
The Rationale for Insulin Therapy in Alzheimer’s Disease

{kind=link}
Abstract
:1. Amyloidogenesis in Alzheimer’s Disease and Diabetes
2. Interactions between Pathological Mechanisms of Alzheimer’s Disease and Diabetes
2.1. Cerebral Blood Flow and Glucose Metabolism
2.1.1. Cerebral Blood Flow and Glucose Metabolism in Alzheimer’s Disease
2.1.2. Cerebral Blood Flow and Glucose Metabolism in Diabetes
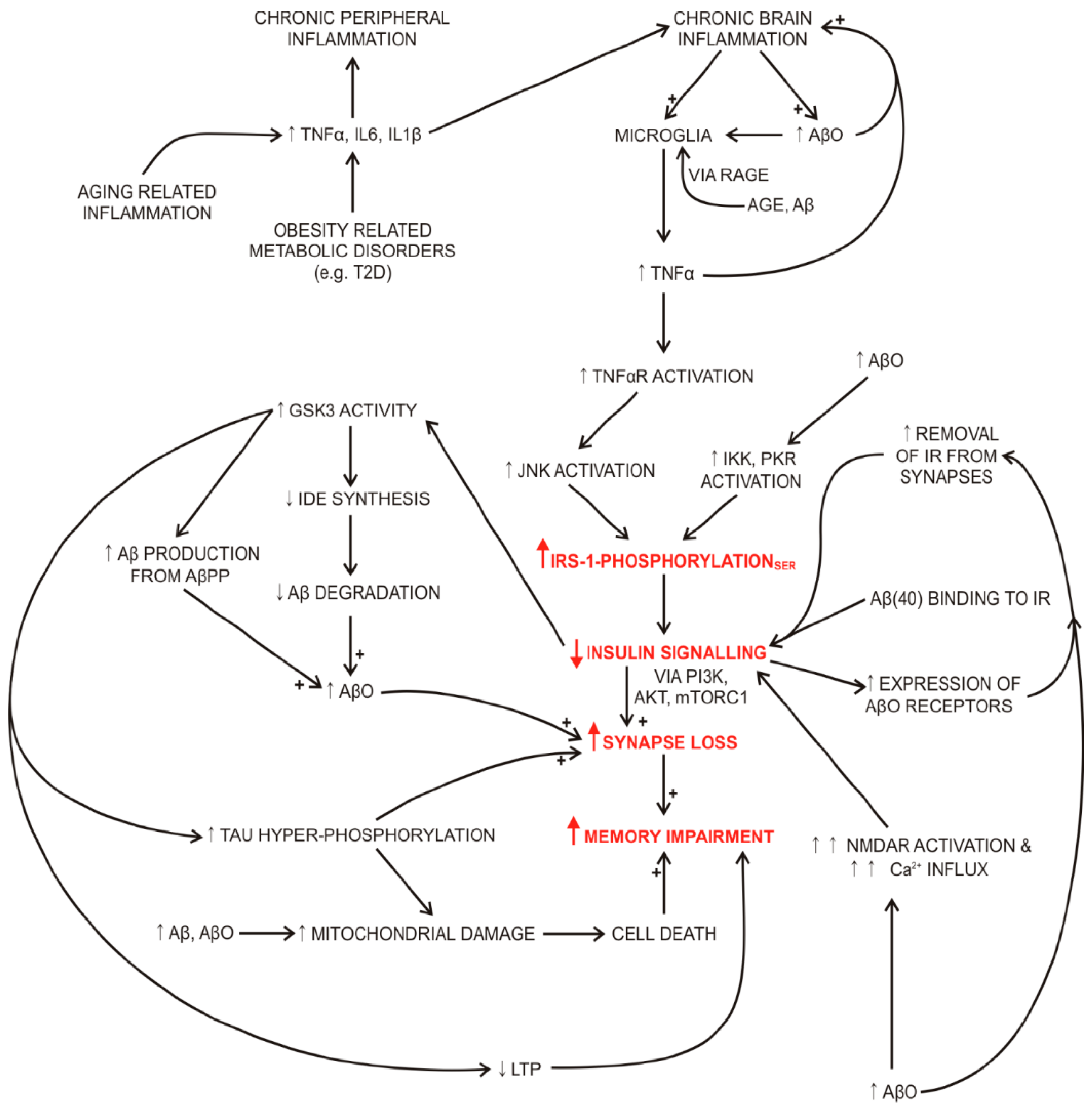
2.2. Impaired Insulin Signalling Links Systemic and Brain Oxidative Stress, Inflammation, Impaired Memory and Insulin Resistance in Diabetes and Alzheimer’s Disease
2.2.1. Impaired Insulin Signalling and Degradation in Alzheimer’s Disease
2.2.2. Impaired Insulin Signalling and Degradation in Diabetes
2.3. Mitochondrial Dysfunction in Alzheimer’s Disease and Diabetes
2.3.1. Mitochondrial Dysfunction in Alzheimer’s Disease
2.3.2. Mitochondrial Dysfunction in Diabetes
2.4. Oxidative Stress
2.4.1. Oxidative Stress in Alzheimer’s Disease
2.4.2. Oxidative Stress in Diabetes
2.5. Advanced Glycation End Products (AGEs)
2.5.1. Advanced Glycation End Products in Alzheimer’s Disease
2.5.2. Advanced Glycation End Products in Diabetes
2.6. Cholesterol Metabolism
2.6.1. Cholesterol Metabolism in Alzheimer’s Disease
2.6.2. Cholesterol Metabolism in Diabetes
2.7. Inflammation
Inflammation in Alzheimer’s Disease
2.8. Cognitive Impairment and Brain Insulin Sensitivity
3. Treatment of Alzheimer’s Disease
4. Antidiabetic Drugs for Treatment of AD
4.1. Peroxisome Proliferator-Activated Receptor-γ Agonists
4.2. Metformin
4.3. Glucagon-Like Peptide-1 Receptor Agonists
4.4. Leptin Analogues
4.5. Amylin Analogues
4.6. Treatment of Alzheimer’s Disease with Intranasal Insulin Application
Clinical Trials of Alzheimer’s Disease Treatment with Intranasal Insulin Application
- (a)
- Single dose trial with regular insulin
- (b)
- Four-months treatment with 20 or 40 IU of regular insulin per day
- (c)
- Twenty-one-days treatment with 20 IU or 40 IU of insulin detemir twice daily
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pahnke, J.; Walker, L.C.; Scheffler, K.; Krohn, M. Alzheimer’s disease and blood-brain barrier function—Why have anti-beta-amyloid therapies failed to prevent dementia progression? Neurosci. Biobehav. Rev. 2009, 33, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Vandal, M.; Bourassa, P.; Calon, F. Can insulin signaling pathways be targeted to transport Aβ out of the brain? Front. Aging Neurosci. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: Therapeutic implications. Adv. Exp. Med. Biol. 2004, 541, 135–152. [Google Scholar] [PubMed]
- Irie, F.; Fitzpatrick, A.L.; Lopez, O.L.; Kuller, L.H.; Peila, R.; Newman, A.B.; Launer, L.J. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: The Cardiovascular Health Study Cognition Study. Arch. Neurol. 2008, 65, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hanninen, T.; Hallikainen, M.; Alhainen, K.; Iivonen, S.; Mannermaa, A.; Tuomilehto, J.; Nissinen, A.; et al. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann. Intern. Med. 2002, 137, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.X.; Manly, J.J.; Mayeux, R. Relation of diabetes to mild cognitive impairment. Arch. Neurol. 2007, 64, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; Pellegrini, S.; Siciliano, G.; Murri, L. Causative and susceptibility genes for Alzheimer’s disease: A review. Brain Res. Bull. 2003, 61, 1–24. [Google Scholar] [CrossRef]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Tong, M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Akisaki, T.; Sakurai, T.; Takata, T.; Umegaki, H.; Araki, A.; Mizuno, S.; Tanaka, S.; Ohashi, Y.; Iguchi, A.; Yokono, K.; Ito, H. Cognitive dysfunction associates with whitematter hyperintensities and subcortical atrophy onmagnetic resonance imaging of the elderly diabetes mellitus Japanese elderly diabetes intervention trial (J-EDIT). Diabetes Metab. Res. Rev. 2006, 22, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Den Heijer, T.; Vermeer, S.E.; van Dijk, E.J.; Prins, N.D.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003, 46, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Manschot, S.M.; Brands, A.M.; van der Grond, J.; Kessels, R.P.; Algra, A.; Kappelle, L.J.; Biessels, G.J. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 2006, 55, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Launer, L.J.; Nilsson, L.G.; Pajak, A.; Sans, S.; Berger, K.; Breteler, M.M.; de Ridder, M.; Dufouil, C.; Fuhrer, R.; et al. Magnetic resonance imaging of the brain in diabetes: The Cardiovascular Determinants of Dementia (CASCADE) Study. Diabetes 2004, 53, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Toth, C.; Martinez, J.; Zochodne, D.W. RAGE, diabetes, and the nervous system. Curr. Mol. Med. 2007, 7, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. 2009, 14, 373–379. [Google Scholar] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, F.; Boulogne, A.; Leys, D.; Fontaine, P. Diabetes mellitus and dementia. Diabetes Metab. 2006, 32, 403–414. [Google Scholar] [CrossRef]
- Nelson, P.T.; Smith, C.D.; Abner, E.A.; Schmitt, F.A.; Scheff, S.W.; Davis, G.J.; Keller, J.N.; Jicha, G.A.; Davis, D.; Wang-Xia, W.; et al. Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim. Biophys. Acta 2009, 1792, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Lyn-Cook, L.E., Jr.; Lawton, M.; Tong, M.; Silbermann, E.; Longato, L.; Jiao, P.; Mark, P.; Wands, J.R.; Xu, H.; de la Monte, S.M. Hepatic ceramide may mediate brain insulin resistance and neurodegeneration in type 2 diabetes and non-alcoholic steatohepatitis. J. Alzheimers Dis. 2009, 16, 715–729. [Google Scholar] [PubMed]
