
Reactions of 5-Indolizyl Lithium Compounds with Some Bielectrophiles



Abstract
: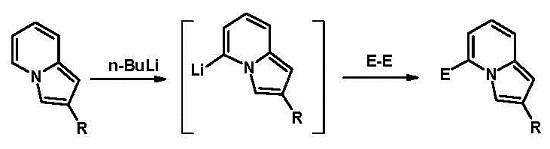
1. Introduction
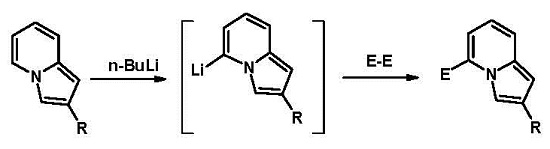
2. Results and Discussion
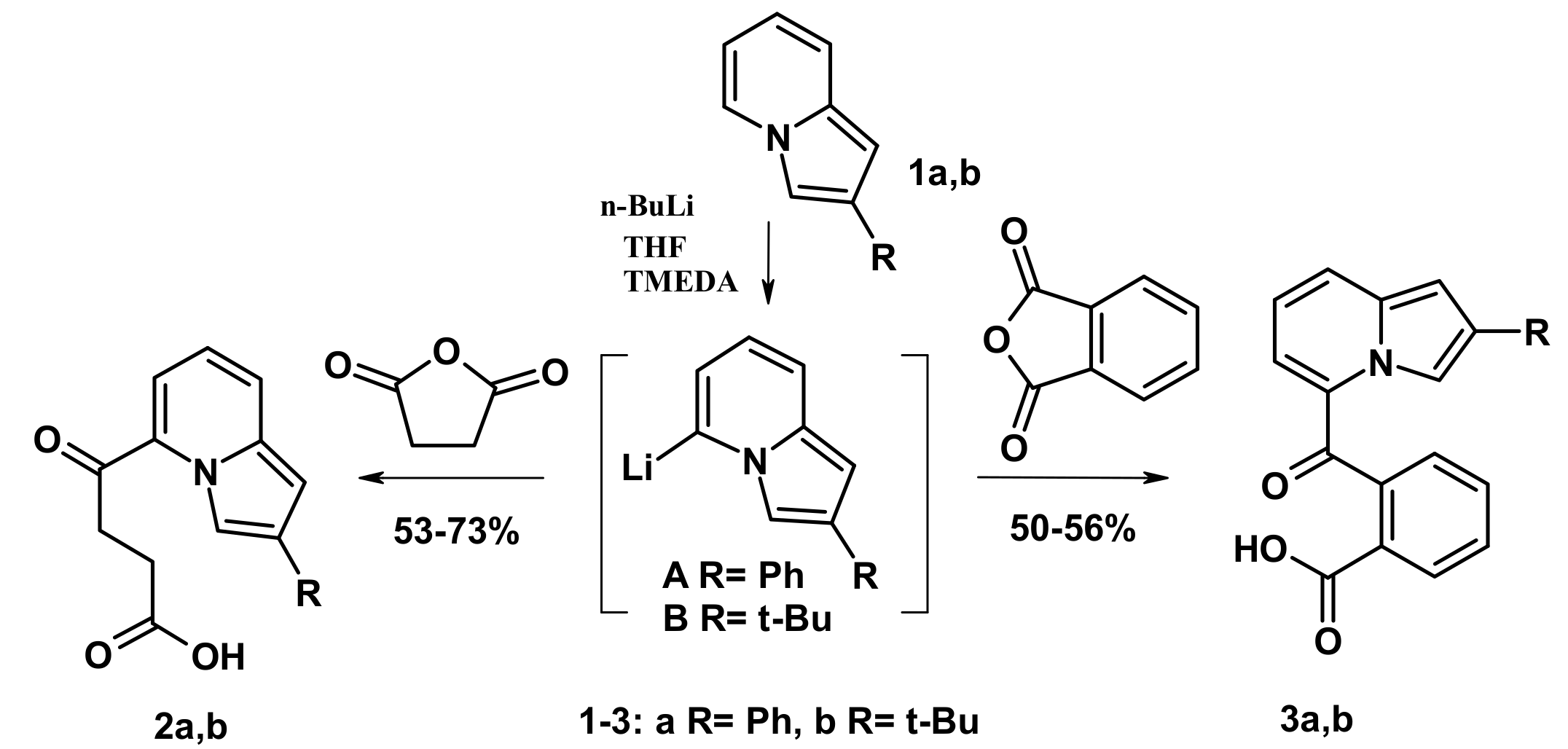
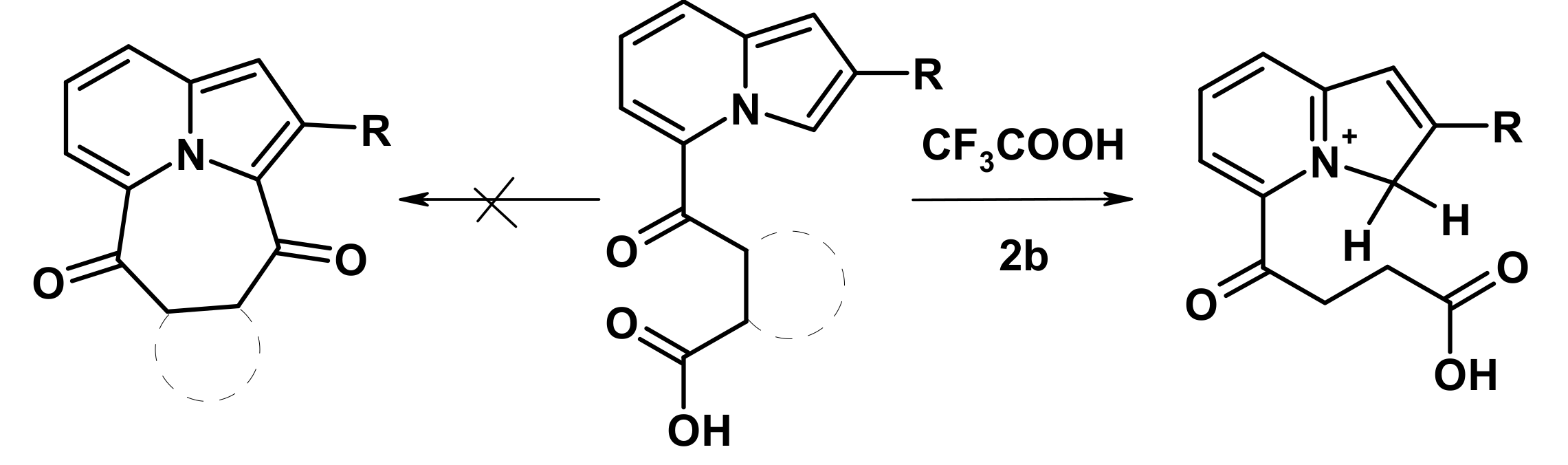
2.1. The Reactions with Succinic and Phtalic Anhydrides
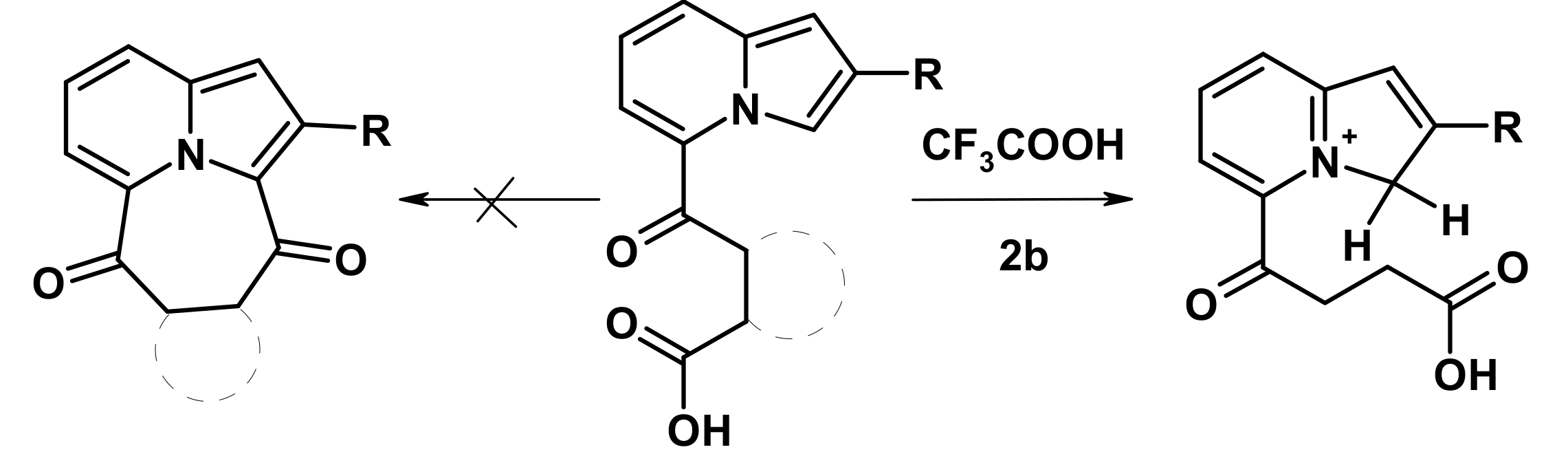
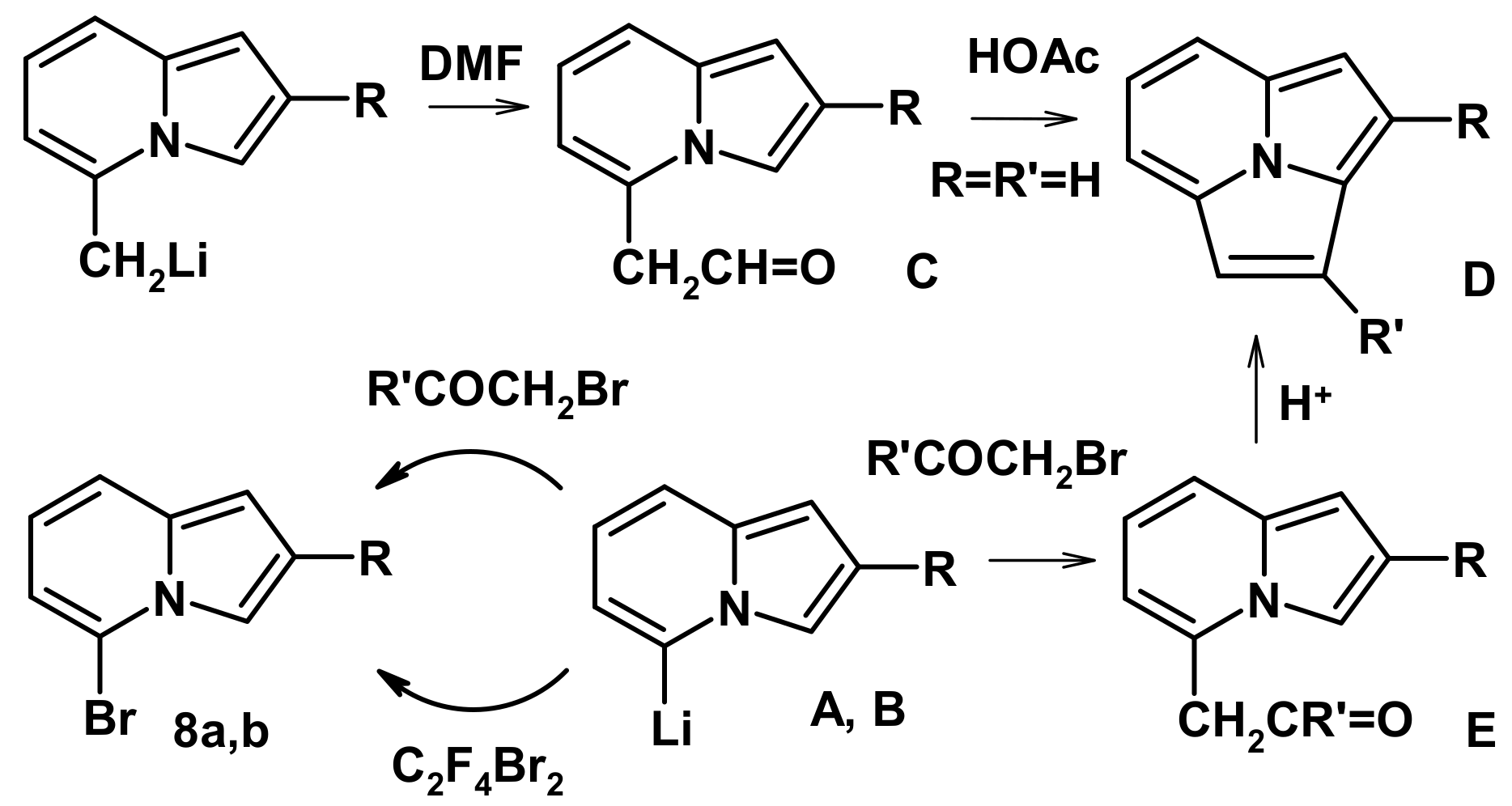
2.2. The Direction of Protonation
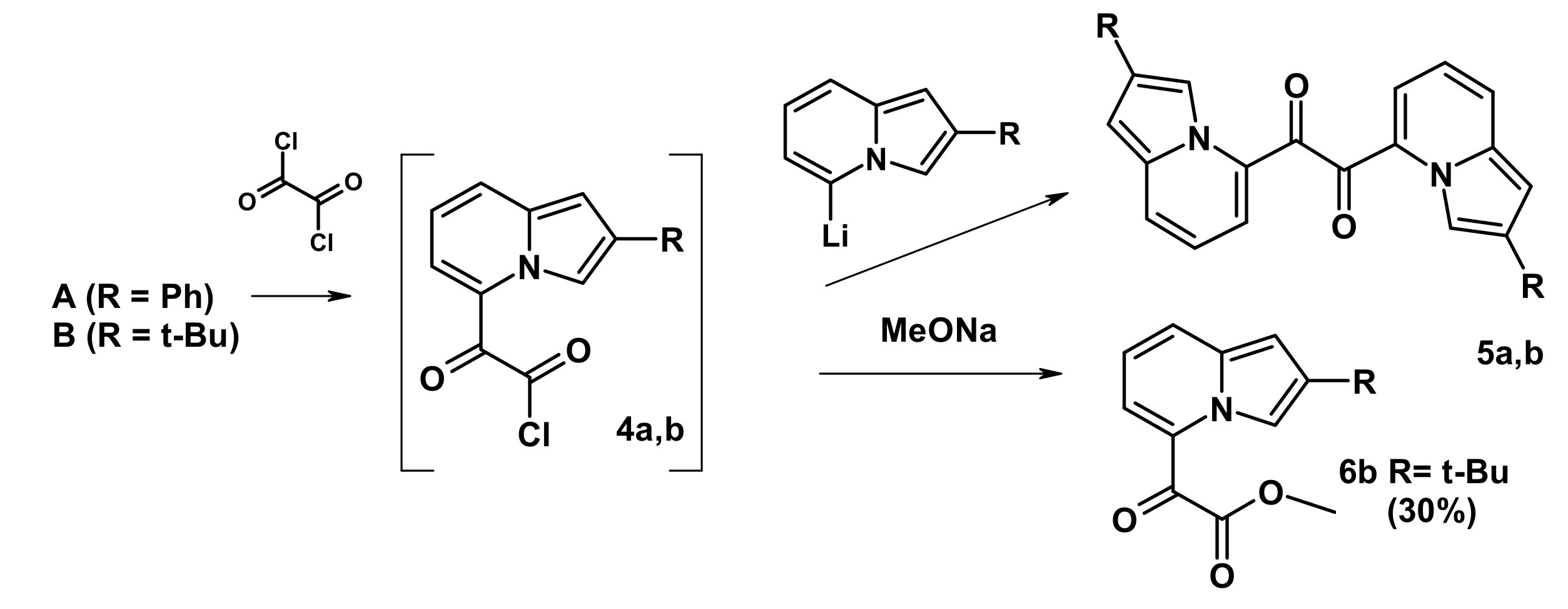
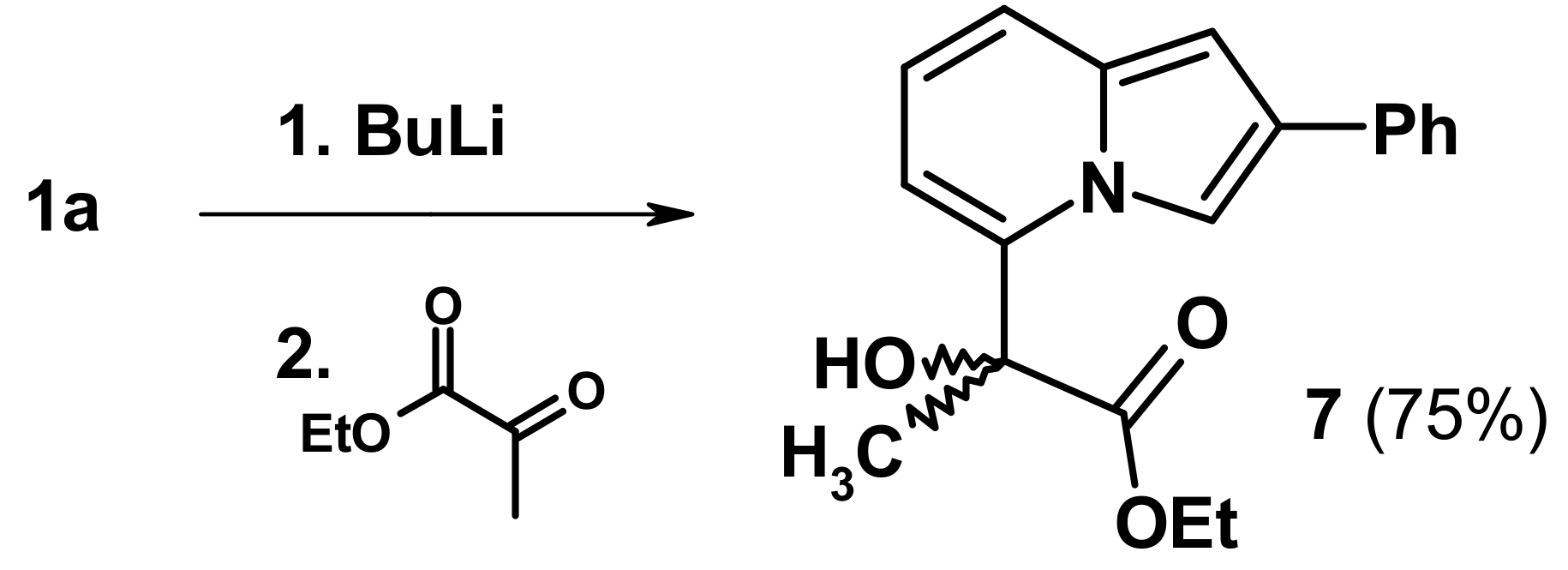
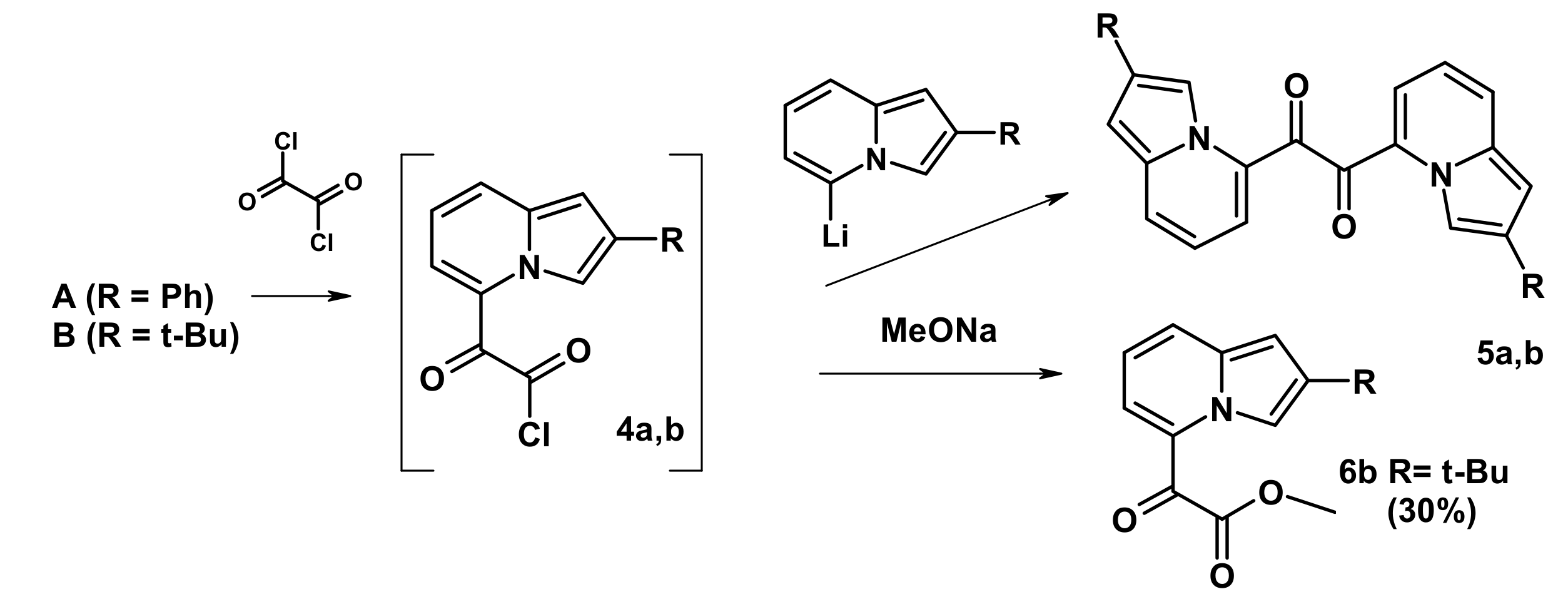
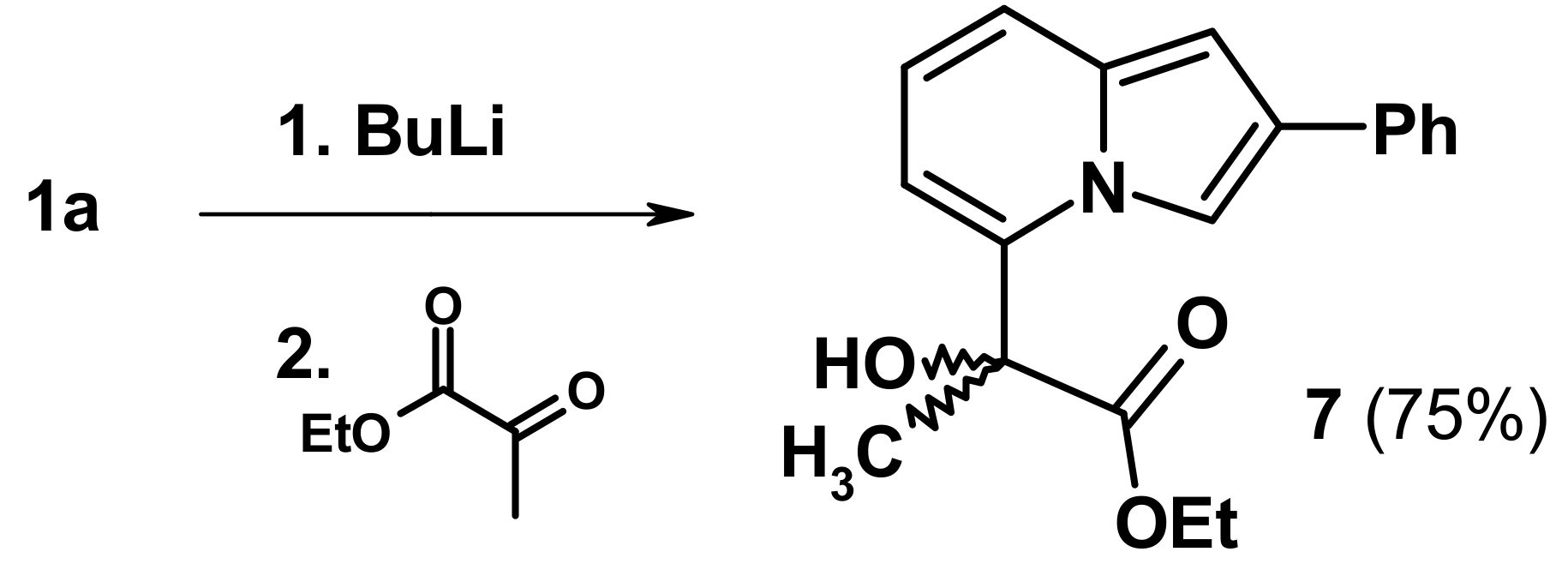
2.3. The Reaction with Oxallyl Chloride and Ethyl Pyruvate
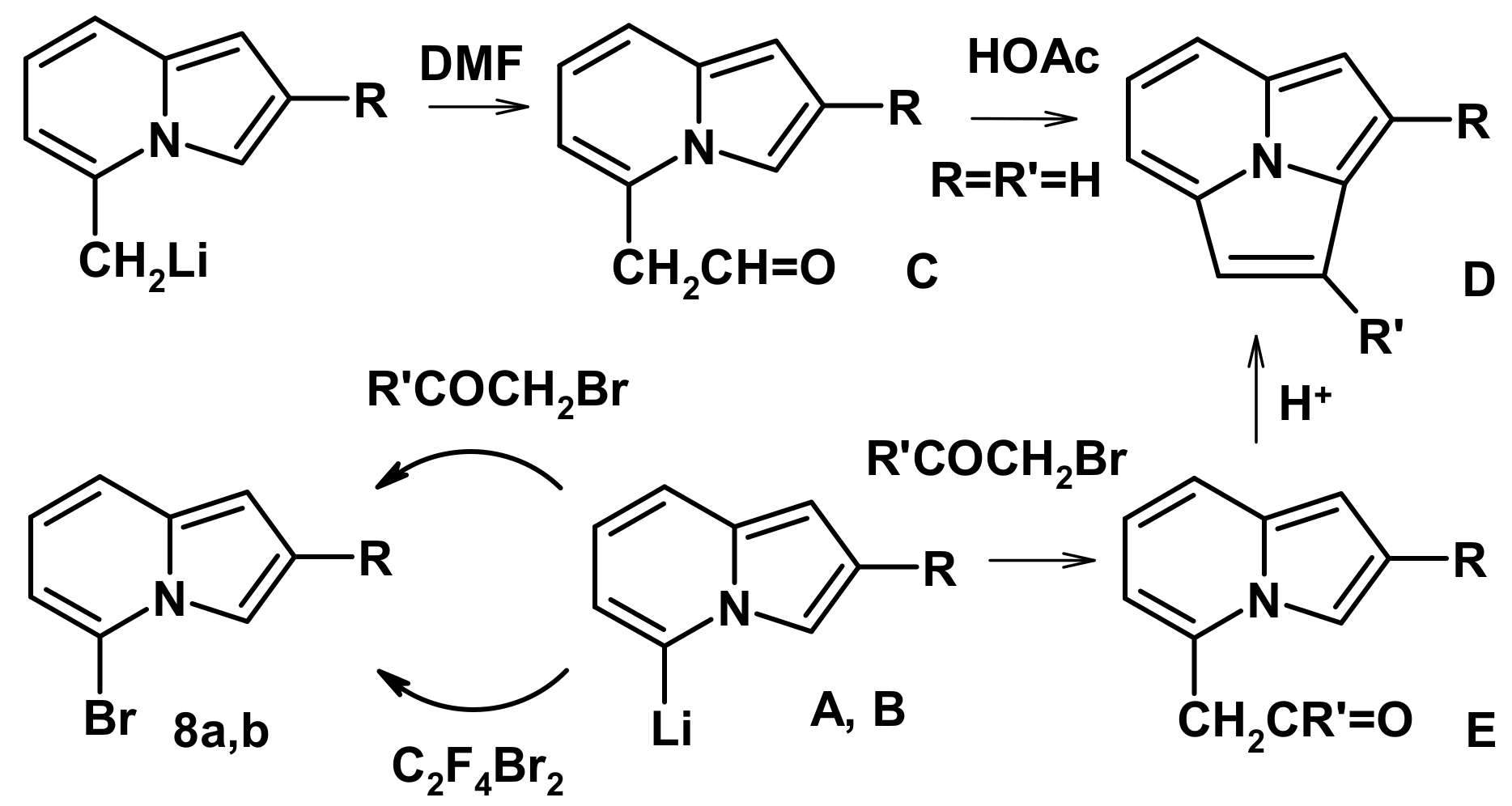
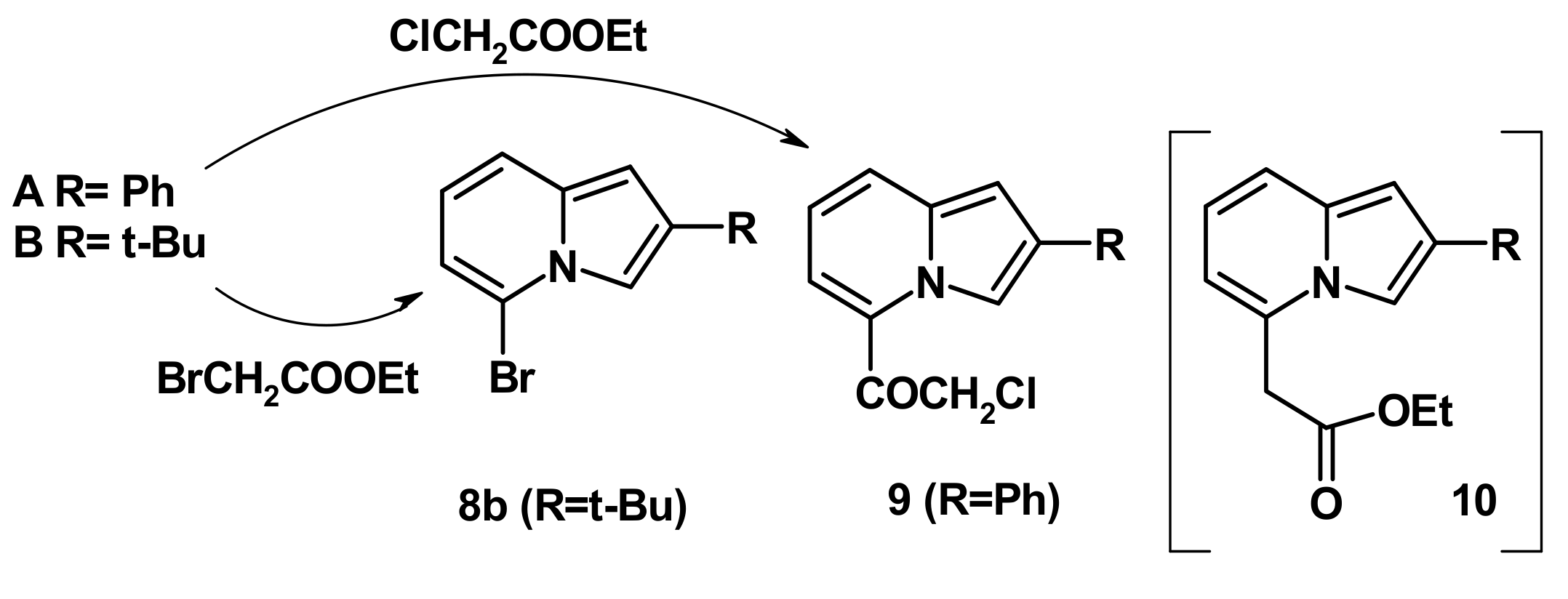
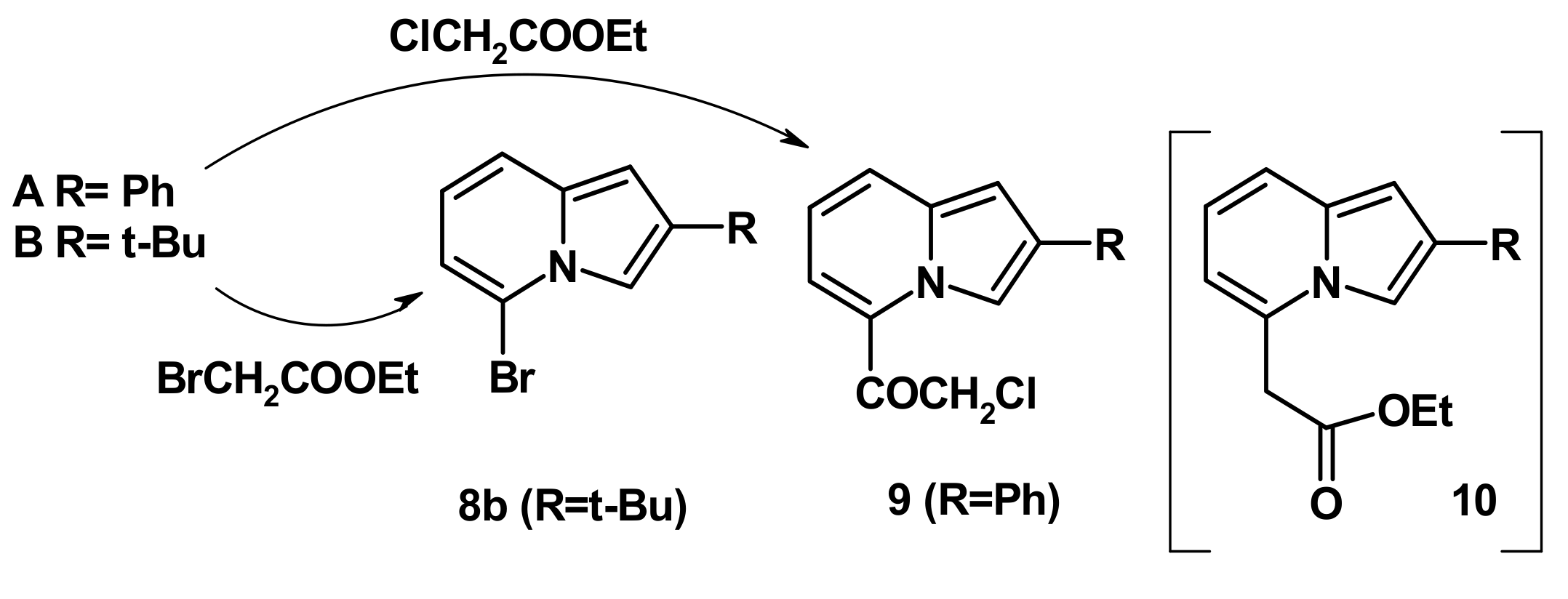
2.4. The Reaction with Phenacyl Bromide and Haloacetic Acid Esters
2.5. Conclusions
