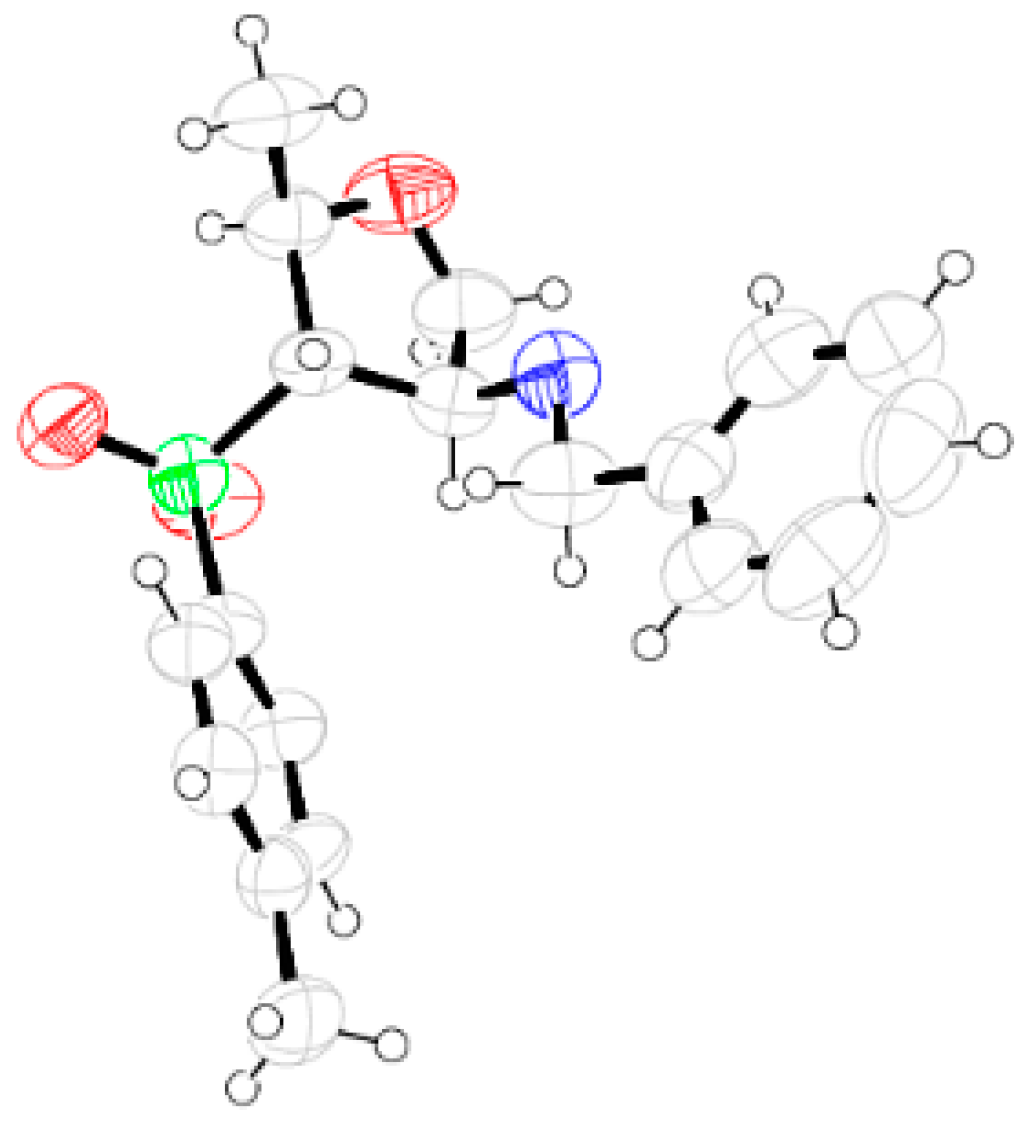
General Methods
All reactions were conducted in a N
2 atmosphere. Melting points were determined in open-end capillary tubes and are uncorrected. Carbohydrates and other fine chemicals were obtained from commercial suppliers and are used without purification. Solvents were dried and distilled following the standard procedures. TLC was carried out on pre-coated plates (Merck silica gel 60, f
254, Merck, Darmstadt, Germany), and the spots were visualized with UV light or by charring the plate dipped in a 5% H
2SO
4–MeOH solution. Column chromatography was performed on silica gel (230–400 mesh).
1H- and
13C-NMR for new compounds were recorded at 200/400 and 50/100 MHz, respectively, using CDCl
3 as the solvent in Bruker (Massachusetts, MA, USA) NMR instrument DEPT experiments had been carried out to identify the methylene carbons. Optical rotations were recorded at 589 nm. Mass spectroscopy data were obtained from a mass analyzer (Xevo G2 QTof) consisting of TOF and quadrupole in either ESI
+ or ESI
− mode. The electronic information of compounds is available in the
Supplementary Materials.
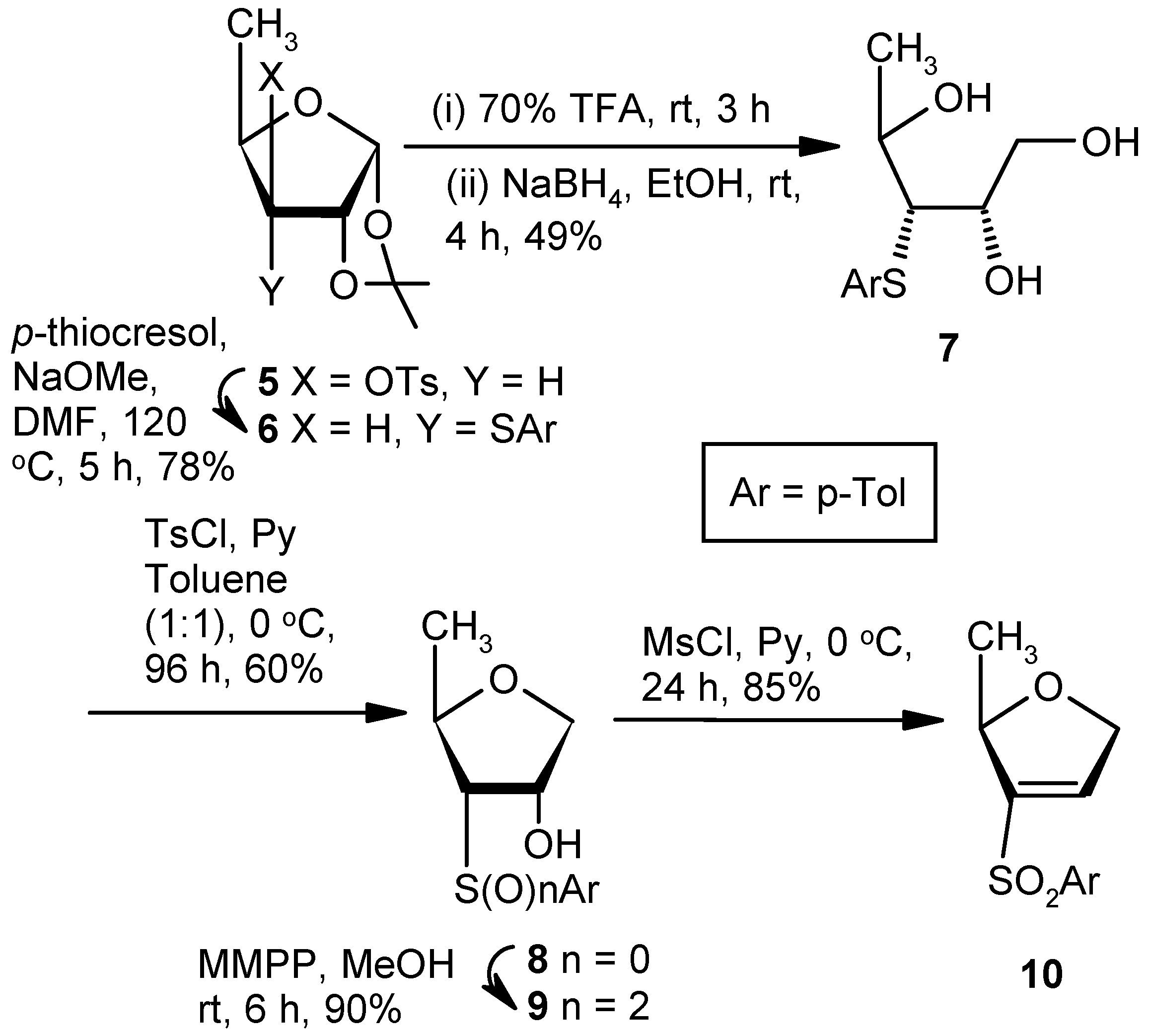
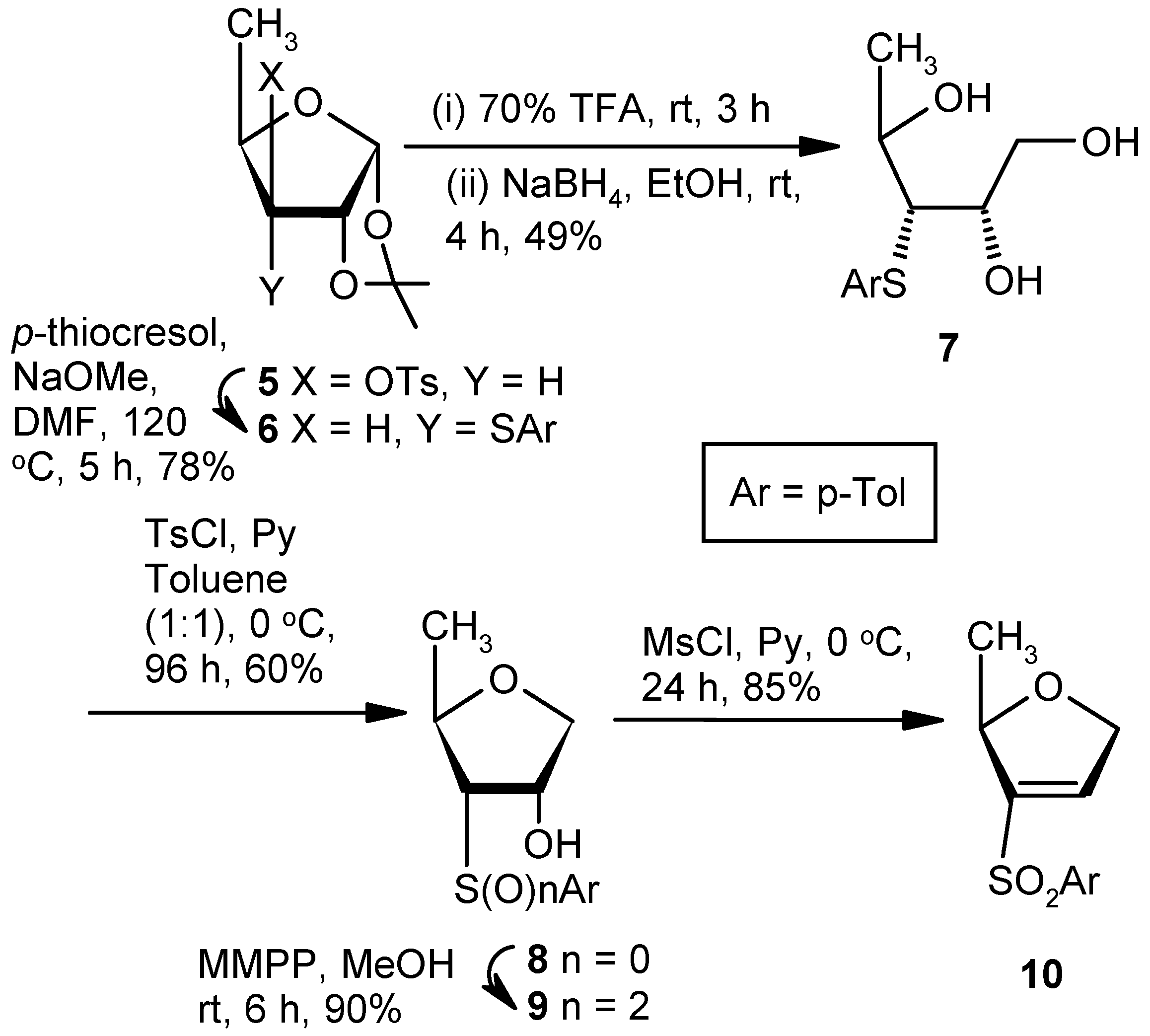
Compound 6: A solution of p-thiocresol (2.83 g, 22.85 mmol) and NaOMe (0.98 g, 18.28 mmol) in anhyd DMF (10 mL) was stirred at room temperature for 0.5 h in a nitrogen atmosphere. A solution of Compound 5 (1.5 g, 4.57 mmol) in anhyd DMF (5 mL) was added, and the final solution was heated at 120 °C After 5 h (tlc), the reaction mixture was cooled to room temperature, poured into cold satd. aqueous solution of NaHCO3, and the product was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The crude material was purified over silica gel to afford 6 (0.99 g, 78%). Eluent: EtOAc: PE (1:9); yellowish gum; = (+) 37.8 (c = 1.02, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.29 (d, J = 6.0 Hz, 3H), 1.38 (s, 3H), 1.58 (s, 3H), 2.34 (s, 3H), 2.88 (dd, J = 4.4, 10.0 Hz, 1H), 4.10–4.14 (m, 1H), 4.77–4.79 (m, 1H), 5.80 (d, J = 4.0 Hz, 1H), 7.12 (d, J = 7.6 Hz, 2H), 7.41 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 17.5, 21.0, 26.3, 26.4, 57.7, 77.5, 81.5, 103.8, 111.8, 129.8, 131.1, 132.3, 137.5; HRMS [ES+, (M + Na)+]: for C15H20O3NaS obsd 303.1056, calcd 303.1031.
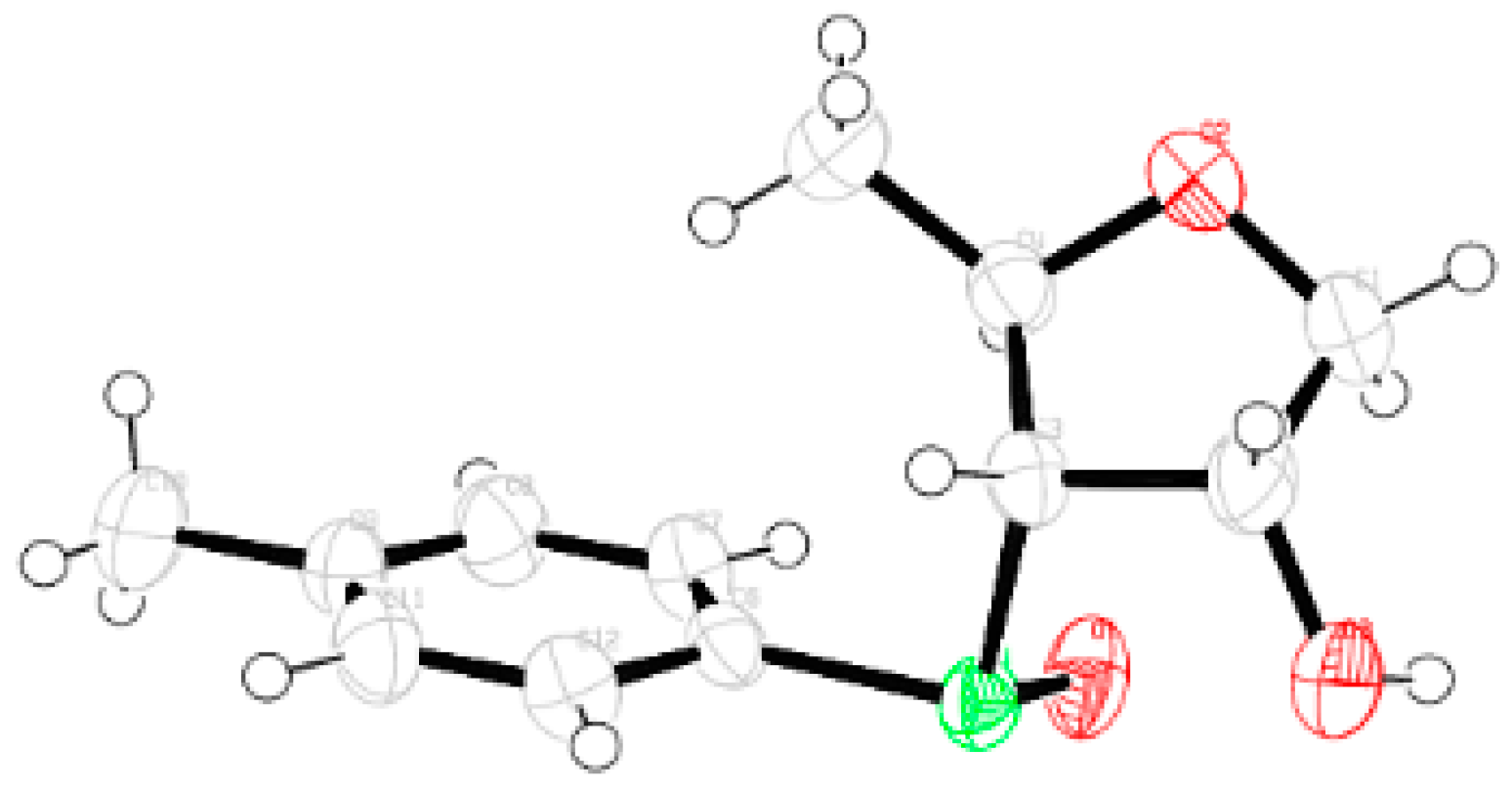
Compound 7: A mixture of Compound 6 (1.0 g, 3.57 mmol) and 70% trifluoroacetic acid in water (10 mL) was stirred at room temperature. After 3 h, (tlc) the reaction mixture was poured into ice-cold satd. Aqueous solution of NaHCO3, and the product was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure to get a residue. The residue was dissolved in EtOH (15 mL), and NaBH4 (0.55 g, 14.28 mmol) was added at 0 °C. After 4 h at room temperature, the reaction mixture was concentrated under reduced pressure to get a residue. The residue was poured into satd. Aqueous solution of NaHCO3, and the product was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The residue was purified over silica gel to get 7 (0.42 g, 49%). Eluent: EtOAc: PE (2:3); yellowish gum; = (−)14.6 (c = 1.02, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.35 (d, J = 6.4 Hz, 3H), 2.32 (s, 3H), 2.50 (bs, 1H), 3.17–3.20 (m, 2H), 3.30 (s, 1H), 3.71–3.75 (m, 1H), 3.88–3.90 (m, 2H), 4.16 (t, J = 6.0 Hz, 1H), 7.11 (d, J = 7.6 Hz, 2H), 7.36 (d, J = 7.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.0, 21.0, 60.6, 64.5 (CH2), 68.4, 72.9, 130.1, 130.7, 132.2, 137.8; HRMS [ES+, (M + Na)+]: for C12H18O3NaS obsd 265.0850, calcd 265.0874.
