Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry

 ,
, 
 ,
, 

Abstract
:1. Introduction
2. Nucleophilic Organometallic Selenium Reagents
3. Water as an Alternative Medium for Organoselenium-Catalyzed Reactions
4. Other “On-Water” Conditions for the Synthesis of Organoselenium Derivatives
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Santoro, S.; Azeredo, J.B.; Nascimento, V.; Sancineto, L.; Braga, A.L.; Santi, C. The green side of the moon: Ecofriendly aspects of organoselenium chemistry. RSC Adv. 2014, 4, 31521–31535. [Google Scholar] [CrossRef]
- Dunn, P. Water as a Green Solvent for Pharmaceutical Applications. In Handbook of Green Chemistry; Anastas, P.T., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Breslow, R. The Principles of and Reasons for Using Water as a Solvent for Green Chemistry. In Handbook of Green Chemistry; Anastas, P.T., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Narayan, S.; Muldoon, J.; Finn, M.G.; Fokin, V.V.; Kolb, H.C.; Sharpless, K.B. “On Water”: Unique Reactivity of Organic Compounds in Aqueous Suspension. Angew. Chem. Int. Ed. 2005, 44, 3275–3279. [Google Scholar] [CrossRef]
- Lindström, U.M. Wiley InterScience (Online service). In Organic Reactions in Water: Principles, Strategies and Applications; Blackwell Pub: Oxford, UK; Ames, IA, USA, 2007. [Google Scholar]
- Breslow, R. Hydrophobic effects on simple organic reactions in water. Acc. Chem. Res. 1991, 24, 159–164. [Google Scholar] [CrossRef]
- Breslow, R. Biomimetic Chemistry and Artificial Enzymes: Catalysis by Design. Acc. Chem. Res. 1995, 28, 146–153. [Google Scholar] [CrossRef]
- Breslow, R. Biomimetic control of chemical selectivity. Acc. Chem. Res. 1980, 13, 170–177. [Google Scholar] [CrossRef]
- Breslow, R. Determining the Geometries of Transition States by Use of Antihydrophobic Additives in Water. Acc. Chem. Res. 2004, 37, 471–478. [Google Scholar] [CrossRef]
- Breslow, R. How do imidazole groups catalyze the cleavage of RNA in enzyme models and in enzymes? Evidence from “negative catalysis”. Acc. Chem. Res. 1991, 24, 317–324. [Google Scholar]
- Breslow, R. Biomimetic Chemistry: A Frontier at the Chemistry/Biology Interface. Chem. Biol. 1998, 5, R27–R28. [Google Scholar] [CrossRef]
