Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments


Abstract
:1. Introduction
2. Canonical Nucleobases’ Thiated Analogs Photophysics
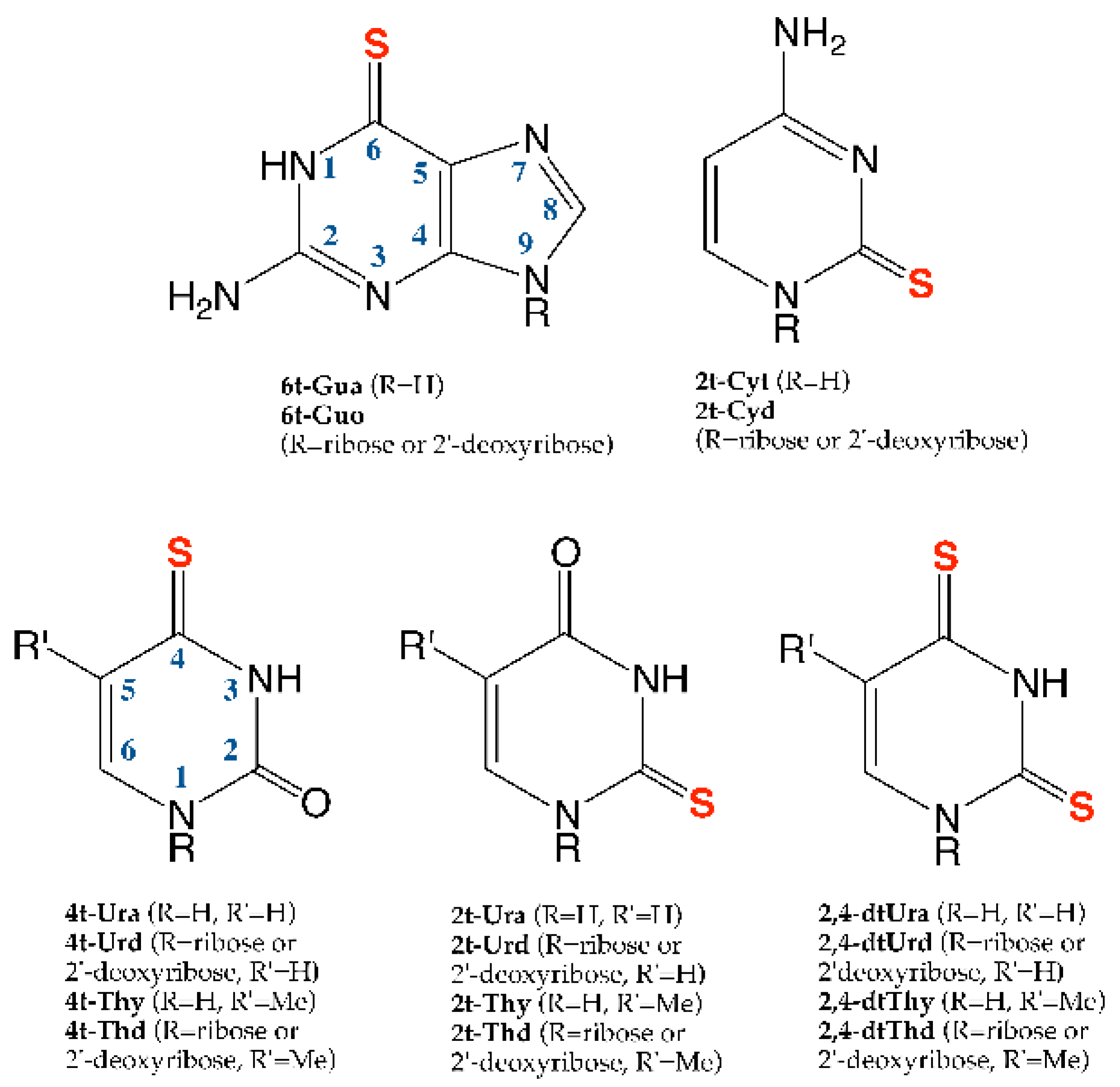
2.1. Thiopurines
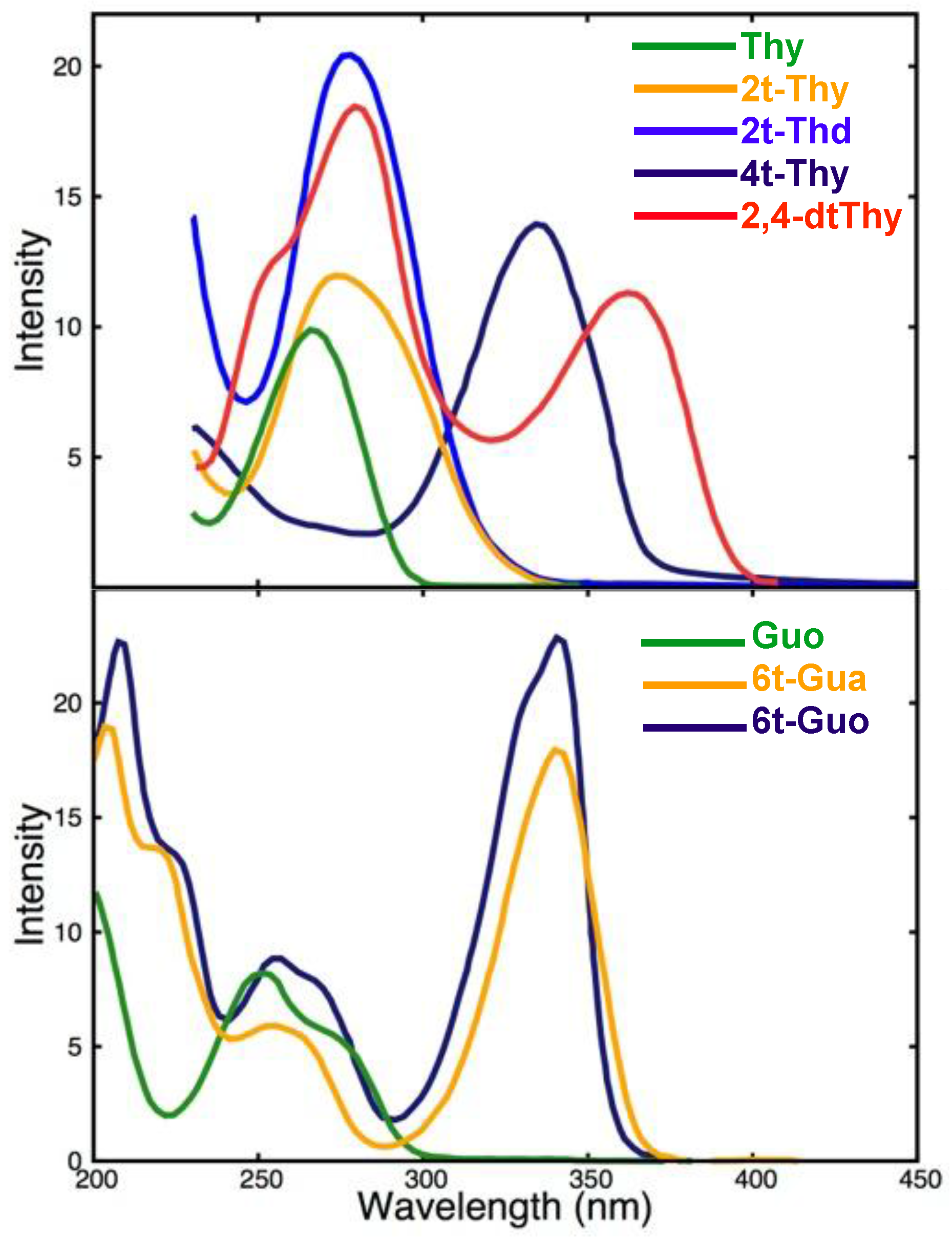
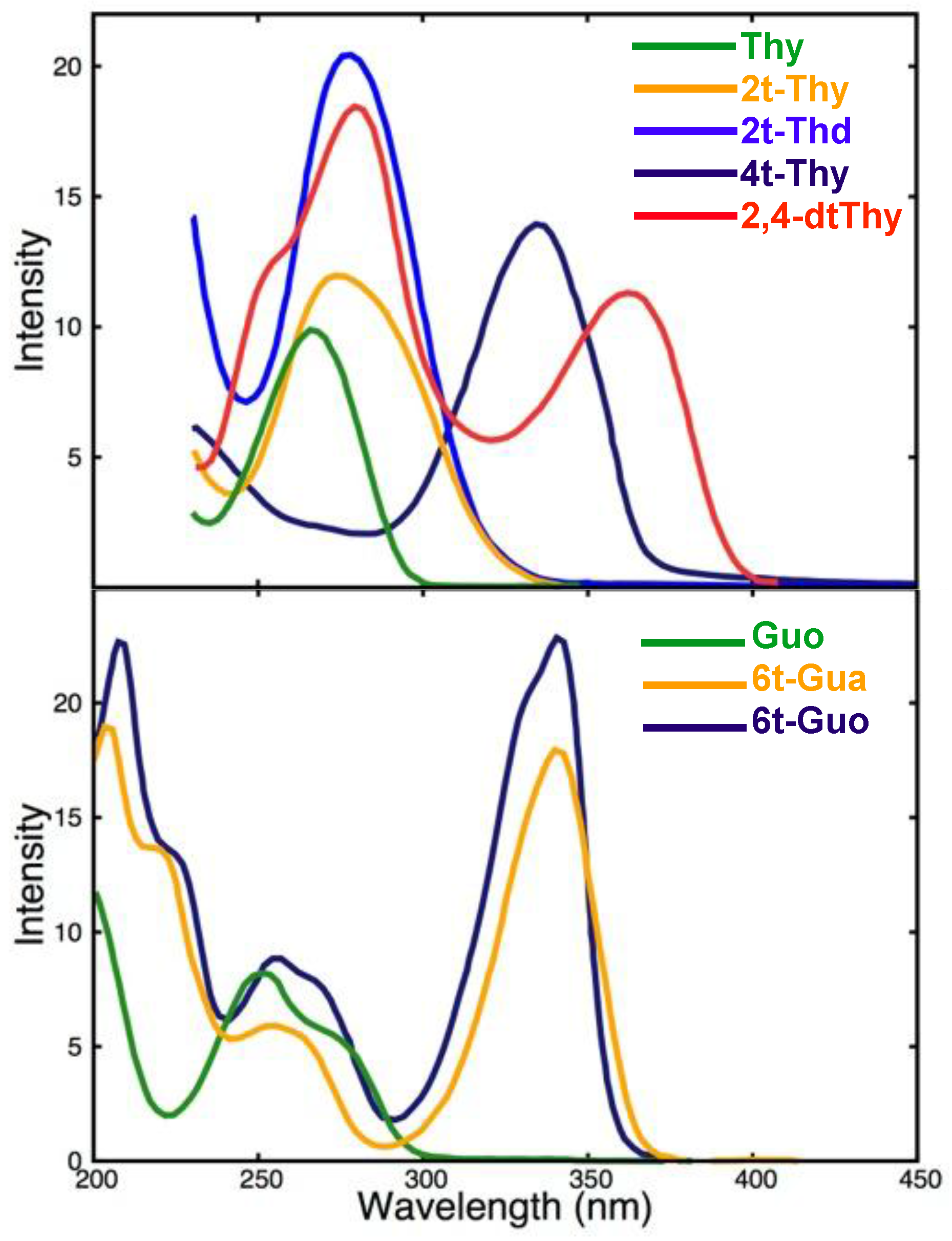
2.1.1. Steady State Absorption and Emission properties
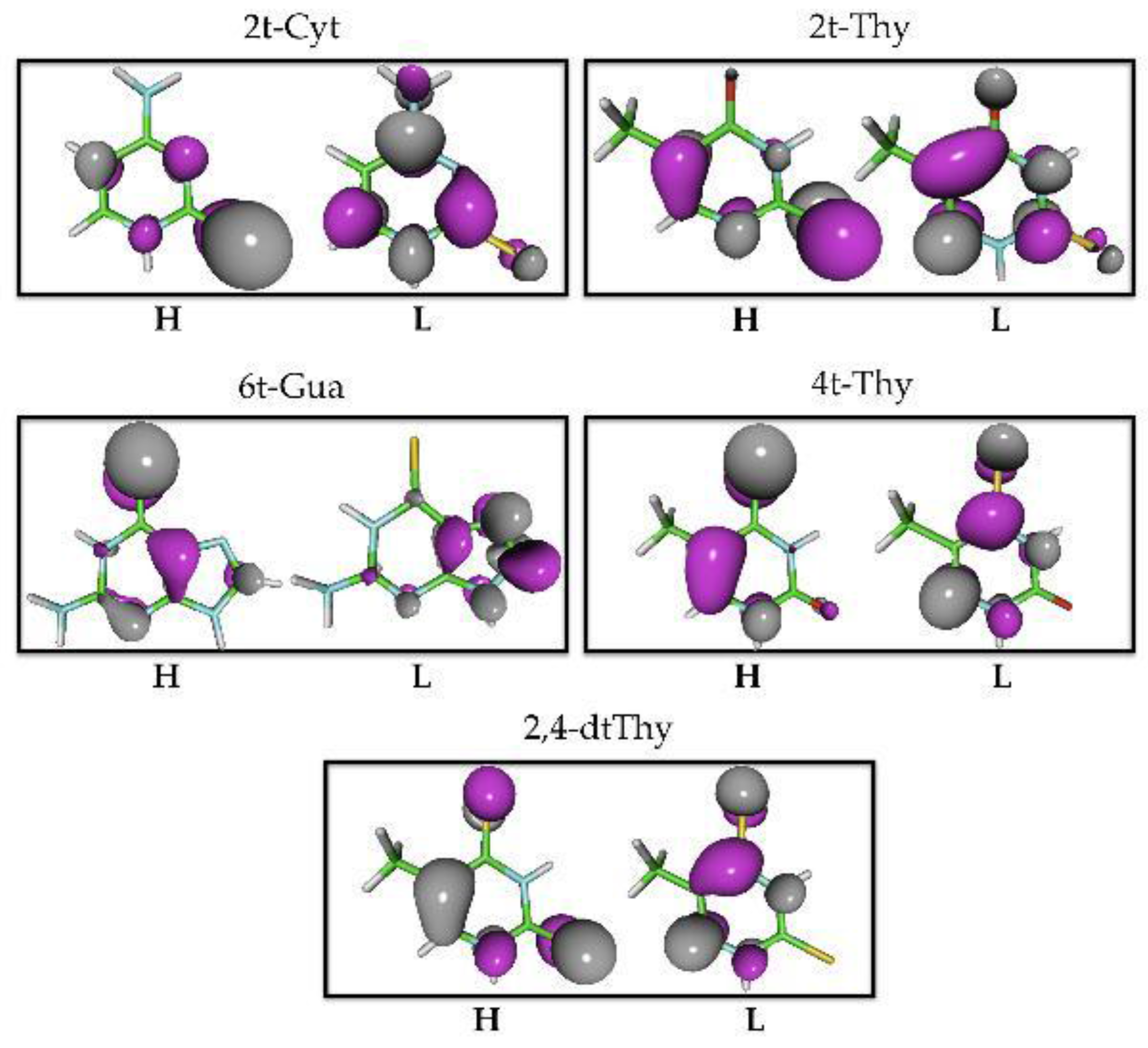
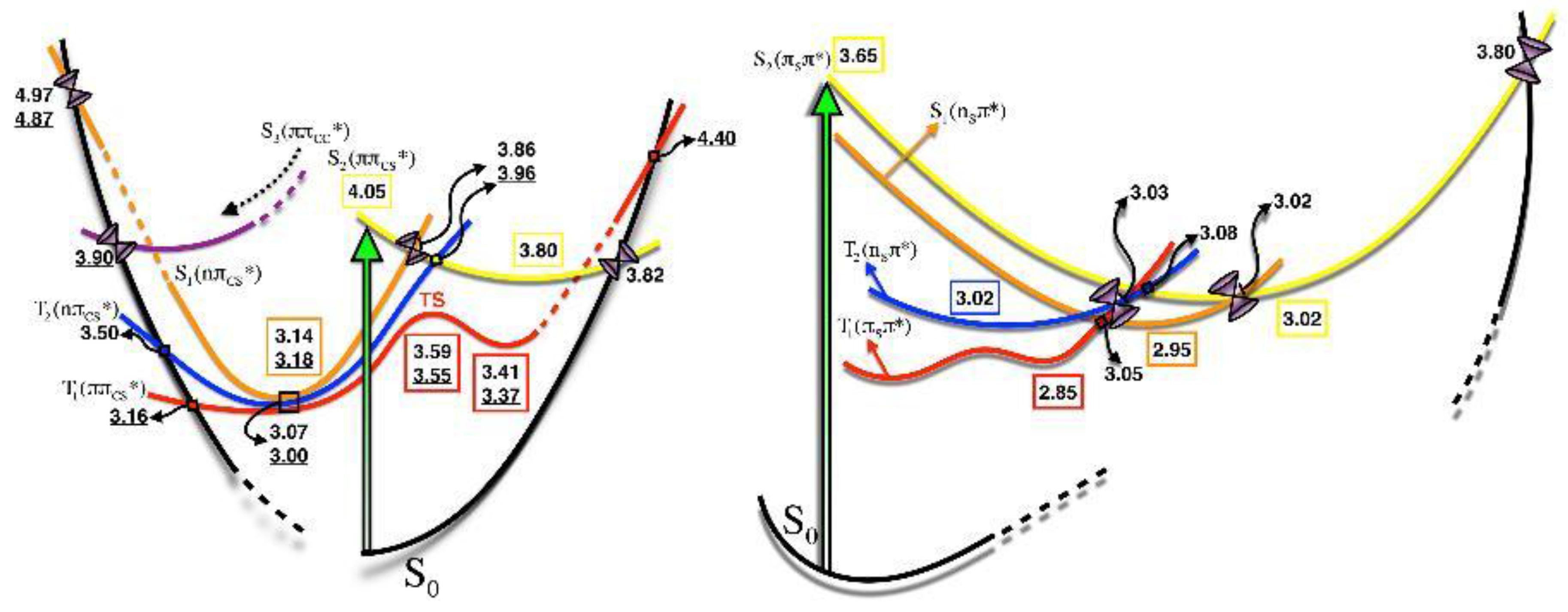
2.1.2. Static Description of the PES
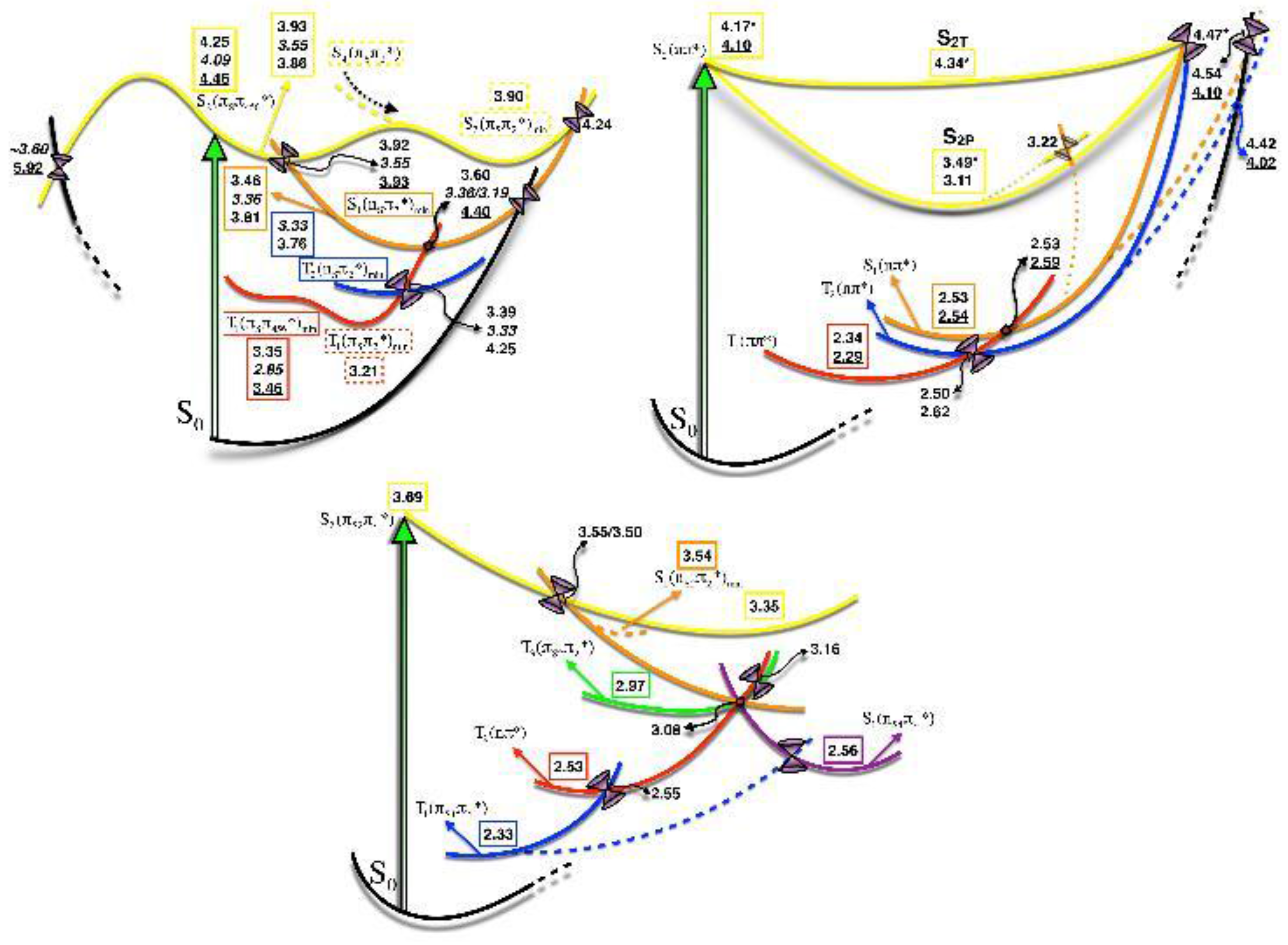
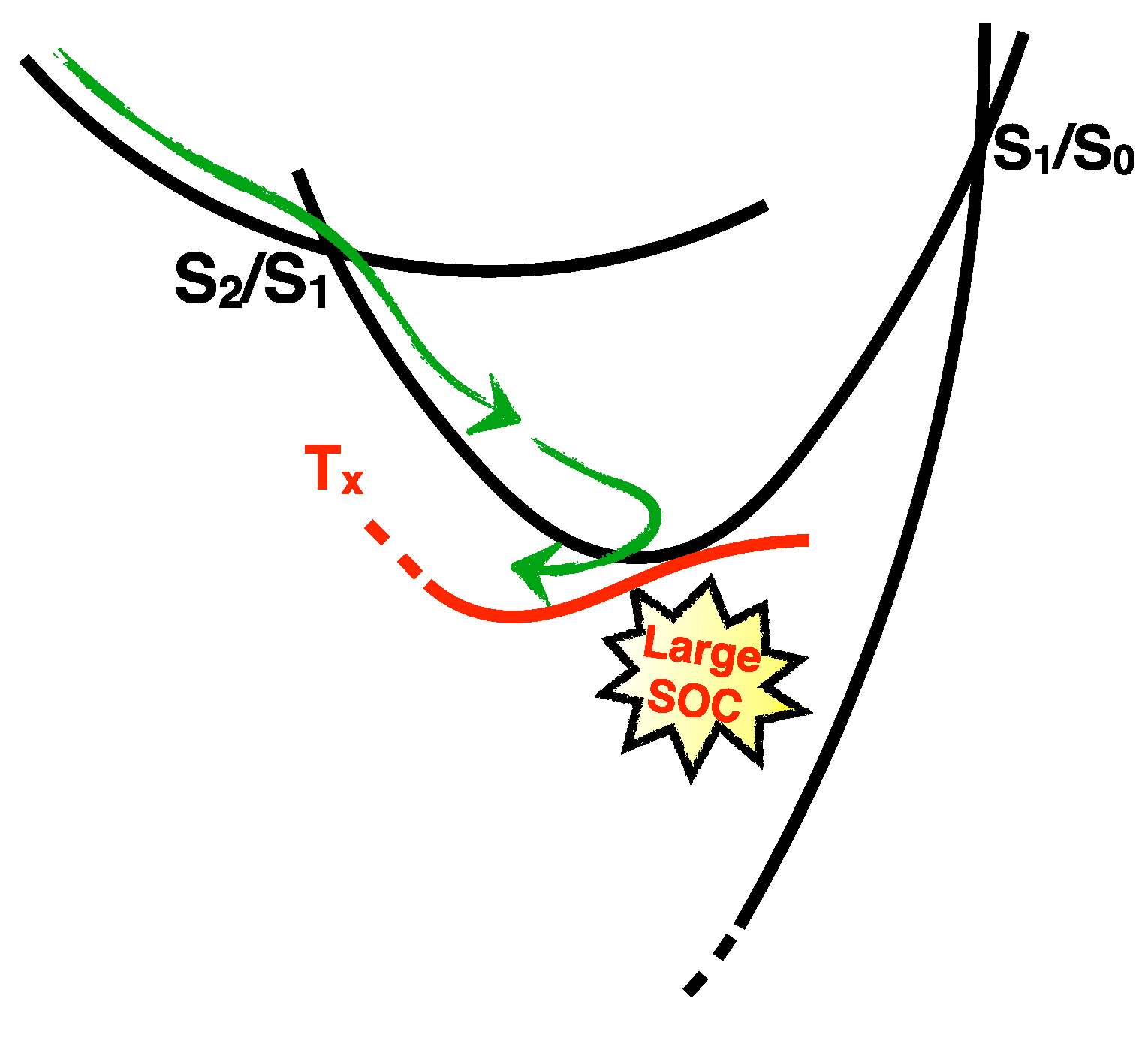
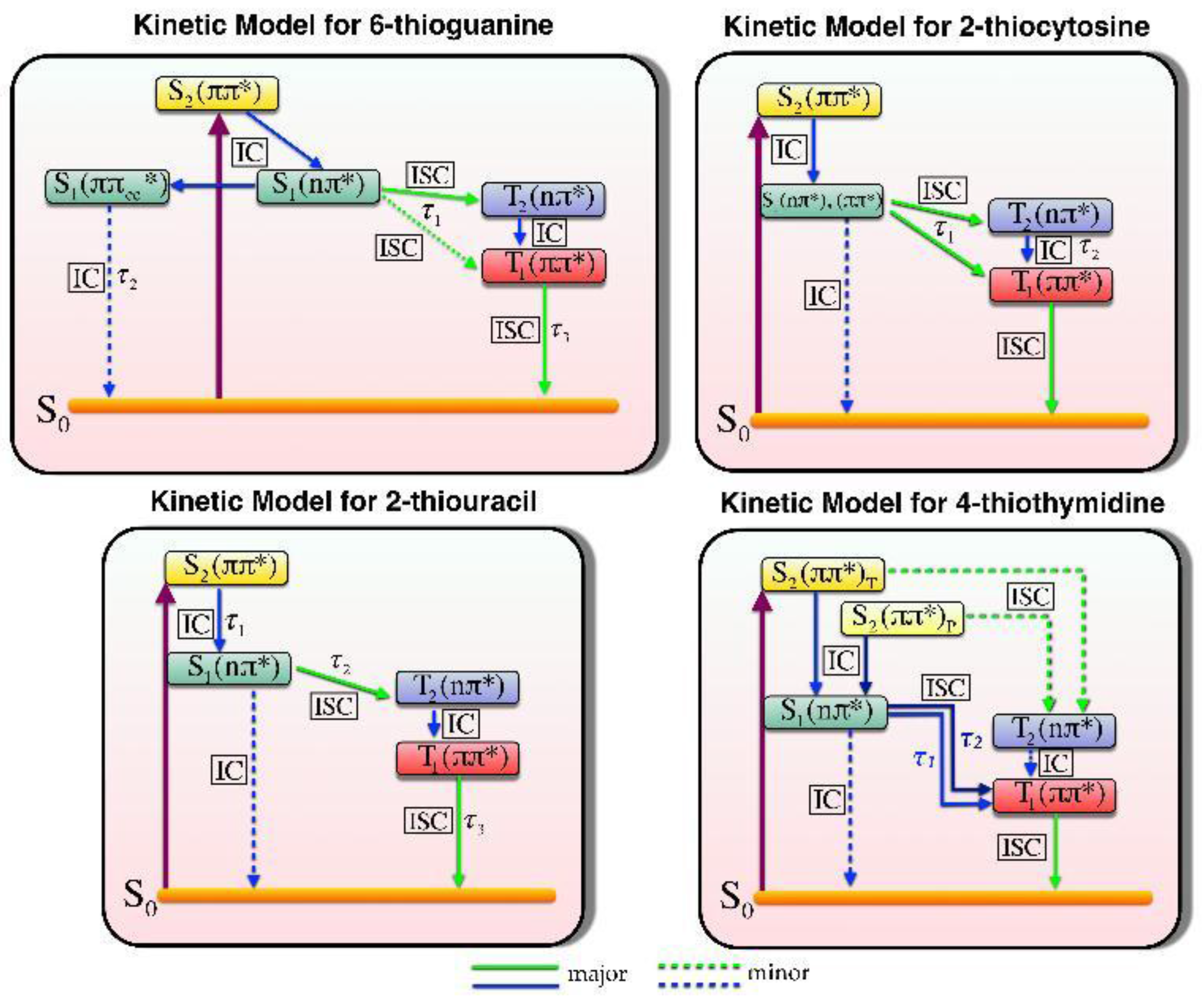
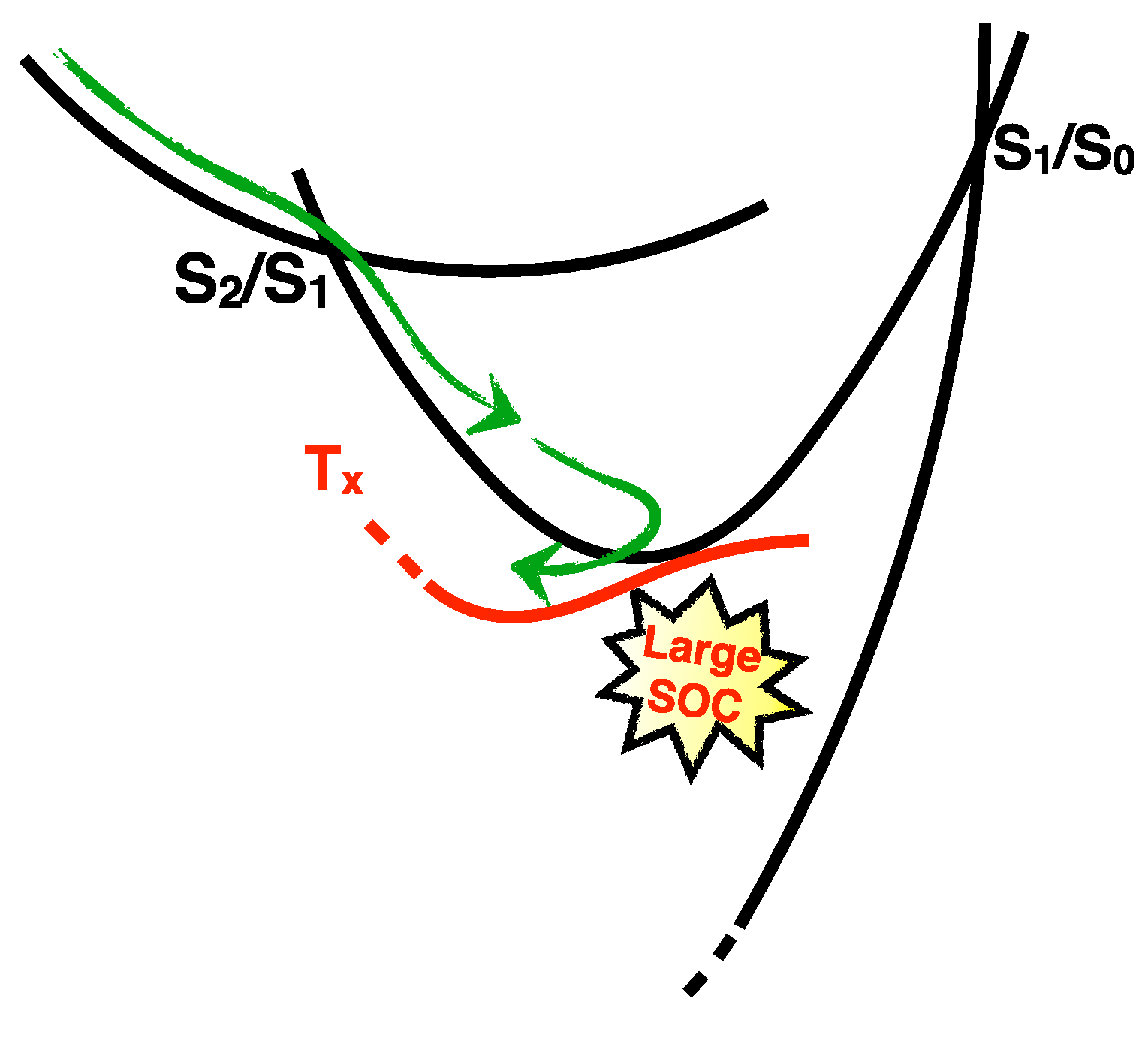
2.1.3. Photodynamics
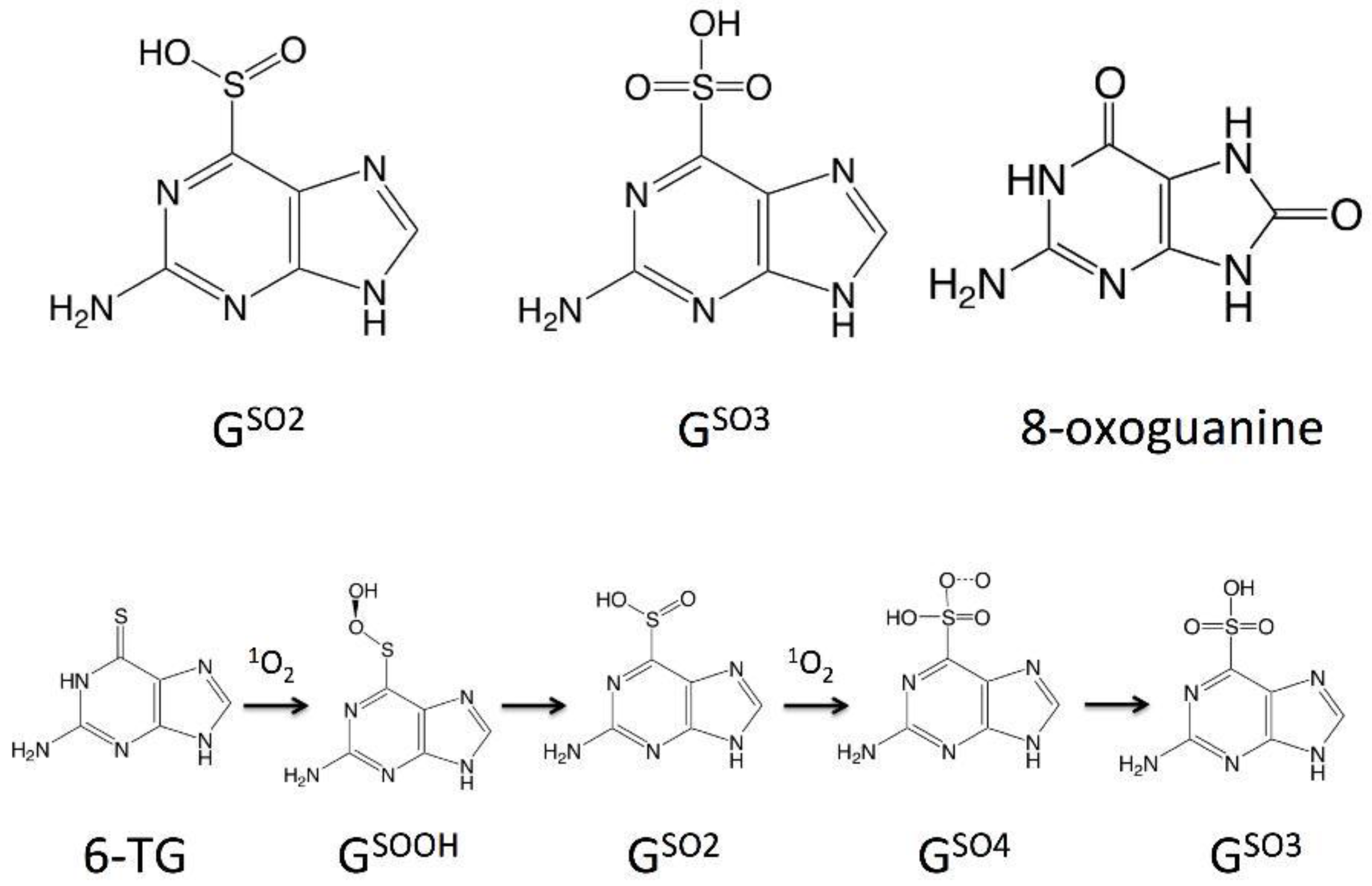
2.1.4. The Fate of the Triplet Excited States of 6t-Gua and 6t-Guo
2.2. Thiopyrimidines
2.2.1. 2-Thiopyrimidines
2t-Cyt/Cyd
2t-Thy/Thd and 2t-Ura/Urd
2.2.2. 4-Thiopyrimidines
4t-Thy/Thd and 4t-Ura/Urd
2.2.3. 2,4-Dithiopyrimidines
Steady State Absorption and Emission properties
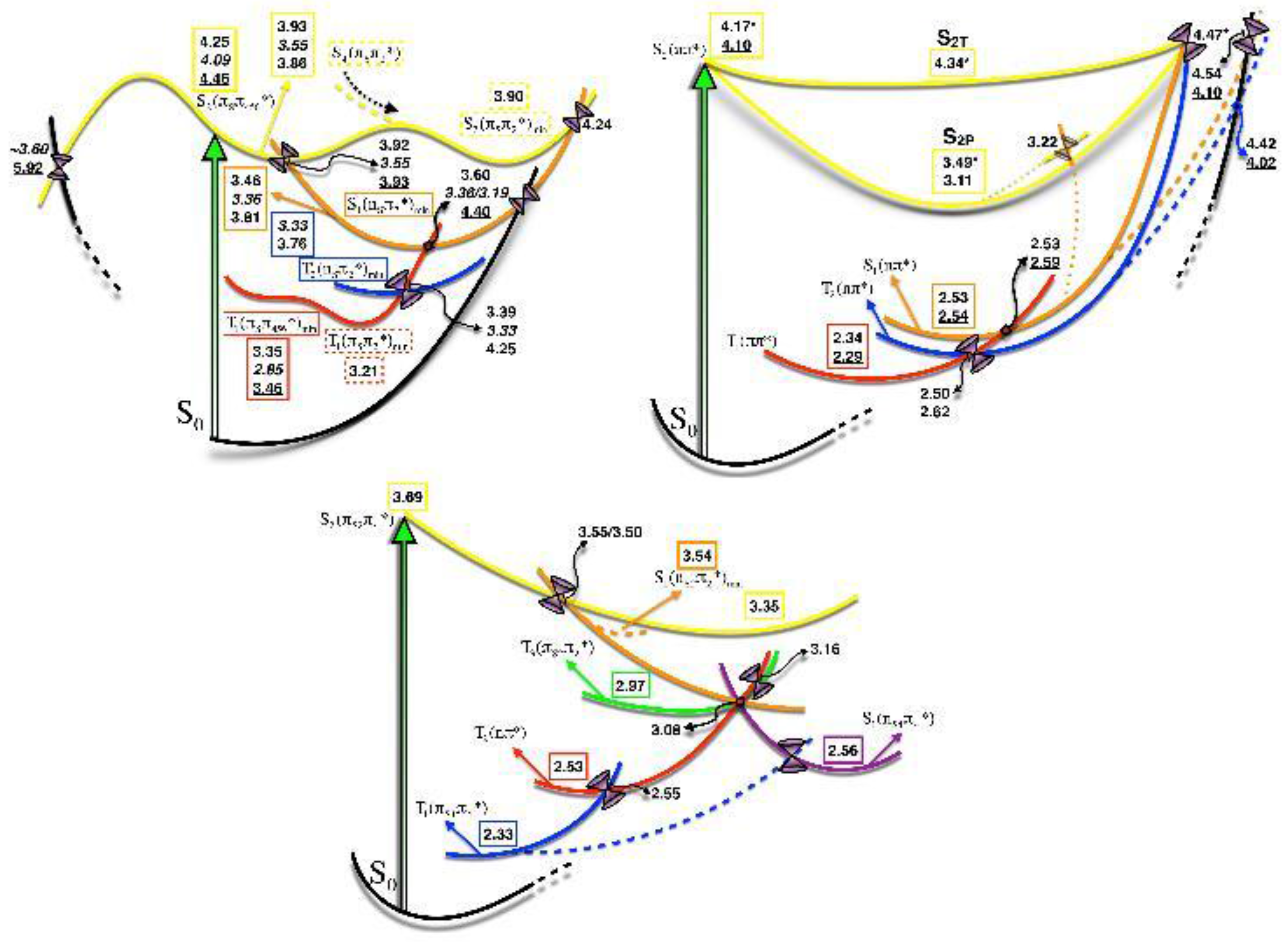
Static description of the PES
Photodynamics
3. General Remarks and Perspectives
Acknowledgments
Conflicts of Interest
References
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 2004, 104, 1977–2020. [Google Scholar] [CrossRef] [PubMed]
- Barbatti, M.; Borin, A.C.; Ullrich, S. Photoinduced phenomena in nucleic acids I, II. In Topics in Current Chemistry; Springer: Berlin, Germany, 2014. [Google Scholar]
- Gustavsson, T.; Improta, R.; Markovitsi, D. DNA/RNA: Building blocks of life under UV irradiation. J. Phys. Chem. Lett. 2010, 1, 2025–2030. [Google Scholar] [CrossRef]
- Pollum, M.; Martínez-Fernández, L.; Crespo-Hernández, C.E. Photochemistry of nucleic acid bases and their thio- and aza-analogues in solution. In Photoinduced Phenomena in Nucleic Acids I: Nucleobases in the Gas Phase and in Solvents; Barbatti, M., Borin, A.C., Ullrich, S., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 245–327. [Google Scholar]
- Crespo-Hernández, C.E.; Martínez-Fernández, L.; Rauer, C.; Reichardt, C.; Mai, S.; Pollum, M.; Marquetand, P.; González, L.; Corral, I. Electronic and structural elements that regulate the excited-state dynamics in purine nucleobase derivatives. J. Am. Chem. Soc. 2015, 137, 4368–4381. [Google Scholar] [CrossRef] [PubMed]
- Matsika, S. Modified nucleobases. In Photoinduced Phenomena in Nucleic Acids I: Nucleobases in the Gas Phase and in Solvents; Barbatti, M., Borin, A.C., Ullrich, S., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 209–243. [Google Scholar]
- Szostak, J.W. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012, 3, 2. [Google Scholar] [CrossRef]
- Zhang, S.; Blain, J.C.; Zielinska, D.; Gryaznov, S.M.; Szostak, J.W. Fast and accurate nonenzymatic copying of an RNA-like synthetic genetic polymer. Proc. Natl. Acad. Sci. USA 2013, 110, 17732–17737. [Google Scholar] [CrossRef] [PubMed]
- Caton-Williams, J.; Huang, Z. Biochemistry of selenium-derivatized naturally occurring and unnatural nucleic acids. Chem. Biodivers. 2008, 5, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Ajitkumar, P.; Cherayil, J.D. Thionucleosides in transfer ribonucleic acid: Diversity, structure, biosynthesis, and function. Microbiol. Rev. 1988, 52, 103–113. [Google Scholar] [PubMed]
- Aarbakke, J.; Janka-Schaub, G.; Elion, G.B. Thiopurine biology and pharmacology. Trends Pharmacol. Sci. 1997, 18, 3–7. [Google Scholar] [CrossRef]
- Relling, M.V.; Dervieux, T. Pharmacogenetics and cancer therapy. Nat. Rev. Cancer 2001, 1, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Warren, D.J.; Andersen, A.; Slørdal, L. Quantitation of 6-thioguanine residues in peripheral blood leukocyte DNA obtained from patients receiving 6-mercaptopurine-based maintenance therapy. Cancer Res. 1995, 55, 1670–1674. [Google Scholar] [PubMed]
- Cuffari, C.; Li, D.Y.; Mahoney, J.; Barnes, Y.; Bayless, T.M. Peripheral blood mononuclear cell DNA 6-thioguanine metabolite levels correlate with decreased interferon-γ production in patients with crohn’s disease on aza therapy. Dig. Dis. Sci. 2004, 49, 133–137. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, P.; Perrett, C.M.; Zhang, X.; Montaner, B.; Xu, Y.-Z.; Harwood, C.A.; McGregor, J.M.; Walker, S.L.; Hanaoka, F.; Karran, P. Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science 2005, 309, 1871–1874. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Karran, P. Multiple forms of DNA damage caused by UVA photoactivation of DNA 6-thioguanine. J. Photochem. Photobiol. 2012, 88, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Favre, A.; Saintomé, C.; Fourrey, J.-L.; Clivio, P.; Laugâa, P. Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid-protein interactions. J. Photochem. Photobiol. B 1998, 42, 109–124. [Google Scholar] [CrossRef]
- Meisenheimer, K.M.; Koch, T.H. Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 101–140. [Google Scholar] [CrossRef] [PubMed]
