
Modulation of Autophagy by a Thioxanthone Decreases the Viability of Melanoma Cells



 ,
,  and
and 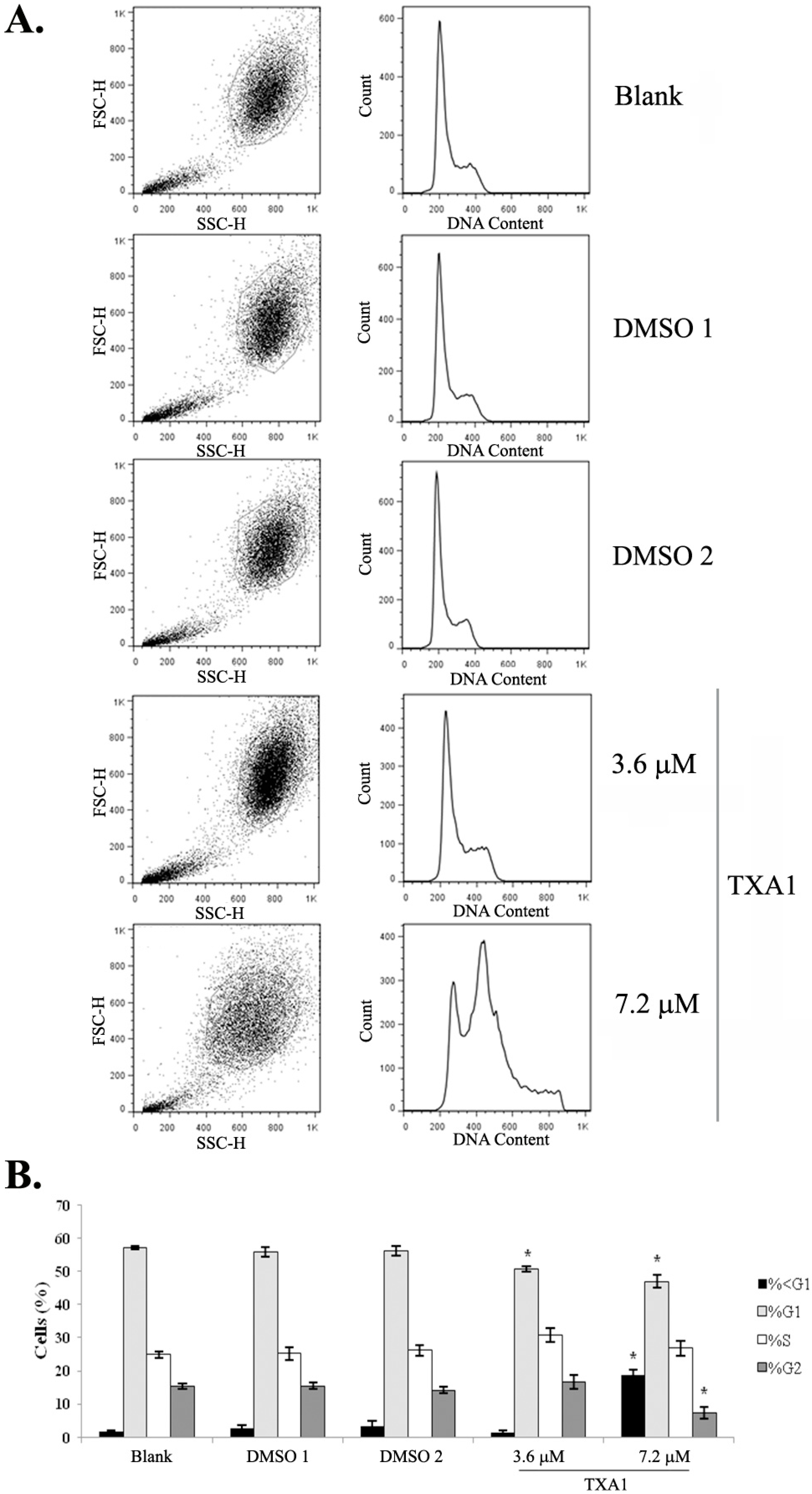{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
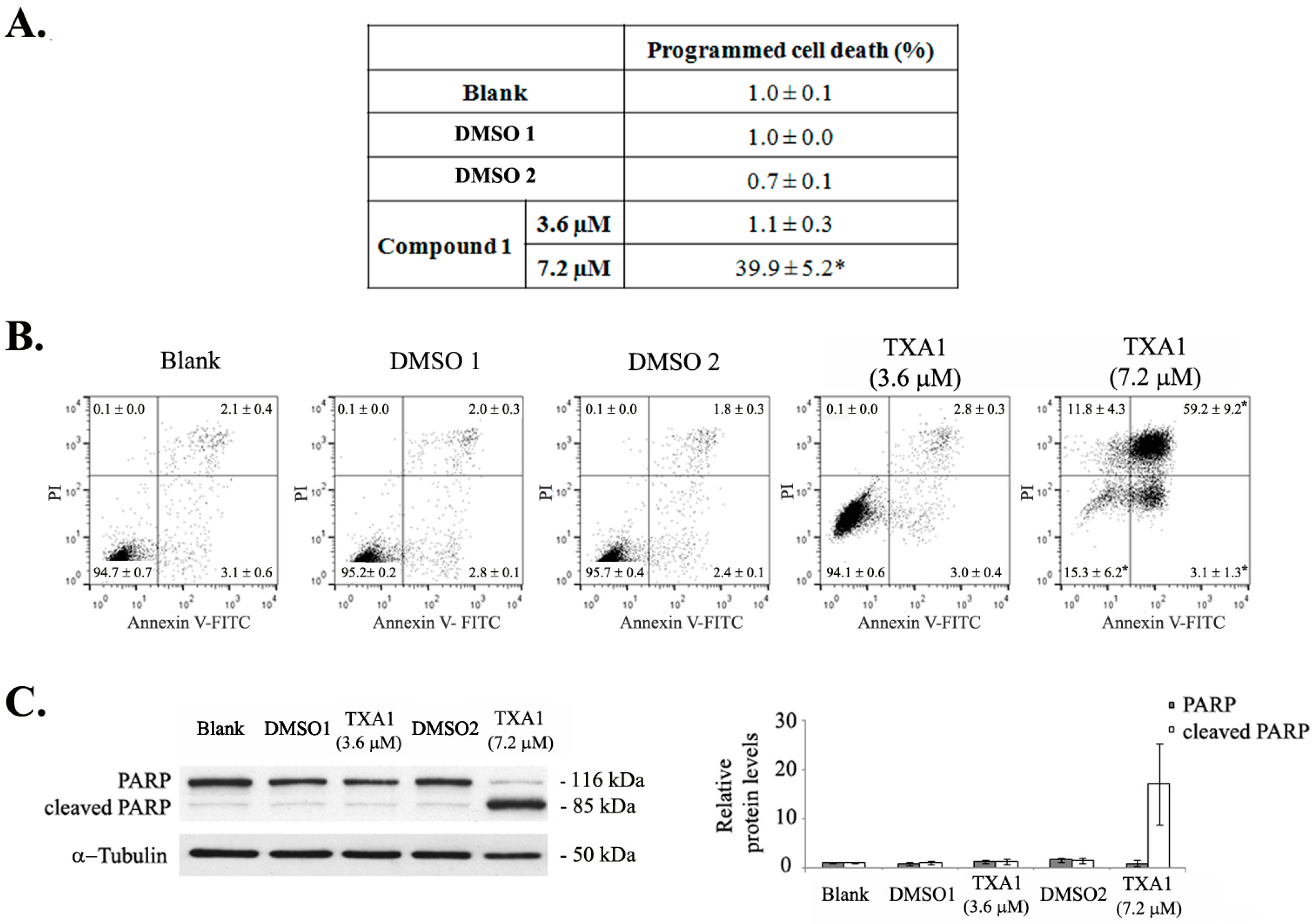
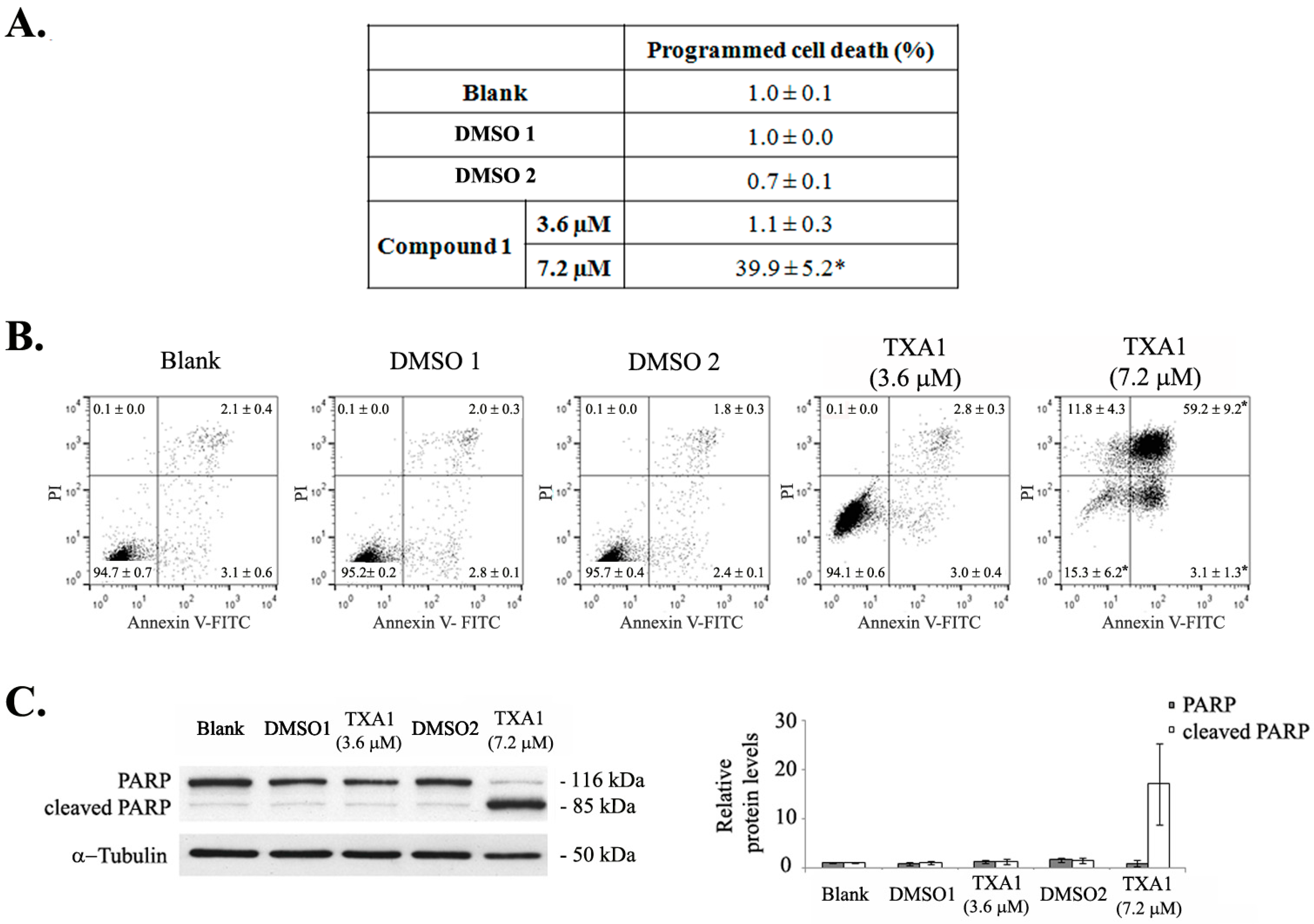
2.1. Treatment of A375-C5 Melanoma Cells with Twice the GI50 Concentration of TXA1 Alters the Cell Cycle Profile and Induces Apoptosis Although No Effect Is Found with the GI50 Concentration
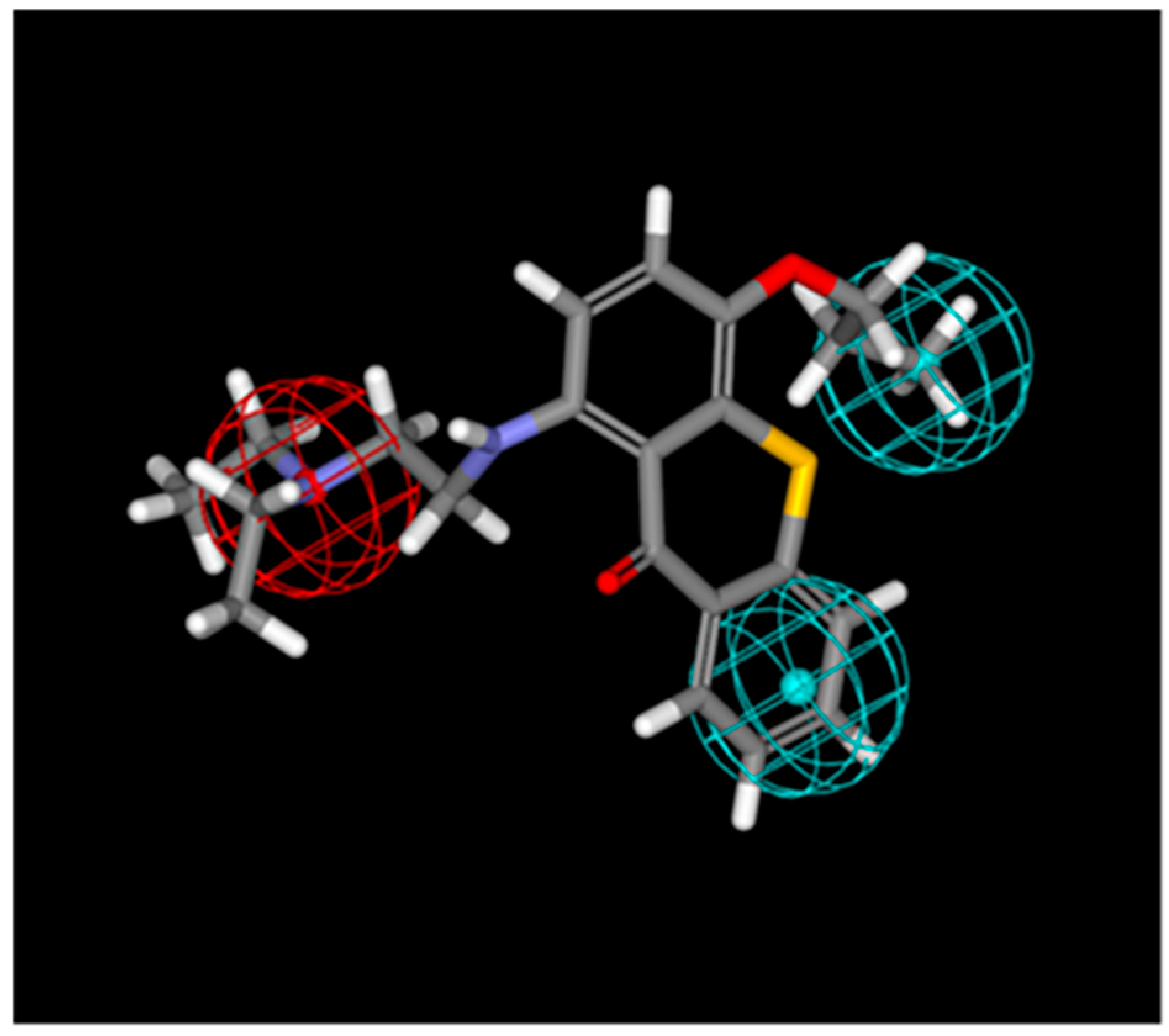
2.2. TXA1 Maps onto a Pharmacophore for Autophagy Induction
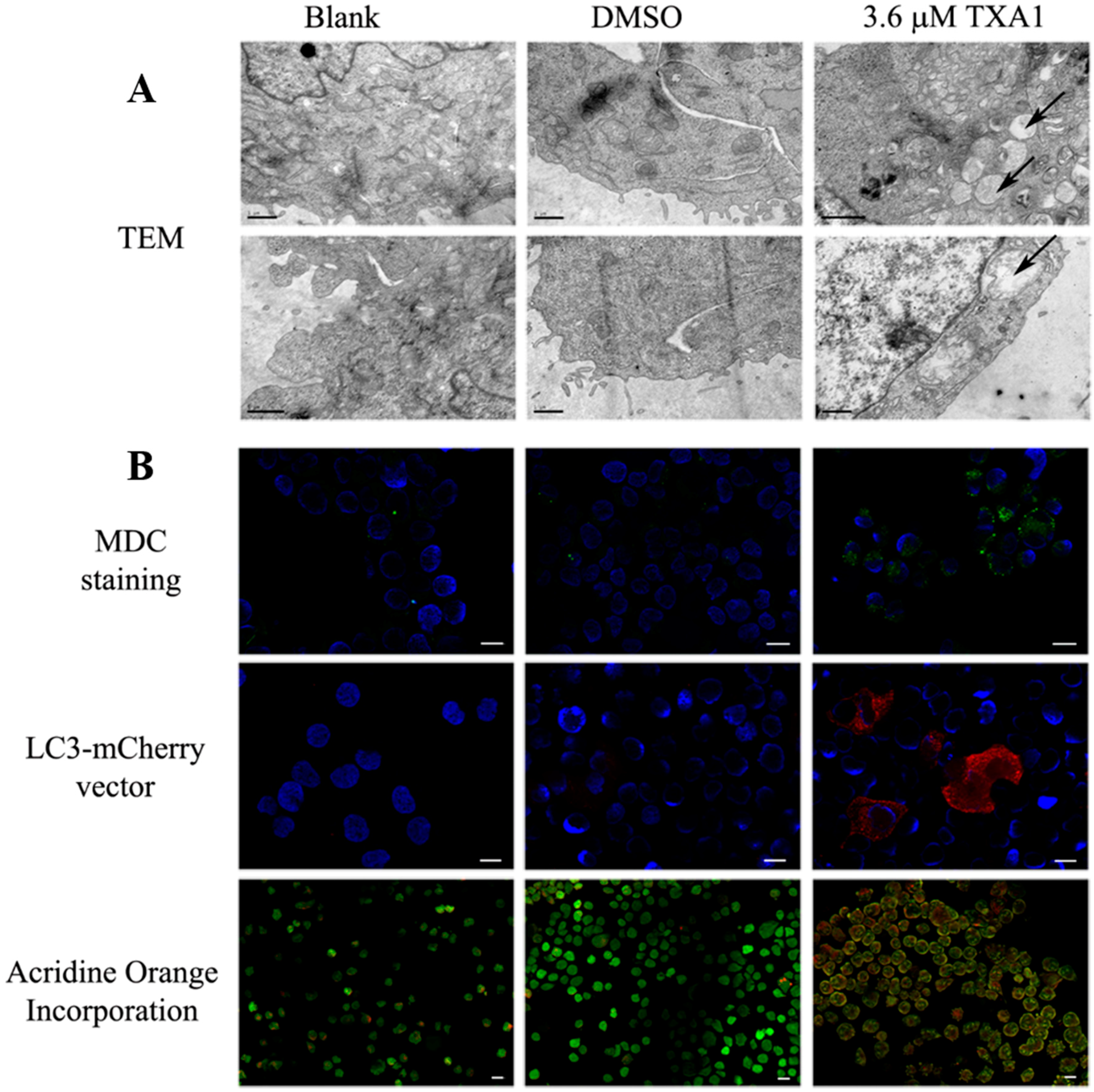