3. Experimental Section
3.1. General Information
3.2. Synthesis
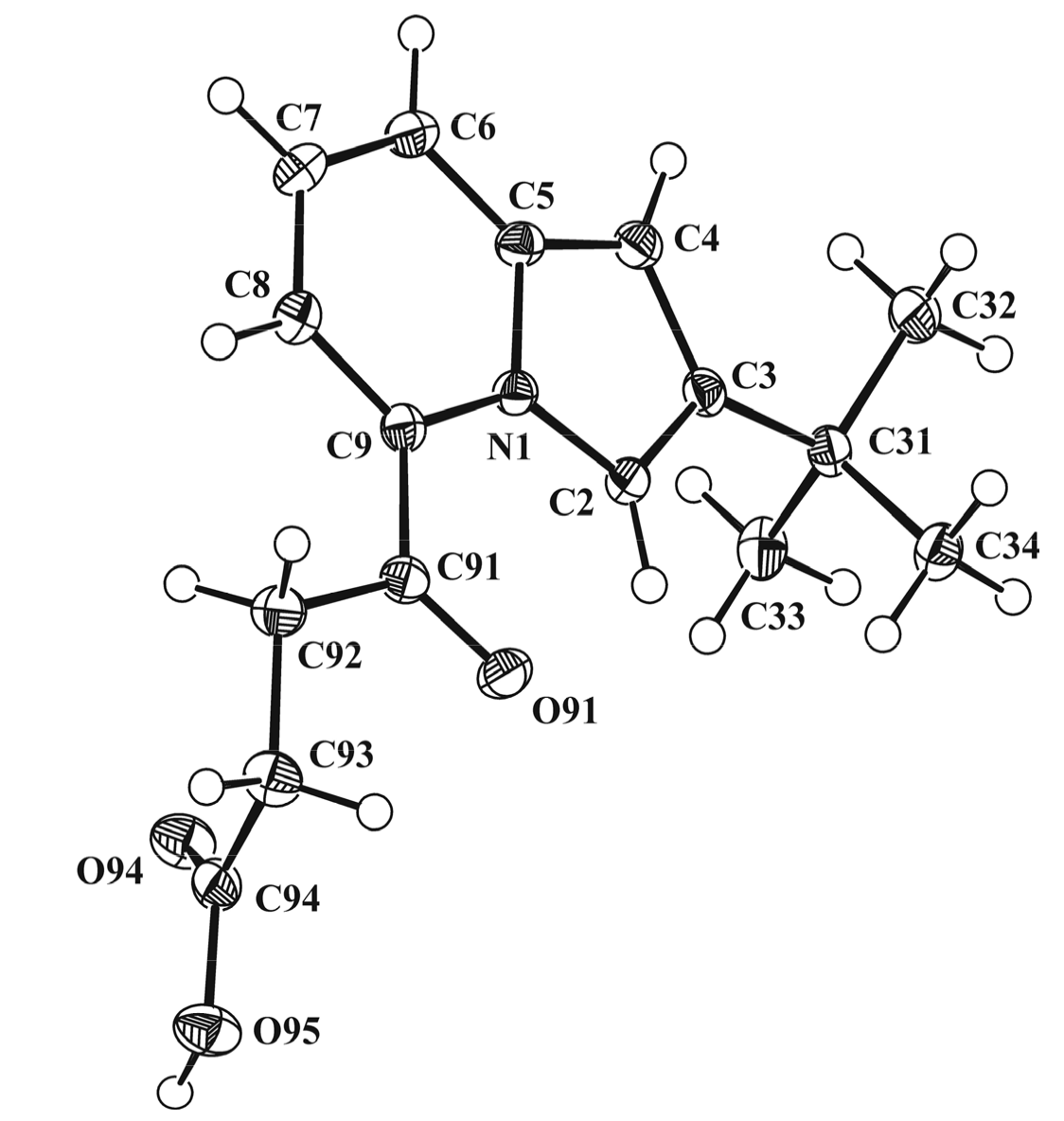
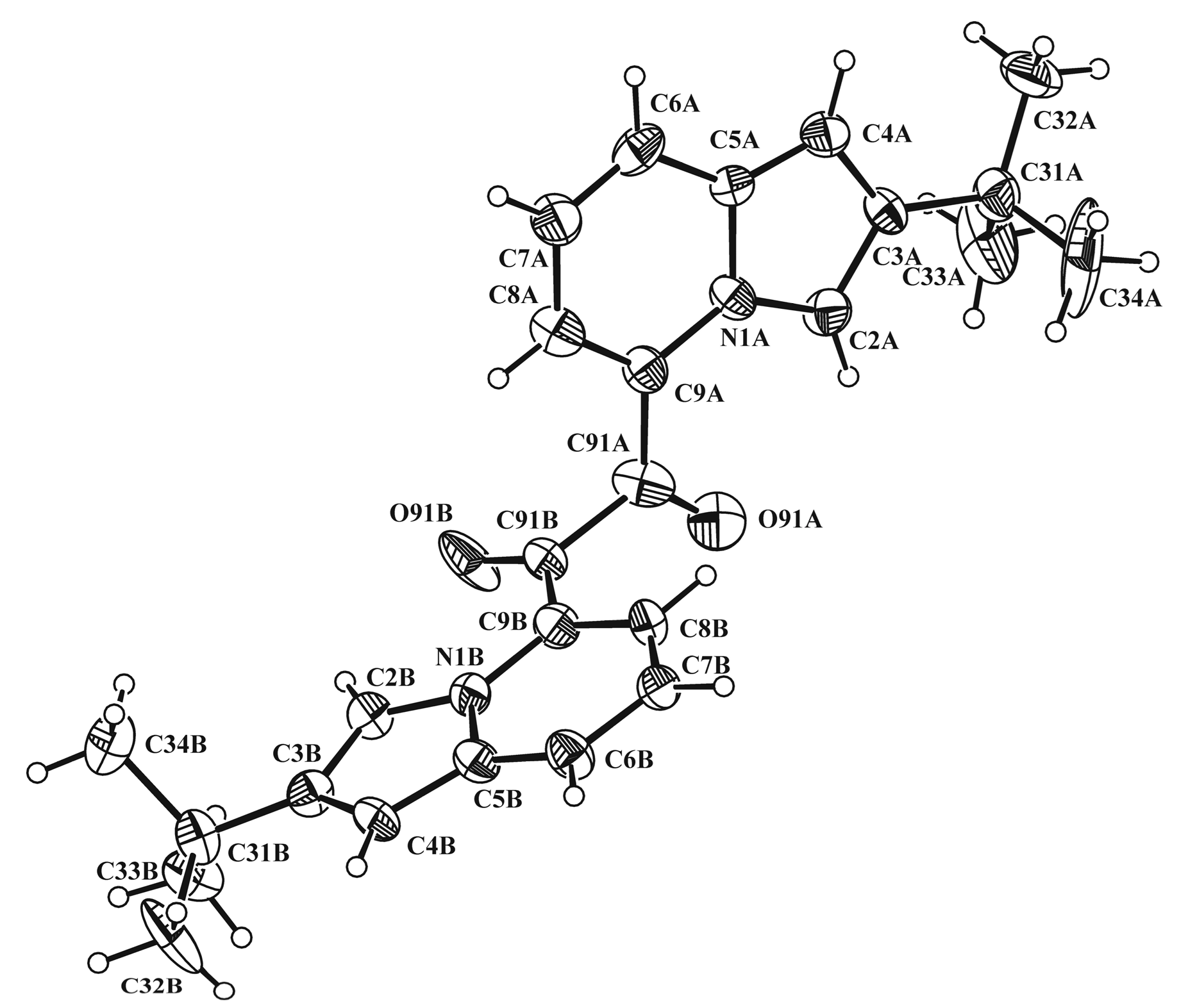
3.3. X-ray Diffraction Study of Compounds 2b and 5b
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, B.; Wang, Z.-J.; Wu, N.; Li, M.; You, J.; Lan, J. Discovery of a full-color-tunable fluorescent core framework through direct C H (hetero) arylation of N-heterocycles. Chem. Eur. J. 2012, 18, 1599–1603. [Google Scholar] [CrossRef] [PubMed]
- Rotaru, A.V.; Druta, I.; Oeser, T.; Mϋller, T.J.J. A novel coupling 1,3-dipolar cycloaddition sequence as a three-component approach to highly fluorescent indolizines. Helv. Chim. Acta 2005, 88, 1798–1812. [Google Scholar] [CrossRef]
- Kim, E.; Lee, S.; Park, S.B. A Seoul-fluor-based bioprobe for lipid droplets and its application in image-based high throughput screening. Chem. Commun. 