Compound 8: A solution of tosylchloride (0.6 g, 3.09 mmol) in anhyd tolune (5 mL) was dropwise added to a solution of 7 (0.5 g, 2.06 mmol) in a mixture of anhyd pyridine and toluene (1:1; 5 mL) at 0 °C. The mixture was stirred for 0.5 h at 0 °C, and the reaction mixture was stored at +4 °C for 96 h. The mixture was filtered through celite, and the filtrate was evaporated to dryness. Residual pyridine was co-evaporated with toluene. The residue was purified over silica gel to afford 8 (0.28 g, 60%). Eluent: EtOAc: PE (1:4); white solid; Mp = 97 °C; = (+) 98.6 (c = 1.01, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.38 (d, J = 6.4 Hz, 3H), 2.33 (s, 3H), 3.0 (s, 1H), 3.10 (dd, J = 4.8, 10.4 Hz, 1H), 3.80–3.86 (m, 2H), 4.10 (dd, J = 4.4, 10.0, 1H), 4.25 (s, 1H), 7.13 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.0, 21.0, 61.4, 70.9, 73.7 (CH2), 76.4, 129.8, 130.1, 131.8, 137.9; HRMS [ES+, (M + H)+]: for C12H17O2S obsd 225.0920, calcd 225.0949.
Compound 9: MMPP (3.3 g, 6.69 mmol) was added to a solution of 8 (0.5 g, 2.23 mmol) in MeOH (8 mL), and the mixture was stirred at room temperature. After 6 h (tlc), the mixture was evaporated under reduced pressure. The solid residue was stirred in a mixture of EtOAc and satd. aqueous NaHCO3 solution for 1 h. The organic part was separated, dried over anhyd Na2SO4, and filtered, and the filtrate was evaporated to dryness. The residue was purified over silica gel to afford 9 (0.51 g, 90%). Eluent: EtOAc: PE (2:3); white solid; Mp = 110 °C; = (+) 26.2 (c = 0.98, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.26 (d, J = 6.4 Hz, 3H), 2.44 (s, 3H), 3.23–3.26 (m, 1H), 3.65 (d, J = 5.2 Hz, 1H), 3.75 (dd, J = 2.8, 10.0 Hz, 1H), 3.94 (dd, J = 4.4, 10.0 Hz, 1H), 4.49–4.58 (m, 2H), 7.37 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.0, 21.6, 72.3, 72.4, 73.8, 73.9 (CH2), 128.3, 130.0, 136.3, 145.4; HRMS [ES+, (M + Na)+]: for C12H16O4NaS obsd 279.0691, calcd 279.0667.
Compound 10: A solution of mesylchloride (0.5 mL, 5.85 mmol) in anhyd pyridine (3 mL) was added dropwise to a solution of 9 (0.5g, 1.95 mmol) in anhyd pyridine (5 mL). The mixture was stirred for 0.5 h and stored at +4 °C. After 24 h (tlc), the reaction mixture was poured into ice-cold water, and the product was extracted with EtOAc. The organic layer was dried over anhyd Na2SO4 and filtered, and the filtrate was evaporated to dryness. The residual pyridine was co-evaporated with toluene. The residue was purified over silica gel to afford 10 (0.39 g, 85%). Eluent: EtOAc: PE (1:4); yellowish gum; = (−) 11.6 (c = 1.36, CHCl3); 1H-NMR (400 MHz, CDCl3): δ =1.31 (d, J = 6.4 Hz, 3H), 2.43 (s, 3H), 4.61–4.78 (m, 2H), 4.89–4.93 (m, 1H), 7.17 (s, 1H), 7.34 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 20.9, 21.6, 73.3 (CH2), 80.5, 127.9, 130.0, 136.6, 138.3, 145.0, 145.3; HRMS [ES+, (M + H)+]: for C12H15O3S obsd 239.0729, calcd 239.0742.
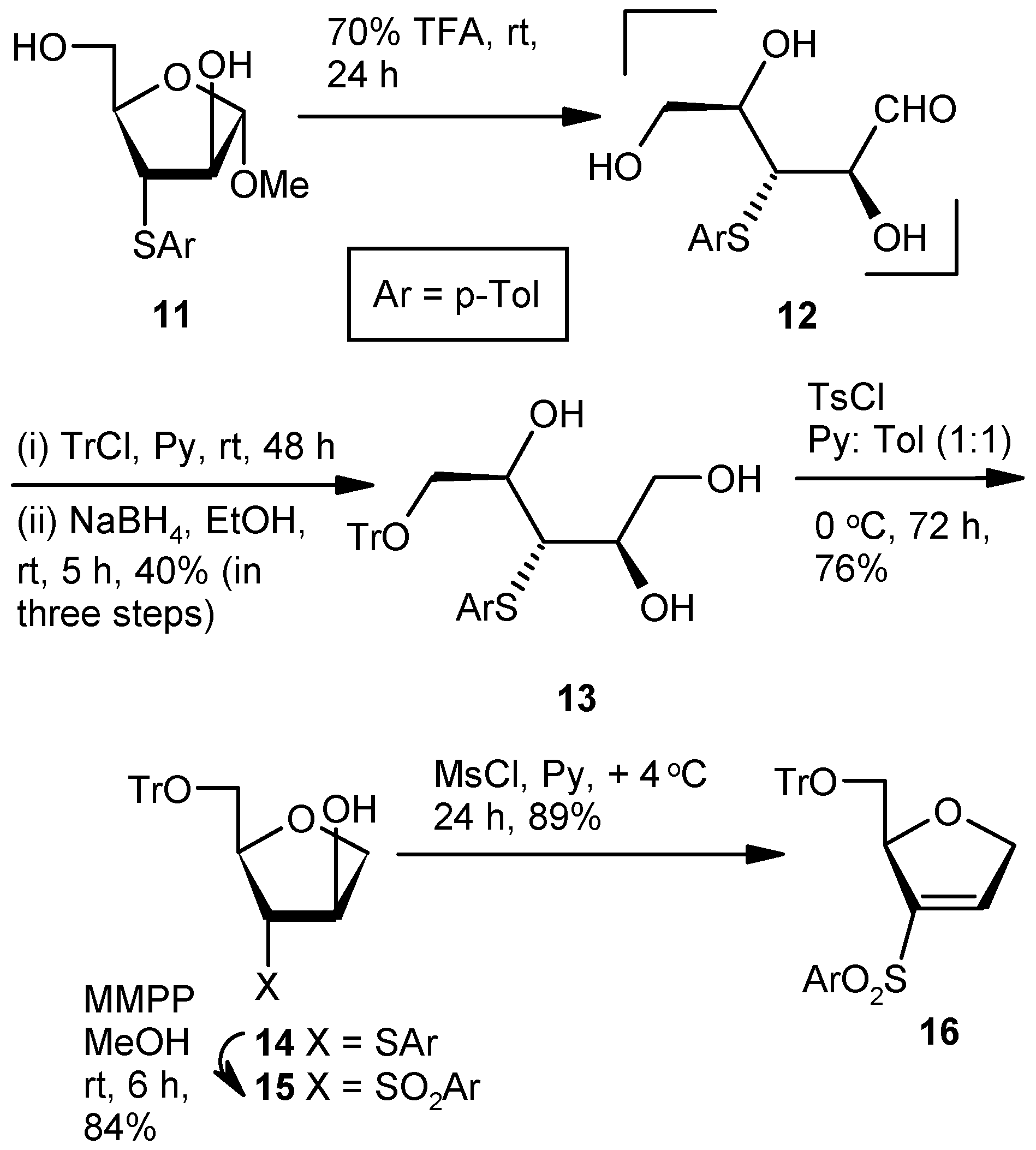
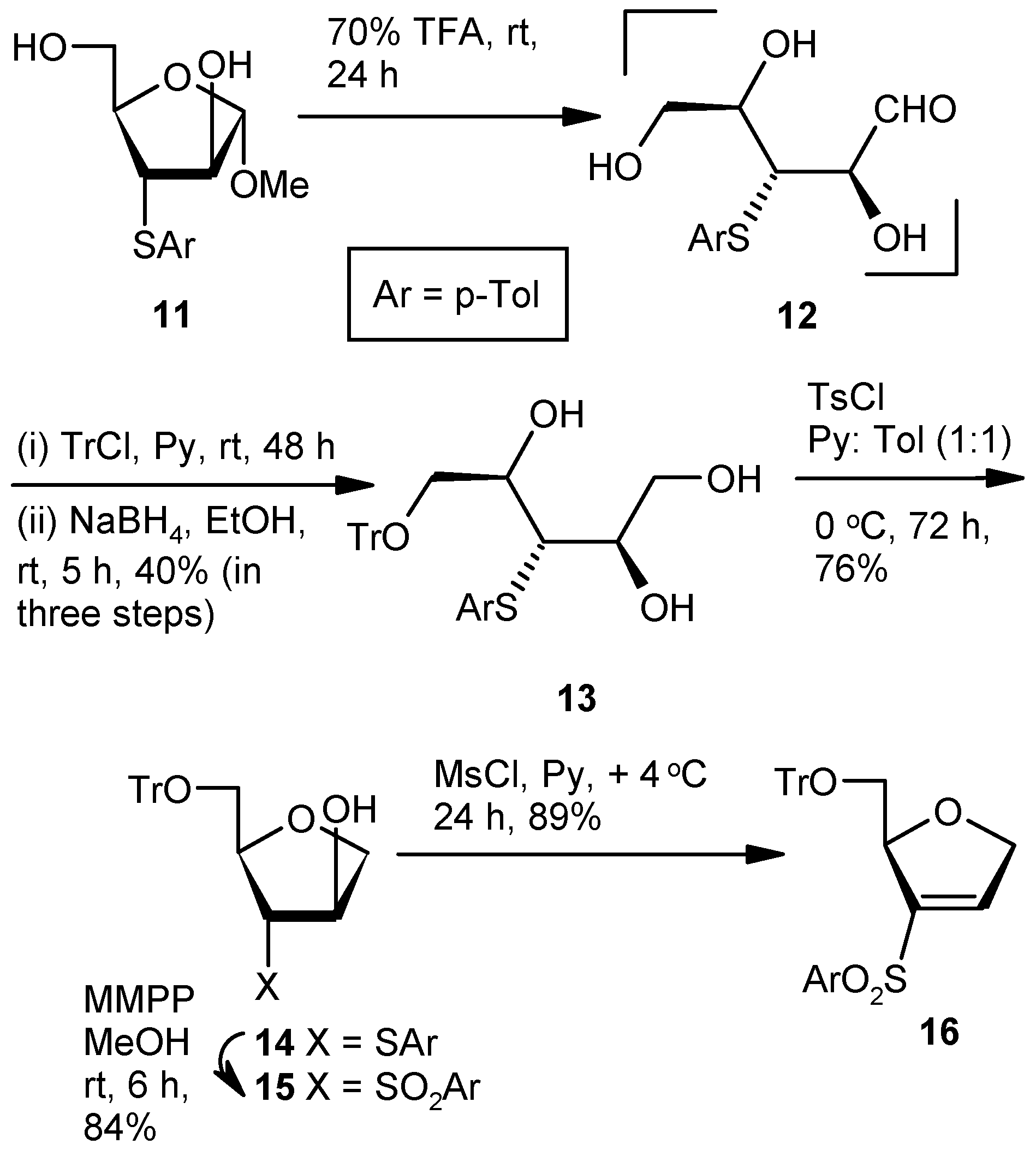
Compound 13: A mixture of Compound 11 (1.5 g, 1.11 mmol) and aqueous 70% trifluoroacetic acid (10 mL) was stirred at room temperature. After 24 h (tlc), the reaction mixture was poured into the ice-cold satd. aqueous solution of NaHCO3, and the product was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure to get the residue 12. The residue was dissolved in pyridine, and tritylchloride (0.93 g, 3.33 mmol) was added. The mixture was stirred at room temperature in an inert atmosphere. After 48 h (tlc), the reaction mixture was poured into ice-cold satd. aqueous solution of NaHCO3, and the product was extracted with EtOAc (3 × 10 mL). Organic layers were pooled, dried over anhyd Na2SO4, and filtered, and the filtrate was concentrated under reduced pressure to get a residue. The residue was dissolved in EtOH (40 mL) and NaBH4 (0.17 g, 4.44 mmol) was added at 0 °C. After 5 h at room temperature, the reaction mixture was concentrated under reduced pressure. The residue was poured into satd. aqueous solution of NaHCO3, and the product was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The residue was purified over silica gel to afford 13 (1.11 g, 40%). Eluent: EtOAc: PE (3:2); yellowish gum; = (−) 7.6 (c = 1.32, CHCl3); 1H-NMR (200 MHz, CDCl3): δ = 2.29 (s, 3H), 3.28–3.34 (m, 2H), 3.40–3.47 (m, 1H), 3.61–3.68 (m, 1H), 3.80–3.90 (m, 1H), 4.00–4.10 (m, 2H), 7.00 (d, J = 15.6 Hz, 2H), 7.17–7.36 (m, 16H); 13C-NMR (50 MHz, CDCl3): δ = 21.3, 54.8, 64.8 (CH2), 65.8 (CH2), 71.9, 72.9, 87.4, 127.4, 128.1, 128.8, 130.1, 131.3, 132.2, 137.5, 143.8 (3 × C); HRMS [ES+, (M + Na)+]: for C31H32O4NaS obsd 523.1964, calcd 523.1919.