- Winocur, G.; Greenwood, C.E. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol. Aging 2005, 26, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Longato, L.; de la Monte, S.M. Early limited nitrosamine exposures exacerbate high fat diet-mediated type2 diabetes and neurodegeneration. BMC Endocr. Dis. 2010, 10. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Kirkitadze, M.D.; Kowalska, A. Molecular mechanisms initiating amyloid beta-fibril formation in Alzheimer’s disease. Acta Biochim. Pol. 2005, 52, 417–423. [Google Scholar] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Golde, T.E.; Ghiso, J.; Cheung, T.T.; Estus, S.; Shaffer, L.M.; Cai, X.D.; McKay, D.M.; Tintner, R.; Frangione, B.; et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotropic and neurotoxic effects of amyloid b protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Choi, J.K.; Surewicz, K.; Surewicz, W.K. Soluble Prion Protein Binds Isolated Low Molecular Weight Amyloid-β Oligomers Causing Cytotoxicity Inhibition. ACS Chem. Neurosci. 2015, 6, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Kuo, Y.M.; Roher, A.E.; Brachova, L.; Shen, Y.; Sue, L.; Beach, T.; Kurth, J.H.; Rydel, R.E.; Rogers, J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999, 155, 853–862. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Pasternak, J.F.; Kuo, H.; Ristic, H.; Lambert, M.P.; Chromy, B.; Viola, K.L.; Klein, W.L.; Stine, W.B.; Krafft, G.A.; et al. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924, 133–140. [Google Scholar] [CrossRef]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Poling, A.; Morgan-Paisley, K.; Panos, J.J.; Kim, E.M.; O’Hare, E.; Cleary, J.P.; Lesne, S.; Ashe, K.H.; Porritt, M.; Baker, L.E. Oligomers of the amyloid-beta protein disrupt working memory: Confirmation with two behavioral procedures. Behav. Brain Res. 2008, 193, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kuroda, Y.; Arispe, N.; Rojas, E. Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 2000, 275, 14077–14083. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, M.; Nowak, R.J.; Lansbury, P.T., Jr. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry 2002, 41, 11338–11343. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.A.; Ittner, L.M.; Lim, Y.L.; Gotz, J. Human but not rat amylin shares neurotoxic properties with Abeta42 in long-term hippocampal and cortical cultures. FEBS Lett. 2008, 582, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.J. Amylin compared with calcitonin gene-related peptide: Structure, biology, and relevance to metabolic disease. Endocr. Rev. 1994, 15, 163–201. [Google Scholar] [CrossRef] [PubMed]
- Green, J.D.; Goldsbury, C.; Kistler, J.; Cooper, G.J.; Aebi, U. Human amylin oligomer growth and fibril elongation define two distinct phases in amyloid formation. J. Biol. Chem. 2004, 279, 12206–12212. [Google Scholar] [CrossRef] [PubMed]
- Porat, Y.; Kolusheva, S.; Jelinek, R.; Gazit, E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry 2003, 42, 10971–10977. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Luca, S.; Yau, W.M.; Leapman, R.; Tycko, R. Peptide conformation and supramolecular organization in amylin fibrils: Constraints from solid-state NMR. Biochemistry 2007, 46, 13505–13522. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Konarkowska, B.; Aitken, J.F.; Kistler, J.; Zhang, S.; Cooper, G.J. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006, 273, 3614–3624. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Kayed, R.; Lin, C.Y.; Gurlo, T.; Haataja, L.; Jayasinghe, S.; Langen, R.; Glabe, C.G.; Butler, P.C. Inhibition of human IAPP fibril formation does not prevent beta-cell death: Evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1317–E1324. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Mirzabekov, T.A.; Lin, M.C.; Kagan, B.L. Pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996, 271, 1988–1992. [Google Scholar] [PubMed]
- Clark, A.; Wells, C.A.; Buley, I.D.; Cruickshank, J.K.; Vanhegan, R.I.; Matthews, D.R.; Cooper, G.J.; Holman, R.R.; Turner, R.C. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988, 9, 151–159. [Google Scholar]
- Kahn, S.E.; Andrikopoulos, S.; Verchere, C.B. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 1999, 48, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Marzban, L.; Trigo-Gonzalez, G.; Verchere, C.B. Processing of pro-islet amyloid polypeptide in the constitutive and regulated secretory pathways of beta cells. Mol. Endocrinol. 2005, 19, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Ling, Z.; Quartier, E.; Foriers, A.; Schuit, F.; Pipeleers, D.; Van Schravendijk, C. Prolonged exposure of pancreatic beta cells to raised glucose concentrations results in increased cellular content of islet amyloid polypeptide precursors. Diabetologia 1999, 42, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Marzban, L.; Rhodes, C.J.; Steiner, D.F.; Haataja, L.; Halban, P.A.; Verchere, C.B. Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes 2006, 55, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Westermark, G.T. Aberrant processing of human proislet amyloid polypeptide results in increased amyloid formation. Diabetes 2005, 54, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Soeller, W.C.; Roche, P.C.; Nelson, R.T.; Torchia, A.J.; Kreutter, D.K.; Butler, P.C. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 7283–7288. [Google Scholar] [CrossRef] [PubMed]
- Verchere, C.B.; D’Alessio, D.A.; Palmiter, R.D.; Weir, G.C.; Bonner-Weir, S.; Baskin, D.G.; Kahn, S.E. Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc. Natl. Acad. Sci. USA 1996, 93, 3492–3496. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Utzschneider, K.M.; Tong, J.; Gerchman, F.; Kahn, S.E. Glucose- and time-dependence of islet amyloid formation in vitro. Biochem. Biophys. Res. Commun. 2007, 354, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Farrall, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.; Castellano, C.A.; Goffaux, P.; Whittingstall, K.; Lepage, M.; Paquet, N.; Bocti, C.; Fulop, T.; Cunnane, S.C. Glucose hypometabolism is highly localized, but lower cortical thickness and brain atrophy are widespread in cognitively normal older adults. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1315–E1321. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Golob, E.J.; Su, M.Y. Vascular volume and blood-brain barrier permeability measured by dynamic contrast enhanced MRI in hippocampus and cerebellum of patients with MCI and normal controls. J. Magn. Reson. Imaging 2006, 24, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Taheri, S.; Gasparovic, C.; Shah, N.J.; Rosenberg, G.A. Quantitative measurement of blood-brain barrier permeability in human using dynamic contrast-enhanced MRI with fast T1 mapping. Magn. Reson. Med. 2011, 65, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; de Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1792, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S.; Nitsch, R. Cerebral excess release of neurotransmitter amino acids subsequent to reduced cerebral glucose metabolism in early-onset dementia of Alzheimer type. J. Neural. Transm. 1989, 75, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007, 1181, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, Q.; Inoue, T.; Polito, V.A.; Tabuchi, K.; Hammer, R.E.; Pautler, R.G.; Taffet, G.E.; Zheng, H. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: Interplay with cerebral blood flow. Mol. Neurodegener. 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ouwens, D.M.; van Duinkerken, E.; Schoonenboom, S.N.; Herzfeld de Wiza, D.; Klein, M.; van Golen, L.; Pouwels, P.J.; Barkhof, F.; Moll, A.C.; Snoek, F.J.; et al. Cerebrospinal fluid levels of Alzheimer’s disease biomarkers in middle-aged patients with type 1 diabetes. Diabetologia 2014, 57, 2208–2214. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Liu, L.P.; Liao, J.M.; Wang, T.S.; Ye, F.Y.; Wu, J.; Wang, Y.Y.; Wang, Y.; Li, Y.Q.; Long, Y.; Xia, Y.Z. Downregulation of LRP1 [correction of LPR1] at the blood-brain barrier in streptozotocin-induced diabetic mice. Neuropharmacology 2009, 56, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.N.; Liu, L.B.; Xue, Y.X.; Wang, P. Effects of insulin combined with idebenone on blood-brain barrier permeability in diabetic rats. J. Neurosci. Res. 2015, 93, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.A.; Knox, R.J.; Smith, T.C.; Wayne, N.L.; Connor, J.A.; Kaczmarek, L.K. Regulation by insulin of a unique neuronal Ca2+ pool and of neuropeptide secretion. Nature 1997, 385, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Xiong, Z.G.; Man, H.Y.; Ackerley, C.A.; Braunton, J.; Lu, W.Y.; Becker, L.E.; MacDonald, J.F.; Wang, Y.T. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 1997, 388, 686–690. [Google Scholar] [PubMed]
- Wang, Y.T.; Linden, D.J. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 2000, 25, 635–647. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Lan, J.; Zheng, X.; Zukin, R.S.; Bennett, M.V. Insulin promotes rapid delivery of N-methyl-d-aspartate receptors to the cell surface by exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, G.; Ju, W.; Liu, L.; Wyszynski, M.; Lee, S.H.; Dunah, A.W.; Taghibiglou, C.; Wang, Y.; Lu, J.; Wong, T.P.; et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 2004, 23, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Alkon, D.L. Role of insulin and insulin receptor in learning and memory. Mol. Cell Endocrinol. 2001, 177, 125–134. [Google Scholar] [CrossRef]
- Kaidanovich, O.; Eldar-Finkelman, H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert Opin. Ther. Targets 2002, 6, 555–561. [Google Scholar] [PubMed]
- Lee, J.; Kim, M.S. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract. 2007, 77, S49–S57. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, Y.; Limaye, A.R.; Levey, A.I.; Srinivasan, S. Glycogen synthase kinase 3beta and Alzheimer’s disease: Pathophysiological and therapeutic significance. Cell Mol. Life Sci. 2006, 63, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Shiiki, T.; Ohtsuki, S.; Kurihara, A.; Naganuma, H.; Nishimura, K.; Tachikawa, M.; Hosoya, K.; Terasaki, T. Brain insulin impairs amyloid-beta(1–40) clearance from the brain. J. Neurosci. 2004, 24, 9632–9637. [Google Scholar] [CrossRef] [PubMed]
- Authier, F.; Posner, B.I.; Bergeron, J.J. Insulin-degrading enzyme. Clin. Investig. Med. 1996, 19, 149–160. [Google Scholar]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Lacor, P.N.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric AB. J. Biol. Chem. 2009, 284, 18742–18753. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Ferreira, S.T. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Invest. 2013, 123, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Clarke, J.R.; Bomfim, T.R.; de Felice, F.G. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement. 2014, 10, S76–S83. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., II; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers Dis. 2008, 13, 323–331. [Google Scholar] [PubMed]
- Craft, S.; Peskind, E.; Schwartz, M.W.; Schellenberg, G.D.; Raskind, M.; Porte, D., Jr. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: Relationship to severity of dementia and apolipoprotein E genotype. Neurology 1999, 50, 164–168. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [PubMed]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [PubMed]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhang-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C.; et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [PubMed]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Kustermann, E.; et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef] [PubMed]
- Grunblatt, E.; Salkovic-Petrisic, M.; Osmanovic, J.; Riederer, P.; Hoyer, S. Brain insulin system dysfunction in streptozotocin intracerebroventricularly treated rats generates hyperphosphorylated tau protein. J. Neurochem. 2007, 101, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.J.; Hernandez, F.; Gomez-Ramos, P.; Moran, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBOJ 2001, 20, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; He, G.; Ly, P.T.; Fox, C.J.; Staufenbiel, M.; Cai, F.; Zhang, Z.; Wei, S.; Sun, X.; Chen, C.H.; et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J. Exp. Med. 2008, 205, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Martins, R.N.; Racchi, M.; Craft, S.; Helmerhorst, E. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. J. Alzheimers. Dis. 2002, 4, 369–374. [Google Scholar] [PubMed]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; van Bronswijk, W.; Martins, R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J. Neurosci. 2002, 22, (RC221), 1–5. [Google Scholar] [PubMed]
- Biessels, G.J.; Kamal, A.; Urban, I.J.; Spruijt, B.M.; Erkelens, D.W.; Gispen, W.H. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: Effects of insulin treatment. Brain Res. 1998, 800, 125–135. [Google Scholar] [CrossRef]
- Li, Z.G.; Zhang, W.; Grunberger, G.; Sima, A.A. Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res. 2002, 946, 221–231. [Google Scholar] [CrossRef]