- Blackmond, D.G.; Armstrong, A.; Coombe, V.; Wells, A. Wasser in organokatalytischen Prozessen: Ein Mythos wird entschleiert. Angew. Chem. Int. Ed. 2007, 119, 3872–3874. [Google Scholar] [CrossRef]
- Perin, G.; Alves, D.; Jacob, R.G.; Barcellos, A.M.; Soares, L.K.; Lenardão, E.J. Synthesis of Organochalcogen Compounds using Non-Conventional Reaction Media. ChemistrySelect 2016, 1, 205–258. [Google Scholar] [CrossRef]
- Sancineto, L.; Palomba, M.; Bagnoli, L.; Marini, F.; Santi, C. Advances in Electrophilic Organochalcogen Reagents. Curr. Org. Chem. 2015, 20, 122–135. [Google Scholar] [CrossRef]
- Grubbs, R.H. Aqueous Organometallic Chemistry and Catalysis; Horváth, I.T., Joó, F., Eds.; Springer: New York, NY, USA, 2012. [Google Scholar]
- Klayman, D.L.; Griffin, T.S. Reaction of selenium with sodium borohydride in protic solvents. A Facile Method for the introduction of selenium into organic molecules. J. Am. Chem. Soc. 1973, 95, 197–199. [Google Scholar] [CrossRef]
- Bird, M.L.; Challenger, F. 113. Potassium alkaneselenonates and other alkyl derivatives of selenium. J. Am. Chem. Soc. 1942, 570–574. [Google Scholar] [CrossRef]
- Lu, G.-L.; Zhang, Y.-M. A novel synthesis of allyl and benzyl selenides via Sm/SbCl3 system in aqueous media. Chin. J. Chem. 2010, 18, 69–71. [Google Scholar]
- Bao, W.; Zheng, Y.; Zhang, Y.; Zhou, J. A novel synthesis of allyl and propargyl selenides in aqueous media promoted by indium. Tetrahedron Lett. 1996, 37, 9333–9334. [Google Scholar] [CrossRef]
- Zheng, Y.; Bao, W.; Zhang, Y. A Novel Synthesis of Allyl and Propargyl Selenides Promoted by Indium. Synth. Commun. 1997, 27, 79–83. [Google Scholar] [CrossRef]
- Galindo, A.C.; Oliveira, J.M.; Barboza, M.A.G.; Gonçalves, S.M.C.; Menezes, P.H. Synthesis of Chalcogenides using Indium Intermediates in Aqueous Media. Phosphorus Sulfur Silicon Relat. Elem. 2001, 172, 129–140. [Google Scholar] [CrossRef]
- Li, C.J.; Chan, T.H. Organic Reactions in Aqueous Media; Wiley-Interscience: New York, NY, USA, 1997; pp. 92–94. [Google Scholar]
- Zheng, Y.; Bao, W.; Zhang, Y. Synthesis of Allyl-Type Selenides and α-Selenoesters in Aqueous Media Promoted by Cadmium. Synth. Commun. 2000, 30, 1731–1736. [Google Scholar] [CrossRef]