- Favre, A.; Yaniv, M.; Michels, A.M. The photochemistry of 4-thiouridine in escherichia coli t-RNAİVal. Biochem. Biophys. Res. Commun. 1969, 37, 266–271. [Google Scholar] [CrossRef]
- Favre, A.; Moreno, G.; Blondel, M.O.; Kliber, J.; Vinzens, F.; Salet, C. 4-thiouridine photosensitized RNA-protein crosslinking in mammalian cells. Biochem. Biophys. Res. Commun. 1986, 141, 847–854. [Google Scholar] [CrossRef]
- Favre, A.; Bezerra, R.; Hajnsdorf, E.; Dubreuil, Y.L.; Expert-Bezançon, A. Substitution of uridine in vivo by the intrinsic photoactivable probe 4-thiouridine in escherichia coli RNA. Eur. J. Biochem. 1986, 160, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, E.J. Site-specific RNA crosslinking with 4-thiouridine. Mol. Biol. Rep. 1994, 20, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.E.; Christian, E.L. Chapter 7—RNA crosslinking methods. In Methods Enzymol.; Academic Press: Amsterdam, The Netherlands, 2009; Volume 468, pp. 127–146. [Google Scholar]
- Kumar, R.K.; Davis, D.R. Synthesis and studies on the effect of 2-thiouridine and 4-thiouridine on sugar conformation and RNA duplex stability. Nucleic Acids Res. 1997, 25, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.S.; Sierzputowska-Gracz, H.; Sochacka, E.; Malkiewicz, A.; Agris, P.F. Chemistry and structure of modified uridine dinucleosides are determined by thiolation. J. Am. Chem. Soc. 1992, 114, 7989–7997. [Google Scholar] [CrossRef]
- Sierzputowska-Gracz, H.; Sochacka, E.; Malkiewicz, A.; Kuo, K.; Gehrke, C.W.; Agris, P.F. Chemistry and structure of modified uridines in the anticodon, wobble position of transfer RNA are determined by thiolation. J. Am. Chem. Soc. 1987, 109, 7171–7177. [Google Scholar] [CrossRef]
- Vormbrock, R.; Morawietz, R.; Gassen, H.G. Codon—Anticodon interaction studied with trinucleoside diphosphates containing 2-thiouridine, 4-thiouridine, 2,4-dithiouridine, or 2-thiocytidine. Biochim. Biophys. Acta 1974, 340, 348–358. [Google Scholar] [CrossRef]
- Park, E.; Baron, R.; Landgraf, R. Higher-order association states of cellular ERBB3 probed with photo-cross-linkable aptamers. Biochemistry 2008, 47, 11992–12005. [Google Scholar] [CrossRef] [PubMed]
- Les, A.; Adamowicz, L. Tautomerism of 2- and 4-thiouracil. Ab initio theoretical study. J. Am. Chem. Soc. 1990, 112, 1504–1509. [Google Scholar] [CrossRef]
- Rostkowska, H.; Szczepaniak, K.; Nowak, M.J.; Leszczynski, J.; KuBulat, K.; Person, W.B. Thiouracils. 2. Tautomerism and infrared spectra of thiouracils. Matrix-isolation and ab initio studies. J. Am. Chem. Soc. 1990, 112, 2147–2160. [Google Scholar] [CrossRef]
- Shukla, M.K.; Leszczynski, J. Electronic transitions of thiouracils in the gas phase and in solutions: Time-dependent density functional theory (TD-DFT) study. J. Phys. Chem. A 2004, 108, 10367–10375. [Google Scholar] [CrossRef]
- Puzzarini, C.; Biczysko, M.; Barone, V.; Peña, I.; Cabezas, C.; Alonso, J.L. Accurate molecular structure and spectroscopic properties for nucleobases: A combined computational—Microwave investigation of 2-thiouracil as a case study. Phys. Chem. Chem. Phys. 2013, 15, 16965–16975. [Google Scholar] [CrossRef] [PubMed]
- Podolyan, Y.; Gorb, L.; Blue, A.; Leszczynski, J. A theoretical investigation of tautomeric equilibria and proton transfer in isolated and hydrated thiocytosine. J. Mol. Struct. THEOCHEM 2001, 549, 101–109. [Google Scholar] [CrossRef]
- Mai, S.; Pollum, M.; Martínez-Fernández, L.; Dunn, N.; Marquetand, P.; Corral, I.; Crespo-Hernández, C.E.; González, L. The origin of efficient triplet state population in sulfur-substituted nucleobases. Nat. Commun. 2016, 7, 13077. [Google Scholar] [CrossRef] [PubMed]
- Leszczynski, J. Tautomers of 6-thioguanine: Structures and properties. J. Phys. Chem. 1993, 97, 3520–3524. [Google Scholar] [CrossRef]
- Civcir, P.U. Tautomerism of 6-thioguanine in the gas and aqueous phases using AM1 and PM3 methods. J. Mol. Struct. THEOCHEM 2001, 536, 161–171. [Google Scholar] [CrossRef]
- Pirillo, J.; De Simone, B.C.; Russo, N. Photophysical properties prediction of selenium- and tellurium-substituted thymidine as potential UVA chemotherapeutic agents. Theor. Chem. Acc. 2016, 135, 8–12. [Google Scholar] [CrossRef]
- Pirillo, J.; Mazzone, G.; Russo, N.; Bertini, L. Photophysical properties of S, Se and Te-substituted deoxyguanosines: Insight into their ability to act as chemotherapeutic agents. J. Chem. Inf. Model. 2017, 57, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Sanchez-Rodriguez, J.A.; Pollum, M.; Crespo-Hernández, C.E.; Mai, S.; Marquetand, P.; González, L.; Ullrich, S. Internal conversion and intersystem crossing pathways in UV excited, isolated uracils and their implications in prebiotic chemistry. Phys. Chem. Chem. Phys. 2016, 18, 20168–20176. [Google Scholar] [CrossRef] [PubMed]