- Sima, A.A.; Li, Z.G. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes 2005, 54, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Lannert, H.; Hoyer, S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav. Neurosci. 1998, 112, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Backus, C.; Oh, S.; Hayes, J.M.; Feldman, E.L. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 2009, 150, 5294–5301. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; Zhang, W.; Sima, A.A. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007, 56, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Seica, R.; Oliveira, C.R. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J. Neurol. Sci. 2007, 257, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Santos, M.S.; Seica, R.; Moreira, P.I. Cortical and hippocampal mitochondria bioenergetics and oxidative status during hyperglycemia and/or insulin-induced hypoglycemia. Biochim. Biophys. Acta. 2010, 1802, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Blake, R.; Duce, J.A.; Ciccotosto, G.D.; Li, Q.X.; Barnham, K.J.; Curtain, C.C.; Cherny, R.A.; Cappai, R.; Dyrks, T.; et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J. Neurosci. 2005, 5, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Du, H.; Yan, S.; Fang, F.; Wang, C.; Lue, L.F.; Guo, L.; Chen, D.; Stern, D.M.; Gunn Moore, F.J.; et al. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Mahr, N.J.; Filley, C.M.; Parks, J.K.; Hughes, D.; Young, D.A.; Cullum, C.M. Reduced platelet cytochrome c oxidase activity in Alzheimer’s disease. Neurology 1994, 44, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural. Transm. 1998, 105, 855–870. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, M.Y.; Tucker, H.M.; Nair, P.; Aksenova, M.V.; Butterfield, D.A.; Estus, S.; Markesbery, W.R. The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer’s disease. Neurochem. Res. 1999, 24, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [PubMed]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Valla, J.; Schneider, L.; Niedzielko, T.; Coon, K.D.; Caselli, R.; Sabbagh, M.N.; Ahern, G.L.; Baxter, L.; Alexander, G.; Walker, G.; et al. Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion 2006, 6, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.M.; Ou, H.C.; Xu, H.; Chen, H.L.; Fowler, C.; Gibson, G.E. Inhibition of alpha-ketoglutarate dehydrogenase complex promotes cytochrome c release from mitochondria, caspase-3 activation, and necrotic cell death. J. Neurosci. Res. 2003, 74, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [PubMed]
- Busciglio, J.; Pelsman, A.; Wong, C.; Pigino, G.; Yuan, M.; Mori, H.; Yankner, B.A. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 2002, 33, 677–688. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Maguire, D.J.; Maguire, R.S.; Bennett, J.P., Jr.; Davis, R.E.; Parker, W.D., Jr. Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 1997, 49, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.L.; Shenoy, D.V.; Thomas, N.; Choudhary, P.K.; Laferla, F.M.; Goodman, S.R.; Breen, G.A. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J. Proteomics 2011, 74, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Calingasan, N.Y.; Yu, F.; Mauck, W.M.; Toidze, M.; Almeida, C.G.; Takahashi, R.H.; Carlson, G.A.; Flint Beal, M.; Lin, M.T.; et al. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 2004, 89, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Seica, R.; Oliveira, C.R. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes 2003, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Restivo, F.; Vercellinatto, I.; Danni, O.; Brignardello, E.; Aragno, M.; Boccuzzi, G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 2005, 187, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Sena, C.; Seica, R.; Oliveira, C.R. Insulin protects against amyloid beta-peptide toxicity in brain mitochondria of diabetic rats. Neurobiol. Dis. 2005, 18, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Fernyhough, P.; Roy Chowdhury, S.K.; Schmidt, R.E. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert. Rev. Endocrinol. Metab. 2010, 5, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.; Carvalho, C.; Santos, R.; Correia, S.; Santos, M.S.; Seica, R.; Oliveira, C.R.; Moreira, P.I. Impact of STZ-induced hyperglycemia and insulin-induced hypoglycemia in plasma amino acids and cortical synaptosomal neurotransmitters. Synapse 2010, 65, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Zhou, W.; Fung, V.; Christensen, M.A.; Qing, H.; Sun, X.; Song, W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J. Neural. Transm. 2005, 112, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Chen, Y.; Liu, H.; Zhang, K.; Zhang, T.; Lin, A.; Jing, N. Hydrogen peroxide promotes Abeta production through JNK-dependent activation of gamma-secretase. J. Biol. Chem. 2008, 283, 17721–17730. [Google Scholar] [CrossRef] [PubMed]
- Tamagno, E.; Guglielmotto, M.; Aragno, M.; Borghi, R.; Autelli, R.; Giliberto, L.; Muraca, G.; Danni, O.; Zhu, X.; Smith, M.A.; et al. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008, 104, 683–695. [Google Scholar] [PubMed]
- Quiroz-Baez, R.; Rojas, E.; Arias, C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem. Int. 2009, 55, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.G.; Arumugam, T.V.; Woo, H.N.; Park, J.S.; Tang, S.C.; Mughal, M.; Hyun, D.H.; Park, J.H.; Choi, Y.H.; Gwon, A.R.; et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Tamaoka, A.; Araki, W. Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J. Neurosci. Res. 2010, 88, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20, S499–S512. [Google Scholar] [PubMed]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Markesbery, W.R.; Zhou, X.Z.; Lu, K.P.; Butterfield, D.A. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol. Aging 2006, 27, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Zelasko, D.A.; Harris, P.L.; Perry, G.; Salomon, R.G.; Smith, M.A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997, 68, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Richey Harris, P.L.; Sayre, L.M.; Beckman, J.S.; Perry, G. Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J. Neurosci. 1997, 17, 2653–2657. [Google Scholar] [PubMed]
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 1998, 71, 2034–2040. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Markesbery, W.R.; Morrow, J.D.; Roberts, L.J., II. Cerebrospinal fluid F2-isoprostane levels are increased in Alzheimer’s disease. Ann. Neurol. 1998, 44, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Nourooz-Zadeh, J.; Liu, E.H.; Yhlen, B.; Anggard, E.E.; Halliwell, B. F4-isoprostanes as specific marker of docosahexaenoic acid peroxidation in Alzheimer’s disease. J. Neurochem. 1999, 72, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Wade, R.; Hirai, K.; Chiba, S.; Smith, M.A. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999, 19, 1959–1964. [Google Scholar] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Ratio of 8-hydroxyguanine in intact DNA to free 8-hydroxyguanine is increased in Alzheimer disease ventricular cerebrospinal fluid. Arch. Neurol. 2001, 58, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Chiba, S.; Lippa, C.F.; Cras, P.; Kalaria, R.N.; Takeda, A.; Honda, K.; Smith, M.A.; Perry, G. Neuronal RNA oxidation is a prominent feature of familial Alzheimer’s disease. Neurobiol. Dis. 2004, 17, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [PubMed]