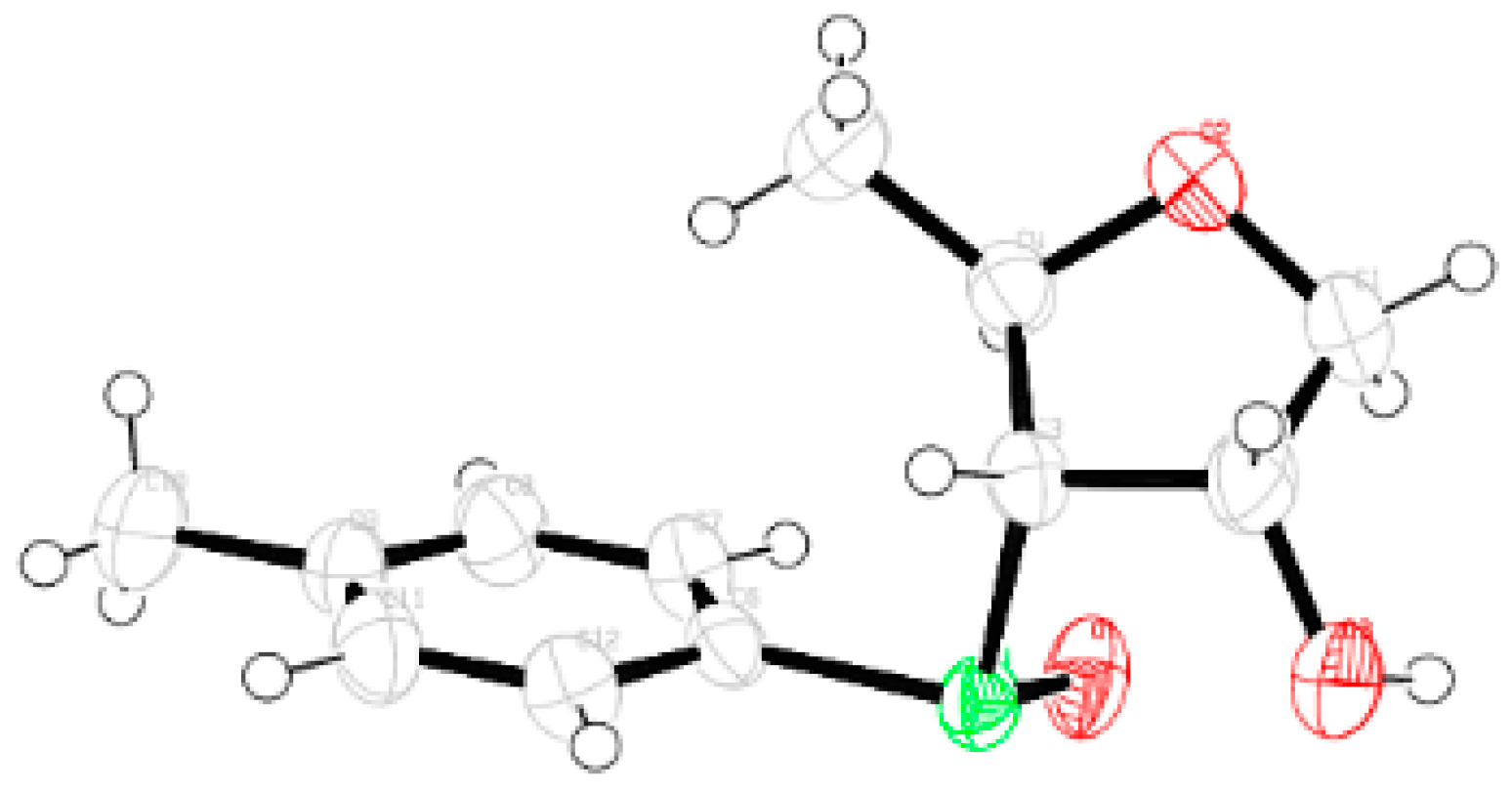
Compound 14: Compound 13 (1.0 g, 2.0 mmol) was converted to 14 (0.73 g, 76%) following the procedure described for the preparation of 8. Eluent: EtOAc: PE (1:4); colorless gum; = (+) 13.8 (c = 1.7, CHCl3); 1H-NMR (200 MHz, CDCl3): δ = 2.29 (s, 3H), 3.10 (dd, J = 3.0, 10.4 Hz, 1H), 3.42 (d, J = 3.8 Hz, 2H), 3.61 (dd, J = 2.6, 10.4 Hz, 1H), 3.89–4.12 (m, 4H), 7.04 (d, J = 7.8, 2H), 7.16–7.46 (m, 18H); 13C-NMR (50 MHz, CDCl3): δ = 21.2, 54.6, 64.8 (CH2), 74.7 (CH2), 77.4, 83.2, 87.9, 127.4, 128.1, 128.9, 130.1, 130.7, 131.4, 137.3, 143.5 (3 × C); HRMS [ES+, (M + H)+]: for C31H31O3S obsd 483.1939, calcd 483.1916.
Compound 15: Compound 14 (0.5 g, 1.04 mmol) was converted to 15 (0.45 g, 84%) following the procedure described for the preparation of 9. Eluent: EtOAc: PE (2:3); white solid; = (+) 26.9 (c = 1.06, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.42 (s, 3H), 2.75 (dd, J = 8.0, 10.8 Hz, 1H), 3.32 (s, 1H), 3.55–3.64 (m, 2H), 4.02–4.03 (m, 2H), 4.40–4.42 (m, 1H), 4.72 (s, 1H), 7.25–7.39 (m, 18H), 7.62 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.9, 64.4 (CH2), 72.6, 73.2, 75.5 (CH2), 77.9, 87.7, 127.5, 128.2, 128.5, 128.8, 130.3, 135.1, 136.5, 143.4 (3 × C), 145.4; HRMS [ES+, (M + Na)+]: for C31H30O5NaS obsd 537.1689, calcd 537.1712.
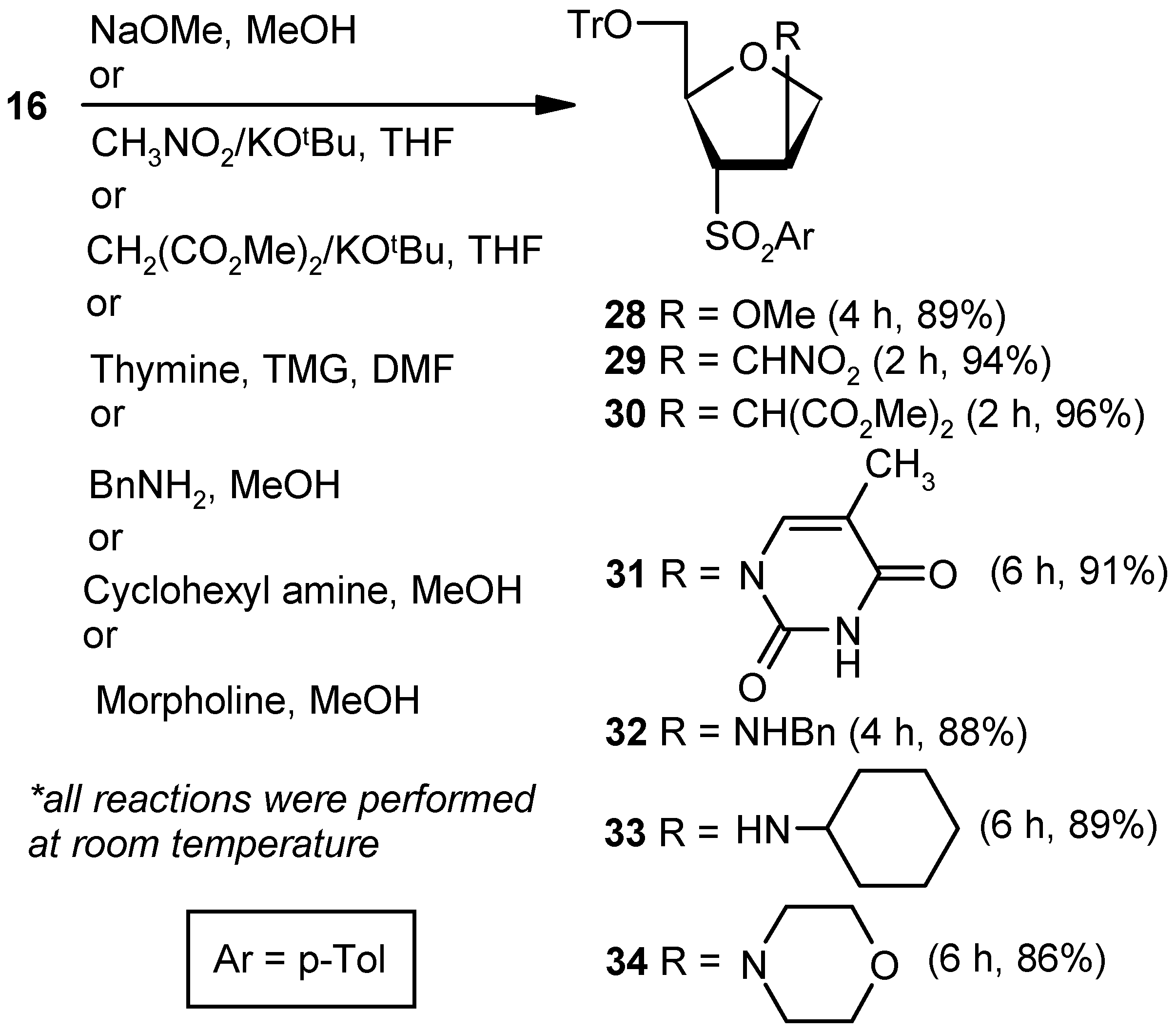
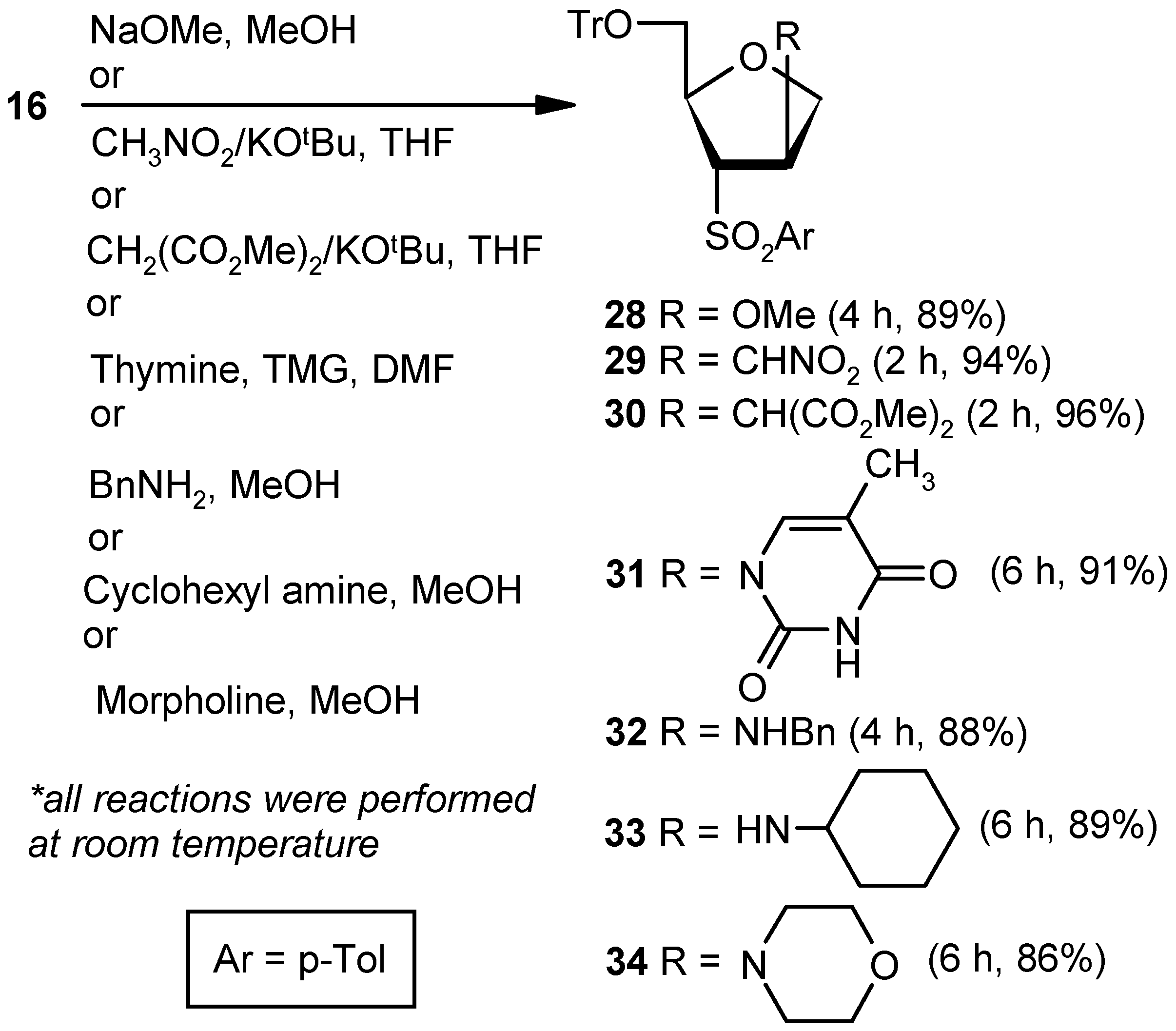
Compound 16: Compound 15 (0.5 g, 0.97 mmol) was converted to 16 (0.43 g, 89%) following the procedure described for the preparation of 10. Eluent: EtOAc: PE (1:4); yellowish gum; = (+) 23.5 (c = 1.06, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.41 (s, 3H), 3.14–3.18 (m, 1H), 3.41 (d, J = 10.4 Hz, 1H), 4.77–4.96 (m, 2H), 5.28 (s, 1H), 7.0 (s, 1H), 7.18–7.38 (m, 18H), 7.61 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.6, 65.2 (CH2), 74.6 (CH2), 84.1, 86.6, 127.0, 127.7, 127.8, 128.7, 129.9, 136.5, 140.8, 141.9, 143.8 (3 × C), 144.7; HRMS [ES+, (M + Na)+]: for C31H28O4NaS obsd 519.1578, calcd 519.1606.