- Ruckenbauer, M.; Mai, S.; Marquetand, P.; González, L. Photoelectron spectra of 2-thiouracil, 4-thiouracil, and 2,4-dithiouracil. J. Chem. Phys. 2016, 144, 074303–074310. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Escudero, D.; Serrano-Andrés, L. Progress and challenges in the calculation of electronic excited states. Chem. Phys. Chem. 2012, 13, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Ruckebusch, C.; Sliwa, M.; Pernot, P.; de Juan, A.; Tauler, R. Comprehensive data analysis of femtosecond transient absorption spectra: A review. J. Photochem. Photobiol. C 2012, 13, 1–27. [Google Scholar] [CrossRef]
- Stolow, A.; Bragg, A.E.; Neumark, D.M. Femtosecond time-resolved photoelectron spectroscopy. Chem. Rev. 2004, 104, 1719–1758. [Google Scholar] [CrossRef] [PubMed]
- Efremov, E.V.; Ariese, F.; Gooijer, C. Achievements in Resonance Raman spectroscopy: Review of a technique with a distinct analytical chemistry potential. Anal. Chim. Acta 2008, 606, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W. Semiempirical quantum–chemical methods. WIREs Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar] [CrossRef]
- Persico, M.; Granucci, G. An overview of nonadiabatic dynamics simulations methods, with focus on the direct approach versus the fitting of potential energy surfaces. Theor. Chem. Acc. 2014, 133, 1–28. [Google Scholar] [CrossRef]
- Lasorne, B.; Worth, G.A.; Robb, M.A. Excited-state dynamics. WIREs Comput. Mol. Sci. 2011, 1, 460–475. [Google Scholar] [CrossRef]
- Barbatti, M. Nonadiabatic dynamics with trajectory surface hopping method. WIREs Comput. Mol. Sci. 2011, 1, 620–633. [Google Scholar] [CrossRef]
- Rubin, Y.V.; Blagoi, Y.P.; Bokovoy, V.A. 6-thioguanine luminescence probe to study DNA and low-molecular-weight systems. J. Fluoresc. 1995, 5, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Santhosh, C.; Mishra, P.C. Electronic structures and spectra of 6-mercaptopurine and 6-thioguanine. Spectrochim. Acta Part A 1993, 49, 985–993. [Google Scholar] [CrossRef]
- Ashwood, B.; Jockusch, S.; Crespo-Hernández, C.E. Excited-state dynamics of the thiopurine prodrug 6-thioguanine: Can N9-glycosylation affect its phototoxic activity? Molecules 2017, 22, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Gomzi, V. TDDFT study of nucleobase thioanalogues and oxo-derivatives excited states. J. Theor. Comput. Chem. 2009, 08, 71–83. [Google Scholar] [CrossRef]
- Reichardt, C.; Guo, C.; Crespo-Hernández, C.E. Excited-state dynamics in 6-thioguanosine from the femtosecond to microsecond time scale. J. Phys. Chem. B 2011, 115, 3263–3270. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; González, L.; Corral, I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: The case of 6-thioguanine. Chem. Commun. 2012, 48, 2134–2136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Smith, J.; Haygood, M.T.; Gao, R. Direct observation and quantitative characterization of singlet oxygen in aqueous solution upon UVA excitation of 6-thioguanines. J. Phys. Chem. B 2011, 115, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Chuan, D.; Wen, Y.; Shaomin, S.; Pin, Y. Determination of thioguanine in pharmaceutical preparations by paper substrate room temperature phosphorimetry. Analyst 2000, 125, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Corral, I.; Granucci, G.; Persico, M. Competing ultrafast intersystem crossing and internal conversion: A time resolved picture for the deactivation of 6-thioguanine. Chem. Sci. 2014, 5, 1336–1347. [Google Scholar] [CrossRef]
- Bai, S.; Barbatti, M. On the decay of the triplet state of thionucleobases. Phys. Chem. Chem. Phys. 2017, 19, 12674–12682. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Crespo-Hernández, C.E. Communication: The dark singlet state as a doorway state in the ultrafast and efficient intersystem crossing dynamics in 2-thiothymine and 2-thiouracil. J. Chem. Phys. 2014, 140, 071101–071105. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, H.; Kobayashi, T.; Suzuki, T.; Ichimura, T. Excited-state dynamics of 6-aza-2-thiothymine and 2-thiothymine: Highly efficient intersystem crossing and singlet oxygen photosensitization. J. Phys. Chem. B 2010, 114, 8782–8789. [Google Scholar] [CrossRef] [PubMed]
- Taras-Goślińska, K.; Burdziński, G.; Wenska, G. Relaxation of the T1 excited state of 2-thiothymine, its riboside and deoxyriboside-enhanced nonradiative decay rate induced by sugar substituent. J. Photochem. Photobiol. A 2014, 275, 89–95. [Google Scholar] [CrossRef]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. 2,4-dithiothymine as a potent UVA chemotherapeutic agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef] [PubMed]
- Milder, S.J.; Kliger, D.S. Spectroscopy and photochemistry of thiouracils: Implications for the mechanism of photocrosslinking in tRNA. J. Am. Chem. Soc. 1985, 107, 7365–7373. [Google Scholar] [CrossRef]