- Resende, R.; Moreira, P.I.; Proenca, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int. J. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628. [Google Scholar] [CrossRef] [PubMed]
- Rösen, P.; Nawroth, P.P.; King, G.; Möller, W.; Tritschler, H.J.; Packer, L. The role of oxidative stress in the onset and progression of diabetes and its complications: A summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab. Res. Rev. 2001, 17, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.W.; Berent-Spillson, A.; Vincent, A.M.; Freimann, C.L.; Sullivan, K.A.; Feldman, E.L. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 2008, 30, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Cerami, A. Advanced glycosylation: Chemistry, biology, and implications for diabetes and aging. Adv. Pharmacol. 1992, 23, 1–34. [Google Scholar] [PubMed]
- Hunt, J.V.; Dean, R.T.; Wolff, S.P. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem. J. 1988, 256, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Mullarkey, C.J.; Edelstein, D.; Brownlee, M. Free radical generation by early glycation products: A mechanism for accelerated atherogenesis in diabetes. Biochem. Biophys. Res. Commun. 1990, 173, 932–939. [Google Scholar] [CrossRef]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, M.D.; Bonay, P.; Colaco, C.; Avila, J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J. Biol. Chem. 1994, 269, 21614–21619. [Google Scholar] [PubMed]
- Yan, S.D.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, C.G.; Nawroth, P.; et al. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef] [PubMed]
- McCance, D.R.; Dyer, D.G.; Dunn, J.A.; Bailie, K.E.; Thorpe, S.R.; Baynes, J.W.; Lyons, T.J. Maillard reaction products and their relation to complications in insulin-dependent diabetes mellitus. J. Clin. Investig. 1993, 91, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Advanced glycation end-products and atherosclerosis. Ann. Med. 1996, 28, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Wells-Knecht, K.J.; Brinkmann, E.; Wells-Knecht, M.C.; Litchfield, J.E.; Ahmed, M.U.; Reddy, S.; Zyzak, D.V.; Thorpe, S.R.; Baynes, J.W. New biomarkers of Maillard reaction damage to proteins. Nephrol. Dial. Transplant. 1996, 11, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Alt, A.; Niwa, T.; Clausen, J.T.; Bretzel, R.G.; Brownlee, M.; Schleicher, E.D. Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia 1999, 42, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.Y.; Cooper, M.E. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Sayre, L.M.; Perry, G. Diabetes mellitus and Alzheimer’s disease: Glycation as a biochemical link. Diabetologia 1996, 39, 247. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Tabaton, M.; Perry, G. Early contribution of oxidative glycation in Alzheimer disease. Neurosci. Lett. 1996, 217, 210–211. [Google Scholar] [CrossRef]
- Wahrle, S.; Das, P.; Nyborg, A.C.; McLendon, C.; Shoji, M.; Kawarabayashi, T.; Younkin, L.H.; Younkin, S.G.; Golde, T.E. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 2002, 9, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Ye, W.; Wang, R.; Wolfe, M.S.; Selkoe, D.J. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 2008, 283, 22529–22540. [Google Scholar] [CrossRef] [PubMed]
- Canevari, L.; Clark, J.B. Alzheimer’s disease and cholesterol: The fat connection. Neurochem. Res. 2007, 32, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.Q.; Xu, Q.; Dee Fish, J.; Wyss-Coray, T.; Buttini, M.; Mucke, L.; et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004, 24, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.R.; Tang, W.; Wang, H.; Vitek, M.P.; Bennett, E.R.; Sullivan, P.M.; Warner, D.S.; Laskowitz, D.T. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J. Biol. Chem. 2003, 278, 48529–48533. [Google Scholar] [CrossRef] [PubMed]
- Van den Elzen, P.; Garg, S.; Leon, L.; Brigl, M.; Leadbetter, E.A.; Gumperz, J.E.; Dascher, C.C.; Cheng, T.Y.; Sacks, F.M.; Illarionov, P.A.; et al. Apolipoprotein-mediated pathways of lipid antigen presentation. Nature 2005, 437, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Thirty years of Alzheimer’s disease genetics: The implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008, 9, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef]
- Puglielli, L.; Konopka, G.; Pack-Chung, E.; Ingano, L.A.; Berezovska, O.; Hyman, B.T.; Chang, T.Y.; Tanzi, R.E.; Kovacs, D.M. Acyl-coenzyme A: Cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat. Cell Biol. 2001, 3, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Niu, Z.; Bryleva, E.Y.; Harris, B.T.; Murphy, S.R.; Kheirollah, A.; Bowen, Z.D.; Chang, C.C.; Chang, T.Y. Acyl-coenzyme A: Cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol. Aging 2015, 36, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Hutter-Paier, B.; Huttunen, H.J.; Puglielli, L.; Eckman, C.B.; Kim, D.Y.; Hofmeister, A.; Moir, R.D.; Domnitz, S.B.; Frosch, M.P.; Windisch, M.; et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 2004, 44, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, R.; Kovacs, D.M. ACAT inhibition and amyloid beta reduction. Biochim. Biophys. Acta 2010, 1801, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Bryleva, E.Y.; Rogers, M.A.; Chang, C.C.; Buen, F.; Harris, B.T.; Rousselet, E.; Seidah, N.G.; Oddo, S.; LaFerla, F.M.; Spencer, T.A.; et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA 2010, 107, 3081–3086. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Keller, P.; de Strooper, B.; Beyreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar] [CrossRef] [PubMed]
- Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Wang, C.; Huang, Y. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-beta (Abeta) and acts in concert with Abeta to elicit neuronal and behavioral deficits in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 4236–4241. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K.; et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Wong, D.; Buttini, M.; Orth, M.; Bellosta, S.; Pitas, R.E.; Mahley, R.W.; Mucke, L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc. Natl. Acad. Sci. USA 1998, 95, 10914–10919. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Wong, D.; Yu, G.Q.; Buttini, M.; Mahley, R.W.; Pitas, R.E.; Mucke, L. Apolipoprotein E and cognitive performance. Nature 2000, 404, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia aging study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Pickup, J.C.; Mattock, M.B.; Chusney, G.D.; Burt, D. NIDDM as a disease of the innate immune system: Association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 1997, 40, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Kroke, A.; Mohlig, M.; Hoffmann, K.; Bergmann, M.M.; Ristow, M.; Boeing, H.; Pfeiffer, A.F. Inflammatory cytokines and the risk to develop type 2 diabetes: Results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam study. Diabetes 2003, 52, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Illig, T.; Rathmann, W.; Martin, S.; Haastert, B.; Muller-Scholze, S.; Holle, R.; Thorand, B.; Koenig, W.; Wichmann, H.E.; et al. Inflammation and type 2 diabetes: Results from KORA Augsburg. Gesundheitswesen 2005, 67, S115–S121. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Brunner, E.J.; Rathmann, W.; Strassburger, K.; Tabak, A.G.; Schloot, N.C.; Witte, D.R. Elevated levels of the anti-inflammatory interleukin-1 receptor antagonist precede the onset of type 2 diabetes: The Whitehall II study. Diabetes Care 2009, 32, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ehses, J.A.; Mathe, Z.; Bosco, D.; Berney, T.; Dayer, J.M.; Reinecke, M.; Halban, P.A.; Donath, M.Y. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc. Natl. Acad. Sci. USA 2004, 101, 8138–8143. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, X.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Lee, S.; Tong, M.; Hang, S.; Deochand, C.; de la Monte, S.M. CSF and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late Alzheimer’s disease. J. Alzheimers Dis. Parkinsonism 2013, 3. [Google Scholar] [CrossRef]
- Blalock, E.M.; Chen, K.C.; Stromberg, A.J.; Norris, C.M.; Kadish, I.; Kraner, S.D.; Porter, N.M.; Landfield, P.W. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer’s disease: Statistical reliability and functional correlation. Ageing Res. Rev. 2005, 4, 481–512. [Google Scholar] [CrossRef] [PubMed]
- Katsel, P.L.; Davis, K.L.; Haroutunian, V. Large-scale microarray studies of gene expression in multiple regions of the brain in schizophrenia and Alzheimer’s disease. Int. Rev. Neurobiol. 2005, 63, 41–82. [Google Scholar] [PubMed]
- Morgan, D.; Gordon, M.N.; Tan, J.; Wilcock, D.; Rojiani, A.M. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: Implications for Alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 2005, 64, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflamm. 2005, 2. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.A.; Thorne, J.E.; Zandi, P.P.; Ek, M.; Messias, E.; Breitner, J.C.; Goodman, S.N. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 2004, 23, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Davis, K.L.; Berg, J.D.; Schafer, K.; Campbell, K.; Thomas, R.G.; Weiner, M.F.; Farlow, M.R.; Sano, M.; Grundman, M.; et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 2000, 54, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J. Effects of rofecoxib or naproxen vs. placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, W.A.; Weinstein, H.C.; Scheltens, P.; Walstra, G.J. Effect of hydroxychloroquine on progression of dementia in early Alzheimer’s disease: An 18-month randomised, double-blind, placebo-controlled study. Lancet 2001, 358, 455–460. [Google Scholar] [CrossRef]
- Reines, S.A.; Block, G.A.; Morris, J.C.; Liu, G.; Nessly, M.L.; Lines, C.R.; Norman, B.A.; Baranak, C.C. Rofecoxib: No effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 2004, 62, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Haj-ali, V.; Mohaddes, G.; Babri, S.H. Intracerebroventricular insulin improves spatial learning and memory in male Wistar rats. Behav. Neurosci. 2009, 123, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.J.; Martinez, J.A.; Liu, W.Q.; Xu, K.; Ayer, A.; Fine, J.; Tour, U.I.; Glazner, G.; Hanson, L.R.; Frey, W.H., II; et al. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain 2008, 131, 3311–3334. [Google Scholar] [PubMed]
- Biessels, G.J.; Deary, I.J.; Ryan, C.M. Cognition and diabetes: A lifespan perspective. Lancet Neurol. 2008, 7, 184–190. [Google Scholar] [CrossRef]
- Brands, A.M.; Biessels, G.J.; de Haan, E.H.; Kappelle, L.J.; Kessels, R.P. The effects of type 1 diabetes on cognitive performance: A meta-analysis. Diabetes Care 2005, 28, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Kodl, C.T.; Seaquist, E.R. Cognitive dysfunction and diabetes melli- tus. Endocr. Rev. 2008, 29, 494–511. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Schmitz, K.; Schultes, B.; Ratter, F.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans: Superiority of insulin aspart. Neuropsychopharmacology 2007, 32, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Krug, R.; Benedict, C.; Born, J.; Hallschmid, M. Comparable sensitivity of postmenopausal and young women to the effects of intranasal insulin on food intake and working memory. J. Clin. Endocrinol. Metab. 2010, 95, E468–E472. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Frey, W.H., II; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef] [PubMed]
- DeJong, R.N. CNS manifestations of diabetes mellitus. Postgrad. Med. 1977, 61, 101–107. [Google Scholar] [PubMed]
- Reske-Nielsen, E.; Lundbaek, K. Diabetic encephalopathy. Diffuse and focal lesions of the brain in long-term diabetes. Acta Neurol. Scand. Suppl. 1963, 39, 273–290. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.V.; Frier, B.M.; Strachan, M.W. The relationship between type 2 diabetes and cognitive dysfunction: Longitudinal studies and their methodological limitations. Eur. J. Pharmacol. 2004, 490, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Roriz-Filho, J.S.; Sa-Roriz, T.M.; Rosset, I.; Camozzato, A.L.; Santos, A.C.; Chaves, M.L.; Moriguti, J.C.; Roriz-Cruz, M. (Pre)diabetes, brain aging, and cognition. Biochim. Biophys. Acta 2009, 1792, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin- like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Kar, S.; Dore, S.; Quirion, R. Insulin-like growth factor-1 (IGF-1): A neuroprotective trophic factor acting via the Akt kinase pathway. J. Neural. Transm. Suppl. 2000, 60, 261–272. [Google Scholar] [PubMed]