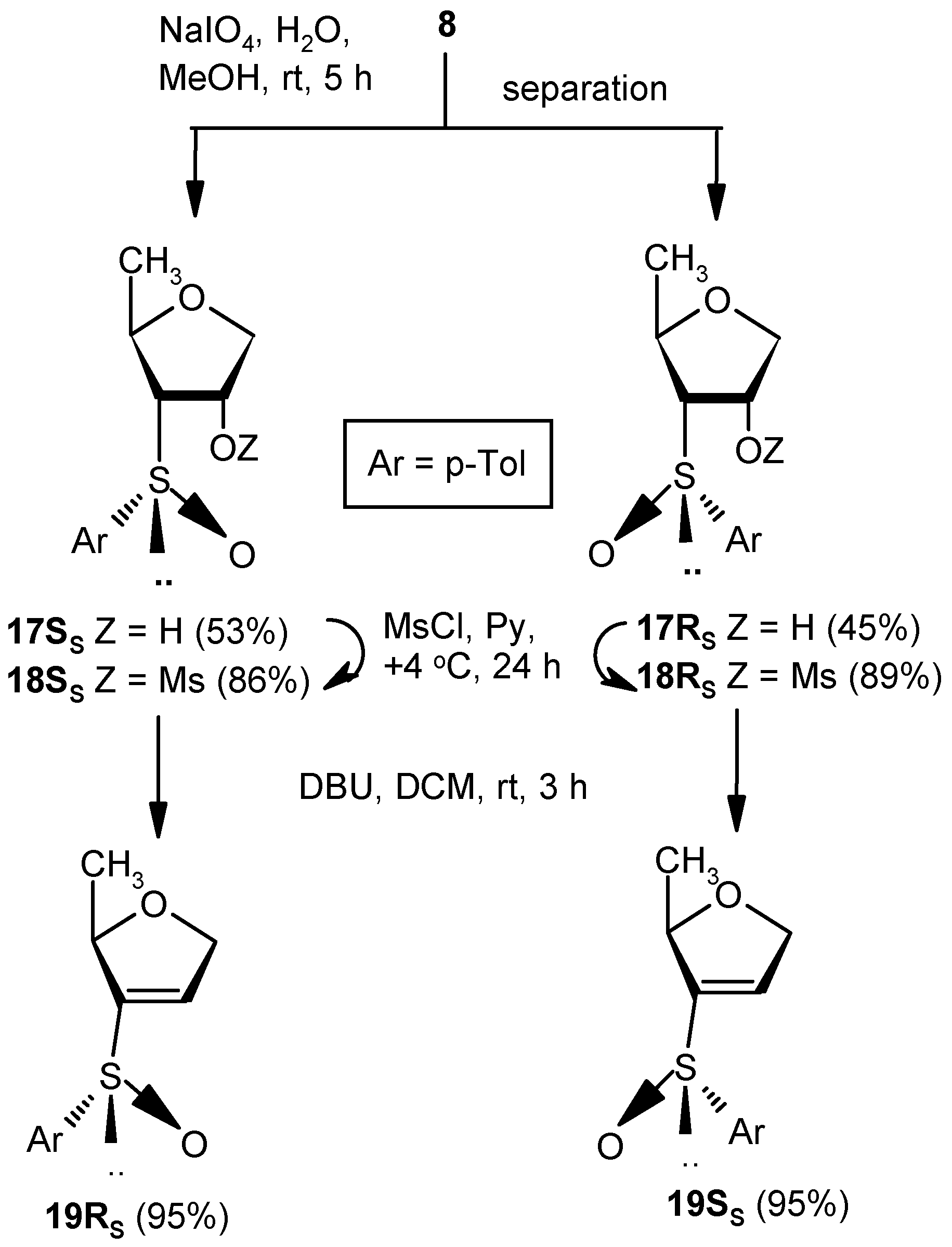
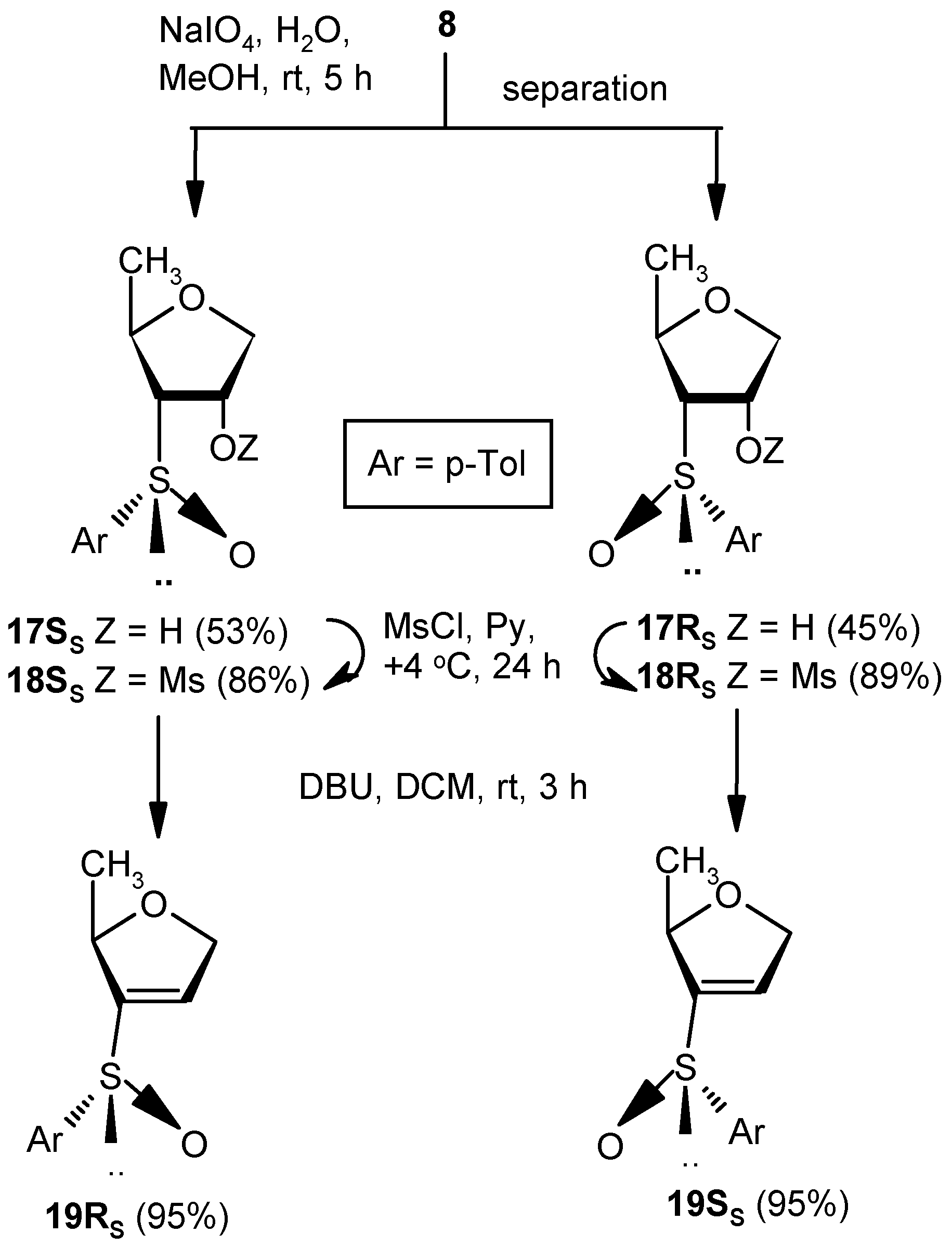
Compounds 17SS and 17RS: A solution of NaIO4 (2.27 g, 10.7 mmol) in water (3 mL) was added to a well-stirred solution of 8 (2.0 g, 8.92 mmol) in MeOH (25 mL), and the mixture was stirred at room temperature. After 5 h, volatile matters were evaporated to dryness under reduced pressure, and the residue was partitioned between satd. aqueous solution of NaHCO3 and EtOAc (3 × 10 mL). The combined organic layer was dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure to get a residue. The residue was purified over silica gel to afford 17SS and 17RS. Compound 17SS: Yield (1.13 g, 53%); Eluent: EtOAc: PE (3:2); white solid; Mp = 127 °C; = (−) 80.7 (c = 0.93, CHCl3); ); 1H-NMR (200 MHz, CDCl3): δ = 0.90 (d, J = 17.2 Hz, 3H), 2.41 (s, 3H), 2.84 (t, J = 7.0 Hz, 1H), 3.71 (dd, J = 4.4, 11.2 Hz, 1H), 3.97 (dd, J = 4.8, 9.6 Hz, 1H), 4.47–4.67 (m, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H); 13C-NMR (50 MHz, CDCl3): δ = 21.4, 21.6, 72.7, 73.1, 73.4, 73.8 (CH2), 124.5, 130.1, 138.5, 141.7; HRMS [ES+, (M + Na)+]: for C12H16O3NaS obsd 263.0688, calcd 263.0718. Compound 17RS: Yield (0.96 g, 45%); Eluent: EtOAc: PE (4:1); white solid; Mp = 98 °C; = (−) 23.1 (c = 1.01, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.78 (d, J = 6.0 Hz, 3H), 2.42 (s, 3H), 2.82–2.85 (m, 1H), 3.83 (dd, J = 2.4, 9.6 Hz, 1H), 3.94 (dd, J = 4.4, 10.8 Hz, 1H), 4.16–4.19 (m, 1H), 4.79 (s, 1H), 5.08 (s, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.63 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.8, 21.7, 73.1, 73.6, 74.3 (CH2), 74.5, 125.7, 130.5, 138.4, 143.1; HRMS [ES+, (M + Na)+]: for C12H16O3NaS obsd 263.0699, calcd 263.0718.
Compound 18SS: Compound 17SS (0.5 g, 2.08 mmol) was converted to 18SS (0.57 g, 86%) following the procedure described for the preparation of 10. Eluent: EtOAc: PE (1:1); colorless gum; = (+) 130.7 (c = 1.01, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.96 (d, J = 6.0 Hz, 3H), 2.43 (s, 3H), 3.00 (t, J = 7.2 Hz, 1H), 3.20 (s, 3H), 3.96 (dd, J = 6.0, 10.0 Hz, 1H), 4.12 (dd, J = 5.6, 10.0 Hz, 1H), 4.60–4.65 (m, 1H), 5.28–5.32 (m, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 4.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.2, 21.4, 38.7, 70.0, 70.2 (CH2), 72.1, 77.3, 124.2, 130.1, 138.2, 141.9; HRMS [ES+, (M + Na)+]: for C13H18O5NaS2 obsd 341.0480, calcd 341.0493.
Compound 18RS: Compound 17RS (1.0 g, 4.16 mmol) was converted to 18RS (1.17 g, 89%) following the procedure described for the preparation of 10. Eluent: EtOAc: PE (3:2); colorless gum; = (+) 188.7 (c = 1.07, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.48 (d, J = 6.4 Hz, 3H), 2.43 (s, 3H), 3.20–3.23 (m, 1H), 3.27 (s, 3H), 3.99–4.04 (m, 2H), 4.22 (d, J = 11.2 Hz, 1H), 4.45 (bs, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.9, 21.8, 38.6, 72.4 (CH2), 72.9, 75.2, 81.8, 125.8, 130.7, 138.4, 143.9; HRMS [ES+, (M + Na)+]: for C13H18O5NaS2 obsd 341.0487, calcd 341.0493.
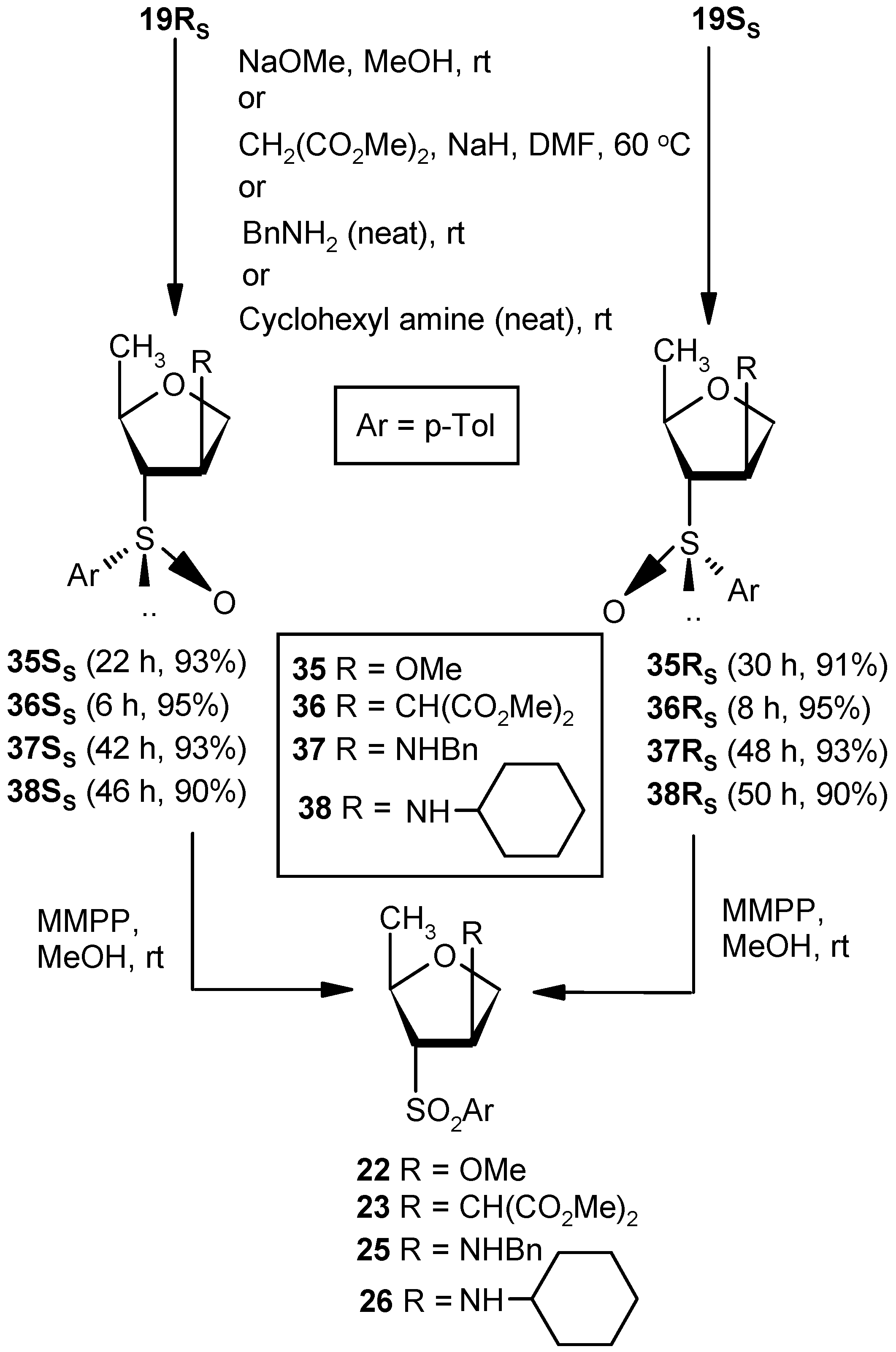
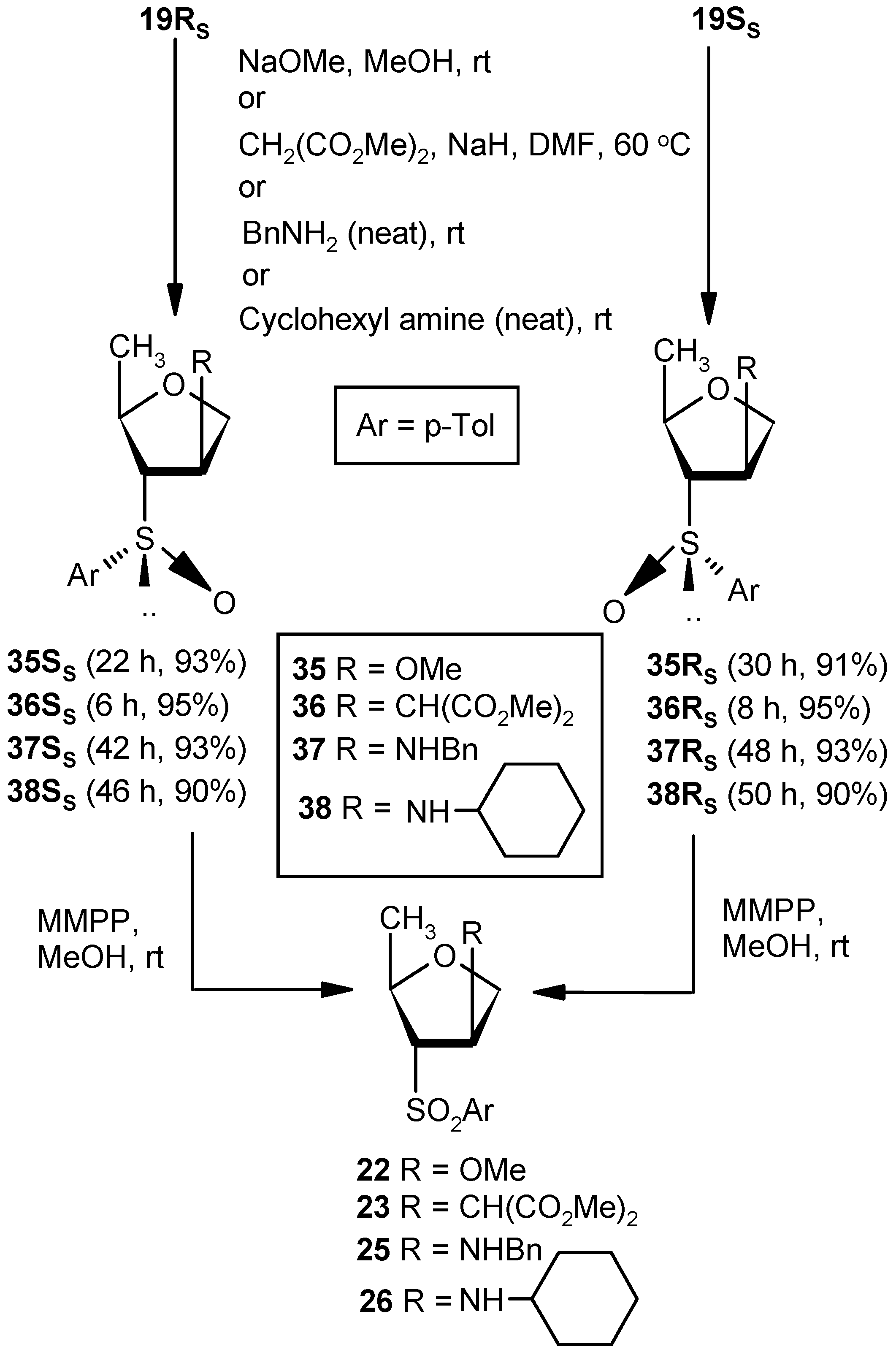
Compound 19RS: Compound 18SS (0.5 g, 1.58 mmol) was treated with DBU (0.5 mL, 3.16 mmol) in DCM (5 mL) at ambient temperature for 3 h. Solvent was evaporated under reduced pressure, and the resulting residue was purified over silica gel to afford 18RS (0.34 g, 95%). Eluent: EtOAc: PE (2:3); colorless gum; = (−) 137.5 (c = 0.98, CHCl3); ); 1H-NMR (400 MHz, CDCl3): δ = 1.21 (d, J = 6.4 Hz, 3H), 2.41 (s, 3H), 4.54–4.55 (m, 1H), 4.69 (d, J = 14.4 Hz, 1H), 4.82 (dd, J = 5.6, 14.0 Hz, 1H), 6.59 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 7.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.2, 21.7, 74.7 (CH2), 80.6, 125.7, 130.5, 130.5, 138.6, 142.9, 148.8; HRMS [ES+, (M + H)+]: for C12H15O2S obsd 223.0784, calcd 223.0793.