- Zhan, Z.; Lu, G.; Zhang, Y. A Novel Synthesis of Allyl Sulfides and Allyl Selenides via Sm-BiCl3 System in Aqueous Media. J. Chem. Res. 1999, 280–281. [Google Scholar] [CrossRef]
- Liao, P.; Bao, W.; Zhang, Y. A Novel Synthesis of Allyl and Prop-2-ynyl Selenides Promoted by Tin in the Presence of Water. J. Chem. Res. 1998, 150–151. [Google Scholar] [CrossRef]
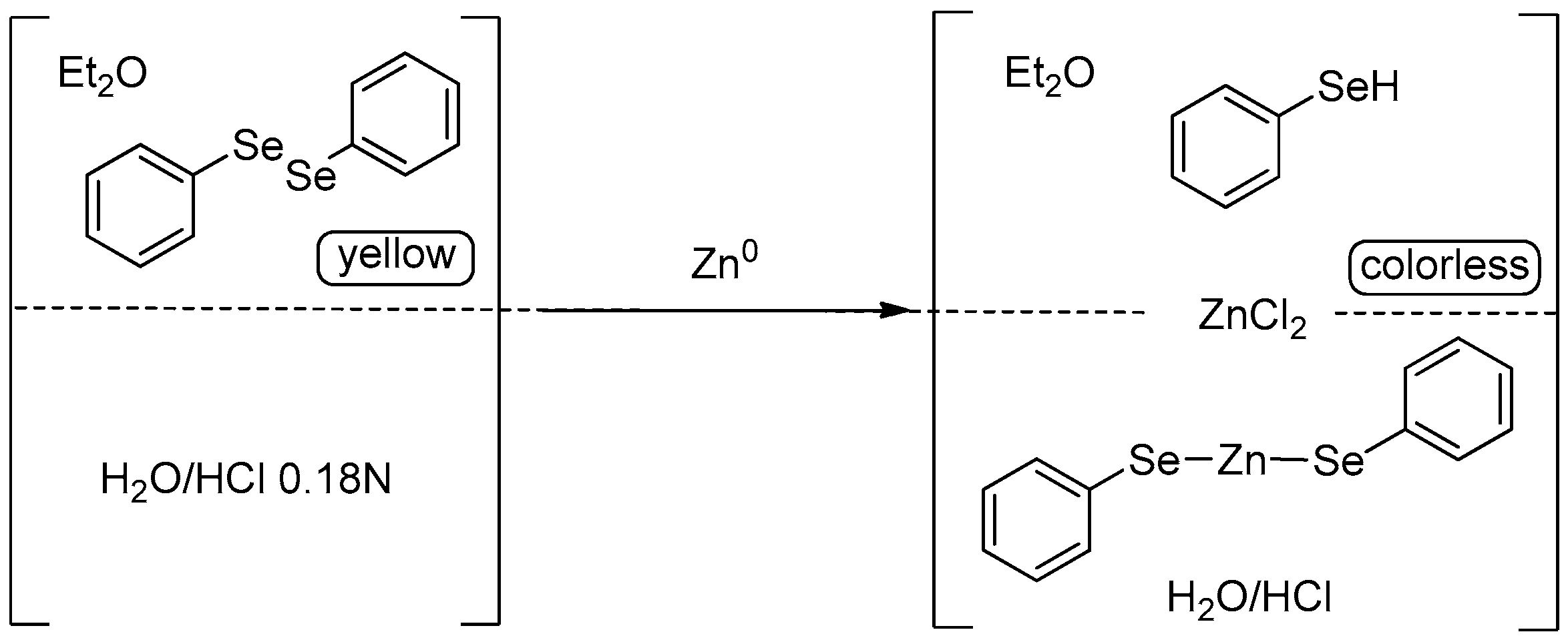
- Santi, C.; Santoro, S.; Testaferri, L.; Tiecco, M. A Simple Zinc-Mediated Preparation of Selenols. Synlett 2008, 2008, 1471–1474. [Google Scholar] [CrossRef]
- Braga, A.L.; Schwab, R.S.; Alberto, E.E.; Salman, S.M.; Vargas, J.; Azeredo, J.B. Ring opening of unprotected aziridines by zinc selenolates in a biphasic system. Tetrahedron Lett. 2009, 50, 2309–2311. [Google Scholar] [CrossRef]
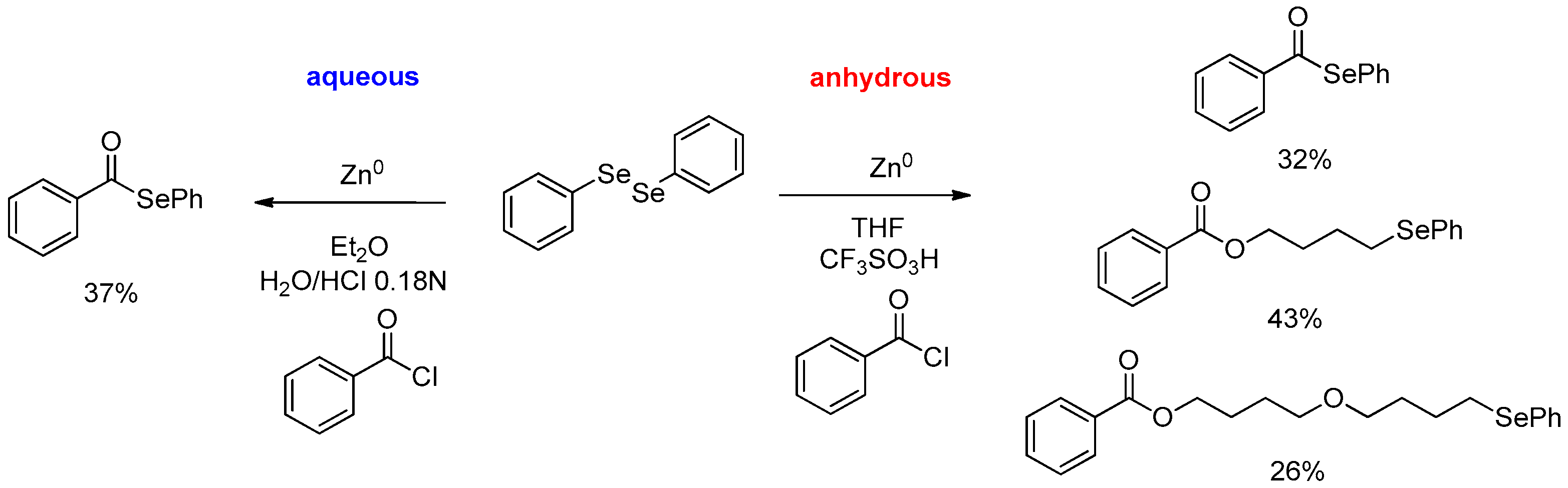
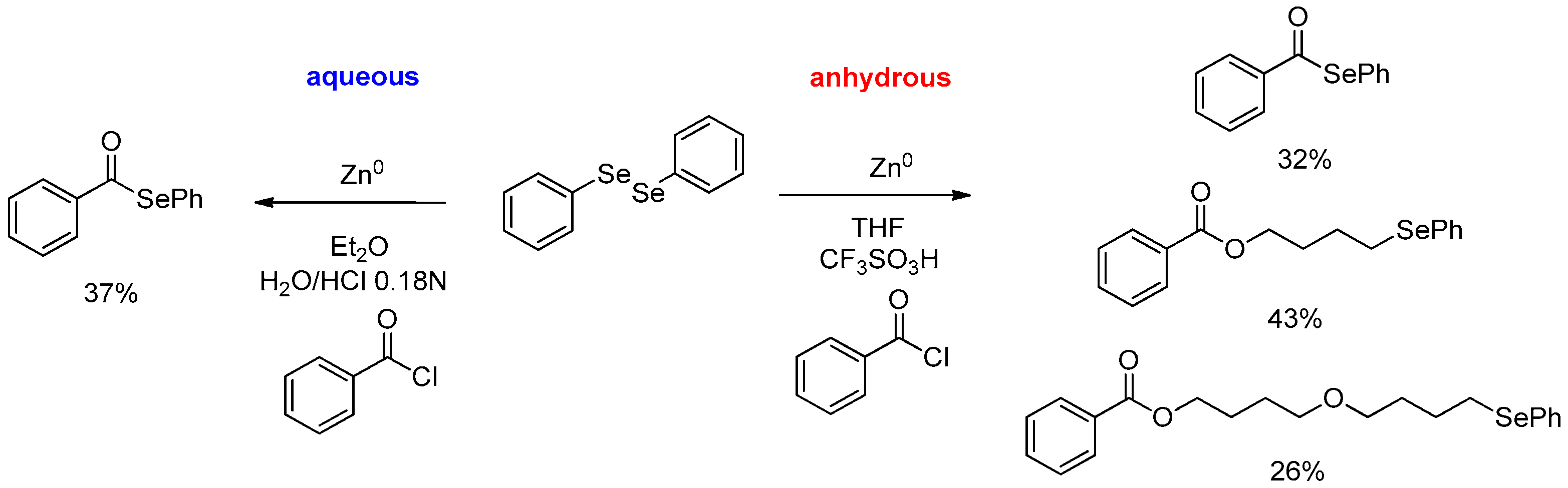
- Bellino, G.; Scisciani, M.; Vargas, J.P.; Sancineto, L.; Bagnoli, L.; Marini, F.; Lüdtke, D.S.; Lenardao, E.J.; Santi, C. Reaction of Acyl Chlorides with In Situ Formed Zinc Selenolates: Synthesis of Selenoesters versus Ring-Opening Reaction of Tetrahydrofuran. J. Chem. 2016, 2016, 2849140. [Google Scholar] [CrossRef]
- Flemer, S., Jr. A comprehensive one-pot synthesis of protected cysteine and selenocysteine SPPS derivatives. Prot. Pept. Lett. 2014, 21, 1257–1264. [Google Scholar]
- Flemer, S.J. Fmoc-Sec(Xan)-OH: Synthesis and utility of Fmoc selenocysteine SPPS derivatives with acid-labile sidechain protection. J. Pept. Sci. 2015, 21, 53–59. [Google Scholar] [CrossRef]
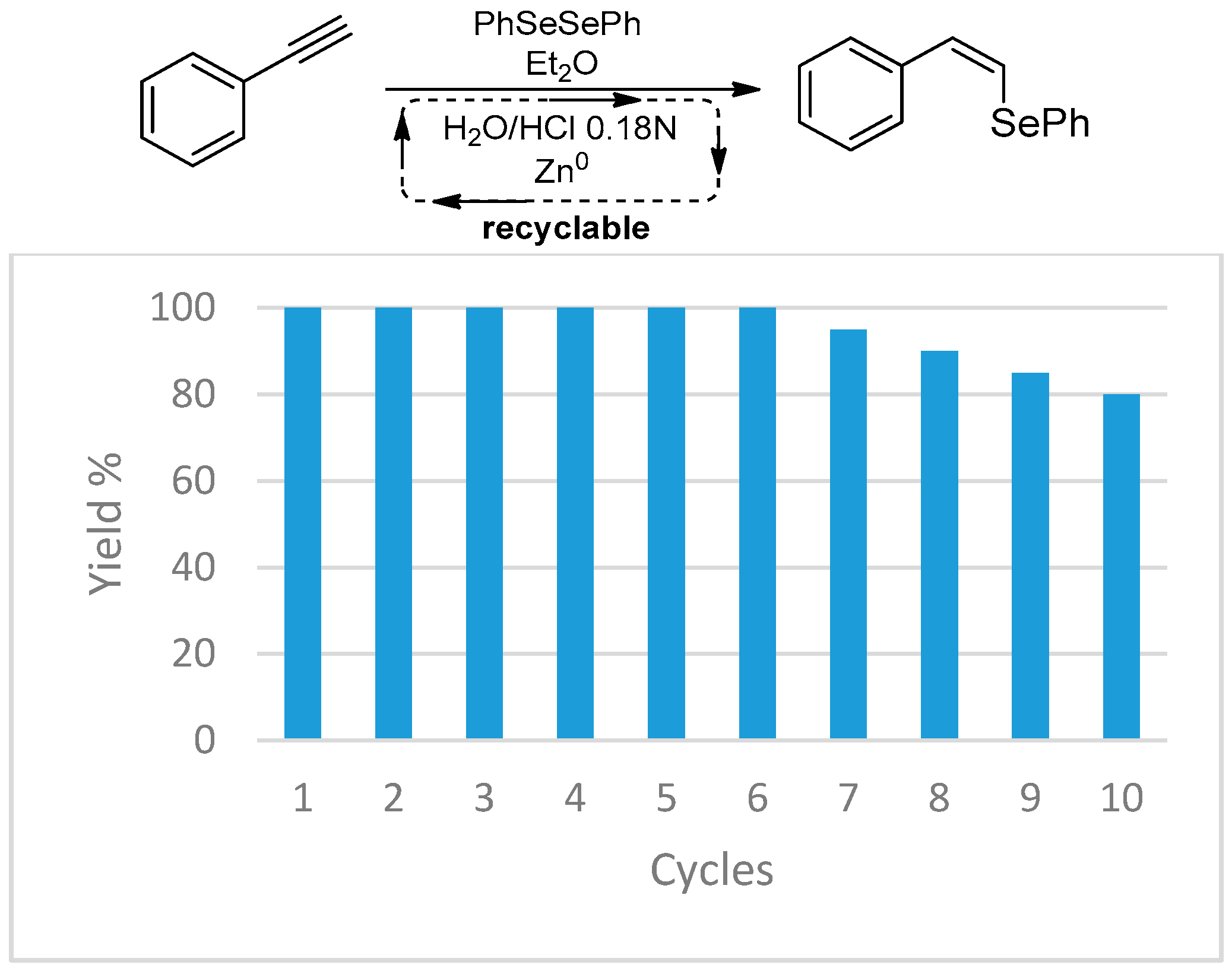
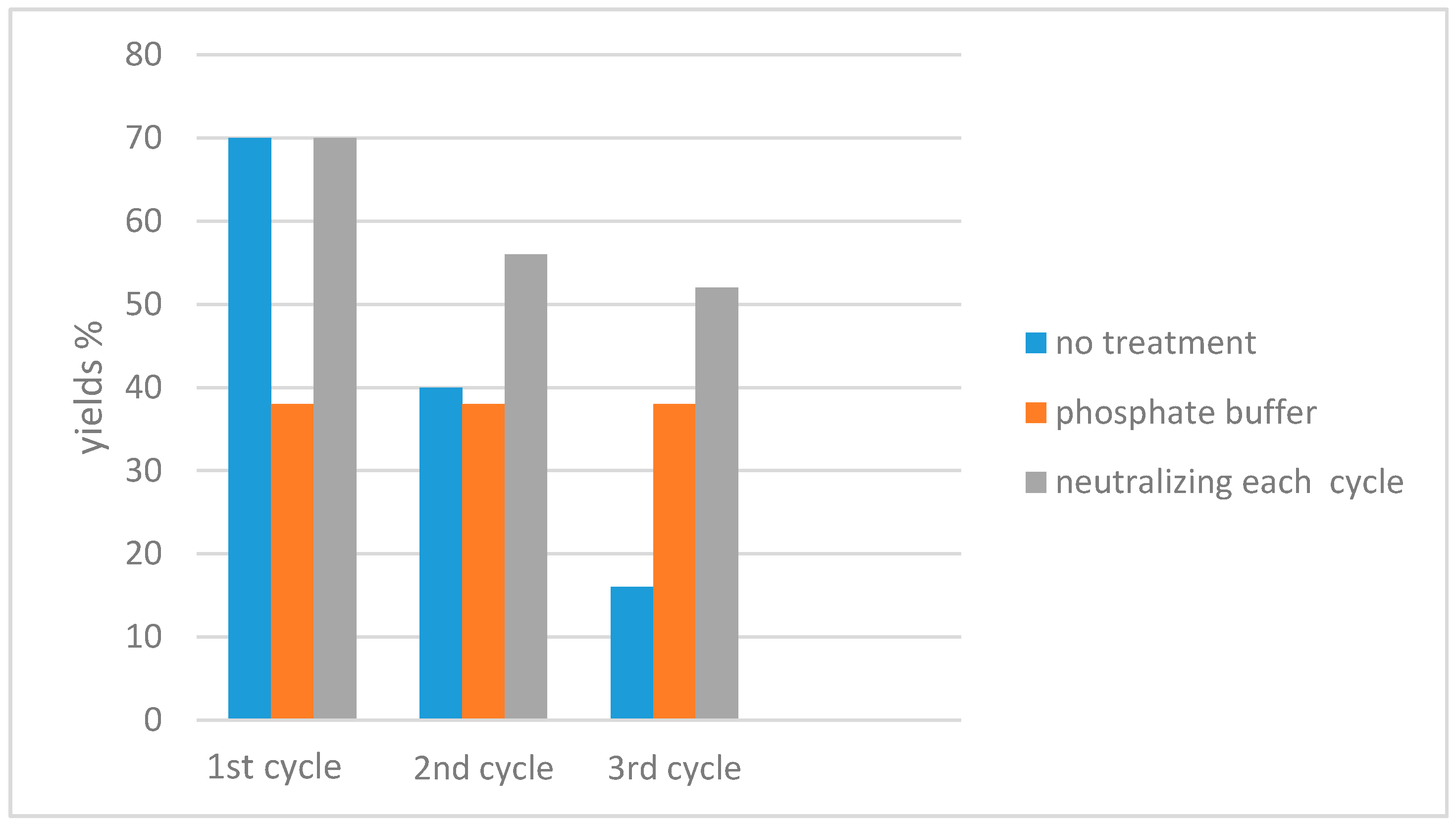
- Tidei, C.; Sancineto, L.; Bagnoli, L.; Battistelli, B.; Marini, F.; Santi, C. A Recyclable Biphasic System for Stereoselective and Easily Handled Hydrochalcogenations: Stereoselective Hydrochalcogenations. Eur. J. Org. Chem. 2014, 2014, 5968–5975. [Google Scholar] [CrossRef]
- Santi, C.; Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M. Preparation of the First Bench-Stable Phenyl Selenolate: An Interesting “On Water” Nucleophilic Reagent. Eur. J. Org. Chem. 2008, 2008, 5387–5390. [Google Scholar] [CrossRef]