- Dore, S.; Bastianetto, S.; Kar, S.; Quirion, R. Protective and rescuing abilities of IGF-I and some putative free radical scavengers against beta-amyloid-inducing toxicity in neurons. Ann. N. Y. Acad. Sci. 1999, 890, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Dore, S.; Kar, S.; Quirion, R. Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc. Natl. Acad. Sci. USA 1997, 94, 4772–4777. [Google Scholar] [CrossRef] [PubMed]
- Evin, G.; Weidemann, A. Biogenesis and metabolism of Alzheimer’s disease Abeta amyloid peptides. Peptides 2002, 23, 1285–1297. [Google Scholar] [CrossRef]
- Tsukamoto, E.; Hashimoto, Y.; Kanekura, K.; Niikura, T.; Aiso, S.; Nishimoto, I. Characterization of the toxic mechanism triggered by Alzheimer’s amyloid-beta peptides via p75 neurotrophin receptor in neuronal hybrid cells. J. Neurosci. Res. 2003, 73, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Pace, V.; Copani, A.; Squatrito, S.; Milardi, D.; Vigneri, R. Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 2012, 154, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Peskind, E.R.; Asthana, S.; Purganan, K.; Wait, C.; Chapman, D.; Schwartz, M.W.; Plymate, S.; Craft, S. Insulin increases CSF Abeta42 levels in normal older adults. Neurology 2003, 60, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Brooks, S.J.; Kullberg, J.; Burgos, J.; Kempton, M.J.; Nordenskjold, R.; Nylander, R.; Kilander, L.; Craft, S.; Larsson, E.M.; et al. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes Care 2012, 35, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease—Associated Abeta oligomers. J. Clin. Investig. 2012, 22, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Kenna, H.; Hoeft, F.; Kelley, R.; Wroolie, T.; Demuth, B.; Reiss, A.; Rasgon, N. Fasting plasma insulin and the default mode network in women at risk for Alzheimer’s disease. Neurobiol. Aging 2013, 34, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Hattori, C.; Iwata, N.; Saido, T.C.; Sasagawa, N.; Szabo, B.; Hashimoto, Y.; Maruyama, K.; Tanuma, S.; Kiso, Y.; Ishiura, S. The novel beta-secretase inhibitor KMI-429 reduces amyloid beta peptide production in amyloid precursor protein transgenic and wild-type mice. J. Neurochem. 2006, 96, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Comery, T.A.; Martone, R.L.; Aschmies, S.; Atchison, K.P.; Diamantidis, G.; Gong, X.; Zhou, H.; Kreft, A.F.; Pangalos, M.N.; Sonnenberg-Reines, J.; et al. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Gravitz, L. Drugs: A tangled web of targets. Nature 2011, 475, S9–S11. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Rovira, M.B.; Forette, F.; et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Saumier, D.; Briand, R.; Laurin, J.; Gervais, F.; Tremblay, P.; Garceau, D. A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology 2006, 67, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Porsteinsson, A.P.; Grossberg, G.T.; Mintzer, J.; Olin, J.T. Memantine treatment in patients with mild to moderate Alzheimer’s disease already receiving a cholinesterase inhibitor: A randomized, double-blind, placebo-controlled trial. Curr. Alzheimer Res. 2008, 5, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Arnold, S.E. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 2014, 63, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Standards of medical care in diabetes–2013. Diabetes Care 2013, 36, S11–S66. [Google Scholar]
- Nicolakakis, N.; Aboulkassim, T.; Ongali, B.; Lecrux, C.; Fernandes, P.; Rosa-Neto, P.; Tong, X.K.; Hamel, E. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J. Neurosci. 2008, 28, 9287–9296. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G.; et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Sawchak, S.; Chiang, C.; Davies, J.; Saunders, A.; Irizarry, M.; Zvartau-Hind, M.; van Dyck, C.; Gold, M. Effects of rosiglitazone-extended release as adjunctive therapy to acetylcholinesterase inhibitors over 48 weeks on cognition in Apoe4-stratified subjects with mild-to-moderate Alzheimer’s disease. Alzheimer Dement. 2009, 5, e17–e18. [Google Scholar] [CrossRef]
- Festuccia, W.T.; Oztezcan, S.; Laplante, M.; Berthiaume, M.; Michel, C.; Dohgu, S.; Denis, R.G.; Brito, M.N.; Brito, N.A.; Miller, D.S.; et al. Peroxisome proliferatoractivated receptor-gamma-mediated positive energy balance in the rat is associated with reduced sympathetic drive to adipose tissues and thyroid status. Endocrinology 2008, 149, 2121–2130. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Sullivan, K.A.; Backus, C.; Feldman, E.L. Cortical neurons develop insulin resistance and blunted Akt signaling: A potential mechanism contributing to enhanced ischemic injury in diabetes. Antioxid. Redox Signal. 2011, 14, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Łabuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopień, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar]
- Nath, N.; Khan, M.; Paintlia, M.K.; Singh, I.; Hoda, M.N.; Giri, S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J. Immunol. 2009, 182, 8005–8014. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, K.; Wang, R.; Liu, Y.; Kwak, Y.D.; Ma, T.; Thompson, R.C.; Zhao, Y.; Smith, L.; Gasparini, L.; et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. USA 2009, 106, 3907–3912. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.A.; Greig, N.H. A new Alzheimer’s disease interventive strategy: GLP-1. Curr. Drug Targets 2004, 5, 565–571. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of longterm potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar]
- Paz-Filho, G.J.; Babikian, T.; Asarnow, R.; Delibasi, T.; Esposito, K.; Erol, H.K.; Wong, M.L.; Licinio, J. Leptin replacement improves cognitive development. PLoS ONE 2008, 3, e3098. [Google Scholar] [CrossRef]
- Greco, S.J.; Bryan, K.J.; Sarkar, S.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N.; Casadesus, G. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 1155–1167. [Google Scholar] [PubMed]