Compound 19SS: Compound 18RS (0.5 g, 1.58 mmol) was converted to 19SS (0.34 g, 95%) following the procedure described for the preparation of 19RS. Eluent: EtOAc: PE (2:3); Yellowish gum; = (+) 55.7 (c = 1.01, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.25 (d, J = 6.0 Hz, 3H), 2.37 (s 3H), 4.58 (d, J = 12.8 Hz, 1H), 4.69–4.77 (m, 2H), 6.52 (s, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.4, 21.7, 73.6 (CH2), 80.4, 124.7, 130.0, 134.4, 138.4, 141.8, 146.9; HRMS [ES+, (M + H)+]: for C12H15O2S obsd 223.0806, calcd 223.0793.
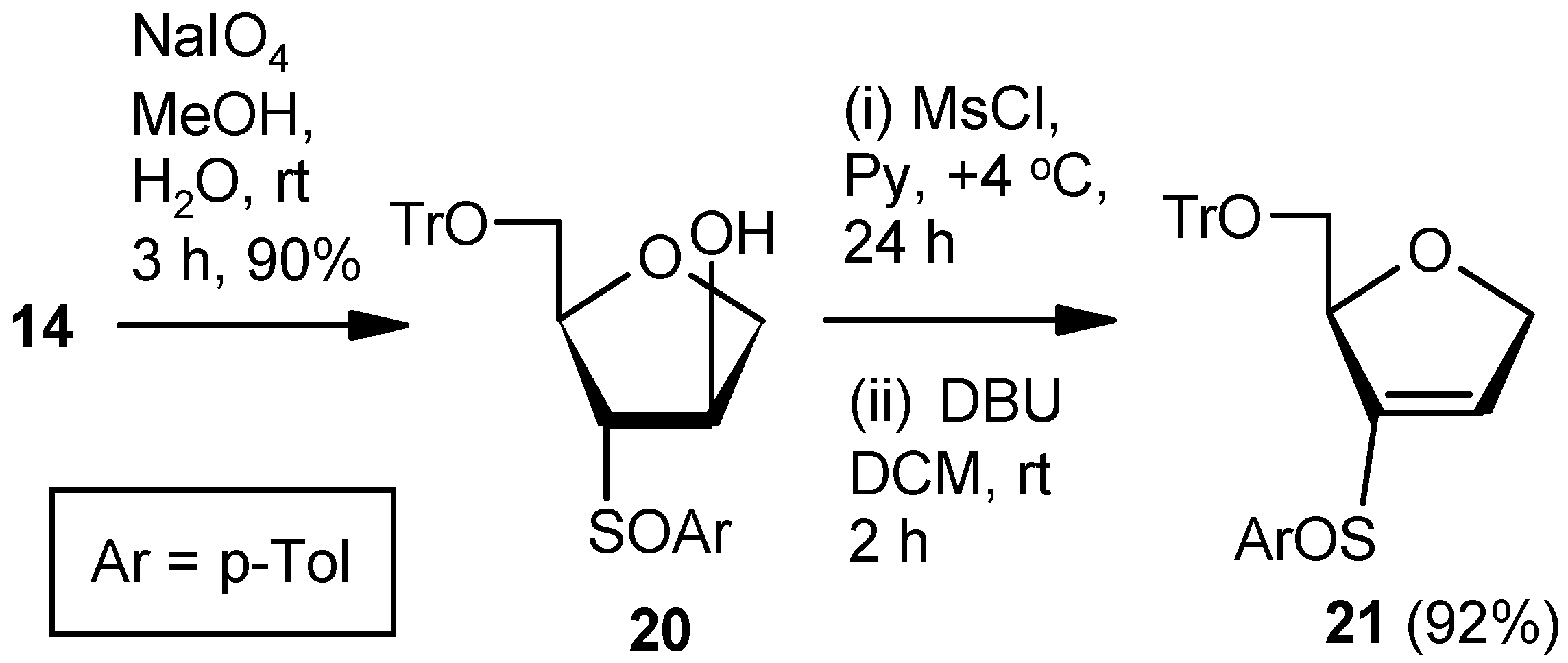
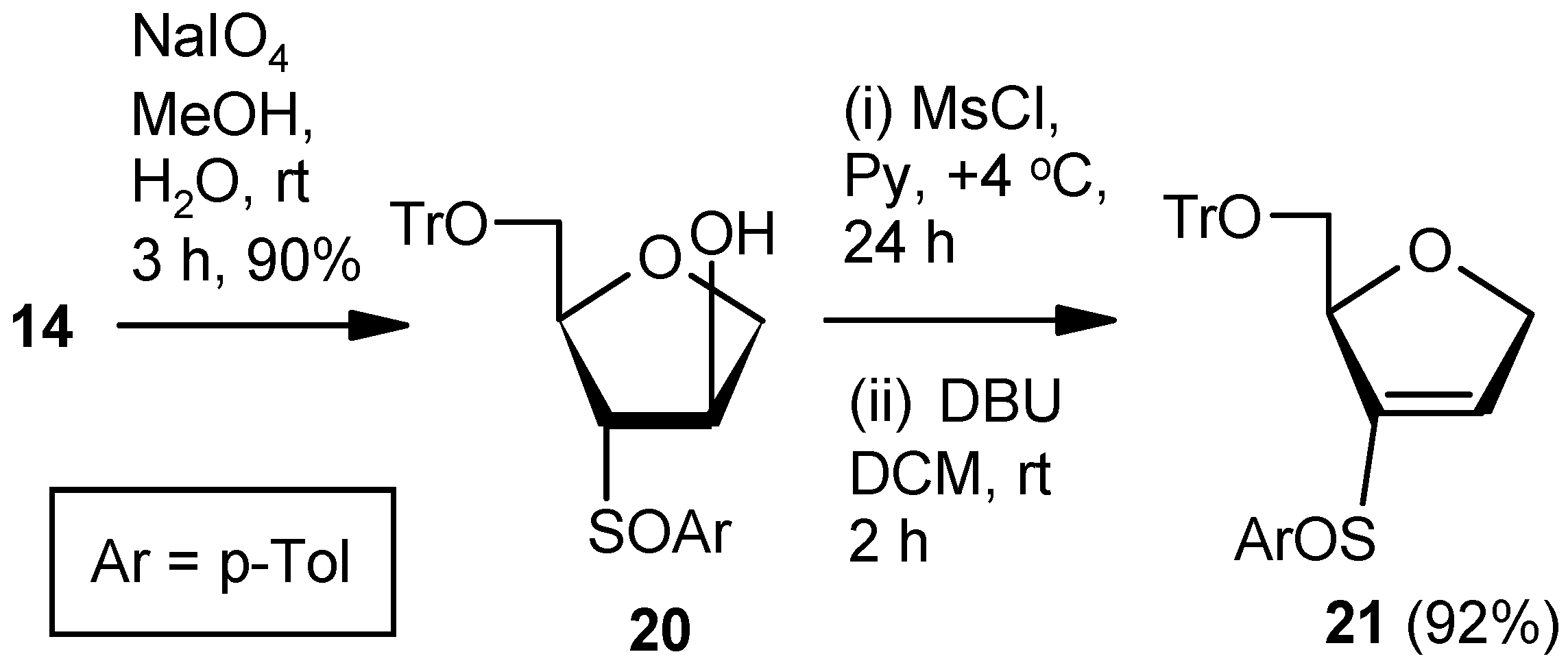
Compound 20: Compound 14 (1.0 g, 2.07 mmol) was converted to the diastereomeric mixture of sulfoxides 20 (0.95 g, 92%) following the procedure described for the preparation of 17SS and 17RS. Colorless gum; 1H-NMR (400 MHz, CDCl3): δ = 2.33, 2.37, 2.81–2.85, 3.28–3.31, 3.42–3.45, 3.96–4.00, 4.08–4.11, 7.09–7.45; HRMS [ES+, (M + Na)+]: for C31H30O4NaS obsd 521.1756, calcd 521.1762.
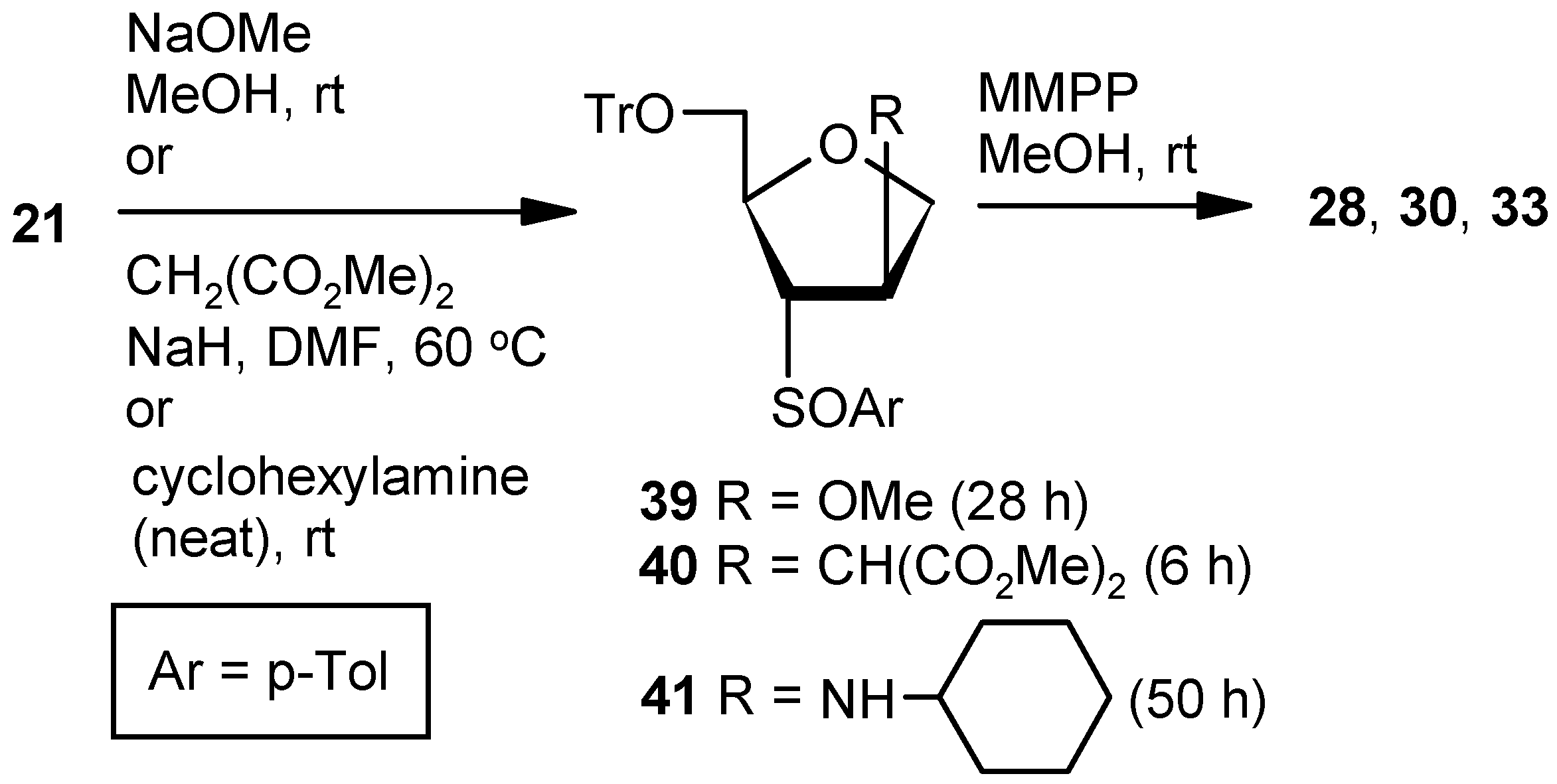
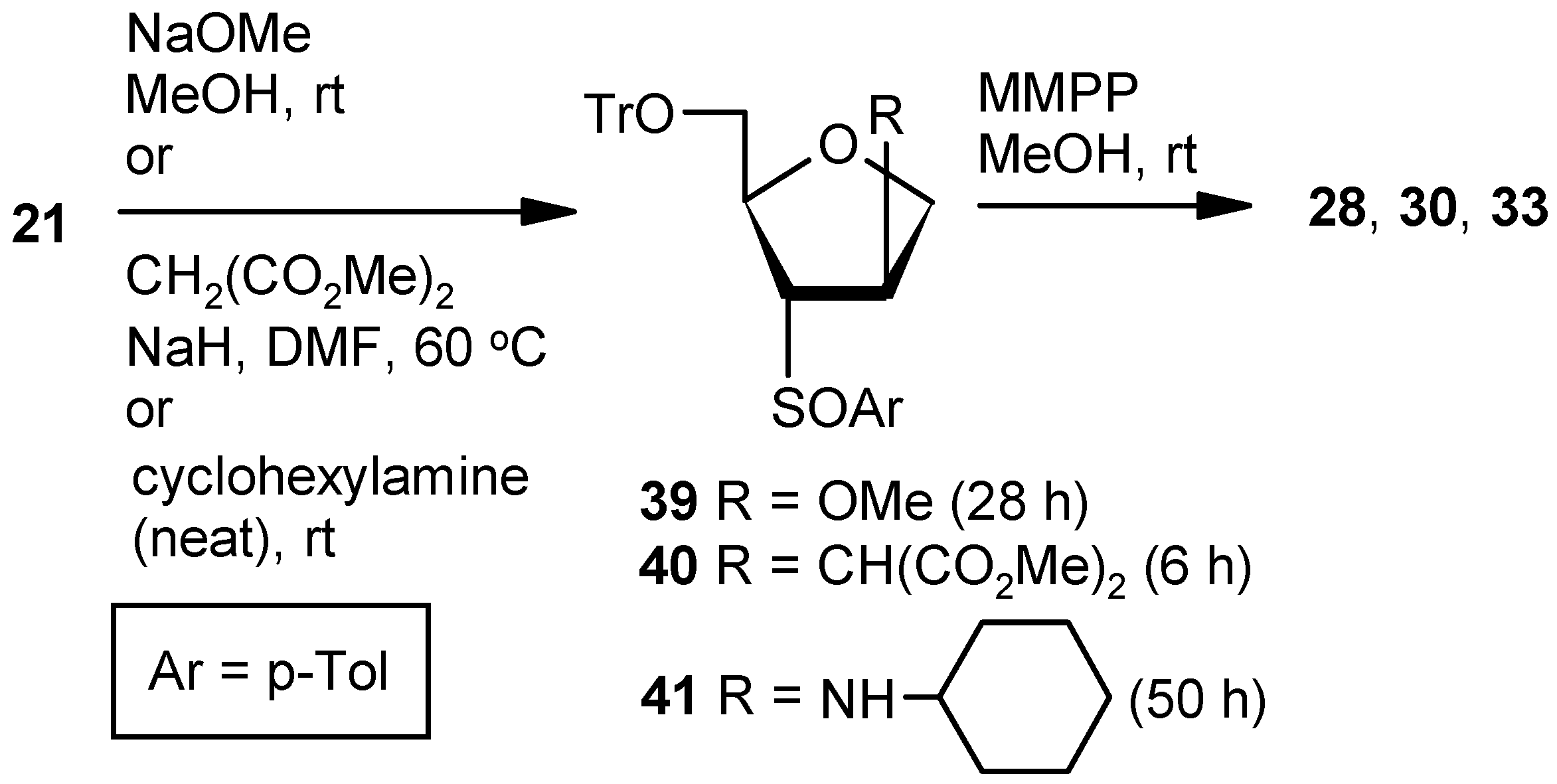
Compound 21: A solution of mesylchloride (0.25 mL, 3.0 mmol) in anhyd pyridine (3 mL) was dropwise added to a stirred solution of 20 (0.5 g, 1.00 mmol) in anhyd pyridine (5 mL) at 0 °C. After 0.5 h, the solution was stored at +4 °C. After 24 h (tlc), the reaction mixture was poured into ice-cold water, and the compound was extracted with EtOAc. The organic layer was separated, dried over anhyd Na2SO4, and filtered, and the filtrate was evaporated to dryness. Residual pyridine was co-evaporated with toluene. The residue was treated with DBU (0.4 mL, 2.5 mmol) in DCM (8 mL) at ambient temperature for 2 h. Solvent was evaporated under reduced pressure, and the resulting residue was purified over silica gel to afford the diastereomeric mixture of vinyl sulfoxides 21 (0.44 g, 92%). Colorless gum; 1H-NMR (400 MHz, CDCl3): δ = 2.25, 2.44, 3.03–3.07, 3.17–3.21, 3.33–3.92, 4.50, 4.74–5.04, 6.50, 6.81, 7.24–7.47; HRMS [ES+, (M + Na)+]: for C31H28O3NaS obsd 503.1664, calcd 503.1657.