- Harada, Y.; Suzuki, T.; Ichimura, T.; Xu, Y.-Z. Triplet formation of 4-thiothymidine and its photosensitization to oxygen studied by time-resolved thermal lensing technique. J. Phys. Chem. B 2007, 111, 5518–5524. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C.; Crespo-Hernández, C.E. Room-temperature phosphorescence of the DNA monomer analogue 4-thiothymidine in aqueous solutions after UVA excitation. J. Phys. Chem. Lett. 2010, 1, 2239–2243. [Google Scholar] [CrossRef]
- Reichardt, C.; Crespo-Hernández, C.E. Ultrafast spin crossover in 4-thiothymidine in an ionic liquid. Chem. Commun. 2010, 46, 5963–5965. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Okabe, C.; Kobayashi, T.; Suzuki, T.; Ichimura, T.; Nishi, N.; Xu, Y.-Z. Ultrafast intersystem crossing of 4-thiothymidine in aqueous solution. J. Phys. Chem. Lett. 2010, 1, 480–484. [Google Scholar] [CrossRef]
- Foote, C.S.; Dobrowolski, D.C. Oxygen Radicals in Chemistry and Biology; Bors, W., Saran, M., Tait, D., Eds.; De Gruyter: Berlin, Germany, 1984; pp. 465–470. [Google Scholar]
- Heihoff, K.; Redmond, R.W.; Braslavsky, S.E.; Rougee, M.; Salet, C.; Favre, A.; Bensasson, R.V. Quantum yields of triplet and O2(1Δg) formation of 4-thiouridine in water and acetonitrile. Phochem. Photobiol. 1990, 51, 635–641. [Google Scholar]
- Salet, C.; Bensasson, R.V.; Favre, A. Studies on the triplet excited state of 4-thiouridine. Photochem. Photobiol. 1983, 38, 521–525. [Google Scholar] [CrossRef]
- Pollum, M.; Ortiz-Rodríguez, L.A.; Jockusch, S.; Crespo-Hernández, C.E. The triplet state of 6-thio-2′-deoxyguanosine: Intrinsic properties and reactivity toward molecular oxygen. Photochem. Photobiol. 2016, 92, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Li, F.; Jeffs, G.; Zhang, X.; Xu, Y.-Z.; Karran, P. Guanine sulphinate is a major stable product of photochemical oxidation of DNA 6-thioguanine by UVA irradiation. Nucleic Acids Res. 2010, 38, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Duarte, T.L.; Cooper, D.; Chen, J.; Nandagopal, S.; Evans, M.D. Combination of azathioprine and UVA irradiation is a major source of cellular 8-oxo-7,8-dihydro-2’-deoxyguanosine. DNA Repair 2008, 7, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Montaner, B.; O’Donovan, P.; Reelfs, O.; Perrett, C.M.; Zhang, X.; Xu, Y.Z.; Ren, X.; Macpherson, P.; Frith, D.; Karran, P. Reactive oxygen-mediated damage to a human DNA replication and repair protein. EMBO Rep. 2007, 8, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhao, H.; Yu, Y.; Su, H. Formation of guanine-6-sulfonate from 6-thioguanine and singlet oxygen: A combined theoretical and experimental study. J. Am. Chem. Soc. 2013, 135, 4509–4515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jeffs, G.; Ren, X.; O’Donovan, P.; Montaner, B.; Perrett, C.M.; Karran, P.; Xu, Y.-Z. Novel DNA lesions generated by the interaction between therapeutic thiopurines and UVA light. DNA Repair 2007, 6, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Daehn, I.; Karran, P. Efficient DNA interstrand crosslinking by 6-thioguanine and UVA radiation. DNA Repair 2011, 10, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Brem, R.; Li, F.; Montaner, B.; Reelfs, O.; Karran, P. DNA breakage and cell cycle checkpoint abrogation induced by a therapeutic thiopurine and UVA radiation. Oncogene 2010, 29, 3953–3963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rana, T.M. RNA−protein interactions in the Tat-trans-activation response element complex determined by site-specific photo-cross-linking. Biochemistry 1998, 37, 4235–4243. [Google Scholar] [CrossRef] [PubMed]
- Gueranger, Q.; Kia, A.; Frith, D.; Karran, P. Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA. Nucleic Acids Res. 2011, 39, 5057–5066. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Granucci, G.; Pollum, M.; Crespo-Hernández, C.E.; Persico, M.; Corral, I. Decoding the molecular basis for the population mechanism of the triplet phototoxic precursors in UVA light-activated pyrimidine anticancer drugs. Chem. Eur. J. 2017, 23, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Igarashi-Yamamoto, N.; Tajiri, A.; Hatano, M.; Shibuya, S.; Ueda, T. Ultraviolet absorption, circular dichroism and magnetic circular dichroism studies of sulfur-containing nucleic acid bases and their nucleosides. Biochim. Biophys. Acta 1981, 656, 1–15. [Google Scholar] [CrossRef]
- Mai, S.; Ashwood, B.; Marquetand, P.; Crespo-Hernández, C.E.; González, L. Solvatochromic effects on the absorption spectrum of 2-thiocytosine. J. Phys. Chem. B 2017, 121, 5187–5196. [Google Scholar] [CrossRef] [PubMed]