- Lieb, W.; Beiser, A.S.; Vasan, R.S.; Tan, Z.S.; Au, R.; Harris, T.B.; Roubenoff, R.; Auerbach, S.; DeCarli, C.; Wolf, P.A.; et al. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 2009, 302, 2565–2572. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.L.; Yarchoan, M.; Hwang, H.M.; Louneva, N.; Blair, J.A.; Palm, R.; Smith, M.A.; Lee, H.G.; Arnold, S.E.; Casadesus, G. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol. Aging 2014, 35, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced weight loss with pramlintide/metreleptin: An integrated neurohormonal approach to obesity pharmacotherapy. Obesity 2009, 17, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Unger, J.W.; Livingston, J.N.; Moss, A.M. Insulin receptors in the central nervous system: Localization, signalling mechanisms and functional aspects. Prog. Neurobiol. 1991, 36, 343–362. [Google Scholar] [CrossRef]
- Baura, G.D.; Foster, D.M.; Porte, D., Jr.; Kahn, S.E.; Bergman, R.N.; Cobelli, C.; Schwartz, M.W. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J. Clin. Invest. 1993, 92, 1824–1830. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M.; Schultes, B.; Marshall, L.; Molle, M.; Kern, W.; Bredthauer, J.; Fehm, H.L.; Born, J. Transcortical direct current potential shift reflects immediate signaling of systemic insulin to the human brain. Diabetes 2004, 53, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.R.; Tucker, K.; Cavallin, M.A.; Mast, T.G.; Fadool, D.A. Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J. Neurosci. 2009, 29, 6734–6751. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, el-S.; Morishita, M.; Onuki, Y.; Takayama, K. Current challenges in non-invasive insulin delivery systems: A comparative review. Adv. Drug. Deliv. Rev. 2007, 59, 1521–1546. [Google Scholar]
- Kupila, A.; Sipila, J.; Keskinen, P.; Simell, T.; Knip, M.; Pulkki, K.; Simell, O. Intranasally administered insulin intended for prevention of type 1 diabetes—a safety study in healthy adults. Diabetes Metab. Res. Rev. 2003, 19, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [PubMed]
- Watson, G.S.; Bernhardt, T.; Reger, M.A.; Cholerton, B.A.; Baker, L.D.; Peskind, E.R.; Asthana, S.; Plymate, S.R.; Frölich, L.; Craft, S. Insulin effects on CSF norepinephrine and cognition in Alzheimer’s disease. Neurobiol. Aging 2006, 27, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Messier, C.; Teutenberg, K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural. Plast. 2005, 12, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D. Insulin and Alzheimer’s disease: An amyloid connection. Neurology 2003, 60, 1886–1887. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Dodt, C.; Hallschmid, M.; Lepiorz, M.; Fehm, H.L.; Born, J.; Kern, W. Immediate but not long-term intranasal administration of insulin raises blood pressure in human beings. Metabolism 2005, 54, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Dhamoon, M.S.; Noble, J.M.; Craft, S. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2009, 72, 292–293; author reply 293–294. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Schioth, H.B.; Craft, S.; Brooks, S.J.; Frey, W.H., II; Benedict, C. Brain insulin signaling and Alzheimer’s disease: Current evidence and future directions. Mol. Neurobiol. 2012, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, E.; Rudich, A.; Harman-Boehm, I.; Cukierman-Yaffe, T. Effect of intranasal insulin on cognitive function: A systematic review. J. Clin. Endocrinol. Metab. 2012, 97, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Xiao, C.; Morgantini, C.; Koulajian, K.; Lewis, G.F. Intranasal insulin suppresses endogenous glucose production in humans compared with placebo in the presence of similar venous insulin concentrations. Diabetes 2005, 64, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, S.; Koliaki, C.; Bierwagen, A.; Nowotny, P.; Heni, M.; Fritsche, A.; Häring, H.U.; Szendroedi, J.; Roden, M. Effects of intranasal insulin on hepatic fat accumulation and energy metabolism in humans. Diabetes 2015, 64, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M.; Benedict, C.; Schultes, B.; Perras, B.; Fehm, H.L.; Kern, W.; Born, J. Towards the therapeutic use of intranasal neuropeptide administration in metabolic and cognitive disorders. Regul. Pept. 2008, 149, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, D.H. Sink hypothesis and therapeutic strategies for attenuating Abeta levels. Neuroscientist 2011, 17, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, C.; Tschritter, O.; Preissl, H.; Heni, M.; Häring, H.U.; Fritsche, A. Insulin sensitivity of the human brain. Diabetes Res. Clin. Pract. 2011, 93, S47–S51. [Google Scholar] [CrossRef]
- Kurtzhals, P. Engineering predictability and protraction in a basal insulin analogue: The pharmacology of insulin detemir. Int. J. Obes. Relat. Metab. Disord. 2004, 28, S23–S28. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, I.; Vague, P.; Selam, J.L.; Skeie, S.; Lang, H.; Draeger, E.; Elte, J.W. Insulin detemir used in basal-bolus therapy in people with type 1 diabetes is associated with a lower risk of nocturnal hypoglycaemia and less weight gain over 12 months in comparison to NPH insulin. Diabetes Obes. Metab. 2005, 7, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.D.; Artru, A.A.; Adkison, K.K. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv. Drug Deliv. Rev. 2004, 56, 1825–1857. [Google Scholar] [CrossRef] [PubMed]
- Monami, M.; Marchionni, N.; Mannucci, E. Long-acting insulin analogues vs. NPH human insulin in type 1 diabetes. A meta-analysis. Diabetes. Obes. Metab. 2009, 11, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Hennige, A.M.; Sartorius, T.; Tschritter, O.; Preissl, H.; Fritsche, A.; Ruth, P.; Haring, H.U. Tissue selectivity of insulin detemir action in vivo. Diabetologia 2006, 49, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M.; Jauch-Chara, K.; Korn, O.; Molle, M.; Rasch, B.; Born, J.; Schultes, B.; Kern, W. Euglycemic infusion of insulin detemir compared with human insulin appears to increase direct current brain potential response and reduces food intake while inducing similar systemic effects. Diabetes 2010, 59, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
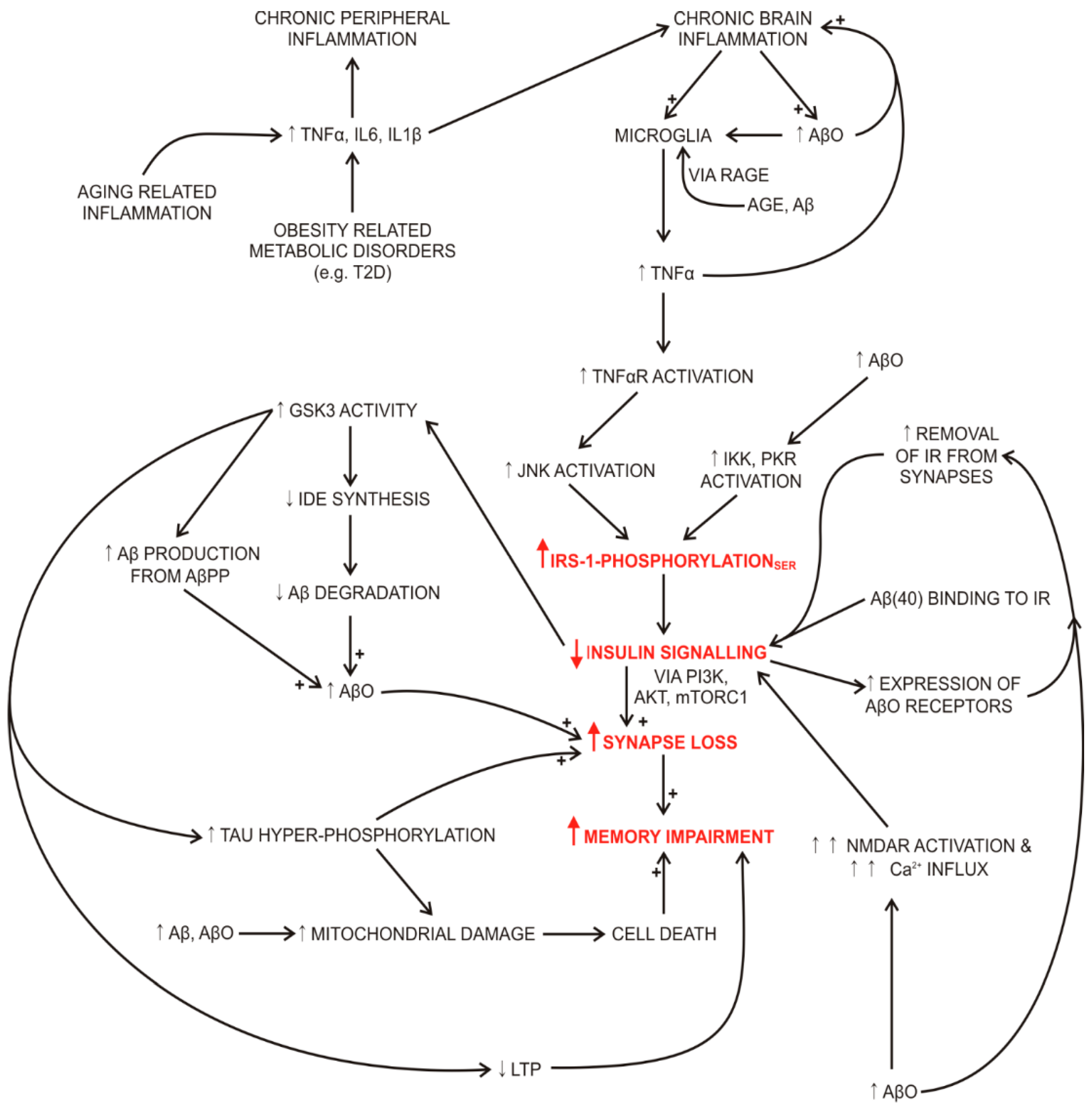
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribarič, S. The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules 2016, 21, 689. https://doi.org/10.3390/molecules21060689
Ribarič S. The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules. 2016; 21(6):689. https://doi.org/10.3390/molecules21060689
Chicago/Turabian StyleRibarič, Samo. 2016. "The Rationale for Insulin Therapy in Alzheimer’s Disease" Molecules 21, no. 6: 689. https://doi.org/10.3390/molecules21060689
APA StyleRibarič, S. (2016). The Rationale for Insulin Therapy in Alzheimer’s Disease. Molecules, 21(6), 689. https://doi.org/10.3390/molecules21060689