2012, 48, 2331–2333. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.S.; Mmatli, E.E. Recent progress in synthesis and bioactivity studies of indolizines. Eur. J. Med. Chem. 2011, 46, 5237–5257. [Google Scholar] [CrossRef] [PubMed]
- Vemula, V.R.; Vurukonda, S.; Bairi, C.K. Indolizine derivatives: Recent advances and potential pharmacological activities. Int. J. Pharm. Sci. Rev. Res. 2011, 11, 159–163. [Google Scholar]
- Huang, W.; Zuo, T.; Luo, X.; Jin, H.; Liu, Z.; Yang, Z.; Yu, X.; Zhang, L.; Zhang, L. Indolizine derivatives as HIV-1 VIF-elongin C interaction inhibitors. Chem. Biol. Drug Des. 2013, 81, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Flitsch, W. Pyrroles with fused six-membered heterocyclic rings: A-fused. In Comprehensive Heterocyclic Chemistry; Katritzky, A., Rees, C.W., Eds.; Pergamon Press: Oxford, UK, 1984; Volume 4, pp. 443–496. [Google Scholar]
- Swinborne, P.-J.; Hunt, J.H.; Klinkert, G. Advances in indolizine chemistry. Adv. Heterocycl. Chem. 1978, 23, 103–167. [Google Scholar]
- Prostakov, N.S.; Baktibaev, O.B. Indolizines. Usp. Khim. 1975, 9, 1649–1687. [Google Scholar] [CrossRef]
- Renard, M.; Gubin, J. Metallation of 2-Phenylindolizine. Tetrahedron Lett. 1992, 33, 4433–4434. [Google Scholar] [CrossRef]
- Kuznetsov, A.G.; Bush, A.A.; Rybakov, V.B.; Babaev, E.V. An Improved Synthesis of Some 5-Substituted Indolizines Using Regiospecific Lithiation. Molecules 2005, 10, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.G.; Bush, A.A.; Babaev, E.V. Synthesis and Reactivity of 5-Br(I)-Indolizines and their Parallel Cross-coupling Reactions. Tetrahedron 2007, 64, 749–756. [Google Scholar] [CrossRef]
- Shadrin, I.A.; Rzhevskii, S.A.; Rybakov, V.B.; Babaev, E.V. Sonogashira Reaction of the Indolizine Ring. Synthesis 2015, 47, 2961–2964. [Google Scholar]