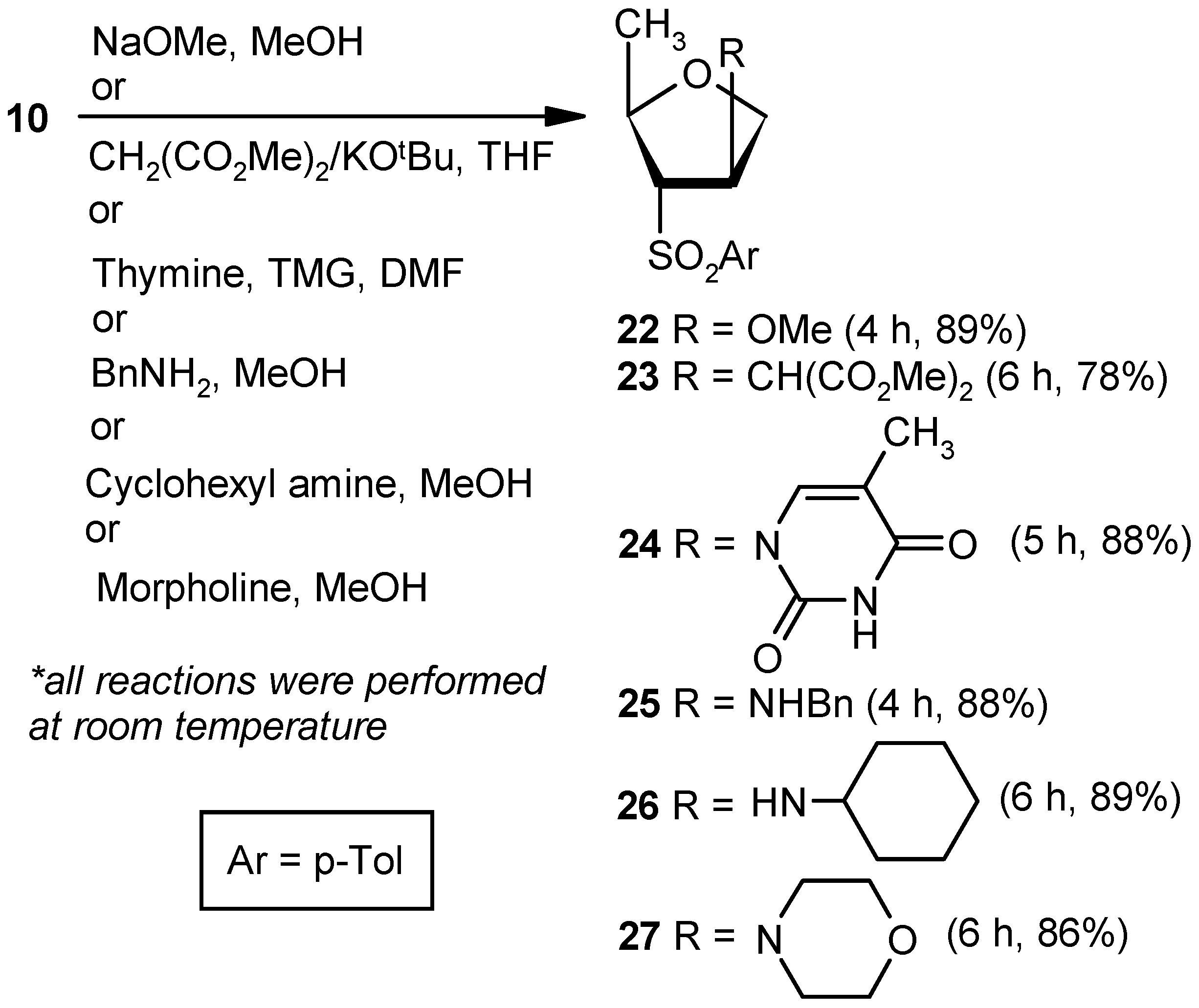
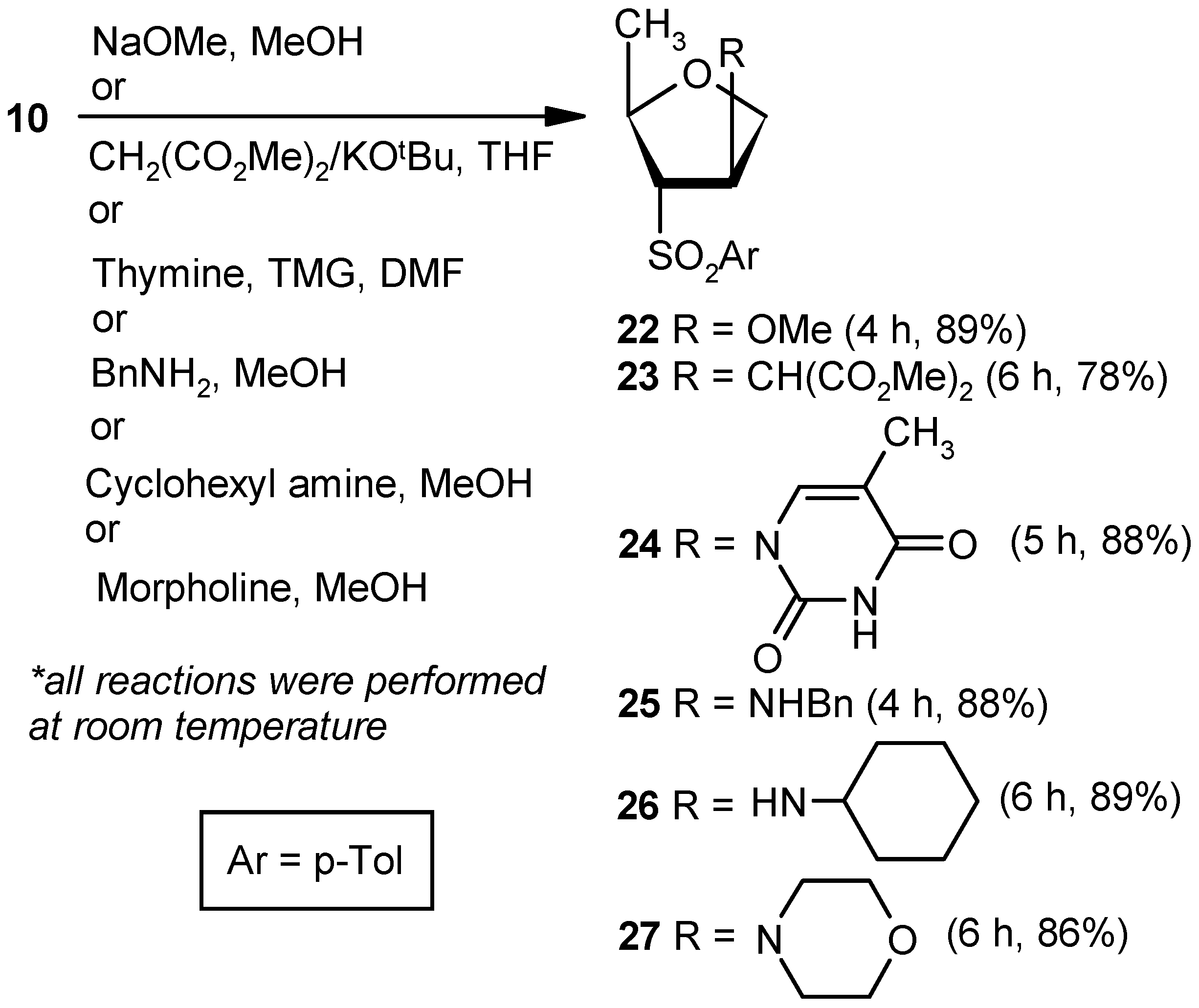
Compound 22: Sodium methoxide (0.02 g, 0.36 mmol) was added to an anhyd methanolic solution (5 mL) of vinyl sulfone 10 (0.043 g, 0.18 mmol), and the mixture was stirred at room temperature. After 4 h (tlc), volatile matters were removed under reduced pressure. The solid residue was stirred in a mixture of EtOAc and satd. aqueous NaHCO3 solution for 1 h. Organic layers were pooled together, dried over anhyd Na2SO4, and filtered, and the filtrate was evaporated. The residue thus obtained was purified over silica gel to afford 22 (0.04 g, 89%). Eluent: EtOAc: PE (1:4); White solid; Mp = 89 °C; = (+) 14.1 (c = 0.42, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.20 (d, J = 6.0 Hz, 3H), 2.50 (s, 3H), 3.20 (s, 3H), 3.21–3.24 (m, 1H), 3.72 (dd, J = 4.4, 10.4 Hz, 1H), 4.01 (d, J = 10.4 Hz, 1H), 4.23–4.32 (m, 2H), 7.42 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 20.4, 21.6, 56.9, 72.5 (CH2), 75.6, 75.6, 82.9, 128.5, 130.1, 135.2, 145.3; HRMS [ES+, (M + H)+]: for C13H19O4S obsd 271.1015, calcd 271.1004.
Compound 23: A mixture of dimethylmalonate (0.28 mL, 2.52 mmol) and KOtBu (0.23 g, 2.1 mmol) in THF (5 mL) was stirred at room temperature in a N2 atmosphere. After 0.5 h, a solution of 10 (0.2 g, 0.84 mmol) in THF (4 mL) was added to the reaction mixture. After stirring for 6 h (tlc), the mixture was evaporated under reduced pressure. The residue was partitioned between a mixture of EtOAc and satd. aqueous NH4Cl. The organic part was separated, dried over anhyd Na2SO4, and filtered, and the filtrate was evaporated to dryness. The residue thus obtained was purified over silica gel coloumn to afford 23 (0.23 g, 78%). Eluent: EtOAc: PE (1:4); Colorless gum; = (+) 45.6 (c = 1.07, CHCl3); 1H-NMR (200 MHz, CDCl3): δ =1.11 (d, J = 6.0 Hz, 3H), 2.47 (s, 3H), 3.33–3.46 (m, 3H), 3.70 (d, J = 14.0 Hz, 6H), 3.86–3.89 (m, 2H), 4.16–4.22 (m, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.78 (d, J = 8.2 Hz, 2H); 13C-NMR (50 MHz, CDCl3): δ = 20.2, 21.8, 42.0, 52.9, 53.9, 70.7 (CH2), 71.5, 76.3, 128.9, 130.3, 135.1, 145.5, 168.2; HRMS [ES+, (M + H)+]: for C17H23O6S obsd 355.1249, calcd 355.1215.
Compound 24: A well-stirred solution of thymine (0.16 g, 1.26 mmol) and TMG (0.11 mL, 0.9 mmol) in DMF (10 mL) was added to 10 (0.043 g, 0.18 mmol), and the mixture was stirred at ambient temperature in a nitrogen atmosphere. After 5 h, the reaction mixture was diluted with EtOAc (20 mL), and the precipitated solid was filtered off. The filtrate was washed with an aqueous satd. aqueous solution of NaHCO3, and the aqueous part was extracted with EtOAc (3 × 10 mL). The combined organic layer was dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure. The residue was purified over silica gel to get 24 (0.05 g, 88%). Eluent: EtOAc: PE (1:3); White solid; Mp = 145 °C; = (−) 69.5 (c = 0.99, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.43 (d, J = 6.4 Hz, 3H), 1.86 (s, 3H), 2.43 (s, 3H), 3.55 (dd, J = 4.8, 7.6 Hz, 1H), 3.94–4.03 (m, 2H), 4.35–4.41 (m, 1H), 5.40–5.43 (m, 1H), 7.06 (s, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.82 (d, J = 8.0 Hz, 2H), 9.12 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 12.5, 20.3, 21.7, 58.8, 71.2 (CH2), 74.1, 76.1, 112.4, 128.7, 130.1, 134.5, 136.9, 145.8, 149.9, 163.3; HRMS [ES+, (M + H)+]: for C17H21N2O5S obsd 365.1156, calcd 365.1171.
Compound 25: Benzylamine (0.2 mL, 1.8 mmol) was added to an anhyd methanolic solution (5 mL) of 10 (0.043 g, 0.18 mmol), and the mixture was stirred at room temperature. After 4 h (tlc), volatile matters were removed under reduced pressure. The residue was partitioned between EtOAc and satd. aqueous NH4Cl solution. Then, the organic part was dried over anhyd Na2SO4 and filtered, and the filtrate was evaporated to dryness. The residue thus obtained was purified over silica gel to afford 25 (0.05 g, 88%). Eluent: EtOAc: PE (1:3); Brown solid; Mp = 139 °C; = (−) 23.1 (c = 0.48, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.22 (d, J = 6.4 Hz, 3H), 2.46 (s, 3H), 3.13–3.16 (m, 1H), 3.72 (q, J = 3.2 Hz, 2H), 3.76–3.85 (m, 3H), 4.22–4.25 (m, 1H), 7.18 (d, J = 8.0 Hz, 2H), 7.23–7.35 (m, 6H), 7.71 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 20.9, 21.6, 51.4 (CH2), 60.8, 72.6 (CH2), 75.3, 75.8, 127.1, 128.0, 128.4, 128.5, 130.0, 135.0, 139.1, 145.1; HRMS [ES+, (M + H)+]: for C19H24NO3S obsd 346.1447, calcd 346.1477.