- Vendrell-Criado, V.; Sáez, J.A.; Lhiaubet-Vallet, V.; Cuquerella, M.C.; Miranda, M.A. Photophysical properties of 5-substituted 2-thiopyrimidines. Photochem. Photobiol. Sci. 2013, 12, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Pepino, A.J.; Segarra-Martí, J.; Banyasz, A.; Garavelli, M.; Improta, R. Computing the absorption and emission spectra of 5-methylcytidine in different solvents: A test-case for different solvation models. J. Chem. Theor. Comput. 2016, 12, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Barone, V. Excited states behavior of nucleobases in solution: Insights from computational studies. In Photoinduced Phenomena in Nucleic Acids I: Nucleobases in the Gas Phase and in Solvents; Barbatti, M., Borin, A.C., Ullrich, S., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 329–357. [Google Scholar]
- Mai, S.; Marquetand, P.; González, L. A static picture of the relaxation and intersystem crossing mechanisms of photoexcited 2-thiouracil. J. Phys. Chem. A 2015, 119, 9524–9533. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Fang, W.-H. State-specific heavy-atom effect on intersystem crossing processes in 2-thiothymine: A potential photodynamic therapy photosensitizer. J. Chem. Phys. 2013, 138, 044315–044323. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, J.P.; Borin, A.C. 2-thiouracil deactivation pathways and triplet states population. Comput. Theor. Chem. 2014, 1040–1041, 195–201. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, T.-S.; Xue, J.-D.; Zheng, X.; Cui, G.; Fang, W.-H. Short-time dynamics of 2-thiouracil in the light absorbing S2(ππ∗) state. J. Chem. Phys. 2015, 143, 175103–175111. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Barbatti, M. Why replacing different oxygens of thymine with sulfur causes distinct absorption and intersystem crossing. J. Phys. Chem. A 2016, 120, 6342–6350. [Google Scholar] [CrossRef] [PubMed]
- Taherian, M.R.; Maki, A.H. Optically detected magnetic resonance study of the phosphorescent states of thiouracils. Chem. Phys. 1981, 55, 85–96. [Google Scholar] [CrossRef]
- Cui, G.; Thiel, W. Intersystem crossing enables 4-thiothymidine to act as a photosensitizer in photodynamic therapy: An ab initio QM/MM study. J. Phys. Chem. Lett. 2014, 5, 2682–2687. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.-B.; Wang, Q.; Guo, W.-W.; Cui, G. The excited-state decay mechanism of 2,4-dithiothymine in the gas phase, microsolvated surroundings, and aqueous solution. Phys. Chem. Chem. Phys. 2017, 19, 7689–7698. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.A. Spin—Orbit coupling and the radiationless processes in nitrogen heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- Mai, S.; Marquetand, P.; González, L. Intersystem crossing pathways in the noncanonical nucleobase 2-thiouracil: A time-dependent picture. J. Phys. Chem. Lett. 2016, 7, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Dai, X.; Liu, K.; Zhao, H.; Song, D.; Su, H. Photophysical and photochemical properties of 4-thiouracil: Time-resolved IR spectroscopy and DFT studies. J. Phys. Chem. B 2014, 118, 5864–5872. [Google Scholar] [CrossRef] [PubMed]
- Wenska, G.; Taras-Goślińska, K.; Skalski, B.; Maciejewski, A.; Burdziński, G.; Karolczak, J. Putative phototautomerization of 4-thiouridine in the S2 excited state revealed by fluorescence study using picosecond laser spectroscopy. J. Photochem. Photobiol. A 2006, 181, 12–18. [Google Scholar] [CrossRef]
- Taras-Goślińska, K.; Wenska, G.; Skalski, B.; Maciejewski, A.; Burdziński, G.; Karolczak, J. Spectral and photophysical properties of the lowest excited triplet state of 4-thiouridine and its 5-halogeno derivatives. J. Photochem. Photobiol. A 2004, 168, 227–233. [Google Scholar] [CrossRef]
- Taras-Goślińska, K.; Wenska, G.; Skalski, B.; Maciejewski, A.; Burdziński, G.; Karolczak, J. Intra- and intermolecular electronic relaxation of the second excited singlet and the lowest excited triplet states of 1,3-dimethyl-4-thiouracil in solution. Photochem. Photobiol. 2002, 75, 448–456. [Google Scholar] [CrossRef]
- Wenska, G.; Taras-Goślińska, K.; Łukaszewicz, A.; Burdzinski, G.; Koput, J.; Maciejewski, A. Mechanism and dynamics of intramolecular triplet state decay of 1-propyl-4-thiouracil and its α-methyl-substituted derivatives studied in perfluoro-1,3-dimethylcyclohexane. Photochem. Photobiol. Sci. 2011, 10, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.K.; Leszczynski, J. Multiconfigurational self-consistent field study of the excited state properties of 4-thiouracil in the gas phase. J. Phys. Chem. A 2004, 108, 7241–7246. [Google Scholar] [CrossRef]
- Wenska, G.; Koput, J.; Burdziński, G.; Taras-Goślińska, K.; Maciejewski, A. Photophysical and photochemical properties of the T1 excited state of thioinosine. J. Photochem. Photobiol. A 2009, 206, 93–101. [Google Scholar] [CrossRef]