- Bieber, L.W.; de Sá, A.C.P.; Menezes, P.H.; Gonçalves, S.M.C. General synthesis of alkyl phenyl selenides from organic halides mediated by zinc in aqueous medium. Tetrahedron Lett. 2001, 42, 4597–4599. [Google Scholar] [CrossRef]
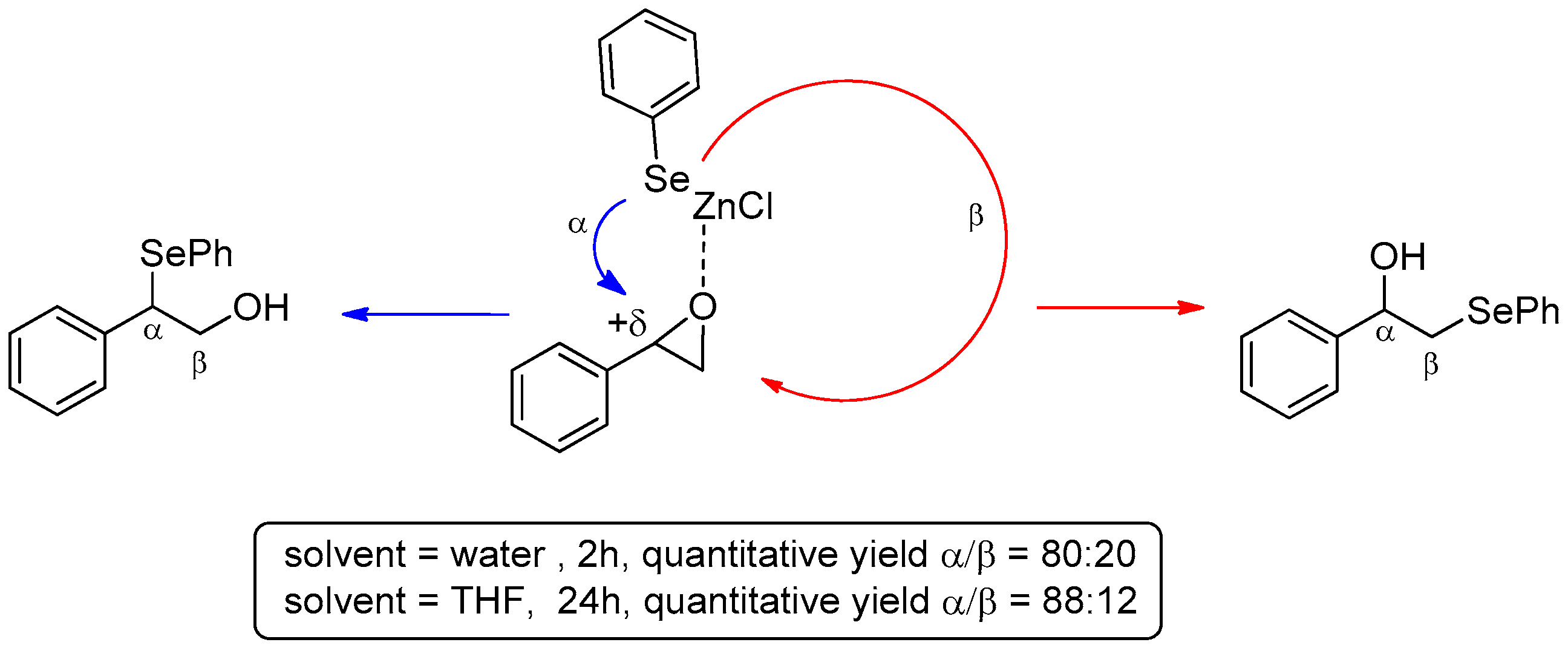
- Sridhar, R.; Srinivas, B.; Surendra, K.; Krishnaveni, N.S.; Rao, K.R. Synthesis of β-hydroxy selenides using benzeneselenol and oxiranes under supramolecular catalysis in the presence of β-cyclodextrin in water. Tetrahedron Lett. 2005, 46, 8837–8839. [Google Scholar] [CrossRef]
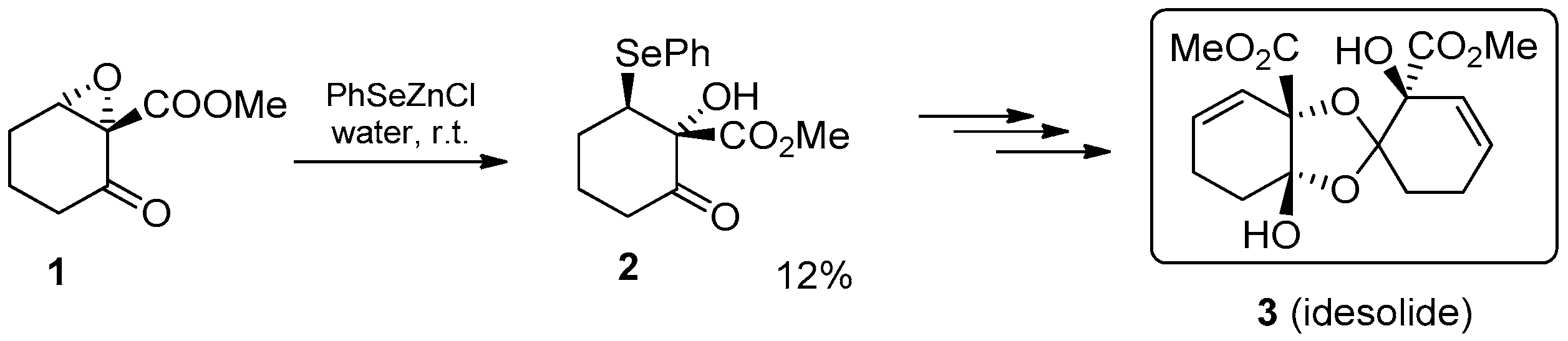
- Nagasawa, T.; Shimada, N.; Torihata, M.; Kuwahara, S. Enantioselective total synthesis of idesolide via NaHCO3-promoted dimerization. Tetrahedron 2010, 66, 4965–4969. [Google Scholar] [CrossRef]
- Jung, M.-H.; Yoo, J.-M.; Kang, Y.-J.; Lee, H.W.; Kim, S.H.; Sung, S.H.; Lee, Y.-J.; Choi, I.; Kim, T.-J. Idesolide, an Isolate of Idesia polycarpa, Inhibits Apoptosis through Induction of Intracellular Heat Shock Protein 70 in C2C12 Muscle Cells. Biol. Pharm. Bull. 2010, 33, 1063–1066. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Lauer, R.F. Mild procedure for the conversion of epoxides to allylic alcohols. First organoselenium reagent. J. Am. Chem. Soc. 1973, 95, 2697–2699. [Google Scholar] [CrossRef]
- Salman, S.; Schwab, R.; Alberto, E.; Vargas, J.; Dornelles, L.; Rodrigues, O.; Braga, A. Efficient Ring Opening of Protected and Unprotected Aziridines Promoted by Stable Zinc Selenolate in Ionic Liquid. Synlett 2011, 2011, 69–72. [Google Scholar] [CrossRef]
- Srinivas, B.; Kumar, V.P.; Sridhar, R.; Reddy, V.P.; Nageswar, Y.V.D.; Rao, K.R. β-Cyclodextrin-Promoted Addition of Benzeneselenol to Conjugated Alkenes in Water. Helv. Chim. Acta 2009, 92, 1080–1084. [Google Scholar] [CrossRef]
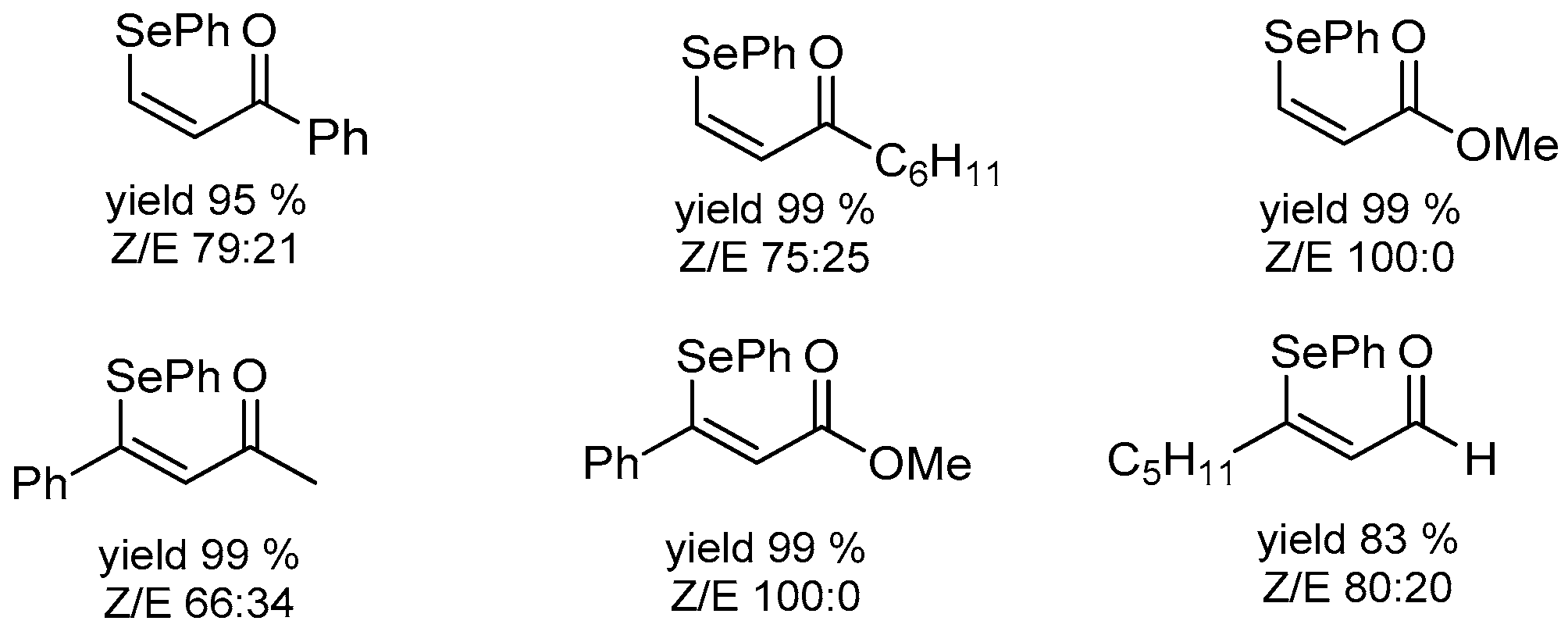
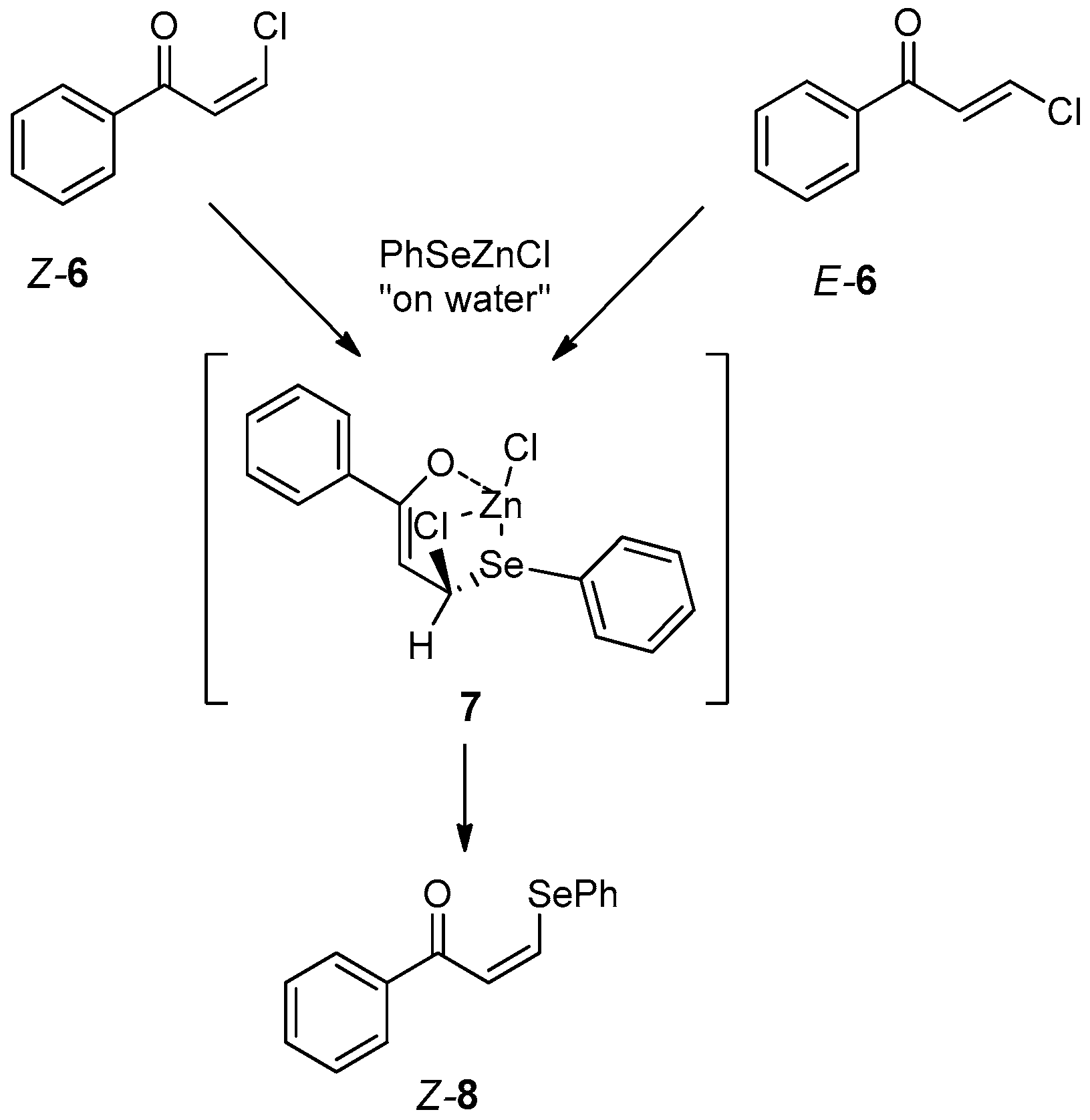
- Battistelli, B.; Lorenzo, T.; Tiecco, M.; Santi, C. “On-Water” Michael-Type Addition Reactions Promoted by PhSeZnCl. Eur. J. Org. Chem. 2011, 2011, 1848–1851. [Google Scholar] [CrossRef]
- Santoro, S.; Battistelli, B.; Testaferri, L.; Tiecco, M.; Santi, C. Vinylic Substitutions Promoted by PhSeZnCl: Synthetic and Theoretical Investigations. Eur. J. Org. Chem. 2009, 2009, 4921–4925. [Google Scholar] [CrossRef]
