The Eukaryotic Elongation Factor 1 Alpha (eEF1α) from the Parasite Leishmania infantum Is Modified with the Immunomodulatory Substituent Phosphorylcholine (PC)

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cultivation of Leishmania infantum Promastigotes
2.2. Immunofluorescence
2.3. Protein Isolation
2.4. Two-Dimensional Gel Electrophoresis and Detection of PC-Modified Proteins
2.5. Tryptic in-Gel Digestion of Proteins, Matrix-Assisted Laser-Desorption Ionization Time-of-Flight Mass Spectrometry (MALDI-TOFMS) and Database Search
2.6. Choline Quantification
2.7. Co-Purification of Leishmania EF1α with Human SHP-1
2.8. Staining of Proteins on Western Blot Membranes
3. Results
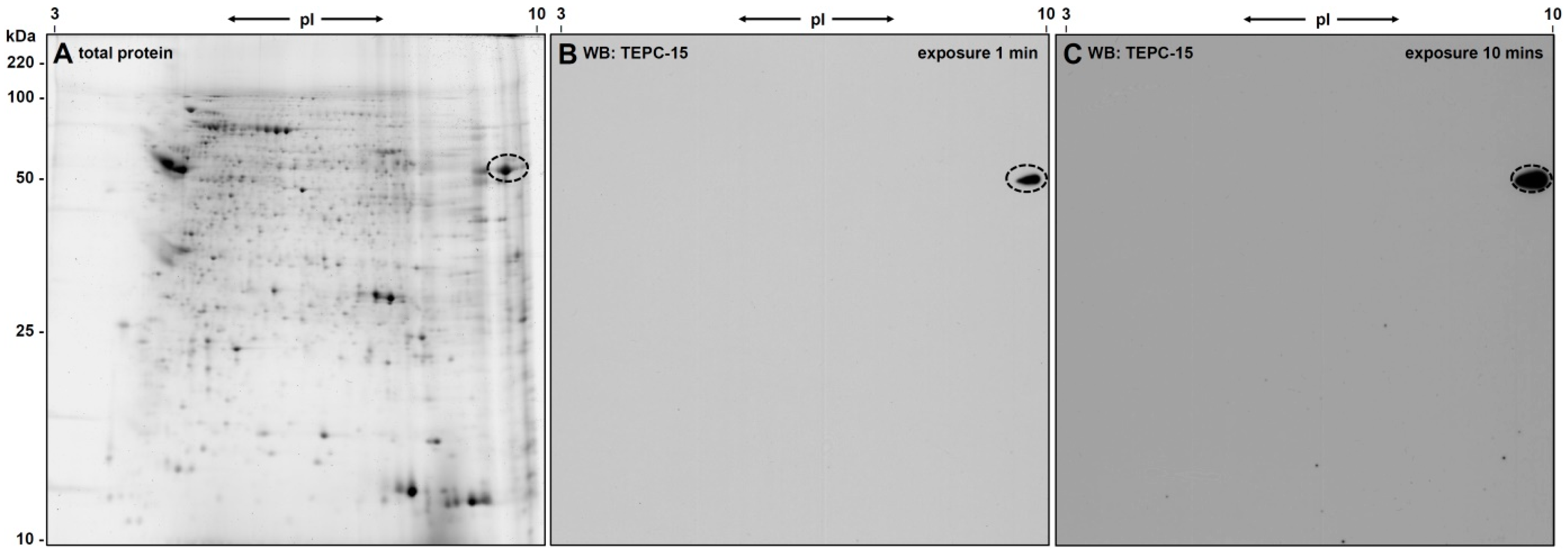
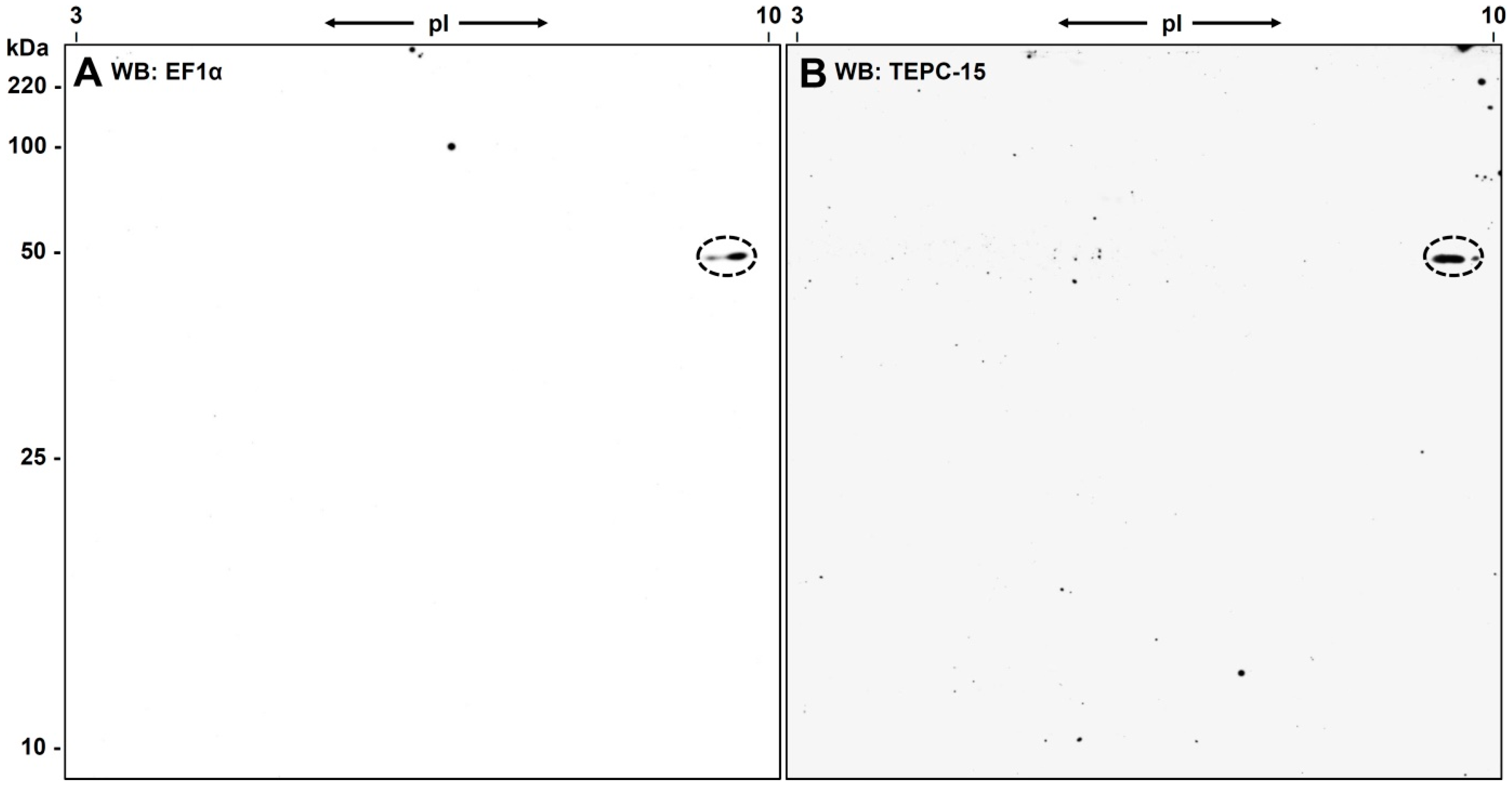
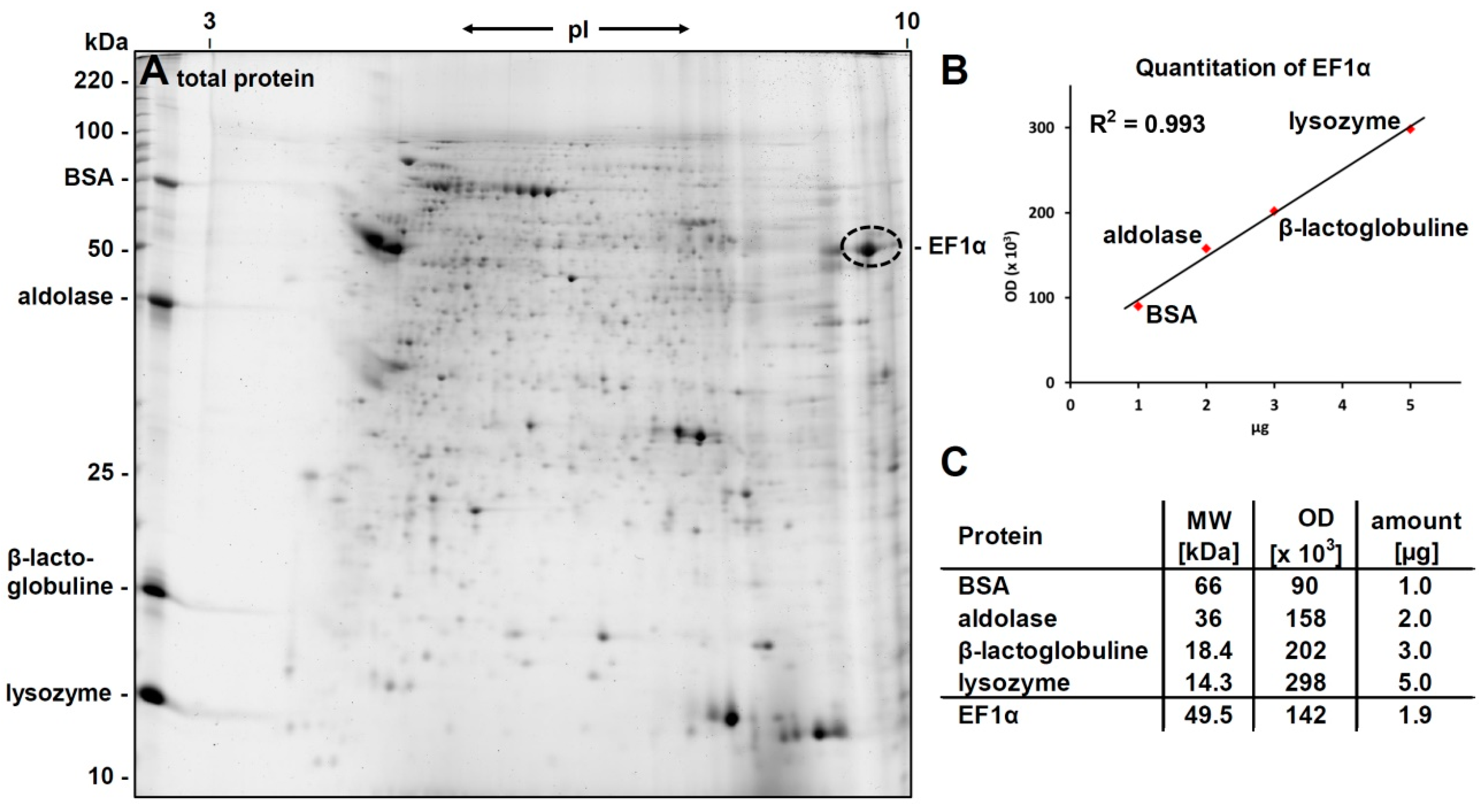
3.1. 2D-Gelelectrophoresis and Western Blot Analyses
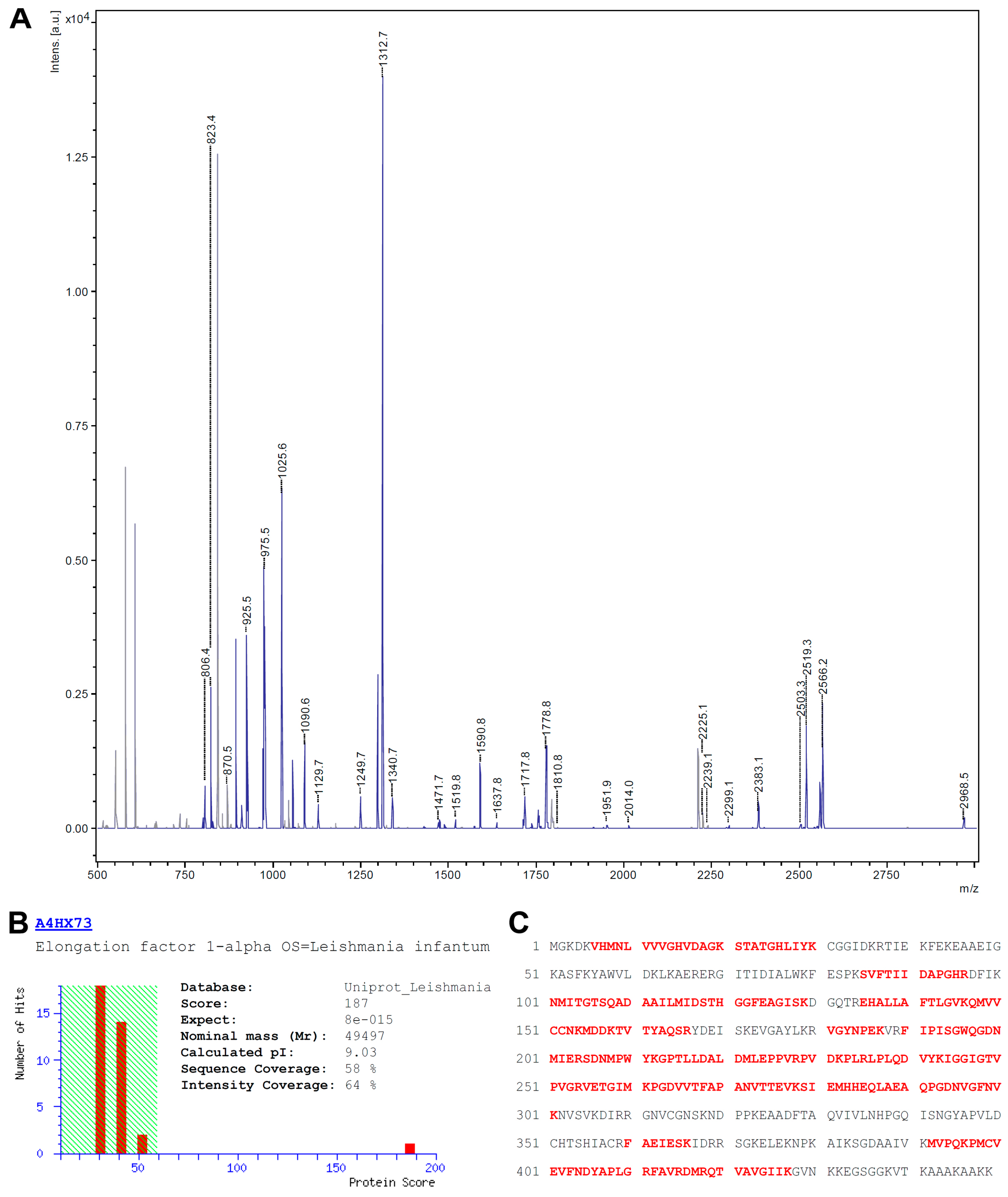
3.2. Identification of eEF1alpha
3.3. Western Blot Analysis with Anti-eEF1α
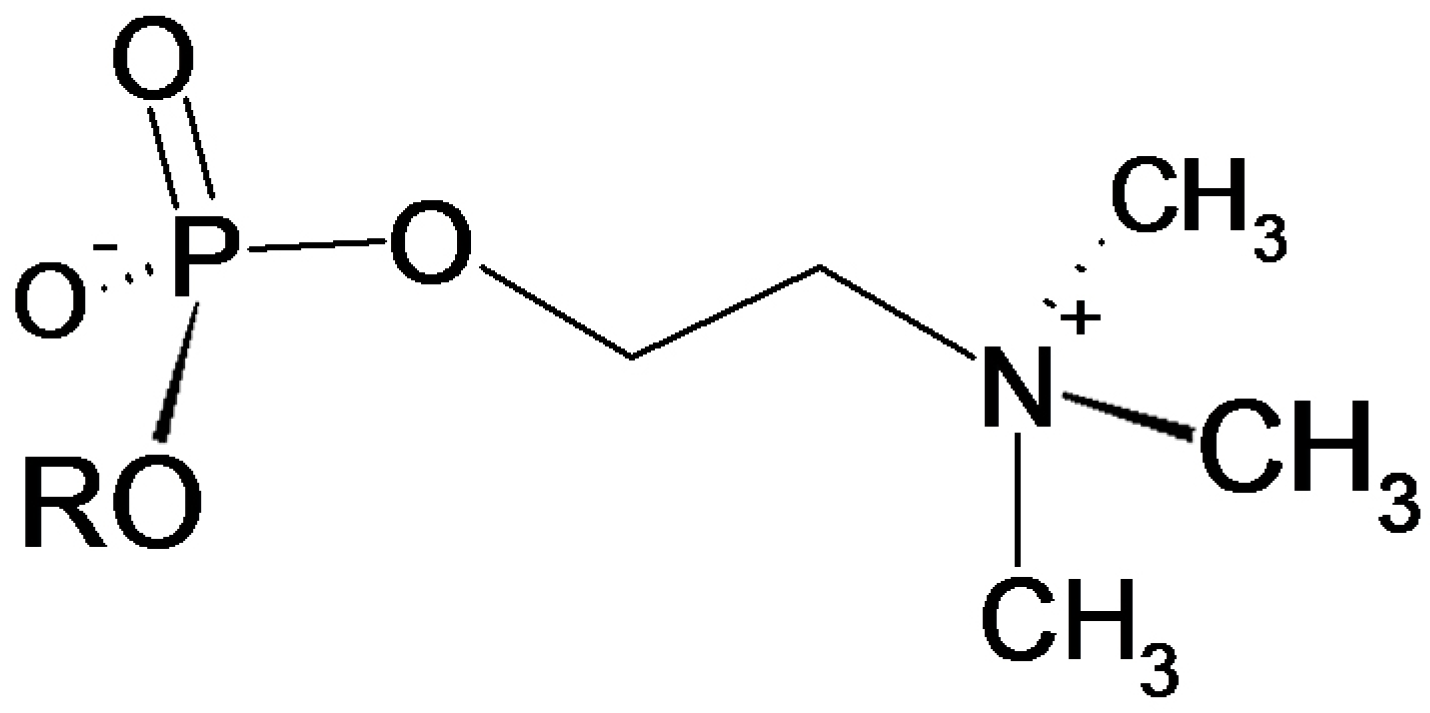
3.4. Verification of the Phosphorylcholine Substitution of eEF1α and Choline Quantitation
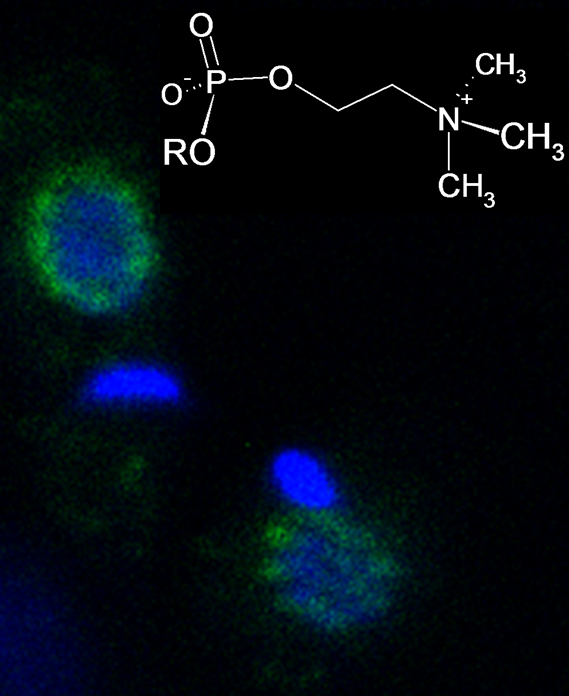
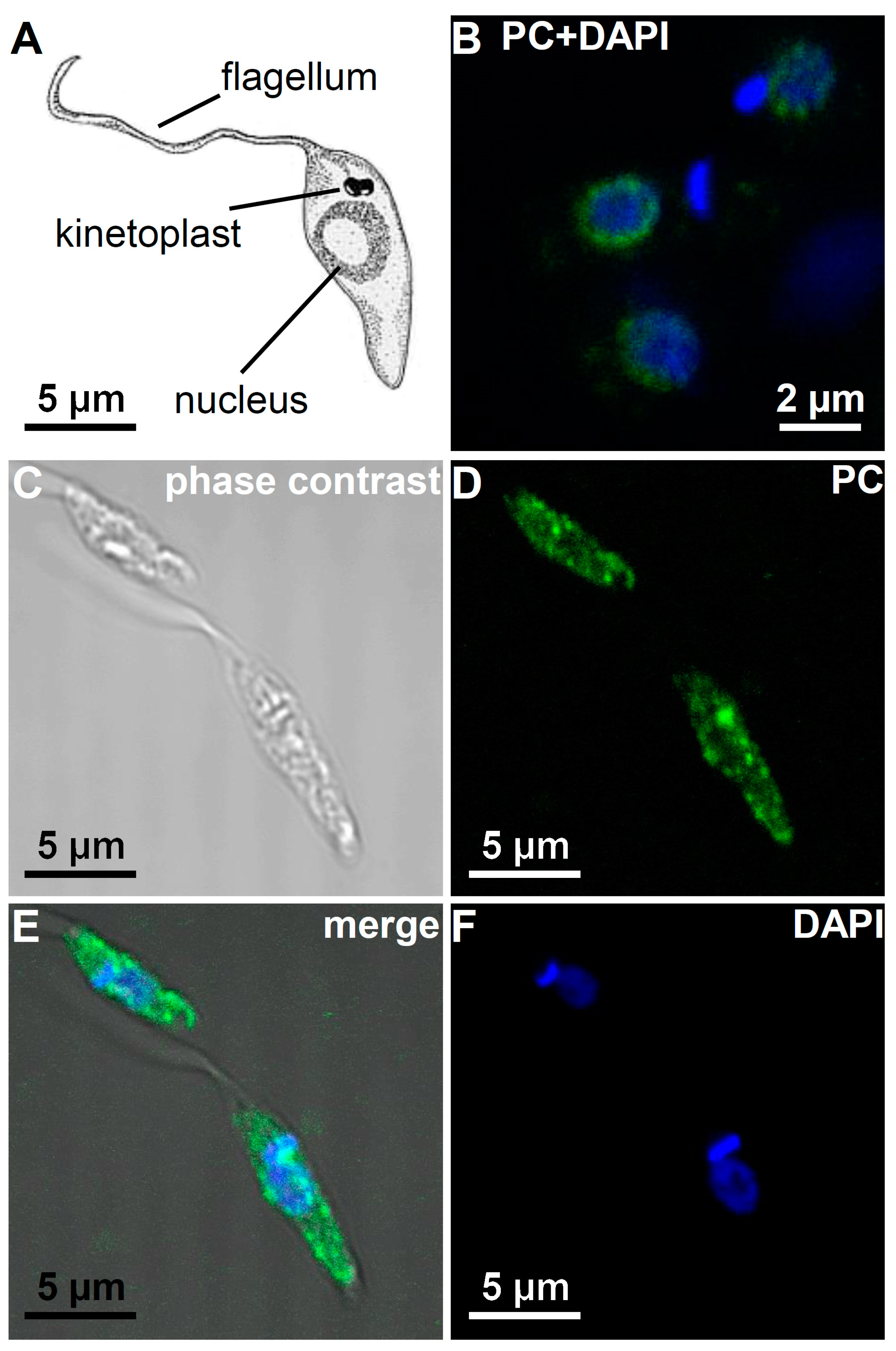
3.5. Immunohistological Localization of PC Epitopes in L. infantum Promastigotes by Confocal Microscopy
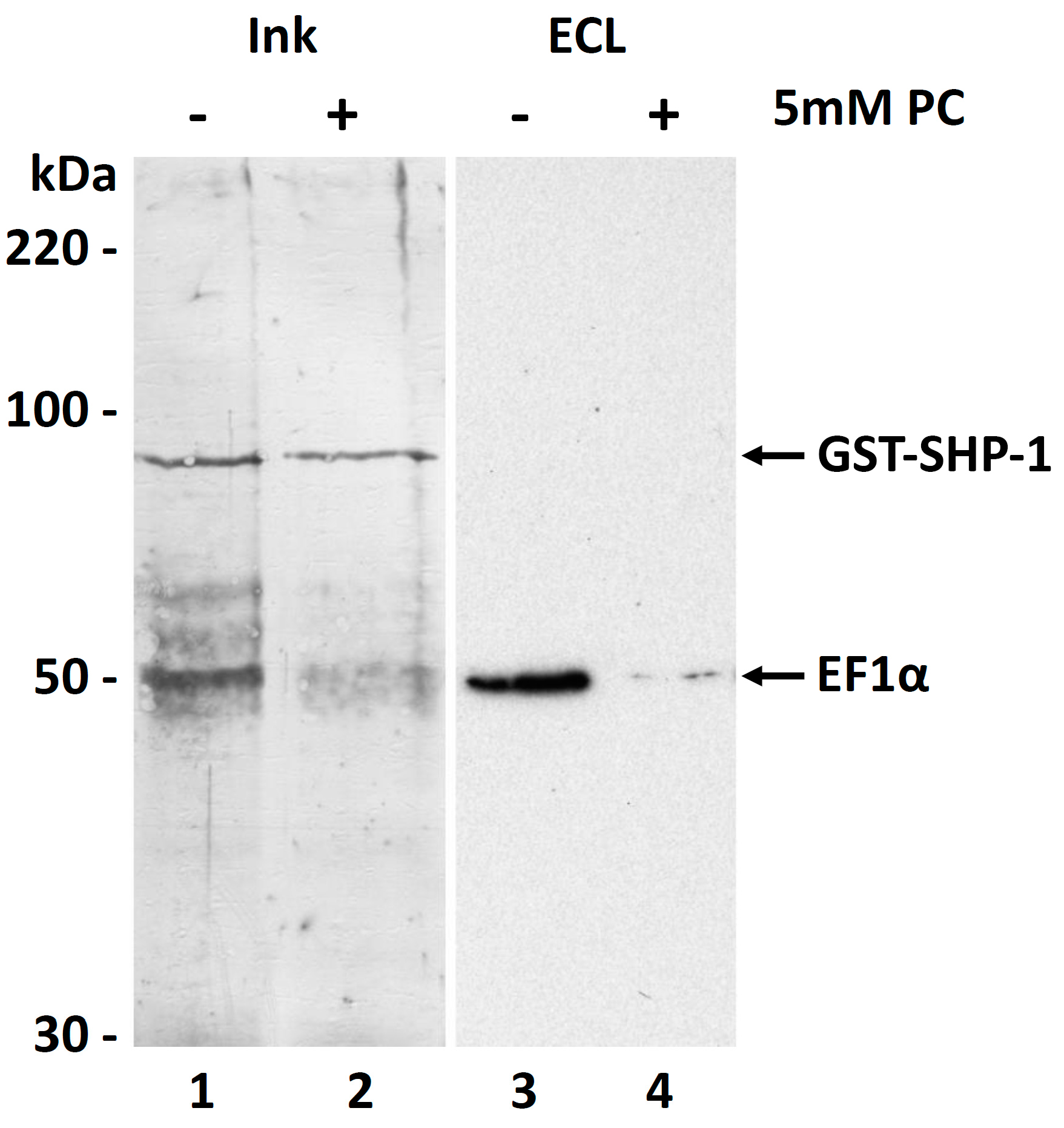
3.6. Molecular Function of the PC Modification of EF1α
4. Discussion
4.1. PC as An Immunomodulatory Substituent
4.2. The Role of iEF1α in Parasitism
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Beverley, S.M.; Turco, S.J. Lipophosphoglycan (LPG) and the identification of virulence genes in the protozoan parasite Leishmania. Trends Microbiol. 1998, 6, 35–40. [Google Scholar] [CrossRef]
- Kaye, P.M.; Aebischer, T. Visceral leishmaniasis: Immunology and prospects for a vaccine. Clin. Microbiol. Infect. 2011, 17, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.A.; Barr, S.C. Canine leishmaniasis in North America: Emerging or newly recognized? Vet. Clin. N. Am. Small Anim. Pract. 2009, 39, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Maroli, M.; Rossi, L.; Baldelli, R.; Capelli, G.; Ferroglio, E.; Genchi, C.; Gramiccia, M.; Mortarino, M.; Pietrobelli, M.; Gradoni, L. The northward spread of leishmaniasis in Italy: Evidence from retrospective and ongoing studies on the canine reservoir and phlebotomine vectors. Trop. Med. Int. Health 2008, 13, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Otranto, D.; Capelli, G.; Genchi, C. Changing distribution patterns of canine vector borne diseases in Italy: Leishmaniosis vs. dirofilariosis. Parasites Vectors 2009, 2 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, J.C. Risk factors in the spread of leishmaniases: Towards integrated monitoring? Trends Parasitol. 