- Pepino, A.J.; Segarra-Martí, J.; Nenov, A.; Improta, R.; Garavelli, M. Resolving ultrafast photoinduced deactivations in water-solvated pyrimidine nucleosides. J. Phys. Chem. Lett. 2017, 8, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Wenska, G.; Filipiak, P.; Taras-Goślińska, K.; Sobierajska, A.; Gdaniec, Z. Orientation-dependent quenching of the triplet excited 6-thiopurine by nucleobases. J. Photochem. Photobiol. A 2011, 217, 55–61. [Google Scholar] [CrossRef]
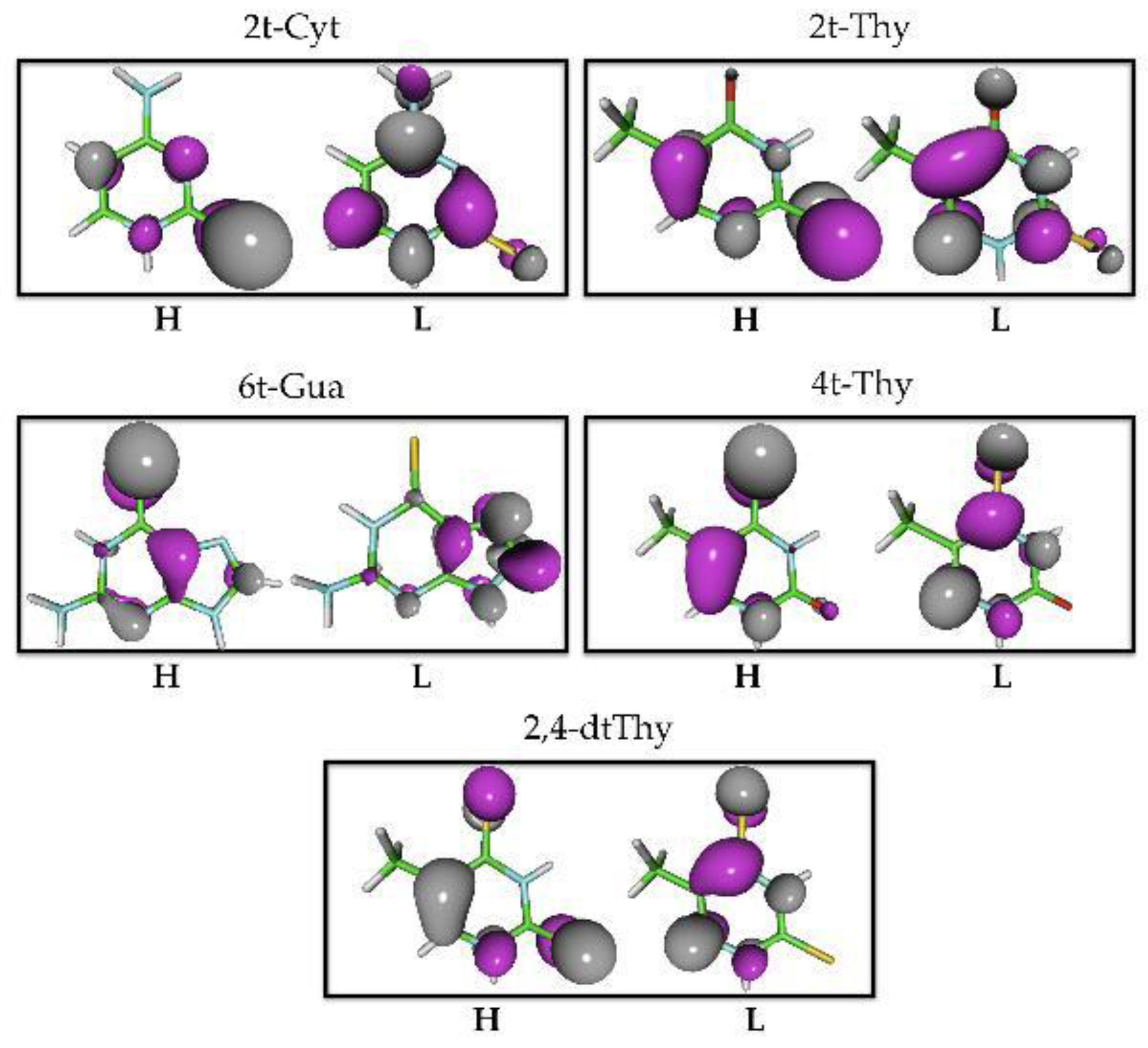
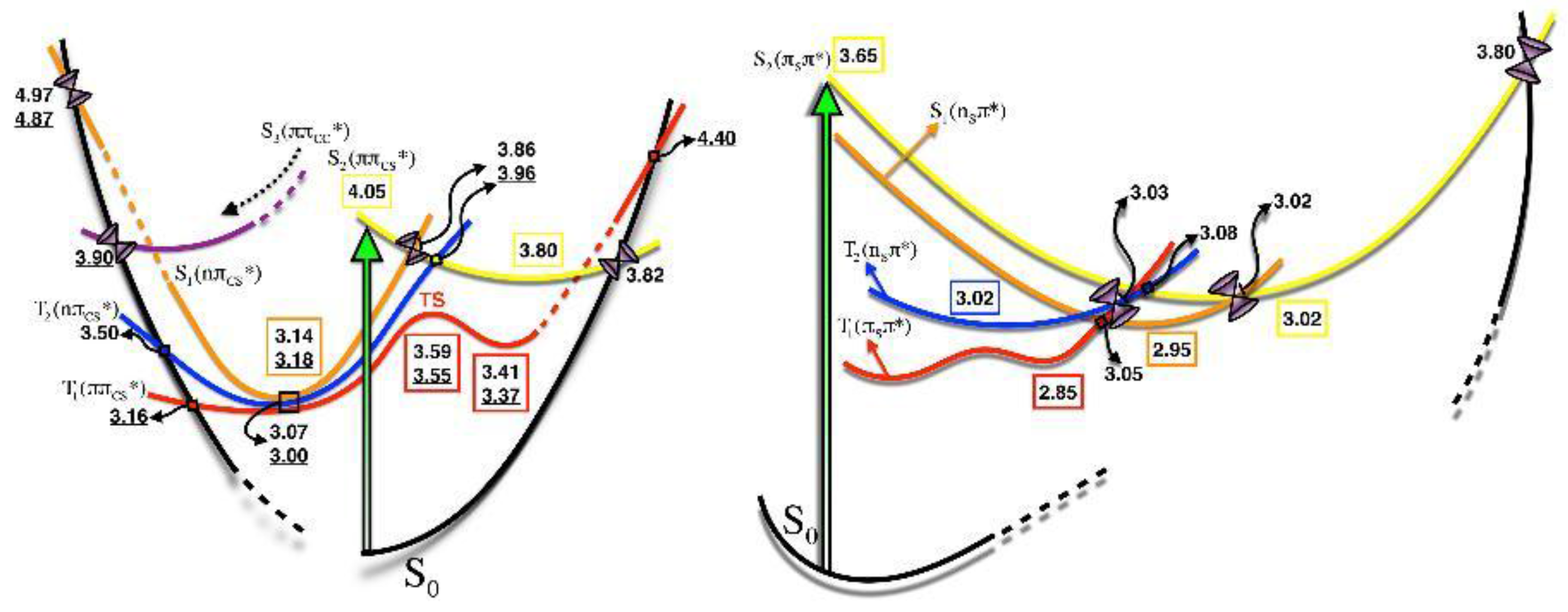

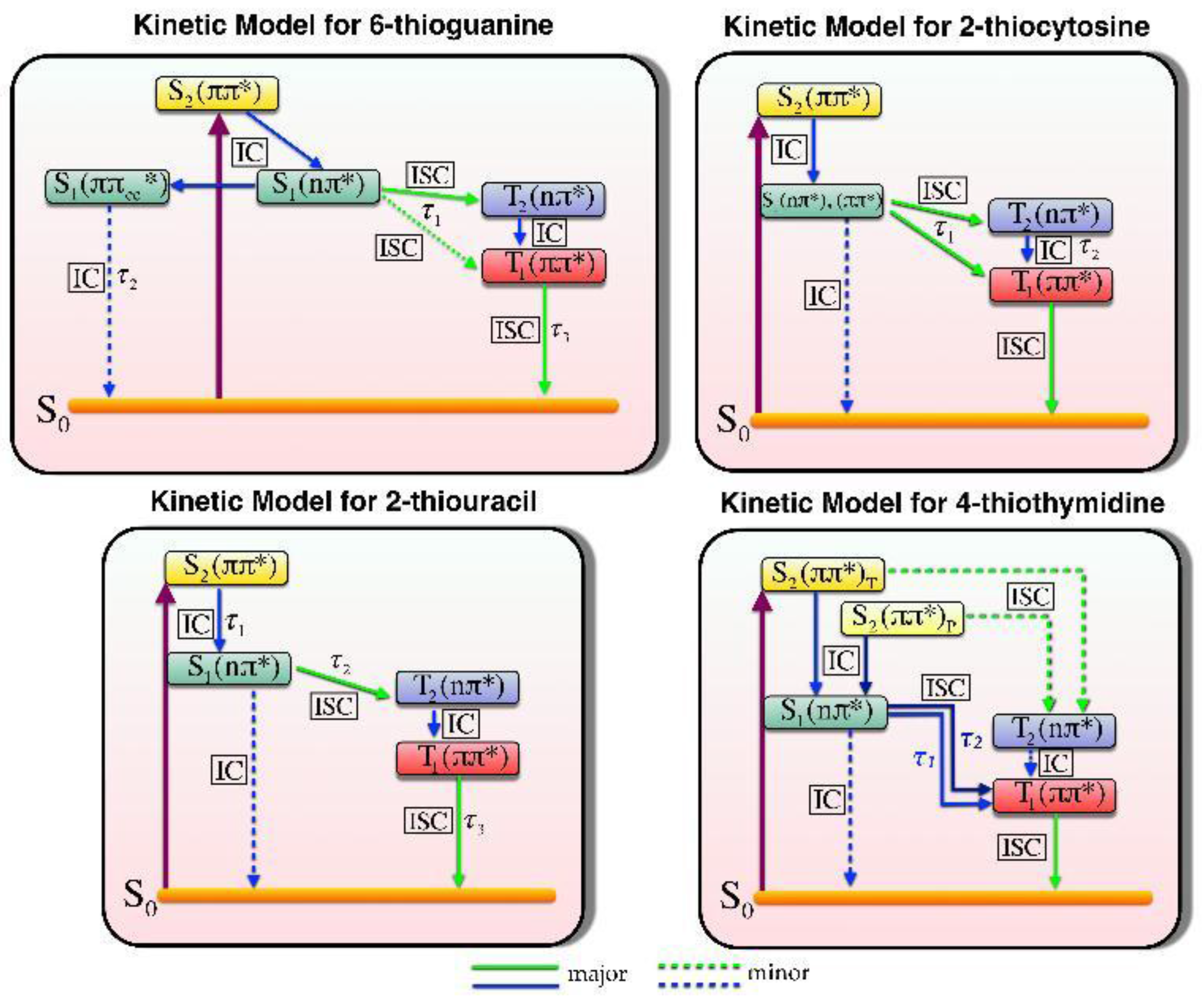
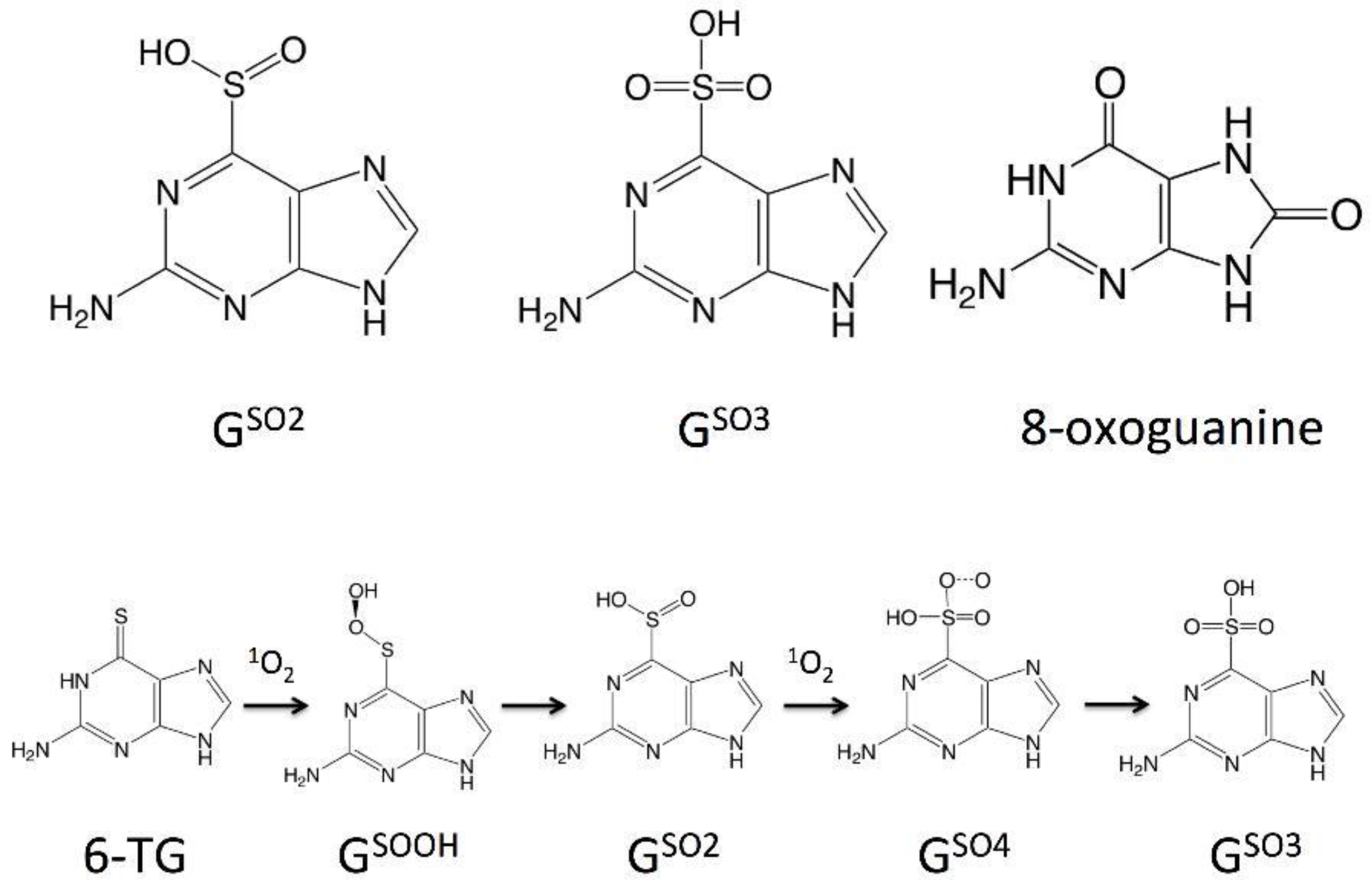

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λmax (nm) | εmax (M−1 cm−1 × 104) | λemission (nm) | Solvent | ||
|---|---|---|---|---|---|
| F | P | ||||
| 6t-Gua | 337 | 1.80 | – | – | NaOH, pH = 10 [56] |
| 340 | 2.10 | – | – | Tris buffer, pH = 7.4 [56] | |
| 340, 255, 220, 205 | – | 400 | – | H2O, pH = 2.3 [51] | |
| 340, 255, 220, 205 | – | 400, 500 1 | – | H2O, pH = 7.8 [51] | |
| 320, 265, 245, 210 | – | 400 | – | H2O, pH = 12.8 [51] | |
| 341, 254, 220 1, 204 | 1.80, 0.60, 1.40, 1.90 | – | – | PBS, pH = 7.4 [52] | |
| 347 2 | 2.08 | – | 478 | pH = 1.0, 77 K [50] | |
| 343 2 | 2.22 | – | 459 | pH = 5.0, 77 K [50] | |
| 323 2 | 1.85 | – | 442 | pH = 10.0, 77 K [50] | |
| 321 2 | 2.12 | – | 442 | 1 N NaOH, 77 K [50] | |
| – | – | 468, 437 1, 494 1 | 430–550 | Tris buffer, pH = 7.4, 77 K [52] | |
| – | – | – | 478 | NaOH, pH = 12.1 [57] | |
| 6t-Guo | 320 | 1.80 | – | – | NaOH, pH = 10 [56] |
| 342 | 2.00 | – | – | Tris buffer, pH = 7.4 [56] | |
| 342 | 2.30 | N.D. | N.D. | PBS, pH = 7 [54] | |
| 346 | 2.20 | N.D. | N.D. | ACN [54] | |
| 341, 258, 227 1, 208 | 2.30, 0.80, 1.20, 2.10 | – | – | PBS, pH = 7.4 [52] | |
| 348 2 | – | – | 481 | pH = 1.0 ,77 K [50] | |
| 344 2 | – | – | 459 | pH = 5.0, 77 K [50] | |
| 316 2 | – | – | 431 | pH = 12.0, 77 K [50] | |
| λmax (nm) | Solvent | Excitation | λemission (nm) | Method | ||
|---|---|---|---|---|---|---|
| F | P | |||||
| 6t-Gua [55] | 369 (0.000) | – | S1 1nπ* | 395 | – | MS-CASPT2// CASSCF(14,12)/ANO-L |
| 306 (0.535) | – | S2 1ππ* | – | – | ||
| 400 | – | T1 3ππ* | – | 404 | ||
| 375 | – | T2 3nπ* | – | – | ||
| 6t-Gua [53] | 397 (0.000) | – | S1 | – | – | TD-B3LYP/6-311++G(d,p) |
| 311 (0.203) | – | S2 | – | – | ||
| 6t-Gua [52] | 381 (0.000) | – | S1 1nπ* | – | – | TD-PBE0/6-311++G(d,p)/ IEFPCM |
| 346 (0.000) | H2O | S1 1nπ* | – | – | ||
| 299 (0.260) | – | S2 1ππ* | – | – | ||
| 310 (0.400) | H2O | S2 1ππ* | – | – | ||
| 6t-Gua [50] | 337 (0.470) | – | S2 1ππ* | – | – | CNDO/S |
| 6t-Gua [51] | 400 (0.000) | – | S1 1nπ* | – | – | CNDO/s-CI |
| 340 (0.290) | – | S2 1ππ* | – | – | ||
| 6t-Guo [54] | 384 (0.000) | – | S1 1nπ* | – | – | TD-PBE0/6-311++G(d,p)/ IEFPCM |
| 341 (0.000) | H2O | S1 1nπ* | – | – | ||
| 342 (0.000) | ACN | S1 1nπ* | – | – | ||
| 305 (0.280) | – | S2 1ππ* | – | – | ||
| 313 (0.450) | H2O | S2 1ππ* | – | – | ||
| 313 (0.450) | ACN | S2 1ππ* | – | – | ||
| 6t-Guo [38] | 359 (0.000) | – | S1 1nπ* | 376 | – | TD-B3LYP/6-31+G* |
| 319 (0.434) | – | S2 1ππ* | – | – | ||
| 457 | – | T1 3ππ* | – | 496 | ||
| 357 (0.000) | – | S1 1nπ* | – | – | TD-M06/6-31+G* | |
| 314 (0.455) | – | S2 1ππ* | – | – | ||
| τ ISC (ps) | Φ ISC | Lifetimes | ΦΔ | |
|---|---|---|---|---|
| 2t-Ura | 0.35 ± 0.06 8 [60] | 0.75 ± 0.20 8 [60] | τ1 < 0.20, τ2 = 0.35 ± 0.06 8, 0.34 ± 0.09 1 [60] | – |
| 0.34 ± 0.09 1 [60] | – | – | ||
| 0.36 ± 0.03 8 [61] | – | – | – | |
| 2t-Thy | 0.62 ± 0.07 8 [60] | 1.00 ± 0.05 3 [62] | τ1 < 0.20, τ2 = 0.62 ± 0.07 8, 0.32 ± 0.09 1 [60], τ3 = 2.7 ± 0.5 (μs) [62] | 0.36 ± 0.02 3 [62] |
| 0.32 ± 0.09 1 [60] | 0.9 ± 0.1 2 [63] | τ1 < 0.10, τ2 = 0.78, τ3 = 203 [39] | – | |
| 2t-Thd | 0.41 ± 0.06 8 [64] | 0.9 ± 0.1 2 [63] | – | – |
| 2t-Cyt | 0.21 ± 0.05 8 [34] | >0.9 8 [34] | τ1 = 0.21 ± 0.05, τ2 = 0.48 ± 0.06 [34] | – |
| dm-4tUra | – | 1.0 ± 0.1 9 [65] | – | – |
| 4t-Ura | 0.24 ± 0.02 8 [61] | 0.9 ± 0.15 8 [61] | – | 0.49 ± 0.02 3 [61] |
| 4t-Thd | 0.24 ± 0.02 8 [64] | 1.0 ± 0.1 2 [66] | – | 0.50 ± 0.10 3 [66] |
| 0.24 ± 0.02 8 [67] | 0.85 ± 0.15 8 [67] | τ1 = 0.24 ± 0.02, τ2 = 84 ± 2 [67] | 0.42 ± 0.02 3 [64] | |
| 0.54 ± 0.01 1 [68] | – | τ1 = 0.54 ± 0.01, τ2 = 45 ± 5 [68], τ1 = 0.54, τ2 = 1.8 [57] | – | |
| ~10 1 [69] | ||||
| 4t-Urd | – | – | – | 0.7 [70] |
| – | 0.9 ± 0.1 9 [65] | – | 0.18 ± 0.04 10 [4,71] | |
| – | 0.02 2 [72] | – | 0.50 ±0.02 3 [4,71] | |
| 2,4-dtUra | 0.22 ± 0.04 8 [61] | 0.9 ± 0.15 8 [61] | – | 0.49 ± 0.02 3 [61] |
| 2,4-dtThy | 0.18 ± 0.02 8 [64] | >0.9 8 [64] | – | 0.46 ± 0.02 3 [64] |
| 6t-Gua | – | – | – | 0.58 ± 0.08 6 [56] |
| – | – | – | 0.56 ± 0.18 5 [56] | |
| 0.56 ± 0.06 8 [52] | ≥0.6± 0.2 8 [52] | τ1 = 0.56 ± 0.06, τ2 = 26 ± 3, τ3 = 830 ± 70 (ns), τ3 = 1420 ± 180 (ns) (N2) [52] | 0.21 ± 0.02 4 [52] | |
| – | – | 0.23 ± 0.02 5 [52] | ||
| 6t-Guo | 0.31 ± 0.05 7 [54] | 0.8 ± 0.2 7 [54] | τ1 = 0.31 ± 0.05, τ2 = 80 ± 15, τ3 = 460 ± 15 (ns), τ3 = 720 ± 10 (ns) (N2) [54] | 0.49 ± 0.09 6 [56] |