- Godoi, M.; Ricardo, E.W.; Botteselle, G.V.; Galetto, F.Z.; Azeredo, J.B.; Braga, A.L. Synthesis of selenol esters from diorganyl diselenides and acyl chlorides under solvent-free conditions and microwave irradiation. Green Chem. 2012, 14, 456–460. [Google Scholar] [CrossRef]
- Santi, C.; Battistelli, B.; Testaferri, L.; Tiecco, M. On water preparation of phenylselenoesters. Green Chem. 2012, 14, 1277–1280. [Google Scholar] [CrossRef]
- Sancineto, L.; Tidei, C.; Bagnoli, L.; Marini, F.; Lippolis, V.; Arca, M.; Lenardão, E.J.; Santi, C. Synthesis of Thiol Esters Using PhSZnBr as Sulfenylating Agent: A DFT-Guided Optimization of Reaction Conditions: Synthesis of Thiol Esters Using PhSZnBr as Sulfenylating Agent: A DFT-Guided Optimization of Reaction Conditions. Eur. J. Org. Chem. 2016, 2016, 2999–3005. [Google Scholar] [CrossRef]
- Umbreit, M.A.; Sharpless, K.B. Allylic oxidation of olefins by catalytic and stoichiometric selenium dioxide with tert-butyl hydroperoxide. J. Am. Chem. Soc. 1977, 99, 5526–5528. [Google Scholar] [CrossRef]
- Gogoi, P.; Das Sharma, S.; Konwar, D. SeO2/H2O2/H2O-Dioxane: A New Catalytic System for Trans Dihydroxylation of Olefins. Lett. Org. Chem. 2007, 4, 249–252. [Google Scholar] [CrossRef]
- Freudendahl, D.M.; Santoro, S.; Shahzad, S.A.; Santi, C.; Wirth, T. Green Chemistry with Selenium Reagents: Development of Efficient Catalytic Reactions. Angew. Chem. Int. Ed. 2009, 48, 8409–8411. [Google Scholar] [CrossRef]
- Toshimitsu, A.; Hayashi, G.; Terao, K.; Uemura, S. Amidoselenation of olefins via hydroxyselenation: Reactions using nitriles in reagent quantity and the synthesis of β-(acrylamido)alkyl phenyl selenides. J. Chem. Soc. Perkin Trans. 1 1986, 343–347. [Google Scholar] [CrossRef]
- Santi, C.; Tiecco, M.; Testaferri, L.; Tomassini, C.; Santoro, S.; Bizzoca, G. Diastereo and Enantioselective Synthesis of 1,2-Diols Promoted by Electrophilic Selenium Reagents. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 956–960. [Google Scholar] [CrossRef]
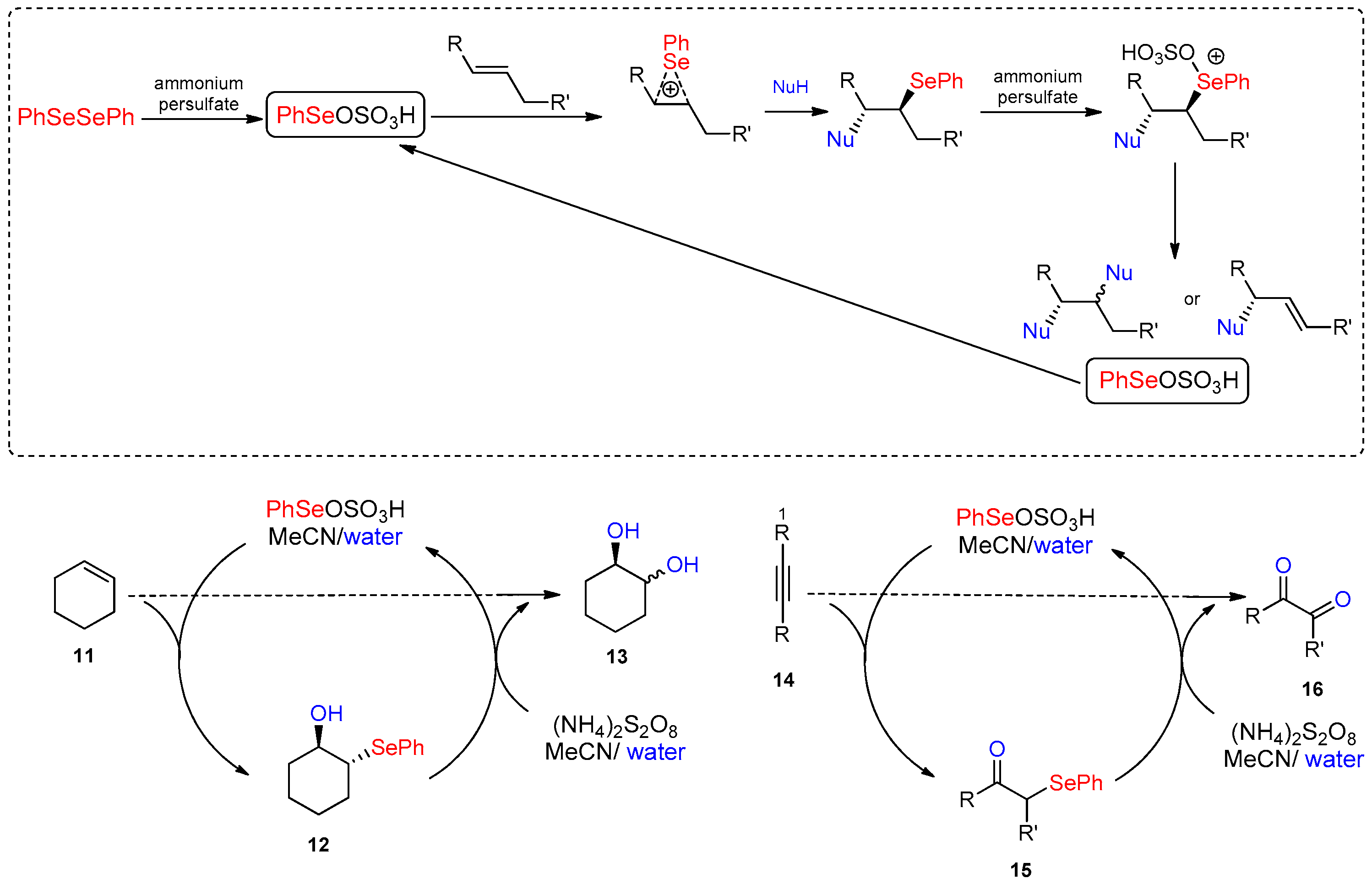
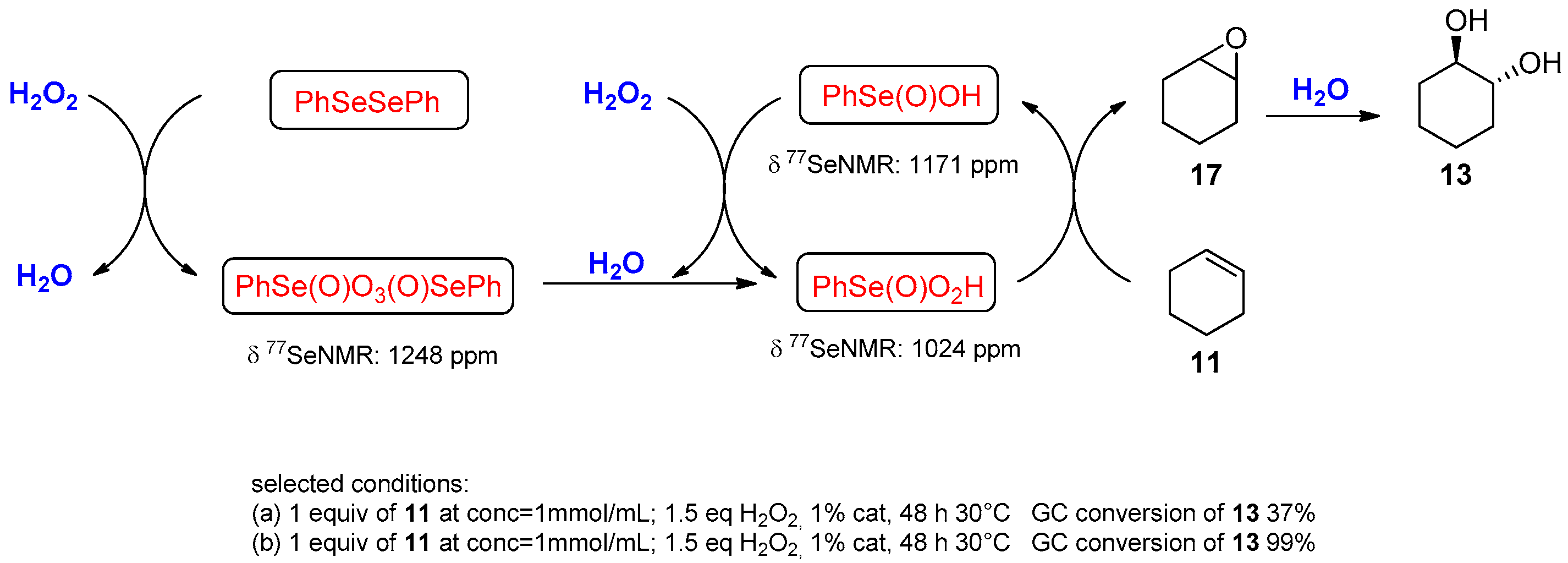