2006, 22, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Seridi, N.; Belkaid, M.; Quispe-Tintaya, W.; Zidane, C.; Dujardin, J.C. Application of PCR-RFLP for the exploration of the molecular diversity of Leishmania infantum in Algeria. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Lochnit, G.; Dennis, R.D.; Geyer, R. Phosphorylcholine substituents in nematodes: Structures, occurrence and biological implications. Biol. Chem. Hoppe Seyler 2000, 381, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Lal, R.B.; Ottesen, E.A. Phosphocholine epitopes on helminth and protozoal parasites and their presence in the circulation of infected human patients. Trans. R. Soc. Trop. Med. Hyg. 1989, 83, 652–655. [Google Scholar] [CrossRef]
- Deehan, M.R.; Goodridge, H.S.; Blair, D.; Lochnit, G.; Dennis, R.D.; Geyer, R.; Harnett, M.M.; Harnett, W. Immunomodulatory properties of Ascaris suum glycosphingolipids—Phosphorylcholine and non-phosphorycholine—Dependent effects. Parasite Immunol. 2002, 24, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Harnett, W.; Harnett, M.M. Modulation of the host immune system by phosphorylcholine-containing glycoproteins secreted by parasitic filarial nematodes. Biochim. Biophys. Acta 2001, 1539, 7–15. [Google Scholar] [CrossRef]
- Harnett, W.; Harnett, M.M. Phosphorylcholine: Friend or foe of the immune system? Immunol. Today 1999, 20, 125–129. [Google Scholar] [CrossRef]
- Harnett, W.; McInnes, I.B.; Harnett, M.M. ES-62, a filarial nematode-derived immunomodulator with anti-inflammatory potential. Immunol. Lett. 2004, 94, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Harnett, W.; Liew, F.Y.; Harnett, M.M. Differential regulation of interleukin-12 p40 and p35 induction via Erk mitogen-activated protein kinase-dependent and -independent mechanisms and the implications for bioactive IL-12 and IL-23 responses. Immunology 2003, 109, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Marshall, F.A.; Grierson, A.M.; Garside, P.; Harnett, W.; Harnett, M.M. ES-62, an immunomodulator secreted by filarial nematodes, suppresses clonal expansion and modifies effector function of heterologous antigen-specific T cells in vivo. J Immunol. 2005, 175, 5817–5826. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Marshall, F.A.; Wilson, E.H.; Houston, K.M.; Liew, F.Y.; Harnett, M.M.; Harnett, W. In vivo exposure of murine dendritic cell and macrophage bone marrow progenitors to the phosphorylcholine-containing filarial nematode glycoprotein ES-62 polarizes their differentiation to an anti-inflammatory phenotype. Immunology 2004, 113, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Melendez, A.J.; Harnett, M.M.; Pushparaj, P.N.; Wong, W.F.; Tay, H.K.; McSharry, C.P.; Harnett, W. Inhibition of FceRI-mediated mast cell responses by ES-62, a product of parasitic filarial nematodes. Nat. Med. 2007, 13, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Cipollo, J.F.; Awad, A.; Costello, C.E.; Robbins, P.W.; Hirschberg, C.B. Biosynthesis in vitro of Caenorhabditis elegans phosphorylcholine oligosaccharides. Proc. Natl. Acad. Sci. USA 2004, 101, 3404–3408. [Google Scholar] [CrossRef] [PubMed]
- Cipollo, J.F.; Awad, A.M.; Costello, C.E.; Hirschberg, C.B. N-glycans of Caenorhabditis elegans are specific to developmental stages. J. Biol. Chem. 2005, 280, 26063–26072. [Google Scholar] [CrossRef] [PubMed]
- Lochnit, G.; Bongaarts, R.; Geyer, R. Searching new targets for anthelminthic strategies: Interference with glycosphingolipid biosynthesis and phosphorylcholine metabolism affects development of Caenorhabditis elegans. Int. J. Parasitol. 2005, 35, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Pöltl, G.; Kerner, D.; Paschinger, K.; Wilson, I.B. N-Glycans of the porcine nematode parasite Ascaris suum are modified with phosphorylcholine and core fucose residues. FEBS J. 2007, 274, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Lochnit, G.; Dennis, R.D.; Ulmer, A.J.; Geyer, R. Structural elucidation and monokine-inducing activity of two biologically active zwitterionic glycosphingolipids derived from the porcine parasitic nematode Ascaris suum. J. Biol. Chem. 1998, 278, 466–474. [Google Scholar] [CrossRef]
- Griffitts, J.S.; Haslam, S.M.; Yang, T.; Garczynski, S.F.; Mulloy, B.; Morris, H.; Cremer, P.S.; Dell, A.; Adang, M.J.; Aroian, R.V. Glycolipids as receptors for Bacillus thuringiensis crystal toxin. Science 2005, 307, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Friedl, C.H.; Lochnit, G.; Zähringer, U.; Bahr, U.; Geyer, R. Structural elucidation of zwitterionic carbohydrates derived from glycosphingolipids of the porcine parasitic nematode Ascaris suum. Biochem. J. 2003, 369, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Dennis, R.D.; Baumeister, S.; Smuda, C.; Lochnit, G.; Waider, T.; Geyer, E. Initiation of chemical studies on the immunoreactive glycolipids of adult Ascaris suum. Parasitology 1995, 110, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.; Dennis, R.D.; Klünder, R.; Schares, G.; Zahner, H.; Geyer, E. Litomosoides carinii: Macrofilariae-derived glycolipids—Chromatography, serology and potential in the evaluation of anthelminthic efficacy. Parasite Immunol. 