| – | – | – | 0.24 ± 0.02 5 [73] | |
| – | – | – | 0.14 ± 0.02 4 [73] | |
| – | – | – | 0.29 ± 0.02 3 [73] | |
| 0.36 ± 0.04 1 [54] | – | τ1 = 0.36 ± 0.04, τ2 = 32 ± 5 [54] | 0.55 ± 0.08 5 [56] |
| λmax (nm) | εmax (M−1 cm−1 × 104) | λemission (nm) | Φ (×10−4) | Solvent | |||
|---|---|---|---|---|---|---|---|
| F | P | F | P | ||||
| 2t-Ura | 274 | 1.45 | – | – | – | – | H2O, pH = 7 [84] |
| 268 | 1.19 | – | – | – | – | ACN [84] | |
| 274, 291 1 | _ | – | – | – | – | MeOH [84] | |
| 271, 290 1 | – | N.D. | 405, 427, 455 | – | 7.0 | EtOH, 77 K [86] | |
| 265 | – | – | – | – | – | PBS, pH = 7.4 [61] | |
| 2t-Urd | 275 | 1.67 | – | – | – | – | H2O, pH = 7 [84] |
| 268 | 1.04 | – | – | – | – | ACN [84] | |
| 2t-Thy | 275, 290 1 | 1.29, 1.33 | N.D. | N.D. | – | – | ACN [62] |
| – | – | – | 454 | – | – | THF, 77 K [62] | |
| 290 | 1.24 | N.D. | 480 | – | 5.0, 9.8 2 | ACN, Ar [63] | |
| – | – | – | 448 | – | – | DCM:MeOH, 77 K [63] | |
| 275, 290 1 | – | N.D. | 425, 451, 483 | – | 7.0 | EtOH, 77 K [86] | |
| 275 | – | – | – | – | – | PBS, pH = 7.4 [64] | |
| 2t-Thd | 285 | 1.41 | N.D. | N.D. | <1 3 | – | ACN, Ar [63] |
| – | – | – | 448 | – | – | DCM:MeOH, 77 K [63] | |
| 277 | – | – | – | – | – | PBS, pH = 7.4 [64] | |
| 2t-Cyt | 270, 242, 220 1 | 1.82 | – | – | – | – | H2O, pH = 7 [84] |
| 269, 241, 219 1 | – | – | – | – | – | PBS, pH = 7.4 [34] | |
| 283, 233 | 1.67, 1.19 | – | – | – | – | ACN [85] | |
| 286 | 1.69 | – | – | – | – | EtOAc [85] | |
| 285 | 1.80 | – | – | – | – | DMSO [85] | |
| 280, 242, 210 1 | 1.79, 1.42, 0.65 | – | – | – | – | EtOH [85] | |
| 278, 242, 212 1 | 1.78, 1.48, 0.71 | – | – | – | – | MeOH [85] | |
| 269, 242, 218 1 | 1.80, 1.80, 0.90 | – | – | – | – | PBS, pH = 7.4 [85] | |
| 5m-2tCyt | 266 | 2.10 | – | – | – | – | H2O, pH = 7 [84] |
| 2t-Cyd | 274, 270 1 | 2.30, 1.40 | – | – | – | – | H2O, pH = 7 [84] |
| λmax (nm) | Solvent | Excitation | λemission (nm) | Method | ||
|---|---|---|---|---|---|---|
| F | P | |||||
| 2t-Ura 1 [89] | 329 (0.000) | – | S1 1nπ* | 358 | – | MS(6/4)-CASPT2/ANO-RCC-VQZQ// SA(6/4)-CASSCF(16,12) |
| 292 (0.350) | – | S2 1ππ* | – | – | ||
| 353 | – | T1 3ππ* | – | 386 | ||
| 322 (0.000) | – | S1 1nπ* | – | – | MRCIS(6,5)/cc-pVDZ// SA(4/2)-CASSCF(12,9) | |
| 215 (0.110) | – | S2 1ππ* | – | – | ||
| 2t-Ura 1 [90] | 323 | – | S1 1nπ* | 325 | – | CASPT2/cc-pVDZ// SA(4/3)-CASSCF(16,11) |
| 278 | – | S2 1ππ* | – | – | ||
| – | – | T1 3ππ* | – | 358 | ||
| 347 | – | S1 1nπ* | – | – | TD-B3LYP/aug-cc-pVDZ | |
| 304 | – | S2 1ππ* | – | – | ||
| 2t-Ura [91] | 340 (0.000) | – | S1 1nπ* | 369 | – | SS-CASPT2/ANO-L-vDZP// SA(6/6)-CASSCF(14,10) |
| 303 (0.236) | – | S2 1ππ* | 349 | – | ||
| 395 | – | T1 3ππ* | – | 435 | ||
| 2t-Ura [31] | 346 (0.000) | – | S1 1nπ* | – | – | TD-B3LYP/6-311++G(d,p) |
| 304 (0.051) | – | S2 1ππ* | – | – | ||
| 317 (0.000) | H2O | S1 1nπ* | – | – | ||
| 286 (0.145) | H2O | S2 1ππ* | – | – | ||
| 318 (0.000) | ACN | S1 1nπ* | – | – | ||
| 287 (0.144) | ACN | S2 1ππ* | – | – | ||
| 2t-Ura [92] | 320 (0.000) | ACN | S1 1nπ* | – | – | TD-B3LYP/6-31+G(d)/PCM |
| 283 (0.108) | ACN | S2 1ππ* | – | – | ||
| 2t-Ura [86] | 332 (0.000) | EtOH | S1 1nπ* | – | – | TD-B3LYP/aug-cc-pVDZ/PCM |
| 298 (0.110) | EtOH | S2 1ππ* | – | – | ||
| 2t-Thy [37] | 325 | H2O | S1 1nπ* | – | – | TD-M06/6-31+G*/PCM |
| 288 | H2O | S2 1ππ* | – | – | ||
| 2t-Thy 1,2 [93] | 286 (0.578) | – | 1ππ* | – | – | MS-CASPT2(14,10)/ANO-RCC-VTZP |
| 295 (0.243) | – | 1ππ* | – | – | DFT/MRCI | |
| 2t-Thy [62] | 315 (0.000) | ACN | S1 1nπ* | – | – | TD-B3LYP/6-31+G(d,p)/PCM |
| 295 (0.145) | ACN | S2 1ππ* | – | – | ||
| 2t-Thy [86] | 323 (0.000) | EtOH | S1 1nπ* | – | – | TD-B3LYP/aug-cc-pVDZ/PCM |
| 301 (0.125) | EtOH | S2 1ππ* | – | – | ||
| 2t-Thy [53] | 339 (0.000) | – | S1 | – | – | TD-B3LYP/6-311++G(d,p) |
| 308 (0.054) | – | S2 | – | – | ||
| 2t-Cyt 1 [34] | 340 (0.001) | – | S1 1nπ* | 420 | – | MS-CASPT2/CASSCF(14,10)/ ANO-RCC-VQZP//RI-MP2/cc-pVQZ |
| 332 (0.097) | – | S2 1ππ* | 410 | – | ||
| 368 | – | T1 3ππ* | – | 435 | ||
| 2t-Cyt [85] | 358 (0.000) | – | S1 1nπ* | – | – | MS-CASPT2(14,10)/cc-pVDZ/PCM//BP86/aug-cc-pVDZ/COSMO |
| 315 (0.000) | EtOAc | S1 1nπ* | – | – | ||
| 309 (0.000) | ACN | S1 1nπ* | – | – | ||
| 309 (0.000) | DMSO | S1 1nπ* | – | – | ||
| 286 (0.000) | EtOH | S1 1nπ* | – | – | ||
| 284 (0.000) | MeOH | S1 1nπ* | – | – | ||
| 276 (0.020) | H2O | S1 1nπ* | – | – | ||
| 357 (0.020) | – | S2 1ππ* | – | – | ||
| 315 (0.010) | EtOAc | S2 1ππ* | – | – | ||
| 311 (0.020) | ACN | S2 1ππ* | – | – | ||
| 307 (0.020) | DMSO | S2 1ππ* | – | – | ||
| 294 (0.030) | EtOH | S2 1ππ* | – | – | ||
| 292 (0.030) | MeOH | S2 1ππ* | – | – | ||
| 293 (0.010) | H2O | S2 1ππ* | – | – | ||
| 2t-Cyt [53] | 403 (0.000) | – | S1 | – | – | TD-B3LYP/6-311++G(d,p) |
| 371 (0.010) | – | S2 | – | – | ||
| λmax (nm) | εmax (M−1 cm−1 × 104) | λemission (nm) | Φ (×10−4) | Solvent | |||
