef]
- Wanniarachchi, Y.A.; Slaughter, L.M. One-step assembly of a chiral palladium bis (acyclic diaminocarbene) complex and its unexpected oxidation to a bis (amidine) complex. Chem. Commun. 2007, 31, 3294–3296. [Google Scholar] [CrossRef] [PubMed]
- Wanniarachchi, Y.A.; Slaughter, L.M. Reversible chelate ring opening of a sterically crowded palladium bis(acyclic diaminocarbene) complex. Organometallics 2008, 27, 1055–1062. [Google Scholar] [CrossRef]
- Miltsov, S.; Karavan, V.; Boyarsky, V.; Gómez-de Pedro, S.; Alonso-Chamarro, J.; Puyol, M. New acyclic Pd–diaminocarbene catalyst for Suzuki arylation of meso-chlorosubstituted tricarboindocyanine dyes. Tetrahedron Lett. 2013, 54, 1202–1204. [Google Scholar] [CrossRef]
- Ryabukhin, D.S.; Sorokoumov, V.N.; Savicheva, E.A.; Boyarskiy, V.P.; Balova, I.A.; Vasilyev, A.V. Catalytic activity of palladium acyclic diaminocarbene complexes in the synthesis of 1,3-Diarylpropynones via Sonogashira reaction: Cross- versus homo-coupling. Tetrahedron Lett. 2013, 54, 2369–2372. [Google Scholar] [CrossRef]
- Balch, A.L.; Parks, J.E. Platinum and palladium complexes formed by chelative addition of amines to isocyanides. J. Am. Chem. Soc. 1974, 96, 4114–4121. [Google Scholar] [CrossRef]
- Zanella, R.; Boschi, T.; Crociani, B.; Belluco, U. Reactions of palladium(II) isocyanide complexes with bifunctional amines. J. Organomet. Chem. 1974, 71, 135–143. [Google Scholar] [CrossRef]
- Zanella, R.; Boschi, T.; Nicolini, M.; Belluco, U. The reactions of bis-isocyanide derivatives of PdII with bidentate ligands. J. Organomet. Chem. 1973, 49, C91–C94. [Google Scholar] [CrossRef]
- Michelin, R.A.; Pombeiro, A.J.L.; Guedes da Silva, M.F.C. Aminocarbene complexes derived from nucleophilic addition to isocyanide ligands. Coord. Chem. Rev. 2001, 218, 75–112. [Google Scholar] [CrossRef]
- Tamm, M.; Hahn, F.E. Reactions of β-functional phenyl isocyanides. Coord. Chem. Rev. 1999, 182, 175–209. [Google Scholar] [CrossRef]
- Crociani, B.; Di Bianca, F.; Fontana, A.; Forsellini, E.; Bombieri, G. Nucleophilic attack at Co-ordinated isocyanides promoted by the 2-pyridyl ligand. J. Chem. Soc. Dalton Trans. 1994, 4, 407–414. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Uguagliati, P.; Crociani, B.; Di Bianca, F. Mechanism of aminocarbene formation by nucleophilic attack on isocyanide ligands in platinum(II) 2-pyrazyl and 4-pyridyl complexes. J. Organomet. Chem. 1997, 535, 69–75. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Uguagliati, P.; Crociani, B. Mechanism of formation of (methoxy)(amino)—And bis(amino) carbene complexes by nucleophilic attack of methoxide ion and amines on platinum(II)-coordinated isocyanide in anhydrous methanol. J. Organomet. Chem. 1997, 543, 145–151. [Google Scholar] [CrossRef]
- Luzyanin, K.V.; Guedes da Silva, M.F.C.; Kukushkin, V.Y.; Pombeiro, A.J.L. First example of an imine addition to coordinated isonitrile. Inorg. Chim. Acta. 2009, 362, 833–838. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef] [PubMed]
- Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; Petersson, H.; et al. Gaussian 09; Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the Pople–Santry–Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Lasri, J.; Kuznetsov, M.L.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. PtII-mediated imine−nitrile coupling leading to symmetrical (1,3,5,7,9-pentaazanona-1,3,6,8-tetraenato)Pt(II) complexes containing the incorporated 1,3-diiminoisoindoline moiety. Inorg. Chem. 2012, 51, 10774–10786. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Avaiable. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsov, M.L.; Kukushkin, V.Y. Metal-Mediated Addition of N-Nucleophiles to Isocyanides: Mechanistic Aspects. Molecules 2017, 22, 1141. https://doi.org/10.3390/molecules22071141
Kuznetsov ML, Kukushkin VY. Metal-Mediated Addition of N-Nucleophiles to Isocyanides: Mechanistic Aspects. Molecules. 2017; 22(7):1141. https://doi.org/10.3390/molecules22071141
Chicago/Turabian StyleKuznetsov, Maxim L., and Vadim Yu. Kukushkin. 2017. "Metal-Mediated Addition of N-Nucleophiles to Isocyanides: Mechanistic Aspects" Molecules 22, no. 7: 1141. https://doi.org/10.3390/molecules22071141
APA StyleKuznetsov, M. L., & Kukushkin, V. Y. (2017). Metal-Mediated Addition of N-Nucleophiles to Isocyanides: Mechanistic Aspects. Molecules, 22(7), 1141. https://doi.org/10.3390/molecules22071141