1994, 16, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Wuhrer, M.; Rickhoff, S.; Dennis, R.D.; Lochnit, G.; Soboslay, P.T.; Baumeister, S.; Geyer, R. Phosphocholine-containing, zwitterionic glycosphingolipids of adult Onchocerca volvulus as highly conserved, antigenic structures of parasitic nematodes. Biochem. J. 2000, 348, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Gerdt, S.; Lochnit, G.; Dennis, R.D.; Geyer, R. Isolation and structural analysis of three neutral glycosphingolipids from a mixed population of Caenorhabditis elegans (Nematoda: Rhabditida). Glycobiology 1997, 7, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Gerdt, S.; Dennis, R.D.; Borgonie, G.; Schnabel, R.; Geyer, R. Isolation, characterization and immunolocalization of phosphocholine-substituted glycolipids in developmental stages of Caenorhabditis elegans. Eur. J. Biochem. 1999, 266, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Harnett, W.; Houston, K.M.; Amess, R.; Worms, M.J. Acanthocheilonema viteae: Phosphorylcholine is attached to the major excretory-secretory product via an N-linked glycan. Exp. Parasitol. 1993, 77, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Harnett, W.; Frame, M.J.; Nor, Z.M.; MacDonald, M.; Houston, K.M. Some preliminary data on the nature/structure of the PC-glycan of the major excretory-secretory product of Acanthocheilonema viteae (ES-62). Parasite 1994, 1, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Haslam, S.M.; Khoo, K.H.; Houston, K.M.; Harnett, W.; Morris, H.R.; Dell, A. Characterisation of the phosphorylcholine-containing N-linked oligosaccharides in the excretory-secretory 62 kDa glycoprotein of Acanthocheilonema viteae. Mol. Biochem. Parasitol. 1997, 85, 53–66. [Google Scholar] [CrossRef]
- Haslam, S.M.; Gems, D.; Morris, H.R.; Dell, A. The glycomes of Caenorhabditis elegans and other model organisms. Biochem. Soc. Symp. 2002, 69, 117–134. [Google Scholar] [CrossRef]
- Haslam, S.M.; Dell, A. Hallmarks of Caenorhabditis elegans N-glycosylation: Complexity and controversy. Biochimie 2003, 85, 25–32. [Google Scholar] [CrossRef]
- Haslam, S.M.; Houston, K.M.; Harnett, W.; Reason, A.J.; Morris, H.R.; Dell, A. Structural studies of N-glycans of filarial parasites. Conservation of phosphorylcholine-substituted glycans among species and discovery of novel chito-oligomers. J. Biol. Chem. 1999, 274, 20953–20960. [Google Scholar] [CrossRef] [PubMed]
- Cipollo, J.F.; Costello, C.E.; Hirschberg, C.B. The fine structure of Caenorhabditis elegans N-glycans. J. Biol. Chem. 2002, 277, 49143–49157. [Google Scholar] [CrossRef] [PubMed]