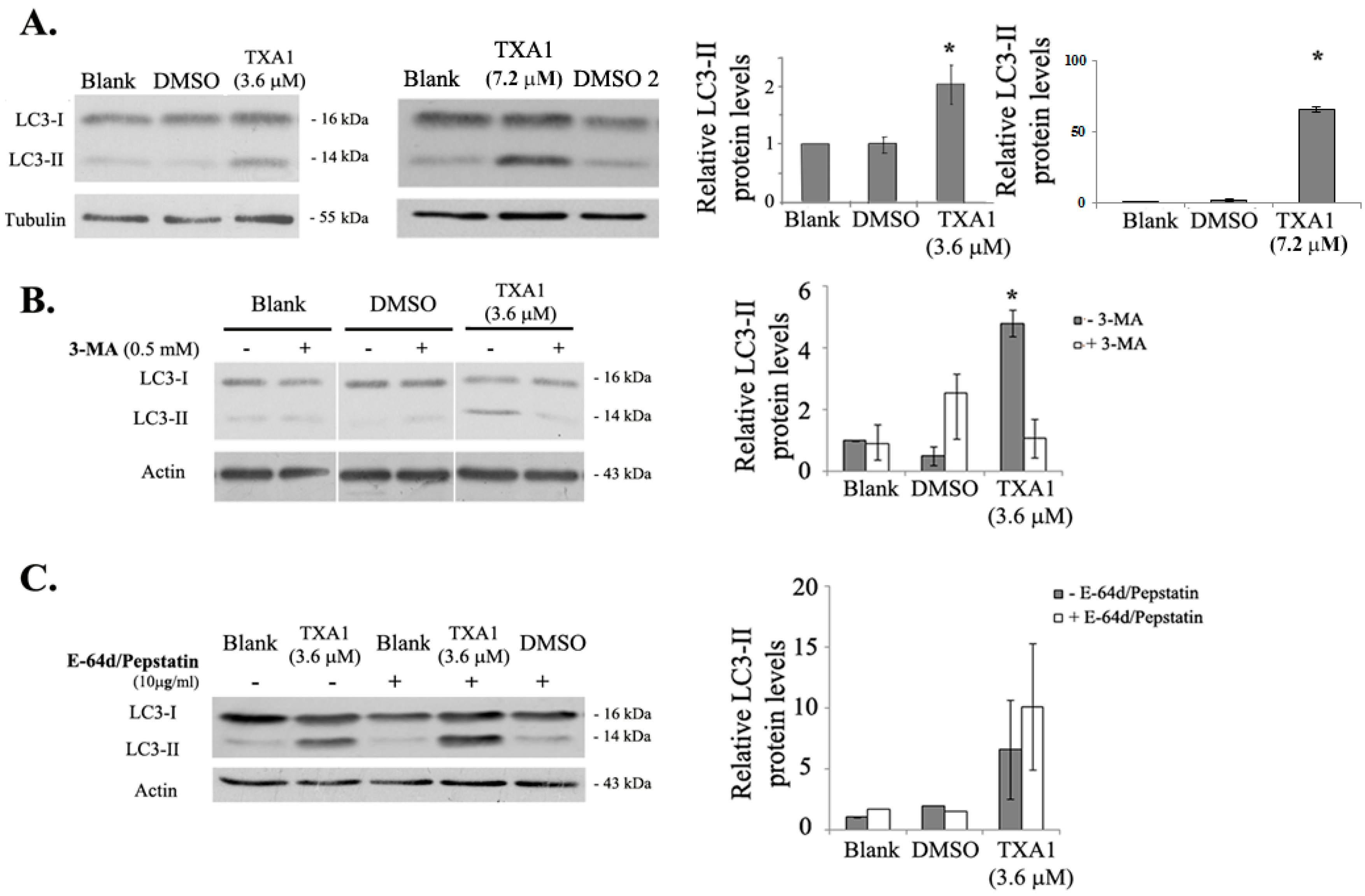
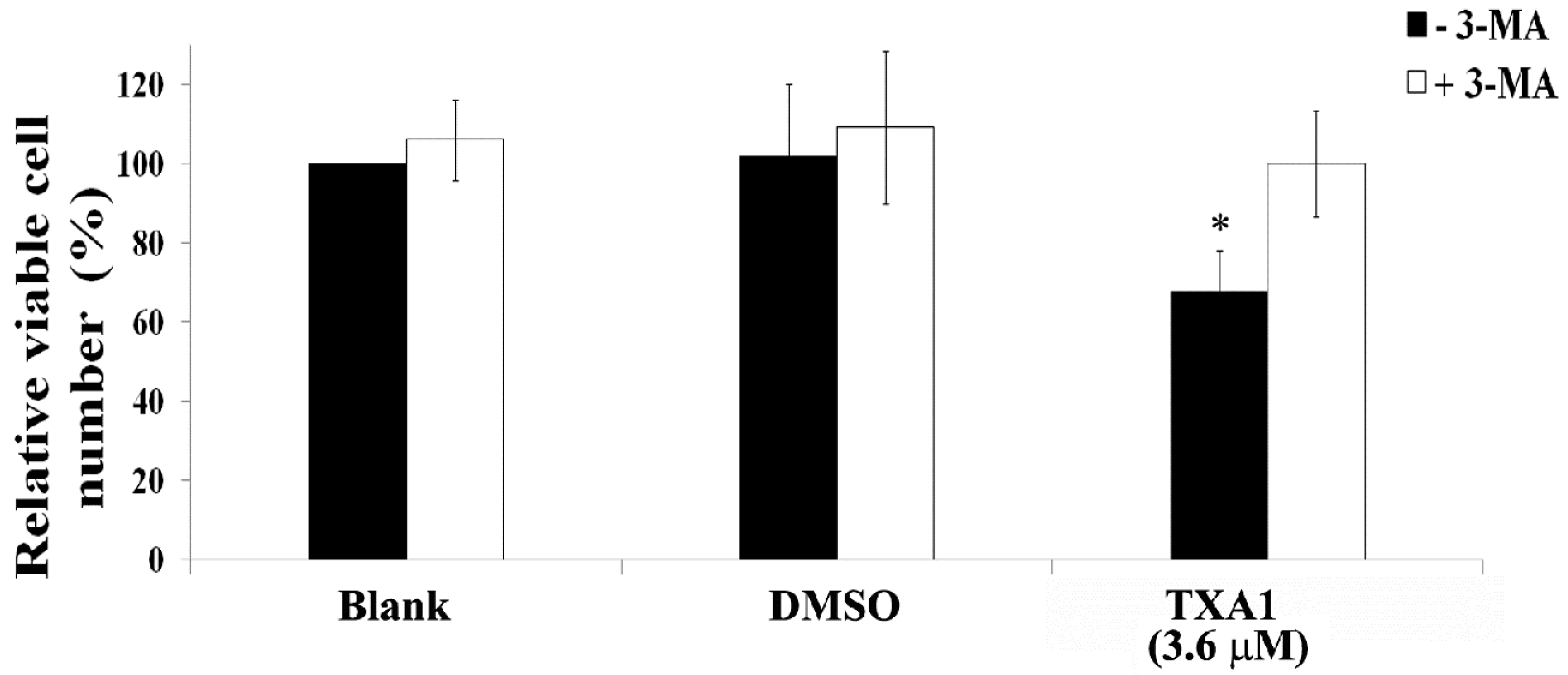
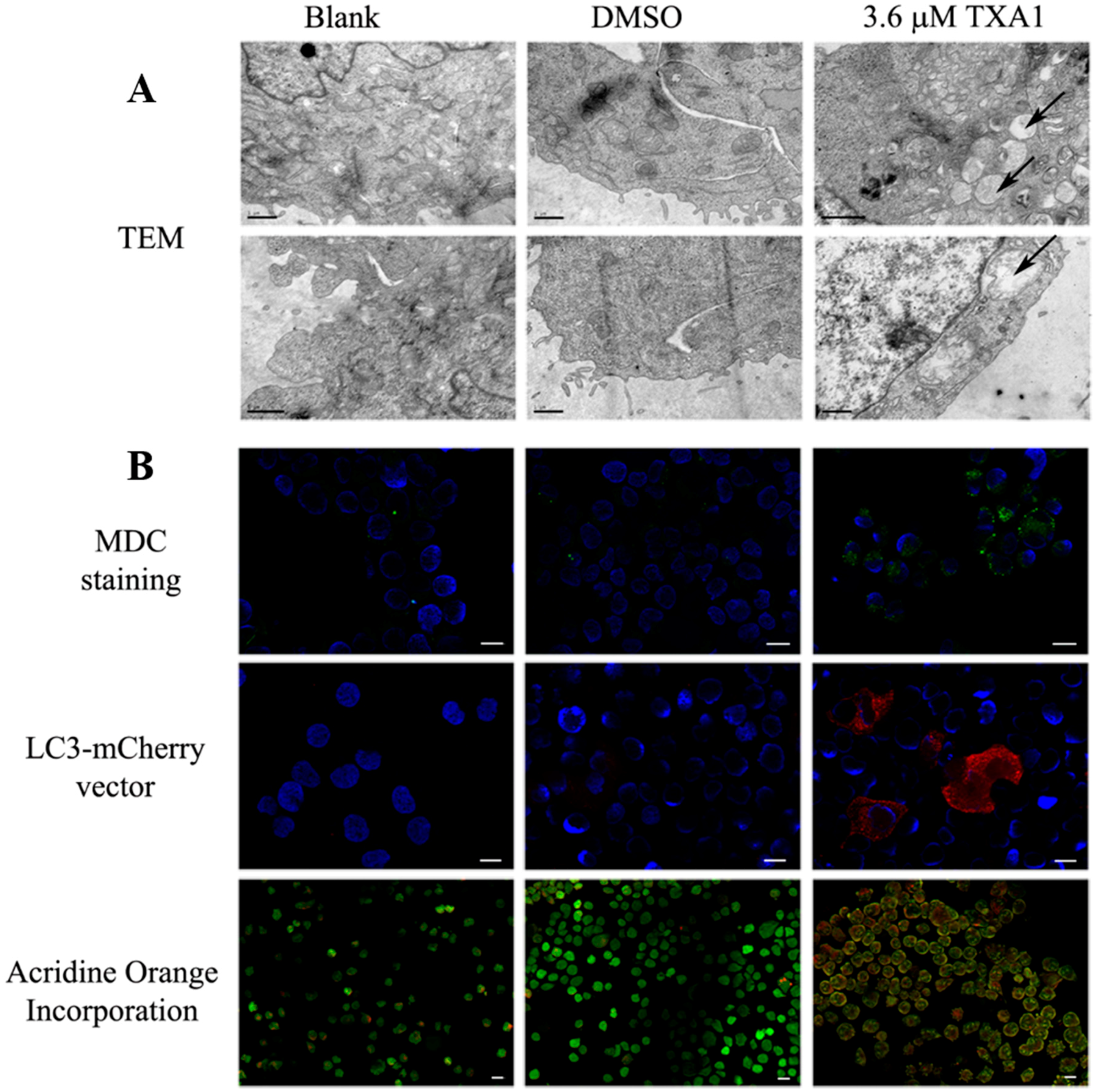
2.3. The GI50 Concentration of TXA1 Induces A375-C5 Cellular Autophagy
3. Discussion
4. Materials and Methods
4.1. Compound
4.2. Mapping of TXA1 onto Pharmacophores for Autophagy Induction
4.3. Cell Culture
4.4. Cell Treatment with TXA1
4.5. Cell Cycle Profile
4.6. Programmed Cell Death
4.7. Expression of Apoptotic and Autophagic Proteins
4.8. Monodansylcadaverine (MDC) and Acridine Orange Staining
4.9. Transfection with LC3-mCherry Expression Vector