- Santoro, S.; Battistelli, B.; Gjoka, B.; Si, C.; Testaferri, L.; Tiecco, M.; Santi, C. Oxidation of Alkynes in Aqueous Media Catalyzed by Diphenyl Diselenide. Synlett 2010, 2010, 1402–1406. [Google Scholar]
- Reich, H.J.; Chow, F.; Peake, S.L. Seleninic acids as catalysts for oxidations of olefins and sulfides using hydrogen peroxide. Synthesis 1978, 1978, 299–301. [Google Scholar] [CrossRef]
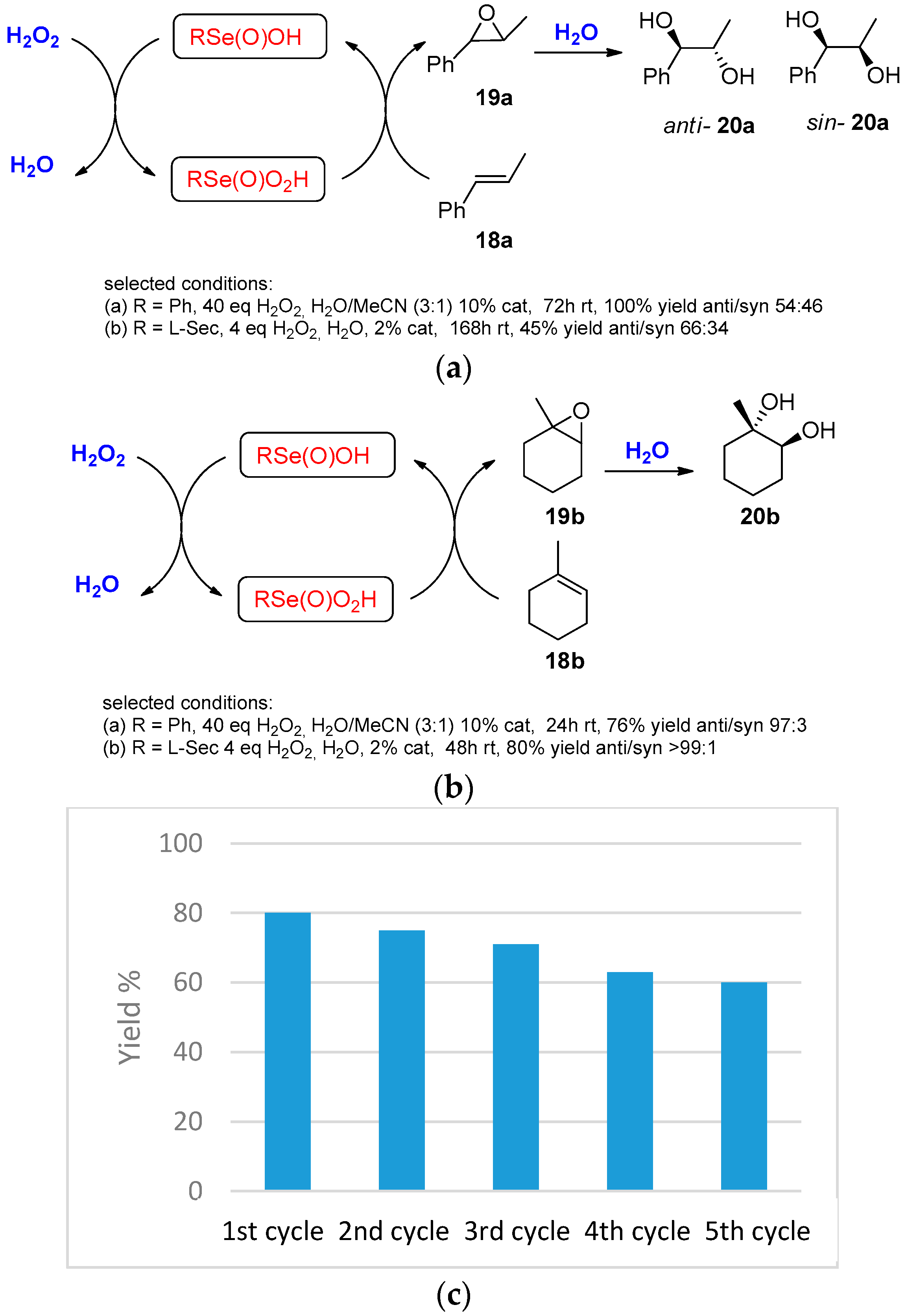
- Ten Brink, G.-J.; Fernandes, B.C.M.; van Vliet, M.C.A.; Arends, I.W.C.E.; Sheldon, R.A. Selenium catalyzed oxidations with aqueous hydrogen peroxide. Part 1: Epoxidation reactions in homogeneous solution. J. Chem. Soc. Perkin Trans. 1 2001, 2001, 224–228. [Google Scholar] [CrossRef]
- Yu, L.; Wang, J.; Chen, T.; Ding, K.; Pan, Y. Access to Cyclohexane-1,2-diol through the Diphenyldiselenide Catalyzed Oxidation of Cyclohexene by Hydrogen Peroxide. Chin. J. Org. Chem. 2013, 33, 1096–1099. [Google Scholar] [CrossRef]
- Yu, L.; Wang, J.; Chen, T.; Wang, Y.; Xu, Q. Recyclable 1,2-bis[3,5-bis(trifluoromethyl)phenyl]diselane-catalyzed oxidation of cyclohexene with H2O2: A practical access to trans-1,2-cyclohexanediol: Organoselenium-catalyzed dihydroxylation of cyclohexene. Appl. Organomet. Chem. 2014, 28, 652–656. [Google Scholar] [CrossRef]
- Santoro, S.; Santi, C.; Sabatini, M.; Testaferri, L.; Tiecco, M. Eco-Friendly Olefin Dihydroxylation Catalyzed by Diphenyl Diselenide. Adv. Synth. Catal. 2008, 350, 2881–2884. [Google Scholar] [CrossRef]
- Santi, C.; Di Lorenzo, R.; Tidei, C.; Bagnoli, L.; Wirth, T. Stereoselective selenium catalyzed dihydroxylation and hydroxymethoxylation of alkenes. Tetrahedron 2012, 68, 10530–10535. [Google Scholar] [CrossRef]
- Ten Brink, G.-J.; Vis, J.-M.; Arends, I.W.C.E.; Sheldon, R.A. Selenium-Catalyzed Oxidations with Aqueous Hydrogen Peroxide. 2. Baeyer-Villiger Reactions in Homogeneous Solution. J. Org. Chem. 2001, 66, 2429–2433. [Google Scholar]
- Ten Brink, G.-J.; Vis, J.M.; Arends, I.W.C.; Sheldon, R.A. Selenium catalysed oxidations with aqueous hydrogen peroxide. Part 3: Oxidation of carbonyl compounds under mono/bi/triphasic conditions. Tetrahedron 2002, 58, 3977–3983. [Google Scholar] [CrossRef]