- Grabitzki, J.; Lochnit, G. Immunomodulation by phosphocholine—Biosynthesis, structures and immunological implications of parasitic PC-epitopes. Mol. Immunol. 2009, 47, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Trimnell, A.R.; Kraemer, S.M.; Mukherjee, S.; Phippard, D.J.; Janes, J.H.; Flamoe, E.; Su, X.Z.; Awadalla, P.; Smith, J.D. Global genetic diversity and evolution of var genes associated with placental and severe childhood malaria. Mol. Biochem. Parasitol. 2006, 148, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fernandez, V.; Sundstrom, A.; Schlichtherle, M.; Datta, S.; Hagblom, P.; Wahlgren, M. Developmental selection of var gene expression in Plasmodium falciparum. Nature 1998, 394, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Shonhai, A.; Boshoff, A.; Blatch, G.L. The structural and functional diversity of Hsp70 proteins from Plasmodium falciparum. Protein Sci. 2007, 16, 1803–1818. [Google Scholar] [CrossRef] [PubMed]
- Nandan, D.; Yi, T.; Lopez, M.; Lai, C.; Reiner, N.E. Leishmania EF-1alpha activates the Src homology 2 domain containing tyrosine phosphatase SHP-1 leading to macrophage deactivation. J. Biol. Chem. 2002, 277, 50190–50197. [Google Scholar] [CrossRef] [PubMed]
- Negrutskii, B.S.; El’skaya, A.V. Eukaryotic translation elongation factor 1 alpha: Structure, expression, functions, and possible role in aminoacyl-tRNA channeling. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 47–78. [Google Scholar] [PubMed]
- Gavin, A.C.; Aloy, P.; Grandi, P.; Krause, R.; Boesche, M.; Marzioch, M.; Rau, C.; Jensen, L.J.; Bastuck, S.; Dümpelfeld, B.; et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 440, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Hermjakob, H.; Montecchi-Palazzi, L.; Lewington, C.; Mudali, S.; Kerrien, S.; Orchard, S.; Vingron, M.; Roechert, B.; Roepstorff, P.; Valencia, A.; et al. IntAct: An open source molecular interaction database. Nucleic Acids Res. 2004, 32, D452–D455. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Reiner, N.E. Leishmania exosomes deliver preemptive strikes to create an environment permissive for early infection. Front. Cell. Infect. Microbiol. 2012, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A. Leishmania: In Vitro Methods for Parasite Cultivation; Taylor, A.E., Baker, J.R., Eds.; Academic Press: Orlando, FL, USA, 1987; pp. 52–75. [Google Scholar]