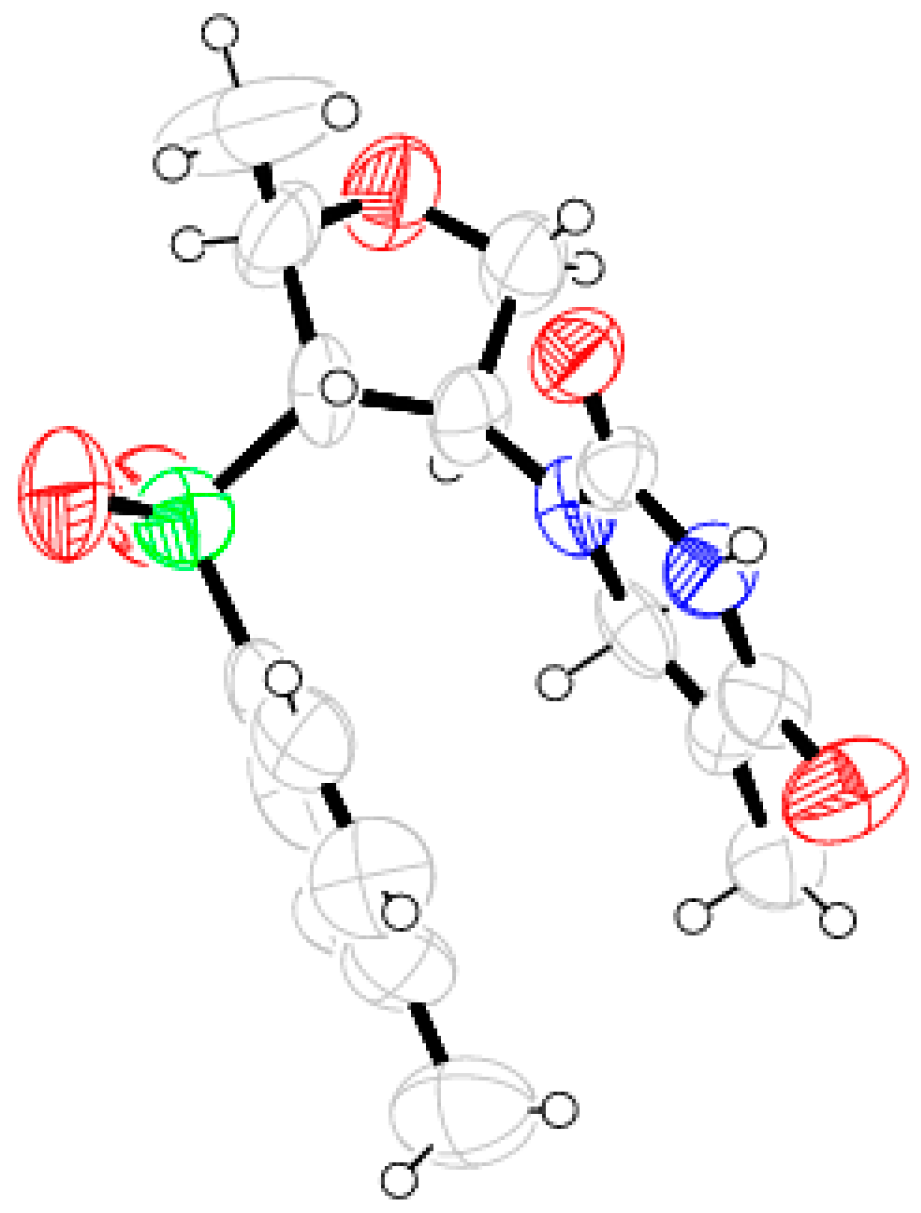
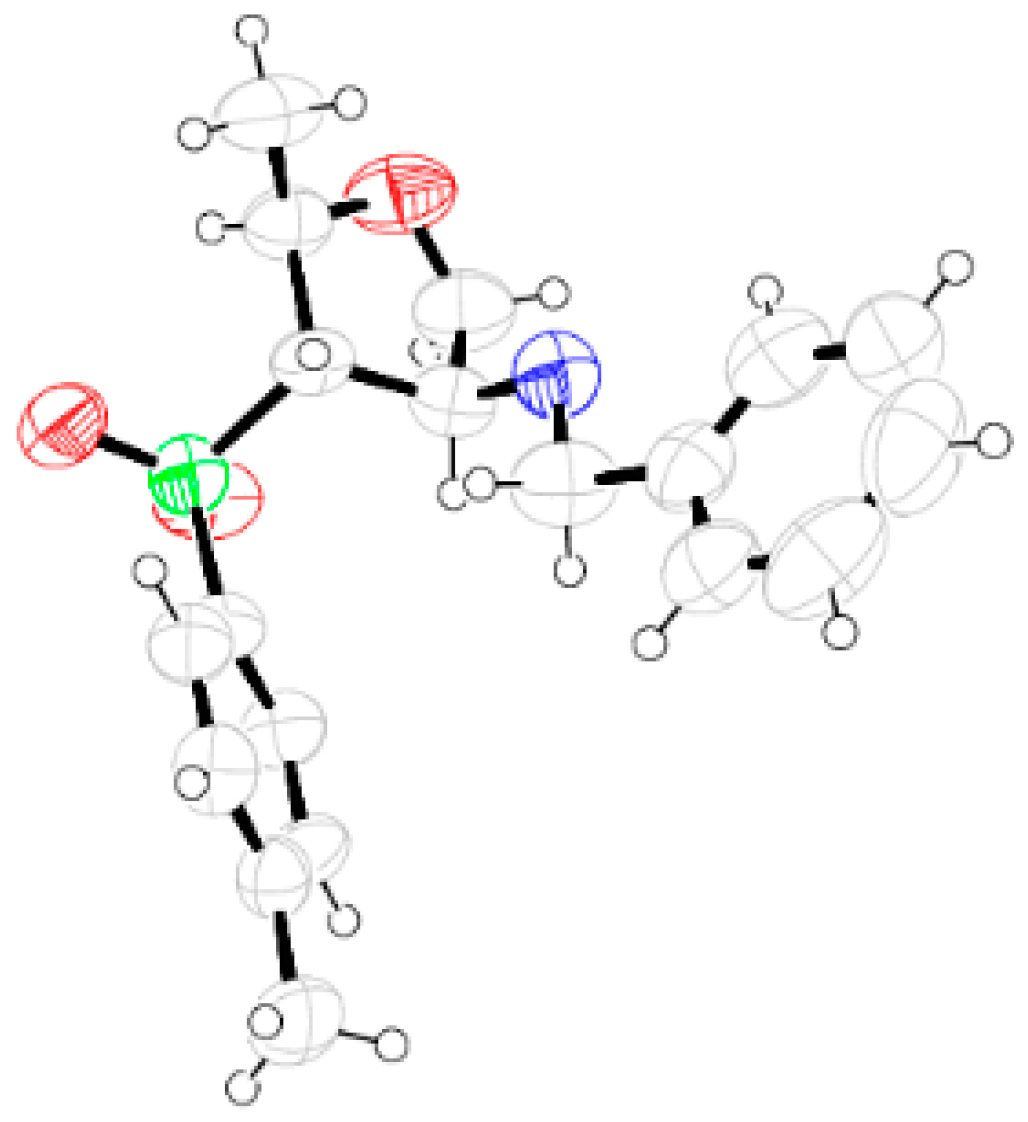
Compound 26: Cyclohexylamine (1.9 mL, 16.80 mmol) was reacted with 10 (0.2 g, 0.84 mmol) following the procedure described for 25 to afford 26 (0.24 g, 89%). Eluent: EtOAc: PE (1:3); Brownish gum; = (−) 37.6 (c = 1.09, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.02–1.16 (m, 3H), 1.21–1.25 (m, 4H), 1.54–1.60 (m, 7H), 2.12–2.18 (m, 1H), 2.46 (s, 3H), 3.08 (dd, J = 3.2, 7.2 Hz, 1H), 3.72–3.82 (m, 3H), 4.25–4.28 (m, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.80 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.2, 21.6, 24.6 (CH2), 24.8 (CH2), 25.8 (CH2), 33.3 (2 × CH2), 54.2, 58.4, 73.7 (CH2), 75.0, 75.9, 128.5, 130.0, 135.3, 145.1; HRMS [ES+, (M + H)+]: for C18H28NO3S obsd 338.1771, calcd 338.1790.
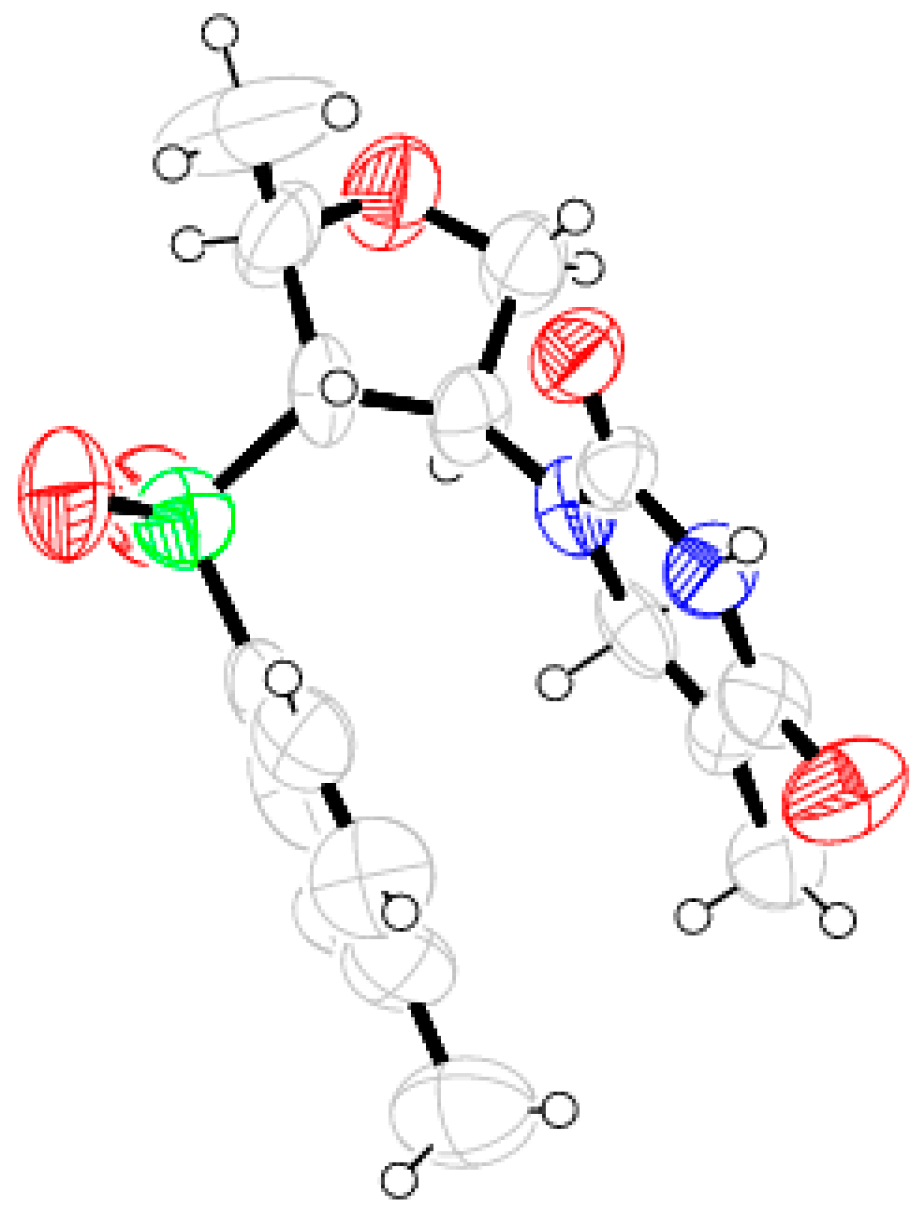
Compound 27: Morpholine (1.4 mL, 16.80 mmol) was reacted with 10 (0.2 g, 0.84 mmol) following the procedure described for 25 to afford 27 (0.22 g, 86%). Eluent: EtOAc: PE (1:4); Brownish solid; Mp = 121 °C; = (−) 33.6 (c = 1.05, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.00 (d, J = 6.0 Hz, 3H), 2.08–2.11 (m, 2H), 2.41 (s, 3H), 3.25 (d, J = 7.2 Hz, 1H), 3.55–3.59 (m, 4H), 3.66–3.71 (m, 1H), 3.82–3.83 (m, 1H), 4.00 (d, J = 10.4 Hz, 1H), 4.11 (t, J = 6.8 Hz, 1H), 7.34 (d, J = 7.6 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.2, 21.8, 49.5 (CH2), 66.8 (CH2), 68.1, 68.8, 71.0 (CH2), 76.2, 128.8, 130.2, 135.3, 145.5; HRMS [ES+, (M + H)+]: for C16H24NO4S obsd 326.1463, calcd 326.1426.
Compound 28: Compound 16 (0.043 g, 0.09 mmol) was converted to 28 (0.04 g, 89%) following the procedure described for the preparation of 22. Eluent: EtOAc: PE (1:4); White sold; Mp = 111 °C; = (+) 56.3 (c = 0.86, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.40 (s, 3H), 2.71 (dd, J = 4.4, 10.0 Hz, 1H), 3.24 (d, J = 3.6 Hz, 1H), 3.27 (s, 3H), 3.84 (d, J = 5.2 Hz, 1H), 3.93 (dd, J = 4.8, 9.6 Hz, 1H), 4.10 (d, J = 10.0 Hz, 1H), 4.33–4.36 (m. 1H), 4.44–4.45 (m, 1H), 7.21–7.28 (m, 12H), 7.37 (d, J = 7.2 Hz, 6H), 7.67 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.6, 56.9, 63.5 (CH2), 69.6, 73.3 (CH2), 78.8, 82.1, 86.4, 126.9, 127.7, 128.3, 128.6, 130.0, 135.0, 143.6 (3 × C), 144.9; HRMS [ES+, (M + Na)+]: for C32H32O5NaS obsd 551.1858, calcd 551.1868.
Compound 29: A mixture of nitromethane (0.03 mL, 0.27 mmol) and KOtBu (0.02 g, 0.22 mmol) in THF (5 mL) was reacted with Compound 16 (0.043 g, 0.09 mmol) in THF (4 mL) following the procedure described for 23 to afford 29 (0.047 g, 94%). Eluent: EtOAc: PE (1:4); Colorless gum; = (+) 89.2 (c = 0.95, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.45 (s, 3H), 2.73 (dd, J = 3.2, 10.8 Hz, 1H), 3.47–3.53 (m, 2H), 3.57–3.60 (m, 1H) 4.00 (dd, J = 3.6, 9.2 Hz, 1H), 4.13–4.17 (m, 1H), 4.39–4.42 (m, 1H), 4.56 (d, J = 7.2 Hz, 2H), 7.26–7.38 (m, 18H), 7.64 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.7, 40.3, 63.6 (CH2), 66.1, 71.3 (CH2), 76.0 (CH2), 79.0, 87.2, 127.2, 127.9, 128.4, 128.5, 130.2, 134.1, 143.2 (3 × C), 145.5; HRMS [ES+, (M + Na)+]: for C32H31NO6NaS obsd 580.1763, calcd 580.1770.
Compound 30: A mixture of dimethylmalonate (0.13 mL, 1.21 mmol) and KOtBu (0.11 g, 1.0 mmol) in THF (5 mL) was reacted with Compound 16 (0.2 g, 0.4 mmol) in THF (4 mL) following the procedure described for 23 to afford 30 (0.24 g, 96%). Eluent: EtOAc: PE (1:4); Colorless gum; = (+) 73.6 (c = 0.59, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.40 (s, 3H), 2.74 (dd, J = 4.4, 10.4 Hz, 1H), 3.26 (dd, J = 2.4, 10.4 Hz, 1H), 3.39–3.43 (m, 1H), 3.61 (s, 3H), 3.69 (s, 3H), 3.83–3.91 (m, 1H), 4.06–4.10 (m, 1H), 4.33–4.36 (m, 1H), 7.20–7.30 (m, 12H), 7.37 (d, J = 7.2 Hz, 6H), 7.63 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.7, 41.1, 52.7, 52.8, 53.2, 63.8 (CH2), 66.2, 71.6 (CH2), 79.3, 86.9, 127.0, 127.7, 128.6, 128.7, 129.9, 134.7, 143.4 (3 × C), 144.9, 167.9, 168.1; HRMS [ES+, (M + Na)+]: for C36H36O8NaS obsd 651.2018, calcd 651.2029.
Compound 31: Compound 16 (0.043 g, 0.09 mmol) was converted to 31 (0.046 g, 88%) following the procedure described under the preparation of 24. Eluent: EtOAc: PE (1:3); White solid; Mp = 110 °C; = (+) 129.6 (c = 0.91, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.43 (s, 3H), 2.42 (s, 3H), 3.07 (dd, J = 3.2, 10.8 Hz, 1H), 3.66 (dd, J = 1.6, 10.8 Hz, 1H), 4.05–4.21 (m, 3H), 4.41–4.46 (m, 1H), 5.73–5.77 (m, 1H), 7.27–7.41 (m, 18H), 7.75 (d, J = 8.0 Hz, 2H), 9.31 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ = 12.3, 21.9, 55.9, 63.5 (CH2), 68.5, 72.4 (CH2), 79.4, 87.6, 112.7, 127.6, 128.1, 128.8, 130.3, 134.5, 136.8, 143.2 (3 × C), 145.8, 150.4, 163.6; HRMS [ES+, (M + Na)+]: for C36H34N2O6NaS obsd 645.2030, calcd 645.2029.
Compound 32: Compound 16 (0.043 g, 0.09 mmol) was converted to 32 (0.046 g, 88%) following the procedure described for 25. Eluent: EtOAc: PE (1:3); Colorless gum; = (+) 91.4 (c = 0.69, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.42 (s, 3H), 2.70 (dd, J = 3.2, 10.4 Hz, 1H), 3.44–3.46 (m, 1H), 3.62 (d, J = 13.6 Hz, 1H), 3.70–3.73 (m, 2H), 3.81–3.82 (m, 1H), 3.95–4.03 (m, 2H), 4.34–4.35 (m, 1H), 7.19–7.29 (m, 16H), 7.37 (d, J = 6.4 Hz, 6H), 7.59 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.7, 51.2 (CH2), 60.3, 63.8 (CH2), 69.6, 73.8 (CH2), 78.3, 86.9, 127.0, 127.0, 127.8, 128.0, 128.3, 128.3, 128.6, 130.0, 134.9, 139.3, 143.4 (3 × C), 144.8; HRMS [ES+, (M + H)+]: for C38H38NO4S obsd 604.2511, calcd 604.2522.
Compound 33: Cyclohexylamine (0.92 mL, 8.06 mmol) was reacted with 16 (0.2 g, 0.40 mmol) following the procedure described for 25 to afford 33 (0.21 g, 89%). Eluent: EtOAc: PE (1:3); Colorless gum; = (+) 103.6 (c = 1.03, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.90–1.37 (m, 7H), 1.52–1.66 (m, 7H), 2.22–2.27 (m, 1H), 2.43 (s, 3H), 2.73 (dd, J = 3.2, 10.4 Hz, 1H), 3.45 (dd, J = 2.8, 10.4 Hz, 1H), 3.61–3.63 (m, 1H), 3.88–4.02 (m, 3H), 4.34–4.37 (m, 1H), 7.25–7.42 (m, 18H), 7.66 (d, J = 8.4 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.6, 24.6 (CH2), 24.8 (CH2), 26.0 (CH2), 32.9 (CH2), 33.3 (CH2), 53.9, 57.7, 63.8 (CH2), 69.8, 74.4 (CH2), 78.1, 86.9, 126.9, 127.0, 127.7, 127.8, 128.3, 128.6, 128.7, 129.9, 135.1, 143.5 (3 × C), 144.8; HRMS [ES+, (M + H)+]: for C37H42NO4S obsd 596.2827, calcd 596.2829.