- Sancineto, L.; Tidei, C.; Bagnoli, L.; Marini, F.; Lenardão, E.; Santi, C. Selenium Catalyzed Oxidation of Aldehydes: Green Synthesis of Carboxylic Acids and Esters. Molecules 2015, 20, 10496–10510. [Google Scholar] [CrossRef]
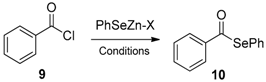
- Saha, A.; Saha, D.; Ranu, B.C. Copper nano-catalyst: Sustainable phenyl-selenylation of aryl iodides and vinyl bromides in water under ligand free conditions. Org. Biomol. Chem. 2009, 7, 1652–1657. [Google Scholar] [CrossRef]
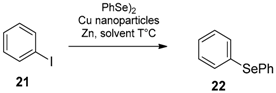
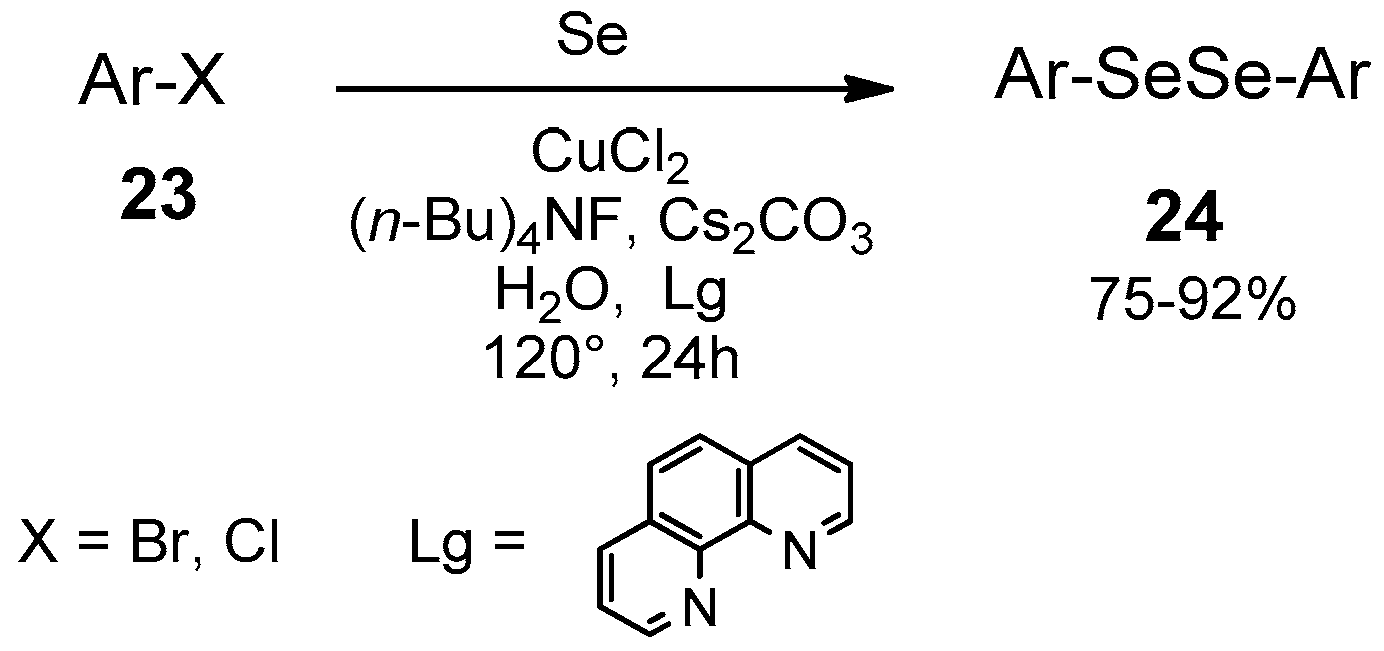
- Li, Z.; Ke, F.; Deng, H.; Xu, H.; Xiang, H.; Zhou, X. Synthesis of disulfides and diselenides by copper-catalyzed coupling reactions in water. Org. Biomol. Chem. 2013, 11, 2943–2946. [Google Scholar] [CrossRef]
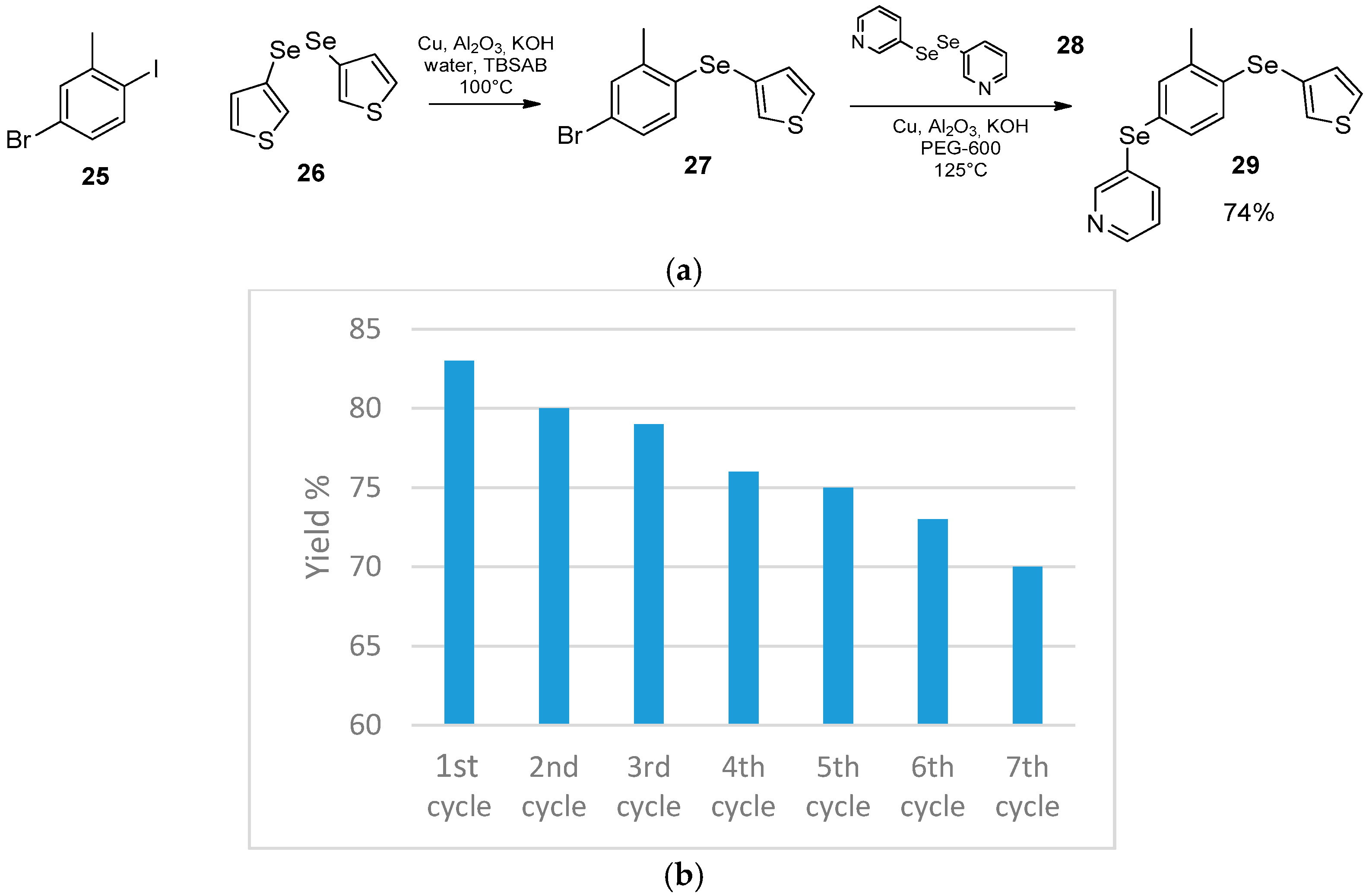
- Chatterjee, T.; Ranu, B.C. Solvent-Controlled Halo-Selective Selenylation of Aryl Halides Catalyzed by Cu(II) Supported on Al2O3. A General Protocol for the Synthesis of Unsymmetrical Organo Mono- and Bis-Selenides. J. Org. Chem. 2013, 78, 7145–7153. [Google Scholar] [CrossRef]
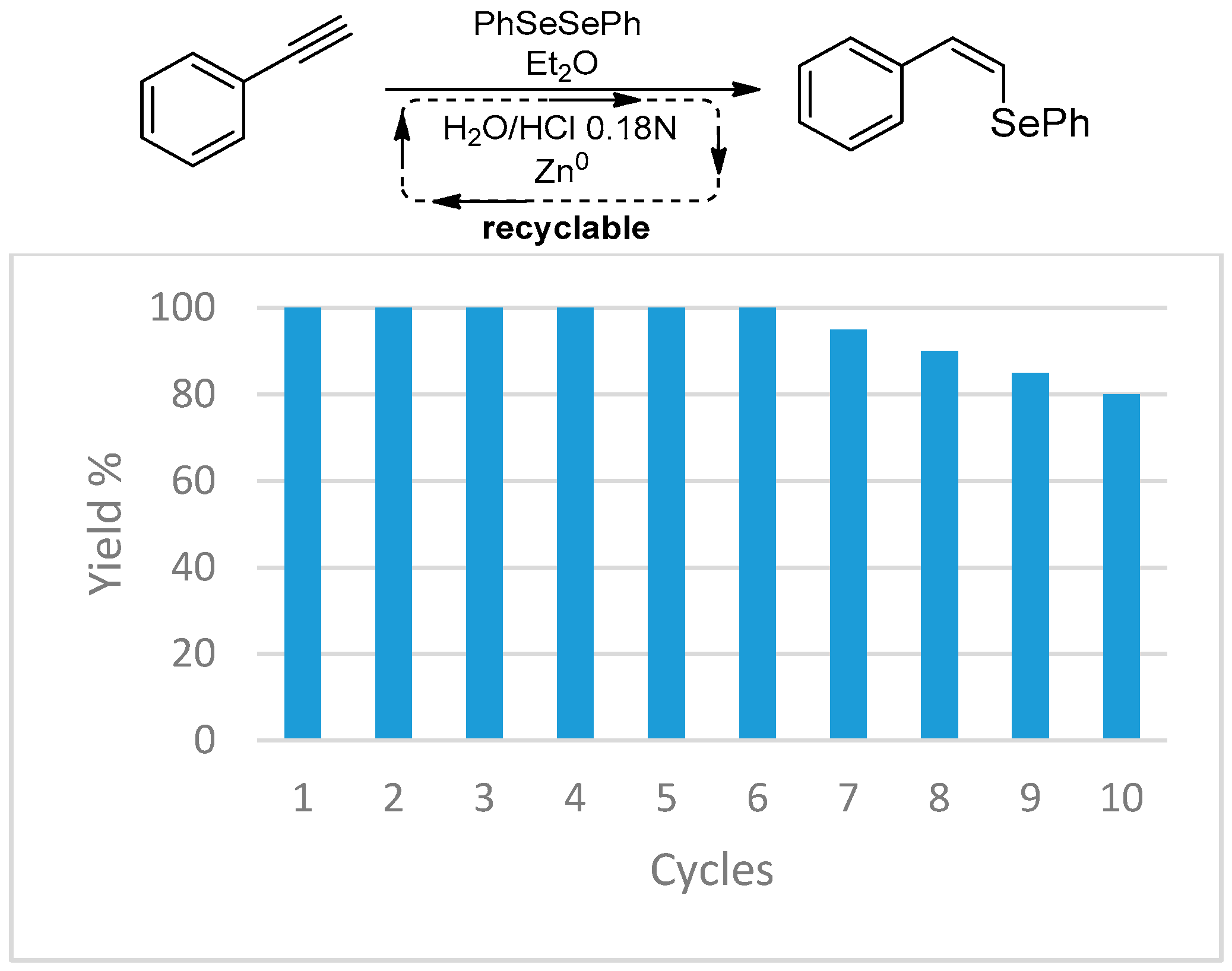
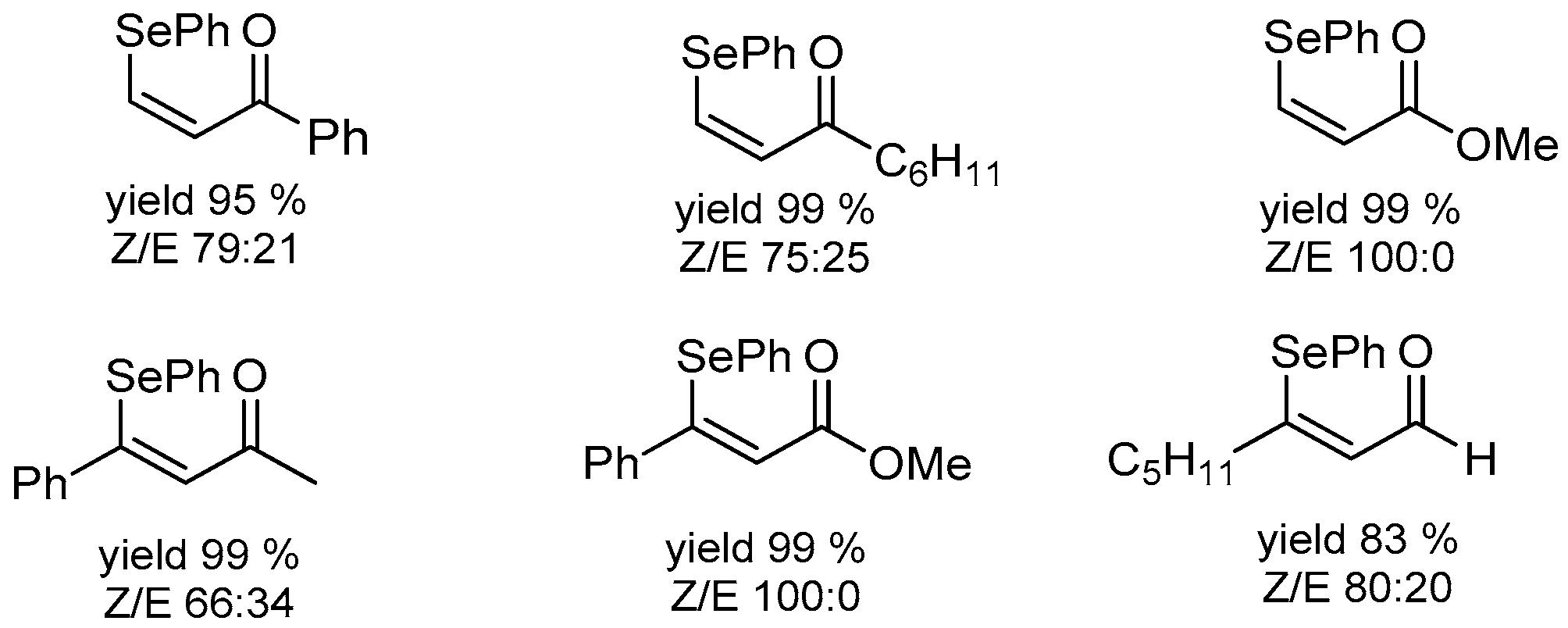
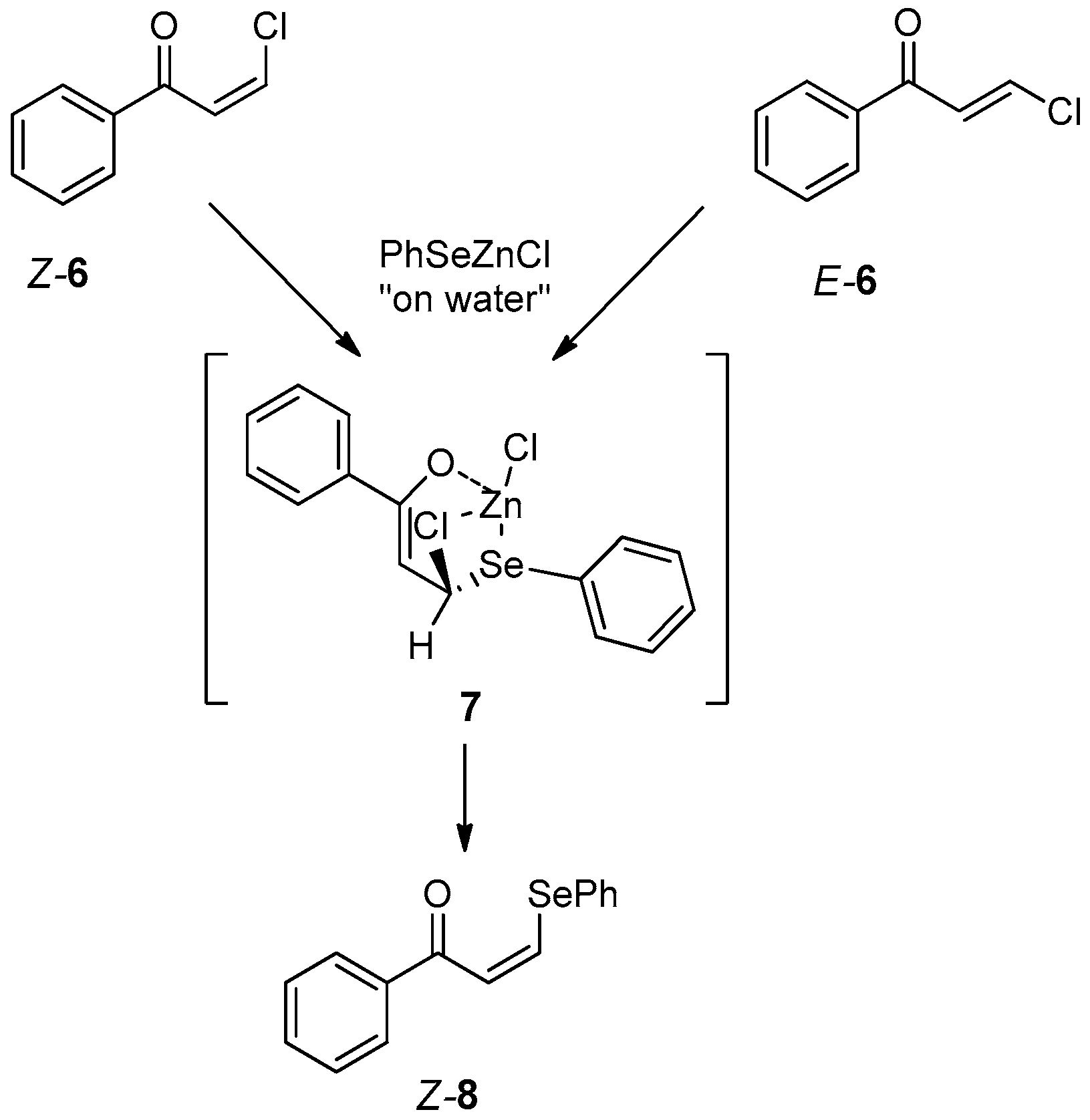
- Luz, E.Q.; Lopes, E.F.; Ricordi, V.G.; Santi, C.; Barcellos, T.; Lenardão, E.J.; Perin, G.; Alves, D. Water-Dependent Selective Synthesis of Mono- or Bis-Selanyl Alkenes from Terminal Alkynes. ChemistrySelect 2016, 1, 4289–4294. [Google Scholar] [CrossRef]
- Sengoden, M.; Vijay, M.; Balakumar, E.; Punniyamurthy, T. Efficient pyrrolidine catalyzed cycloaddition of aziridines with isothiocyanates, isoselenocyanates and carbon disulfide “on water”. RSC Adv. 2014, 4, 54149–54157. [Google Scholar] [CrossRef]
- Palomba, M.; Rossi, L.; Sancineto, L.; Tramontano, E.; Corona, A.; Bagnoli, L.; Santi, C.; Pannecouque, C.; Tabarrini, O.; Marini, F. A new vinyl selenone-based domino approach to spirocyclopropyl oxindoles endowed with anti-HIV RT activity. Org. Biomol. Chem. 2016, 14, 2015–2024. [Google Scholar] [CrossRef]
- Ribeiro Neto, P.B.; Santana, S.O.; Levitre, G.; Galdino, D.; Oliveira, J.L.; Ribeiro, R.T.; Barros, M.E.S.B.; Bieber, L.W.; Menezes, P.H.; Navarro, M. Electrochemical synthesis of organochalcogenides in aqueous medium. Green Chem. 2016, 18, 657–661. [Google Scholar] [CrossRef]