- Timm, T.; Grabitzki, J.; Severcan, C.; Muratoglu, S.; Ewald, L.; Yilmaz, Y.; Lochnit, G. The PCome of Ascaris suum as a model system for intestinal nematodes: Identification of phosphorylcholine-substituted proteins and first characterization of the PC-epitope structures. Parasitol. Res. 2016, 115, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Mohr, F.; Zimmermann, M.; Klein, J. Mice heterozygous for AChE are more sensitive to AChE inhibitors but do not respond to BuChE inhibition. Neuropharmacology 2013, 67, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Dennis, R.D.; Lochnit, G.; Geyer, R. Strategies for preliminary characterization of novel amphoteric glycosphingolipids. Methods Mol. Biol. 1998, 76, 197–212. [Google Scholar] [PubMed]
- Sorci, G.; Cornet, S.; Faivre, B. Immune evasion, immunopathology and the regulation of the immune system. Pathogens 2013, 2, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Weiser, J.N. Microbial modulation of host immunity with the small molecule phosphorylcholine. Infect. Immun. 2013, 81, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Swords, W.E.; Buscher, B.A.; Ver Steeg Ii, K.; Preston, A.; Nichols, W.A.; Weiser, J.N.; Gibson, B.W.; Apicella, M.A. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol. Microbiol. 2000, 37, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Lochnit, G.; Nispel, S.; Dennis, R.D.; Geyer, R. Structural analysis and immunohistochemical localization of two acidic glycosphingolipids from the porcine, parasitic nematode, Ascaris suum. Glycobiology 1998, 8, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Weiser, J.N.; Pan, N. Adaptation of Haemophilus influenzae to acquire and innate humoral immunity based on phase variation of lipopolysaccharide. Mol. Microbiol. 1998, 30, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Risberg, A.; Masoud, H.; Martin, A.; Richards, J.C.; Moxon, E.R.; Schweda, E.K. Structural analysis of the lipopolysaccharide oligosaccharide epitopes expressed by a capsule-deficient strain of Haemophilus influenzae Rd. Eur. J. Biochem. 1999, 261, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Schweda, E.K.; Brisson, J.R.; Alvelius, G.; Martin, A.; Weiser, J.N.; Hood, D.W.; Moxon, E.R.; Richards, J.C. Characterization of the phosphocholine-substituted oligosaccharide in lipopolysaccharides of type B Haemophilus influenzae. Eur. J. Biochem. 2000, 267, 3902–3913. [Google Scholar] [CrossRef] [PubMed]
- Mansson, M.; Bauer, S.H.; Hood, D.W.; Richards, J.C.; Moxon, E.R.; Schweda, E.K. A new structural type for Haemophilus influenzae lipopolysaccharide. Structural analysis of the lipopolysaccharide from nontypeable Haemophilus influenzae strain 486. Eur. J. Biochem. 2001, 268, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Mansson, M.; Hood, D.W.; Li, J.; Richards, J.C.; Moxon, E.R.; Schweda, E.K. Structural analysis of the lipopolysaccharide from nontypeable Haemophilus influenzae strain 1003. Eur. J. Biochem. 