- Babaev, E.V.; Torocheshnikov, V.N.; Bobrovsky, S.I. NMR spectra of indolizines and their sigma-complexes. Chem. Heterocycl. Comp. 1995, 31, 1079–1087. [Google Scholar] [CrossRef]
- Fraser, M.; Mc Kenzie, S.; Reid, D.H. Nuclear magnetic resonance. Part IV. The protonation of indolizines. J. Chem. Soc. (B) 1966, 1, 44–48. [Google Scholar] [CrossRef]
- Armarego, W.L.F. C-1 and C-3 protonation of indolizines. J. Chem. Soc. (B) 1966, 2, 191–194. [Google Scholar] [CrossRef]
- Windgassen, R.J.; Saunders, W.H.; Boekelheide, V. Cyclazines. A new class of aromatic heterocycles. J. Am. Chem. Soc. 1959, 81, 1459–1465. [Google Scholar] [CrossRef]
- Bruker. APEX2 and SAINT-Plus; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Stoe & Cie. X-AREA; Stoe & Cie GmbH: Darmstadt, Germany, 2012. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for WI: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Groom, C.R.; Allen, F.H. The Cambridge Structural Database in Retrospect and Prospect. Angew. Chem. Int. Ed. 2014, 53, 662–671. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–3, 5, 8 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Compound Code | 2b | 5b |
|---|---|---|
| Empirical formula | C16H19NO3 | C26H28N2O2 |
| Formula weight | 273.32 | 400.50 |
| Temperature, K | 100(2) | 295(2) |
| Wavelength, Å | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | Pbca | P21/c |
| a, Å | 8.4621(5) | 14.219(2) |
| b, Å | 10.9254(6) | 9.9954(13) |
| c, Å | 30.2222(18) | 16.642(3) |
| α, deg | 90 | 90 |
| β, deg | 90 | 107.735(13) |
| γ, deg | 90 | 90 |
| Volume, Å3 | 2794.1(3) | 2252.8(6) |
| Z | 8 | 4 |
| Density (calculated), Mg/m3 | 1.299 | 1.181 |
| Absorption coefficient, mm−1 | 0.090 | 0.075 |