|---|---|---|---|---|---|---|---|
| F | P | F | P | ||||
| 4t-Ura | 327 | 1.28 | – | – | – | – | ACN [84] |
| 328 | 1.67 | – | – | – | – | H2O, pH = 7 [84] | |
| 327 | 1.94 | – | – | – | – | ACN [99] | |
| 330 | – | – | 550 | – | 3.0 1 | H2O [17] | |
| dm-4tUra | 328 | 1.98 | 415 | 528 | 0.6 1 | 14.0 1 | CCl4, He [102] |
| 317 | – | 416 | 535 | 1.5 1 | 25.0 1 | PFDMCH, He [102] | |
| 329 | 1.88 | 420 | 535 | 0.4 | – | H2O [102] | |
| 4t-Urd | 331 | 2.30 | – | – | – | – | H2O, pH = 7 [84] |
| 331 | 1.19 | – | – | – | – | ACN [84] | |
| Ac-4tUrd | 328 | 2.06 | 420 | 550 | 1.0 | 2.0 | ACN [100] |
| 330 | 2.06 | 420 | 540 | – | 123 1 | CCl4, He [101] | |
| – | – | – | – | – | 25 1 | ACN, Ar [105] | |
| 4t-Thd | 337 | – | 410 | 550 | – | – | ACN [68] |
| 335 | – | 400 | 542 | – | – | PBS, pH = 7.4 [67] | |
| 337 | 1.94 | N.D. | N.D. | – | – | ACN [66] | |
| – | – | – | 494, 512, 560 2 | – | – | EtOH, 77 K [66] | |
| λmax (nm) | Solvent | Excitation | λemission (nm) | Method | ||
|---|---|---|---|---|---|---|
| F | P | |||||
| 4t-Ura [31] | 446 (0.000) | – | S1 1nπ* | – | – | TD-B3LYP/6-311++G(d,p) |
| 295 (0.298) | – | S2 1ππ* | – | – | ||
| 387 (0.000) | H2O | S1 1nπ* | – | – | ||
| 298 (0.437) | H2O | S2 1ππ* | – | – | ||
| 389 (0.000) | ACN | S1 1nπ* | – | – | ||
| 299(0.440) | ACN | S2 1ππ* | – | – | ||
| 4t-Ura [104] | 401 | – | S1 1nπ* | – | – | CASSCF |
| 259 | – | S2 1ππ* | – | – | ||
| 440 | – | S1 1nπ* | – | – | MCQDPT2 | |
| 318 | – | S2 1ππ* | – | – | ||
| 4t-Ura [99] | 444 (0.000) | – | S1 1nπ* | – | – | TD-B3LYP/6-311++G(d,p) |
| 294 (0.299) | – | S2 1ππ* | – | – | ||
| dm-4tUra [103] | 408 | – | S1 1nπ* | – | – | EOM-CC2/aug-cc-pVDZ |
| 296 | – | S2 1ππ* | – | – | ||
| 4t-Thy 1 [93] | 310 (0.662) | – | 1ππ* | – | – | MS-CASPT2 (14,10)/ANO-RCC/VTZP |
| 312 (0.427) | – | 1ππ* | – | – | DFT/MRCI | |
| 4t-Thy [53] | 438 (0.000) | – | S1 | – | – | TD-B3LYP/6-311++G(d,p) |
| 298 (0.282) | – | S2 | – | – | ||
| 4t-Thy [37] | 403 | H2O | S1 1nπ* | – | – | TD-M06/6-31+G*/PCM |
| 312 | H2O | S2 1ππ* | – | – | ||
| 4t-Thy [83] | 419 (0.000) | H2O (PCM + 7 H2O ) | S1 1nπ* | – | – | MS-CASPT2 (12,9)/ANO-L |
| 326 (0.546) | H2O (PCM + 7 H2O ) | S2 1ππ* | 430 | – | ||
| 448 | – | T1 3ππ* | – | 570 | ||
| 4t-Thd 1 [67] | 385 (0.000) | H2O (PCM + 2 H2O ) | S1 1nπ* | – | – | TD-PBE0/IEFPCM/6-311++G(d,p) |
| 310 (0.509) | H2O (PCM + 2 H2O ) | S2 1ππ* | – | – | ||
| 478 | H2O (PCM + 2 H2O ) | T1 3ππ* | – | 557 2 | ||
| 4t-Thd [66] | 292 (0.379) | – | S2 1ππ* | – | – | TD-B3LYP/6-31G(d,p) |
| 4t-Thd [69] | ~435 | – | S1 1nπ* | – | – | TD-B3LYP/6-31G(d,p) |
| ~294 | – | S2 1ππ* | – | – | ||
| 4t-Thd [95] | 413 | H2O (MM) | S1 1nπ* | – | – | QM(CASPT2//CASSCF(10,8))/6-31G*/MM |
| 302 | H2O (MM) | S2 1ππ* | – | – | ||
| λmax (nm) | εmax (M−1 cm−1 × 104) | λemission (nm) | Solvent | ||
|---|---|---|---|---|---|
| F | P | ||||
| 2,4-dtUra | 351 | 0.46 | – | – | H2O, pH = 7 [84] |
| 351 | 0.10 | – | – | ACN [84] | |
| 2,4-dtUrd | 345 | 1.14 | – | – | H2O, pH = 7 [84] |
| 351 | 1.00 | – | – | ACN [84] | |
| 2,4-dtThy | 363 | 0.97 | – | – | PBS [64] |
| λmax (nm) | Solvent | Excitation | λemission (nm) | Method | ||
|---|---|---|---|---|---|---|
| F | P | |||||
| 2,4-dtUra [31] | 449 (0.000) | – | S1 1nπ* | – | – | TD-B3LYP/6-311++G(d,p) |
| 349 (0.014) | – | S2 1ππ* | – | – | ||
| 396 (0.000) | H2O | S1 1nπ* | – | – | ||
| 335 (0.063) | H2O | S2 1ππ* | – | – | ||
| 399 (0.000) | ACN | S1 1nπ* | – | – | ||
| 336 (0.063) | ACN | S2 1ππ* | – | – | ||
| 2,4-dtThy 1 [93] | 333 (0.216) | – | 1ππ* | – | – | MS-CASPT2(14,10)/ANO-RCC-VTZP |
| 343 (0.078) | – | 1ππ* | – | – | DFT/MRCI | |
| 2,4-dtThy 1 [96] | 400 | H2O | S1 1nπ* | 446 | – | QM(MS-CASPT2)/MM |
| 334 | H2O | S2 1ππ* | – | – | ||
| 445 | H2O | T1 1ππ* | – | 474 | ||
| 2,4-dtThy [53] | 443 (0.000) | – | S1 | – | – | TD-B3LYP/6-311++G(d,p) |
| 361 (0.014) | – | S2 | – | – | ||
| 2,4-dtThy [37] | 409 | H2O | S1 | – | – | TD-M06/6-31+G*/PCM |
| 340 | H2O | S2 | – | – | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arslancan, S.; Martínez-Fernández, L.; Corral, I. Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules 2017, 22, 998. https://doi.org/10.3390/molecules22060998
Arslancan S, Martínez-Fernández L, Corral I. Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules. 2017; 22(6):998. https://doi.org/10.3390/molecules22060998
Chicago/Turabian StyleArslancan, Serra, Lara Martínez-Fernández, and Inés Corral. 2017. "Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments" Molecules 22, no. 6: 998. https://doi.org/10.3390/molecules22060998
APA StyleArslancan, S., Martínez-Fernández, L., & Corral, I. (2017). Photophysics and Photochemistry of Canonical Nucleobases’ Thioanalogs: From Quantum Mechanical Studies to Time Resolved Experiments. Molecules, 22(6), 998. https://doi.org/10.3390/molecules22060998