2002, 269, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Fudala, R.; Kondakova, A.N.; Bednarska, K.; Senchenkova, S.N.; Shashkov, A.S.; Knirel, Y.A.; Zähringer, U.; Kaca, W. Structure and serological characterization of the O-antigen of Proteus mirabilis O18 with a phosphocholine-containing oligosaccharide phosphate repeating unit. Carbohydr. Res. 2003, 338, 1835–1842. [Google Scholar] [CrossRef]
- Landerholm, M.K.; Li, J.; Richards, J.C.; Hood, D.W.; Moxon, E.R.; Schweda, E.K. Characterization of novel structural features in the lipopolysaccharide of nondisease associated nontypeable Haemophilus influenzae. Eur. J. Biochem. 2004, 271, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Arnold, R.J.; Luo, Z.Q. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc. Natl. Acad. Sci. USA 2011, 108, 21212–21217. [Google Scholar] [CrossRef] [PubMed]
- Lochnit, G.; Grabitzki, J.; Henkel, B.; Tavernarakis, N.; Geyer, R. First identification of a phosphorylcholine-substituted protein from Caenorhabditis elegans. Isolation and characterization of the aspartyl protease ASP-6. Biol. Chem. Hoppe Seyler 2006, 387, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Lovell, T.M.; Woods, R.J.; Butlin, D.J.; Brayley, K.J.; Manyonda, I.T.; Jarvis, J.; Howell, S.; Lowry, P.J. Identification of a novel mammalian post-translational modification, phosphocholine, on placental secretory polypeptides. J. Mol. Endocrinol. 2007, 39, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, P.J.; Alonso, A.; Larraga, V. Proteome profiling of Leishmania infantum promastigotes. J. Eukaryot Microbiol. 2011, 58, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Nandan, D.; Cherkasov, A.; Sabouti, R.; Yi, T.; Reiner, N.E. Molecular cloning, biochemical and structural analysis of elongation factor-1 alpha from Leishmania donovani: Comparison with the mammalian homologue. Biochem. Biophys. Res. Commun. 2003, 302, 646–652. [Google Scholar] [CrossRef]
- Houston, K.; Lochnit, G.; Geyer, R.; Harnett, W. Investigation of the nature of potential phosphorylcholine donors for filarial nematode glycoconjugates. Mol. Biochem. Parasitol. 2002, 123, 55–66. [Google Scholar] [CrossRef]
- Harnett, W.; Rzepecka, J.; Houston, K.M. How do nematodes transfer phosphorylcholine to carbohydrates? Trends Parasitol. 2010, 26, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Billaut-Mulot, O.; Fernandez-Gomez, R.; Loyens, M.; Ouaissi, A. Trypanosoma cruzi elongation factor 1-alpha: Nuclear localization in parasites undergoing apoptosis. Gene 1996, 174, 19–26. [Google Scholar] [CrossRef]