Compound 34: Morpholine (0.70 mL, 8.06 mmol) was reacted with 16 (0.2 g, 0.40 mmol) following the procedure described for 25 to afford 34 (0.0.20 g, 86%). Eluent: EtOAc: PE (1:4); Brownish gum; = (+) 96.2 (c = 0.87, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 2.40 (s, 3H), 2.45–2.49 (m, 3H), 2.84 (dd, J = 5.2, 10.4 Hz, 1H), 3.14 (d, J = 9.6 Hz, 1H), 3.58 (s, 3H), 3.77–3.90 (m, 3H), 4.14–4.16 (m, 1H), 4.28–4.30 (m, 1H), 7.19–7.37 (m, 18H), 7.65 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.9, 50.0 (CH2), 63.7 (CH2), 64.6, 66.9 (CH2), 67.6, 70.5 (CH2), 79.3, 87.2, 127.3, 127.9, 128.2, 128.6, 128.9, 130.1, 130.5, 131.3, 135.3, 143.6 (3 × C), 145.2; HRMS [ES+, (M + H)+]: for C35H38NO5S obsd 584.2498, calcd 584.2471.
Compound 35SS: Compound 19RS (0.07 g, 0.31 mmol) was converted to 35SS (0.07 g, 93%) following the procedure described for the preparation of 22. Eluent: EtOAc: PE (1:2); Colorless gum; = (−) 35.6 (c = 1.01, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.89 (d, J = 6.0 Hz, 3H), 2.42 (s, 3H), 2.78 (d, J = 6.0 Hz, 1H), 3.27 (s, 3H), 3.59–3.63 (m, 1H), 3.96 (d, J = 10.4 Hz, 1H), 4.18–4.23 (m, 2H), 7.34 (d, J = 7.6 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 20.8, 21.4. 57.0, 71.4 (CH2), 72.1, 73.1, 83.5, 124.2, 130.0, 137.8, 141.9; HRMS [ES+, (M + H)+]: for C13H19O3S obsd 255.1071, calcd 255.1055.
Compound 35RS: Compound 19SS (0.07 g, 0.31 mmol) was converted to 35RS (0.069 g, 91%) following the procedure described for the preparation of 22. Eluent: EtOAc: PE (1:2); Colorless gum; = (+) 126.8 (c = 0.99, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.22 (d, J = 6.4 Hz, 3H), 2.41 (s, 3H), 2.78 (d, J = 7.6 Hz, 1H), 2.97 (s, 3H), 3.65–3.69 (m, 1H), 3.96–4.02 (m, 2H), 4.23 (s, 1H), 7.34 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.7, 21.4, 56.5, 72.8 (CH2), 75.0, 75.6, 80.8, 124.5, 130.0, 138.5, 142.1; HRMS [ES+, (M + H)+]: for C13H19O3S obsd 255.1069, calcd 255.1055.
Compound 22 from 35RS or 35SS: Compound 35RS (0.1 g, 0.39 mmol) was converted to 22 (0.09 g, 85%) following the procedure described for the preparation of 9. Compound 35SS was converted to 22 in a similar fashion.
Compound 36SS: Dimethylmalonate (0.5 mL, 4.05 mmol) was added to a well stirred solution of NaH (0.05 g, 3.37 mmol) in DMF (5 mL), and the mixture was stirred for 0.5 h at ambient temperature in an inert atmosphere. Vinyl sulfoxide 19RS (0.3 g, 1.35 mmol) was added, and the whole mixture was stirred at 60 °C. After 6 h (tlc), the reaction mixture was partitioned between aqueous satd. solution of NaHCO3 and EtOAc (3 × 10 mL). The combined organic layer was dried over anhyd Na2SO4 and filtered, and the filtrate was concentrated under reduced pressure to get a residue. The residue was purified over silica gel to get 36SS (0.45 g, 95%). Eluent: EtOAc: PE (1:3); Colorless gum; = (−) 69.3 (c = 0.65, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.24 (d, J = 6.0 Hz, 3H), 2.50–2.60 (m, 2H), 3.61 (s, 3H), 3.68 (s, 3H), 3.76–3.87 (m, 2H), 3.92–4.02 (m, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.7, 21.5, 39.7, 52.8, 54.0, 67.1 (CH2), 72.0, 76.3, 124.6, 130.0, 138.2, 141.9, 167.9, 168.1; HRMS [ES+, (M + Na)+]: for C17H22O6NaS obsd 377.1058, calcd 377.1035.
Compound 36RS: A mixture of dimethylmalonate (0.5 mL, 4.05 mmol) and NaH (0.05 g, 3.37 mmol) in DMF (5 mL) was reacted with Compound 19SS (0.3 g, 1.35 mmol) in DMF (4 mL) following the procedure described for 36SS to afford Compound 36RS (0.45 g, 95%). Eluent: EtOAc: PE (1:3); Colorless gum compound; = (+) 112.5 (c = 0.98, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.15 (d, J = 6.0 Hz, 3H), 2.41 (s, 3H), 2.94–2.97 (m, 1H), 3.02 (d, J = 6.4 Hz, 1H), 3.27–3.32 (m, 1H), 3.62 (s, 3H), 3.68 (s, 3H), 3.76–3.82 (m, 2H), 3.80 (dd, J = 2.8, 9.6 Hz, 1H), 4.00 (q, J = 6.4 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 19.5, 21.4, 39.4, 52.6, 53.8, 70.6 (CH2), 71.6, 75.3, 124.5, 130.1, 138.2, 142.1, 168.1, 168.2; HRMS [ES+, (M + Na)+]: for C17H22O6NaS obsd 377.1064, calcd 377.1035.
Compound 26 from 36RS or 36SS: Compound 36RS (0.06 g, 0.16 mmol) was converted to 26 (0.048 g, 77%) following the procedure described for the preparation of 9. Compound 36SS was converted to 26 in a similar fashion.
Compound 37SS: A mixture of vinyl sulfoxide 19RS (0.3 g, 1.35 mmol) and benzylamine (2.9 mL, 27.0 mmol) was stirred at room temperature for 42 h. After completion of reaction, the mixture was partitioned between EtOAc and satd. aqueous NH4Cl solution. Then, the organic part was separated, dried over anhyd Na2SO4, and filtered, and the filtrate was evaporated to dryness. The residue thus obtained was purified over silica gel to afford Compound 37SS (0.41 g, 93%). Eluent: EtOAc: PE (2:3); Brownish gum; = (−) 59.6 (c = 0.88, CHCl3); ); 1H-NMR (400 MHz, CDCl3): δ = 1.70 (d, J = 6.4 Hz, 3H), 1.88–2.10 (m, 3H), 2.44 (s, 3H), 2.58–2.79 (m, 1H), 4.00 (d, J = 10.4 Hz, 2H), 4.39 (t, J = 8.0 Hz, 1H), 7.38–7.60 (m, 7H), 7.68 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.3, 21.4, 52.2 (CH2), 58.8, 71.6, 74.7 (CH2), 76.6, 123.7, 126.9, 128.0, 128.0, 130.2, 138.9, 139.3, 142.3; HRMS [ES+, (M + H)+]: for C19H24NO2S obsd 330.1554, calcd 330.1528.
Compound 37RS: Benzylamine (2.9 mL, 27.0 mmol) was reacted with 19SS (0.3 g, 1.35 mmol) following the procedure described for 37SS to afford 37RS (0.41 g, 93%). Eluent: EtOAc: PE (2:3); Brownish gum; = (−) 103.7 (c = 0.76, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 1.19 (d, J = 6.0 Hz, 3H), 2.42 (s, 3H), 2.68–2.70 (m, 1H), 3.49 (t, J = 15.2 Hz, 2H), 3.77–3.85 (m, 3H), 4.00–4.06 (m, 1H), 7.09 (d, J = 6.8 Hz, 2H), 7.21–7.34 (m, 6H), 7.52 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 20.0, 21.4, 51.9 (CH2), 59.0, 73.0 (CH2), 74.8, 76.0, 124.4, 127.1, 128.0, 128.3, 130.1, 138.6, 139.3, 142.1; HRMS [ES+, (M + H)+]: for C19H24NO2S obsd 330.1563, calcd 330.1528.
Compound 25 from 37RS or 37SS: Compound 37RS (0.1 g, 0.30 mmol) was converted to 25 (0.031 g, 30%) following the procedure described for the preparation of 9. Compound 37SS was converted to 25 in a similar fashion. The oxidation of 37RS/37SS was quenched within 0.5 h to avoid over-oxidation.
Compound 38SS: Cyclohexylamine (3.1 mL, 27.0 mmol) was reacted with 19RS (0.3 g, 1.35 mmol) following the procedure described for 37SS to afford 38SS (0.39 g, 90%). Eluent: EtOAc: PE (2:3); Brownish gum; = (−) 39.5 (c = 0.86, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.94–1.01 (m, 5H), 1.08–1.19 (m, 3H), 1.57–1.74 (m, 7H), 2.28 (t, J = 10.4 Hz, 1H), 2.41 (s, 3H), 2.61–2.63 (m, 1H), 3.62 (d, J = 3.6 Hz, 1H), 3.74 (d, J = 3.2 Hz, 2H), 4.22 (t, J = 6.4 Hz, 1H), 7.33 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 21.4, 21.4, 24.8 (CH2), 25.8, 33.2 (CH2), 33.6 (CH2), 54.7, 58.5, 72.9, 72.9 (CH2), 74.6, 124.4, 130.0, 138.4, 141.9; HRMS [ES+, (M + H)+]: for C18H28NO2S obsd 322.1875, calcd 322.1841.
Compound 38RS: Cyclohexylamine (3.1mL, 27.0 mmol) was reacted with 19SS (0.3 g, 1.35 mmol) following the procedure described for 37SS to afford 38RS (0.39 g, 90%). Eluent: EtOAc: pet ether (2:3); Brownish gum; = (−) 120.3 (c = 0.91, CHCl3); 1H-NMR (400 MHz, CDCl3): δ = 0.46–0.58 (m, 2H), 0.95–1.03 (m, 3H), 1.22–1.35 (m, 4H), 1.69–1.75 (m, 4H), 2.44 (s, 3H), 2.92–2.95 (m, 1H), 3.05 (t, J = 5.6 Hz, 1H), 3.30–3.36 (m, 1H), 4.08 (t, J = 8.0 Hz, 1H), 4.39 (t, J = 6.4 Hz, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 7.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 16.5, 21.4, 24.7 (CH2), 24.8 (CH2), 25.7 (CH2), 33.0 (CH2), 33.8 (CH2), 55.1, 57.0, 73.7 (CH2), 75.5, 76.3, 126.3, 130.1, 138.8, 142.9; HRMS [ES+, (M + H)+]: for C18H28NO2S obsd 322.1825, calcd 322.1841.
Compound 26 from 38RS or 38SS: Compound 38RS (0.1 g, 0.31 mmol) was converted to 26 (0.029 g, 28%) following the procedure described for the preparation of 9. Compound 38SS was converted to 26 in a similar fashion. The oxidation of 38RS/38SS was quenched within 0.5 h to avoid over-oxidation.
Compound 28 from 21: Compound 21 (0.3 g, 0.62 mmol) was converted to 39 following the procedure described for the preparation of 22. The inseparable mixture 39 was converted to 28 following the procedure described for the preparation of 9.
Compound 30 from 21: Compound 21 (0.3 g, 0.62 mmol) was converted to 40 following the procedure described under the preparation of 36SS. The inseparable mixture 40 was converted to 30 following the procedure described for the preparation of 9.
Compound 33 from 21: Compound 21 (0.3 g, 0.62 mmol) was converted to 41 following the procedure described for the preparation of Compound 37SS. The inseparable mixture 41 was converted to 33 following the procedure described for the preparation of 9. The oxidation of 41 was quenched within 0.5 h to avoid over-oxidation.


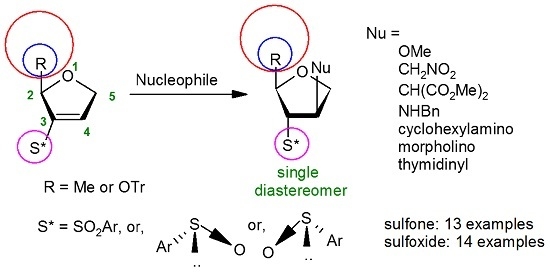
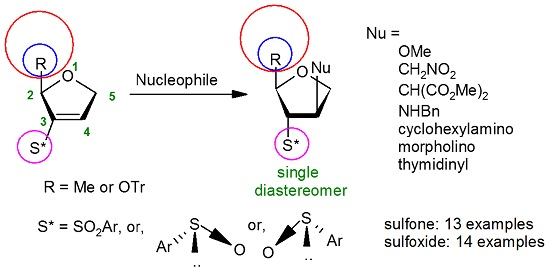{kind=link}
{kind=link}
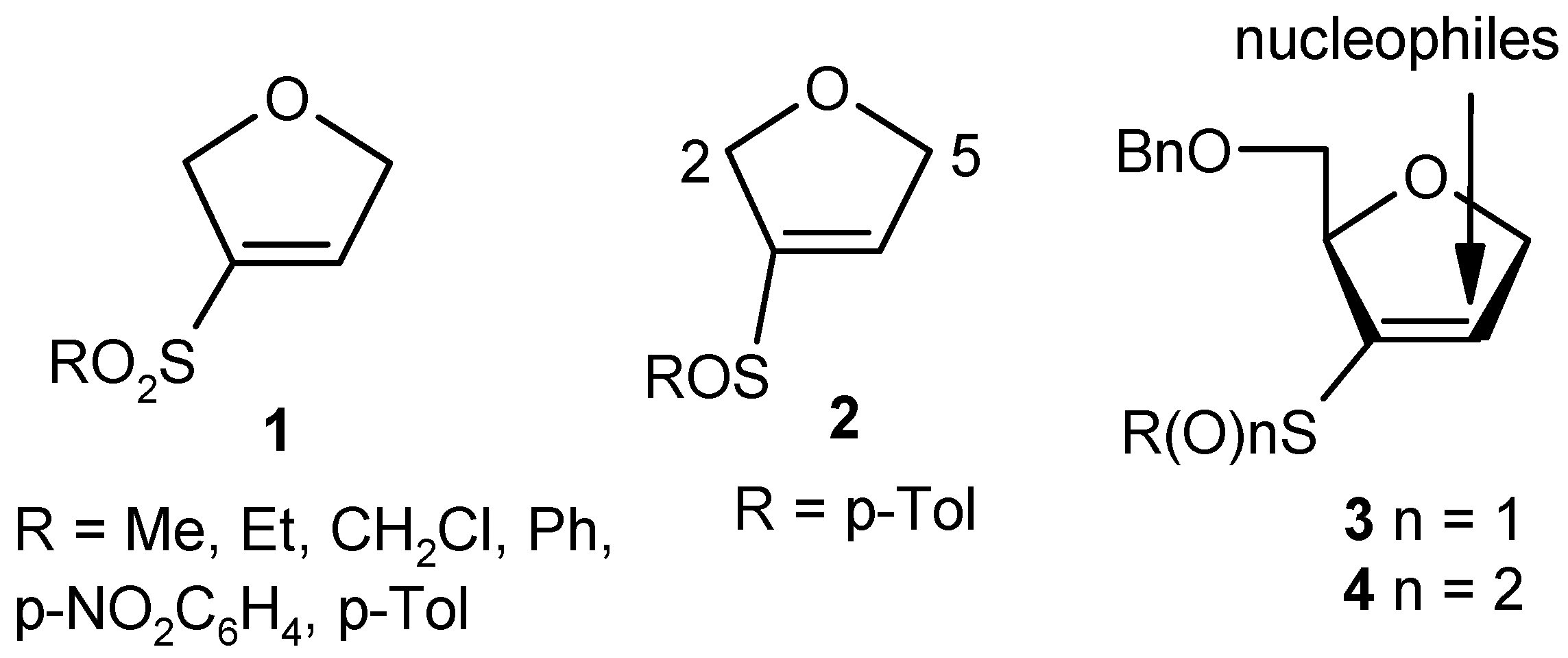{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}