| F(000) | 1168 | 856 |
| Crystal size, mm | 0.30 × 0.15 × 0.15 | 0.10 × 0.10 × 0.10 |
| Theta range for data collection, deg. | 2.70–30.02 | 2.41–25.24 |
| Index ranges | −11 ≤ h ≤ 11, −15 ≤ k ≤ 15, −42 ≤ l ≤ 42 | −17 ≤ h ≤ 8, −12 ≤ k≤ 12, −20 ≤ l ≤ 20 |
| Reflections collected | 30,104 | 34,200 |
| Independent reflections [Rint] | 4074 [0.0411] | 11014 [0.0261] |
| Max. and min. transmission | ------------------------ | --------------------------- |
| Data/restraints/parameters | 4074/0/188 | 34,200/0/274 |
| Goodness-of-fit on F2 | 1.053 | 0.843 |
| R1/wR2, I > 2δ(I) | 0.0537/0.1382 | 0.0448/0.0841 |
| R1/wR2, all data | 0.0682/0.153 | 0.1694/0.1156 |
| Δрmax/Δрmin, e/Å3 | 0.512/−0.242 | 0.203/−0.178 |
| Extinction coefficient | ------------------------ | 0.0053(5) |
| Diffractometer model | Bruker APEX-II CCD | STADI-VARY Pilatus-100K |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rzhevskii, S.A.; Rybakov, V.B.; Khrustalev, V.N.; Babaev, E.V. Reactions of 5-Indolizyl Lithium Compounds with Some Bielectrophiles. Molecules 2017, 22, 661. https://doi.org/10.3390/molecules22040661
Rzhevskii SA, Rybakov VB, Khrustalev VN, Babaev EV. Reactions of 5-Indolizyl Lithium Compounds with Some Bielectrophiles. Molecules. 2017; 22(4):661. https://doi.org/10.3390/molecules22040661
Chicago/Turabian StyleRzhevskii, Sergey A., Victor B. Rybakov, Victor N. Khrustalev, and Eugene V. Babaev. 2017. "Reactions of 5-Indolizyl Lithium Compounds with Some Bielectrophiles" Molecules 22, no. 4: 661. https://doi.org/10.3390/molecules22040661
APA StyleRzhevskii, S. A., Rybakov, V. B., Khrustalev, V. N., & Babaev, E. V. (2017). Reactions of 5-Indolizyl Lithium Compounds with Some Bielectrophiles. Molecules, 22(4), 661. https://doi.org/10.3390/molecules22040661