- Alcolea, P.J.; Alonso, A.; Gomez, M.J.; Moreno, I.; Dominguez, M.; Parro, V.; Larraga, V. Transcriptomics throughout the life cycle of Leishmania infantum: High down-regulation rate in the amastigote stage. Int. J. Parasitol. 2010, 40, 1497–1516. [Google Scholar] [CrossRef] [PubMed]
- Deehan, M.R.; Harnett, W.; Harnett, M.M. A filarial nematode-secreted phosphorylcholine-containing glycoprotein uncouples the B cell antigen receptor from extracellular signal-regulated kinase-mitogen-activated protein kinase by promoting the surface ig-mediated recruitment of src homology 2 domain-containing tyrosine phosphatase-1 and pac-1 mitogen-activated kinase-phosphatase. J. Immunol. 2001, 166, 7462–7468. [Google Scholar] [PubMed]
- Deehan, M.R.; Harnett, M.M.; Harnett, W. A filarial nematode secreted product differentially modulates expression and activation of protein kinase C isoforms in B lymphocytes. J. Immunol. 1997, 159, 6105–6111. [Google Scholar] [PubMed]
- Goodridge, H.S.; Deehan, M.R.; Harnett, W.; Harnett, M.M. Subversion of immunological signalling by a filarial nematode phosphorylcholine-containing secreted product. Cell Signal. 2005, 17, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Harnett, W.; Harnett, M.M. Inhibition of murine B cell proliferation and down-regulation of protein kinase C levels by a phosphorylcholine-containing filarial excretory-secretory product. J. Immunol. 1993, 151, 4829–4837. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timm, T.; Annoscia, G.; Klein, J.; Lochnit, G. The Eukaryotic Elongation Factor 1 Alpha (eEF1α) from the Parasite Leishmania infantum Is Modified with the Immunomodulatory Substituent Phosphorylcholine (PC). Molecules 2017, 22, 2094. https://doi.org/10.3390/molecules22122094
Timm T, Annoscia G, Klein J, Lochnit G. The Eukaryotic Elongation Factor 1 Alpha (eEF1α) from the Parasite Leishmania infantum Is Modified with the Immunomodulatory Substituent Phosphorylcholine (PC). Molecules. 2017; 22(12):2094. https://doi.org/10.3390/molecules22122094
Chicago/Turabian StyleTimm, Thomas, Giada Annoscia, Jochen Klein, and Günter Lochnit. 2017. "The Eukaryotic Elongation Factor 1 Alpha (eEF1α) from the Parasite Leishmania infantum Is Modified with the Immunomodulatory Substituent Phosphorylcholine (PC)" Molecules 22, no. 12: 2094. https://doi.org/10.3390/molecules22122094
APA StyleTimm, T., Annoscia, G., Klein, J., & Lochnit, G. (2017). The Eukaryotic Elongation Factor 1 Alpha (eEF1α) from the Parasite Leishmania infantum Is Modified with the Immunomodulatory Substituent Phosphorylcholine (PC). Molecules, 22(12), 2094. https://doi.org/10.3390/molecules22122094