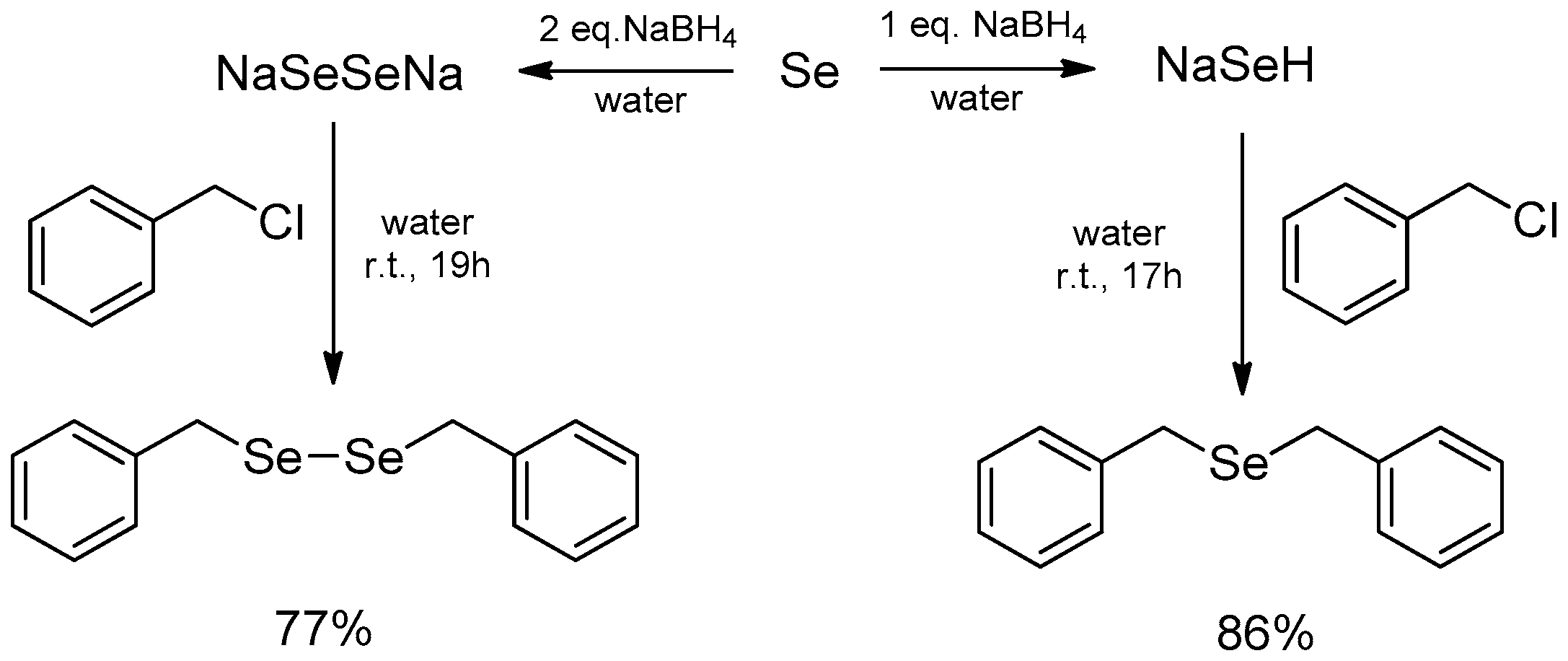
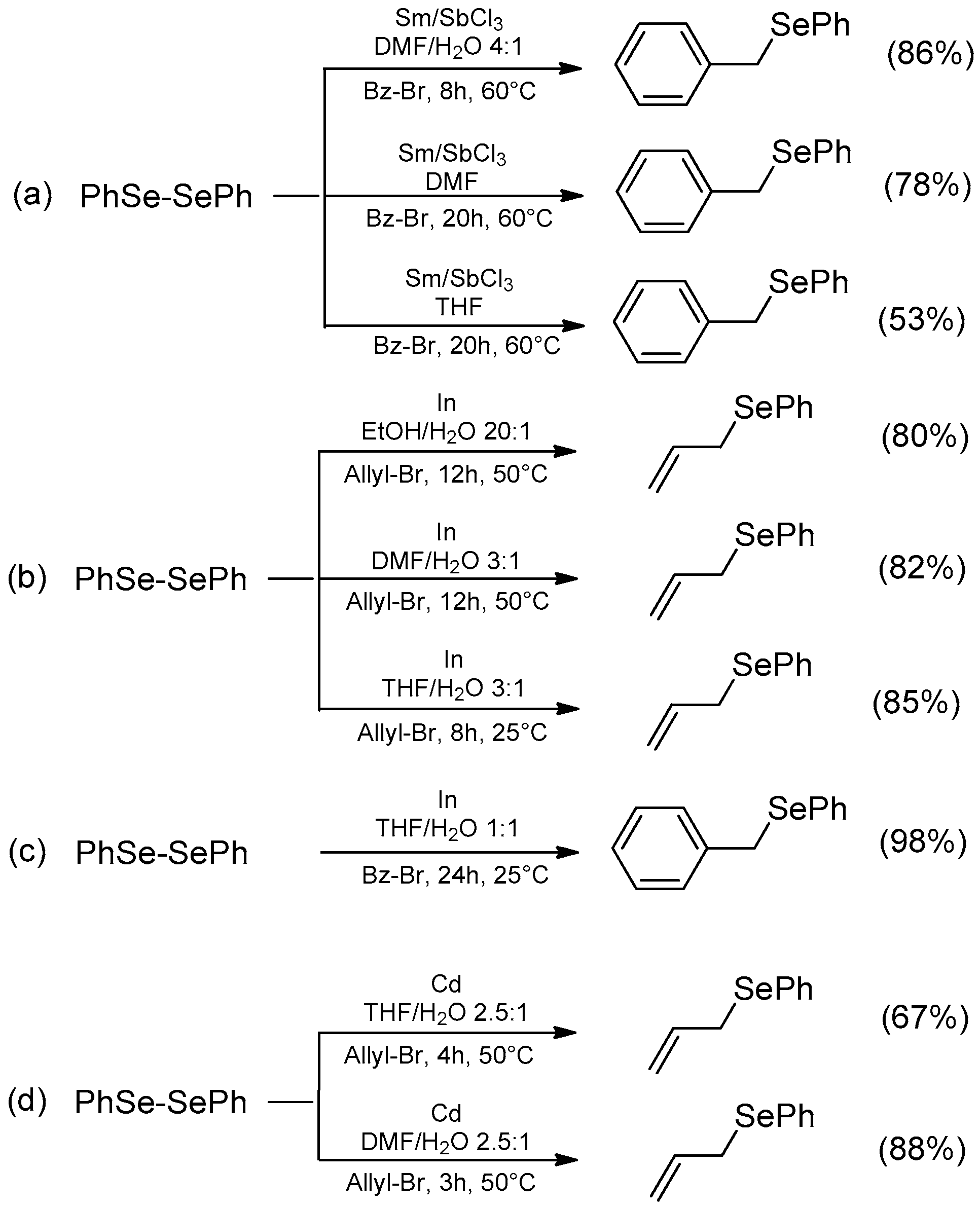
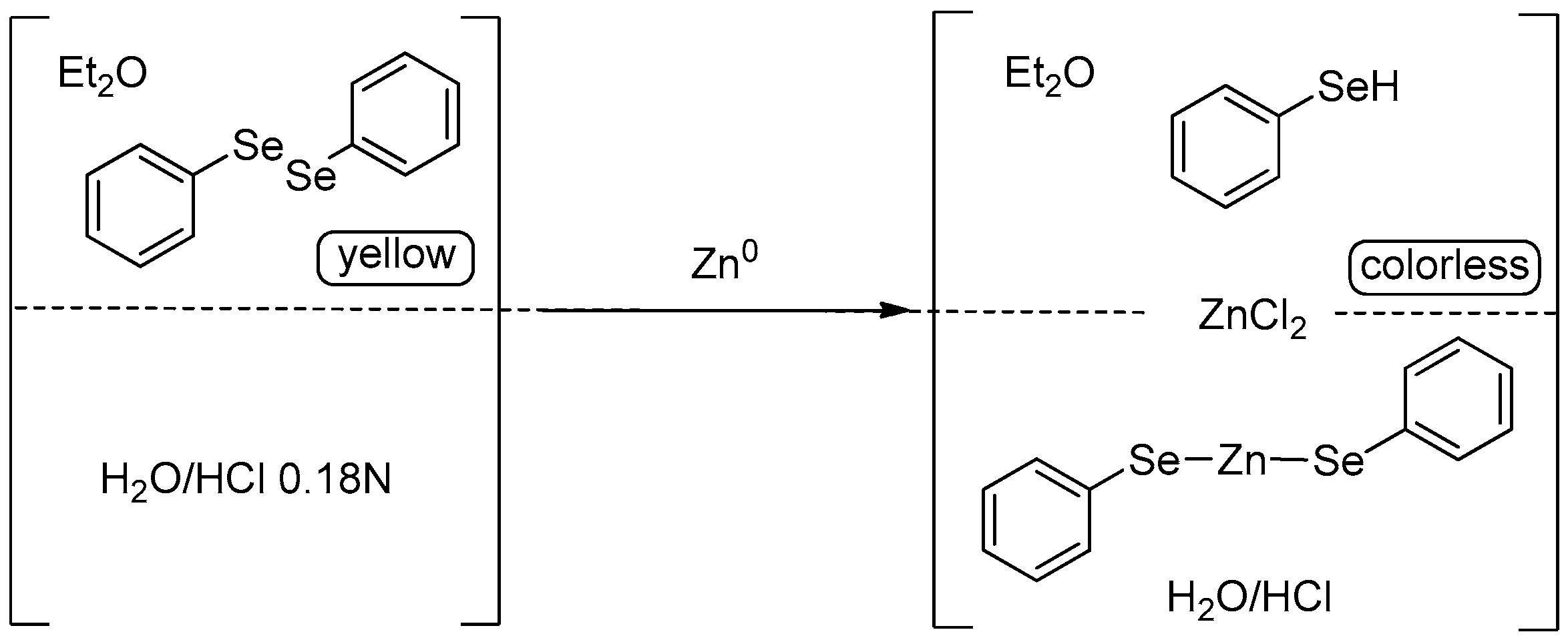
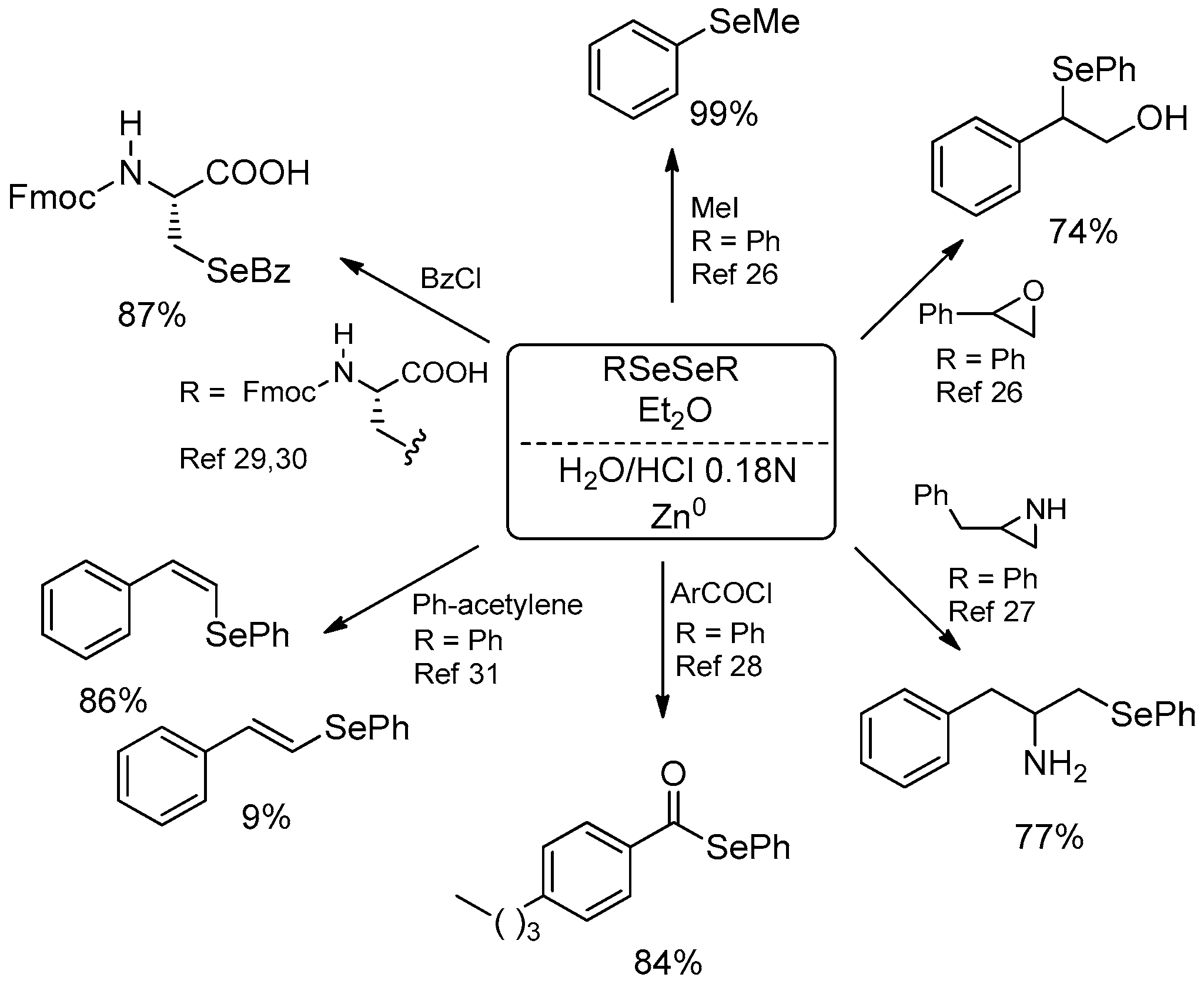


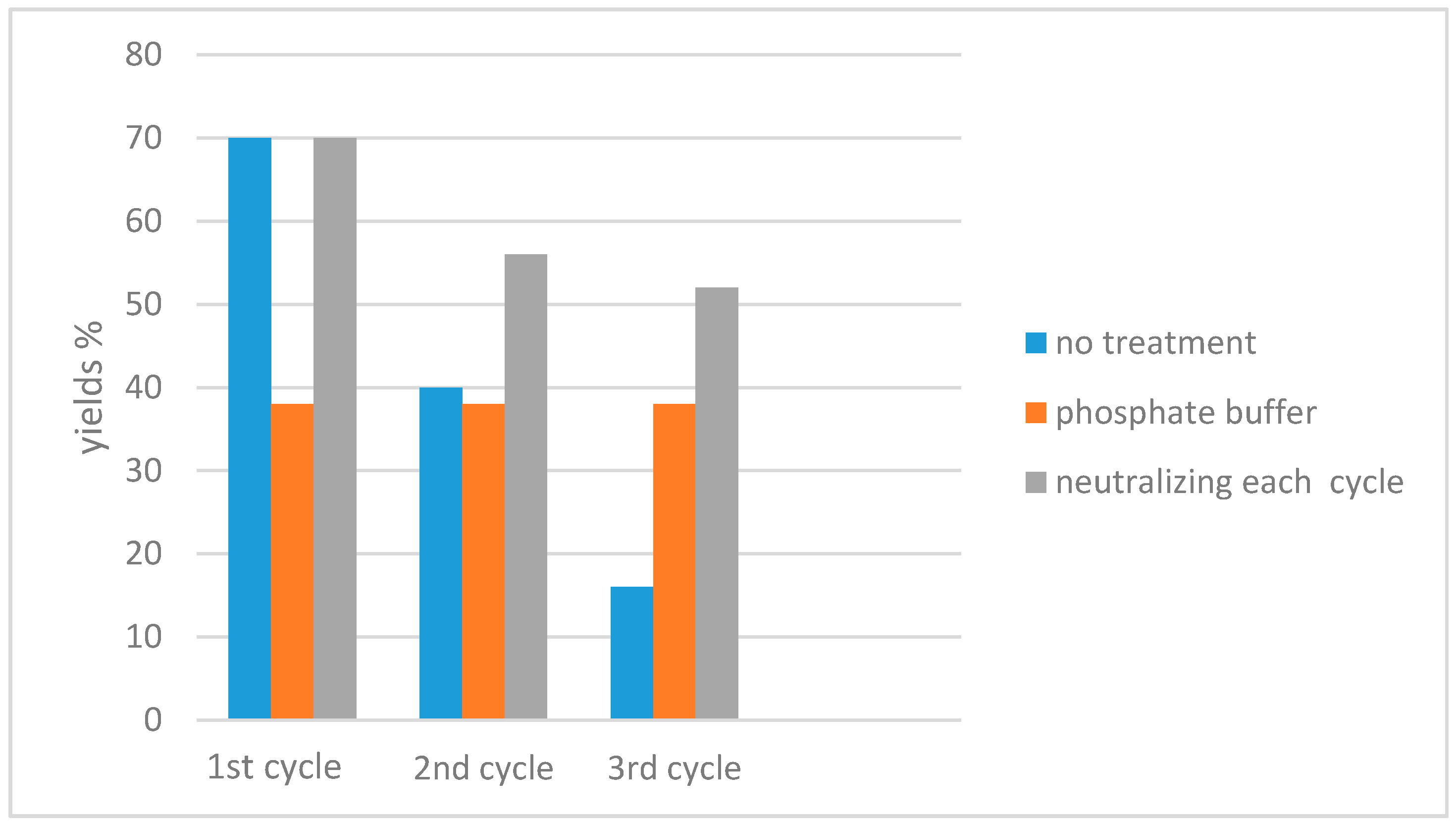

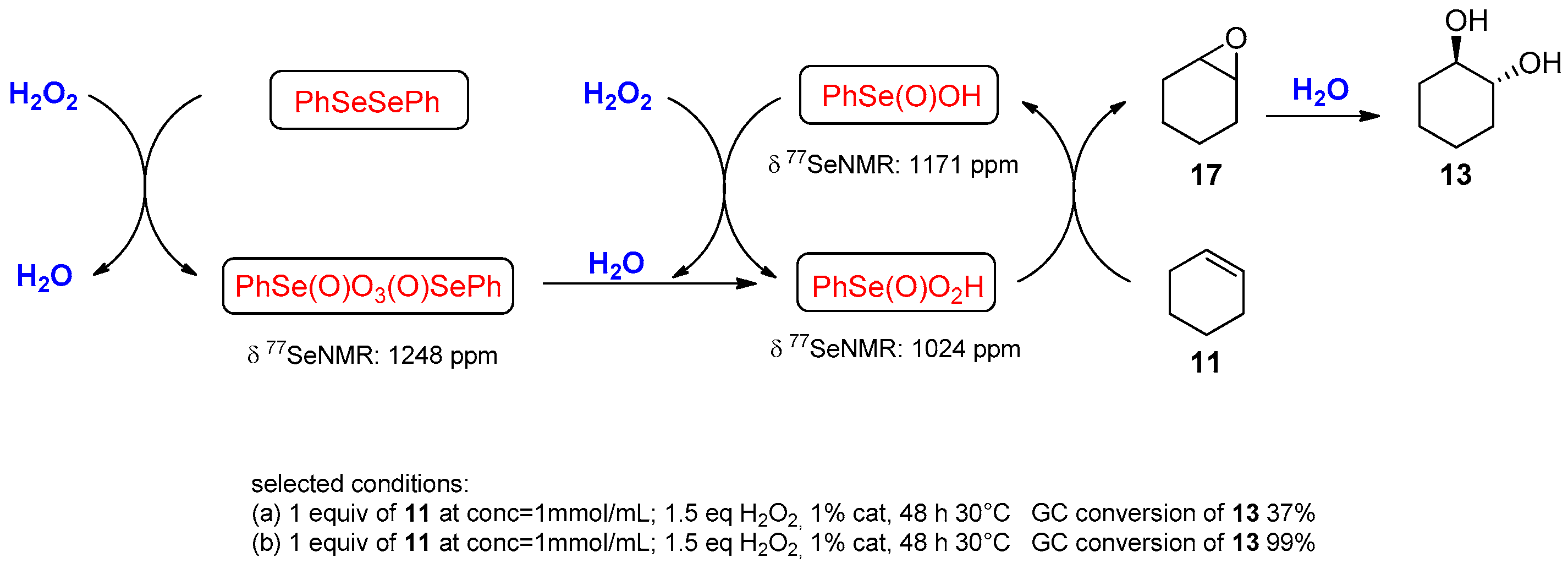
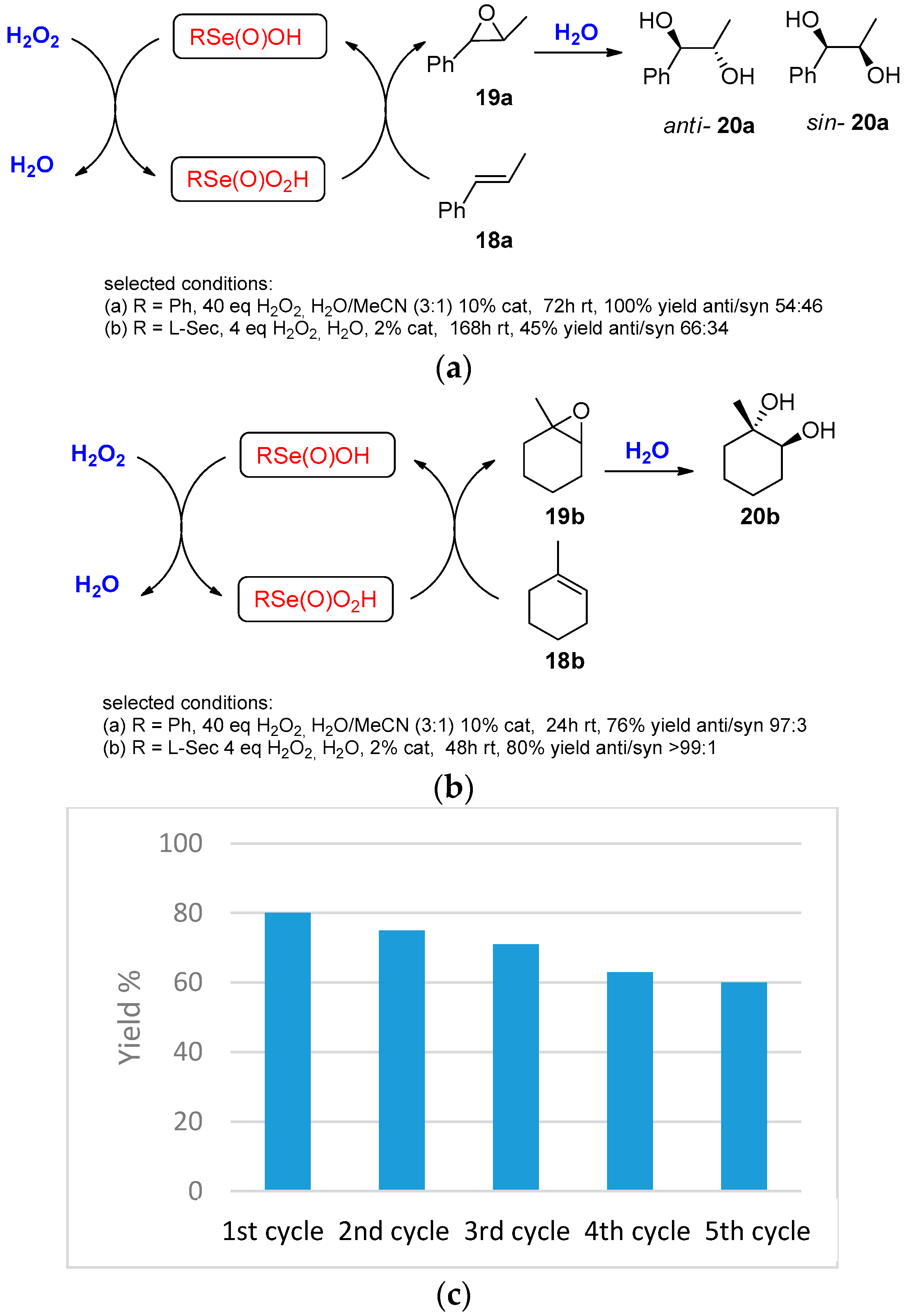
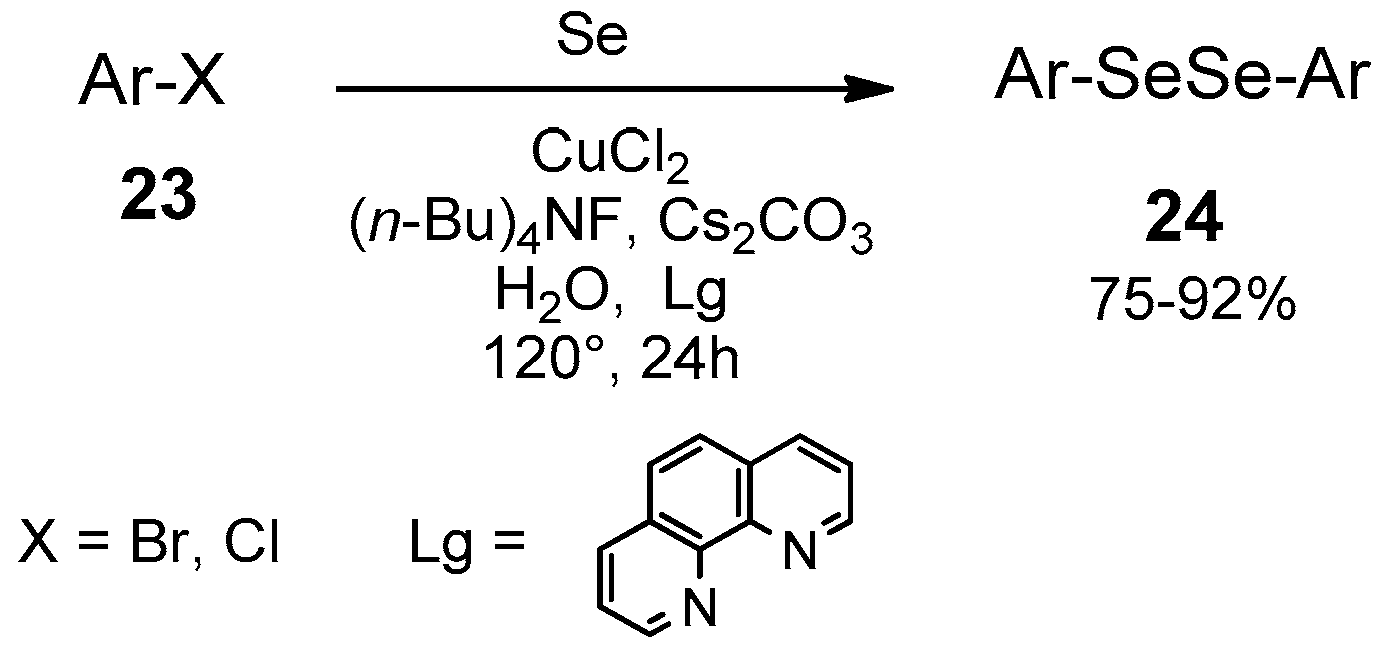
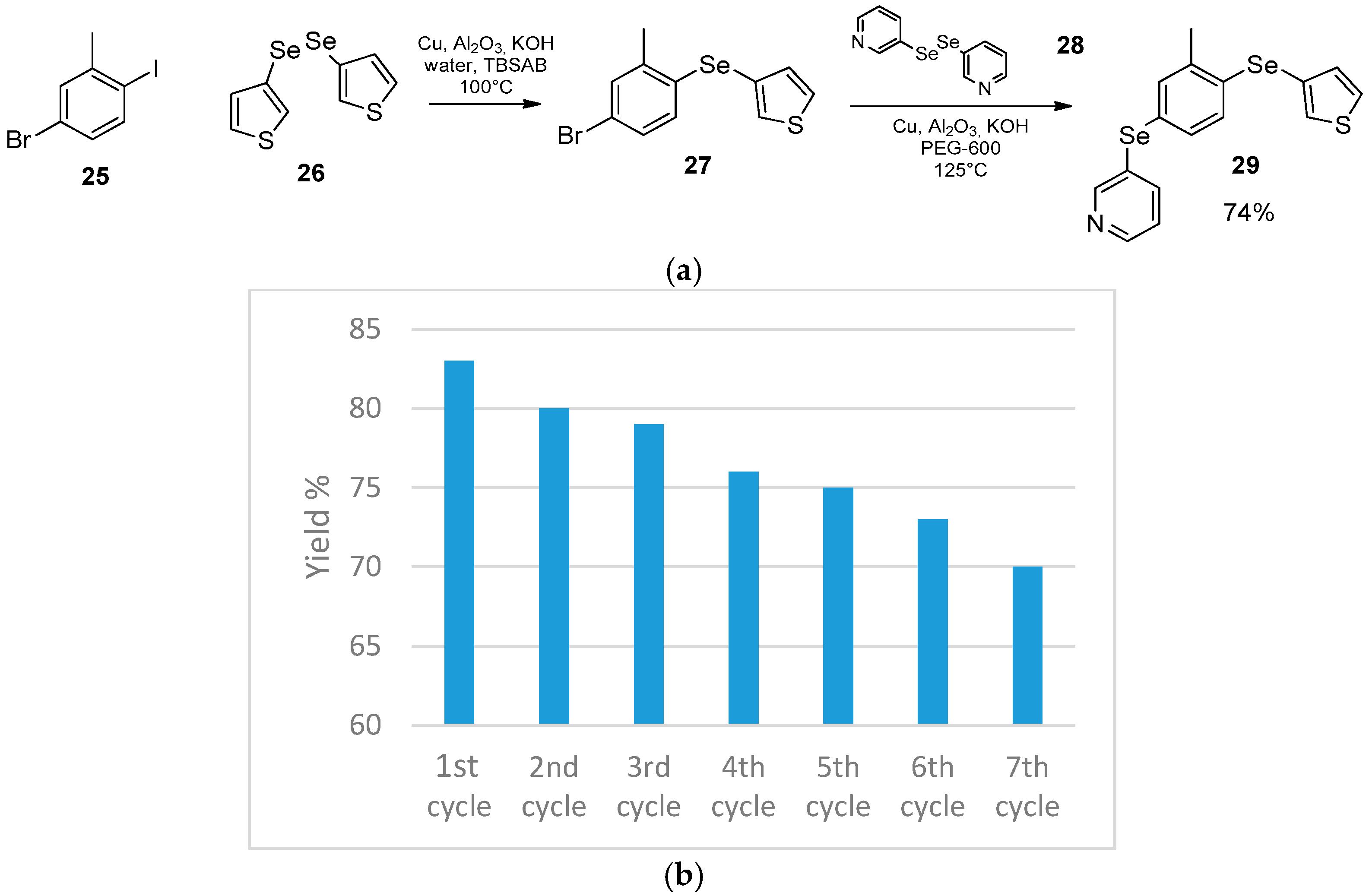

| Conditions | Reaction Time | Yield % | Reference |
|---|---|---|---|
| PhSeH, water, β-CD, r.t. | 45 min | 82 | [39] |
| PhSeZnCl, THF, r.t. | 24 h | 90 | [40] |
| PhSeZnCl, water, r.t. | 140 h | 53 | [40] |

| Entry | X | Reaction Time | T °C | Solvent | Activation | Yield % | Reference |
|---|---|---|---|---|---|---|---|
| 1 | SePh | 5‘ | 80 | none | MW 100 watt | 90 | [42] |
| 2 | SePh | 90‘ | 80 | none | heating | 67 | [42] |
| 3 | Cl | 25h | 80 | THF | none | 25 | [43] |
| 4 | Br | 25h | 80 | THF | none | 30 | [43] |
| 5 | Cl | 3h | rt | water | none | 60 | [43] |
| 6 | Br | 3h | rt | water | none | 70 | [43] |
| 7 | Cl | 5‘ | rt | none | mortar | 25 | [44] |
| 8 | Br | 5‘ | rt | none | mortar | 30 | [44] |
| Solvent | Temperature | Yield % |
|---|---|---|
| water | 100 °C | 88 |
| DMF | 120 °C | 78 |
| THF | 66 °C | 41 |
| EtOH | 78 °C | 38 |
| MeOH | 65 °C | 32 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santi, C.; Jacob, R.G.; Monti, B.; Bagnoli, L.; Sancineto, L.; Lenardão, E.J. Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules 2016, 21, 1482. https://doi.org/10.3390/molecules21111482
Santi C, Jacob RG, Monti B, Bagnoli L, Sancineto L, Lenardão EJ. Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules. 2016; 21(11):1482. https://doi.org/10.3390/molecules21111482
Chicago/Turabian StyleSanti, Claudio, Raquel G. Jacob, Bonifacio Monti, Luana Bagnoli, Luca Sancineto, and Eder J. Lenardão. 2016. "Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry" Molecules 21, no. 11: 1482. https://doi.org/10.3390/molecules21111482
APA StyleSanti, C., Jacob, R. G., Monti, B., Bagnoli, L., Sancineto, L., & Lenardão, E. J. (2016). Water and Aqueous Mixtures as Convenient Alternative Media for Organoselenium Chemistry. Molecules, 21(11), 1482. https://doi.org/10.3390/molecules21111482