4.10. Transmission Electron Microscopy
4.11. Treatment with Autophagy Inhibitors
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Boil. Ther. 2011, 11, 127–137. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Buttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Kimchi, A. Life in the balance—A mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci. 2012, 125, 5259–5268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.H.; Park, J.; Shin, I. Autophagy-regulating small molecules and their therapeutic applications. Chem. Soc. Rev. 2012, 41, 3245–3263. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Boil. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Kwon, H.J. Control of autophagy with small molecules. Arch. Pharm. Res. 2010, 33, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.W., Jr.; Tan, L.; Boitano, A.E.; Sorenson, D.R.; Aurora, A.; Liu, J.R. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004, 64, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Maffei, R.; Beau, I.; Codogno, P.; Ghidoni, R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell. Death Differ. 2008, 15, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Qin, Z.; Li, F.; Zhang, H.; Fang, Z.; Hao, E. Apoptotic Cell Death Induced by Resveratrol Is Partially Mediated by the Autophagy Pathway in Human Ovarian Cancer Cells. PLoS ONE 2015, 10, e0129196. [Google Scholar] [CrossRef] [PubMed]
- Ertmer, A.; Huber, V.; Gilch, S.; Yoshimori, T.; Erfle, V.; Duyster, J.; Elsasser, H.P.; Schatzl, H.M. The anticancer drug imatinib induces cellular autophagy. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund UK 2007, 21, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investing. 2009, 119, 1359–1372. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kismali, G.; Aggarwal, B.B. Curcumin, a component of turmeric: From farm to pharmacy. BioFactors 2013, 39, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.Z.; Xu, S.L.; Sun, G.C.; Chen, X.B. Novel curcumin analogue IHCH exhibits potent antiproliferative effects by inducing autophagy in A549 lung cancer cells. Mol. Med. Rep. 2014, 10, 441–446. [Google Scholar] [PubMed]
- Qu, W.; Xiao, J.; Zhang, H.; Chen, Q.; Wang, Z.; Shi, H.; Gong, L.; Chen, J.; Liu, Y.; Cao, R.; et al. B19, a novel monocarbonyl analogue of curcumin, induces human ovarian cancer cell apoptosis via activation of endoplasmic reticulum stress and the autophagy signaling pathway. Int. J. Biol. Sci. 2013, 9, 766–777. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.Z.; Zhang, S.N.; Zhang, L.; Sun, G.C.; Chen, X.B. A synthetic curcumin derivative hydrazinobenzoylcurcumin induces autophagy in A549 lung cancer cells. Pharm. Boil. 2014, 52, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, S.; Chan, D.A.; Sutphin, P.D.; Hay, M.P.; Denny, W.A.; Giaccia, A.J. A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 2008, 14, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, A.; Hahm, E.R.; Xiao, D.; Powolny, A.A.; Fisher, A.L.; Jiang, Y.; Singh, S.V. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009, 69, 3704–3712. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of P-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharm. 2012, 83, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.; Lima, R.T.; Sousa, D.; Gomes, A.S.; Palmeira, A.; Seca, H.; Choosang, K.; Pakkong, P.; Bousbaa, H.; Pinto, M.M.; et al. Screening a Small Library of Xanthones for Antitumor Activity and Identification of a Hit Compound which Induces Apoptosis. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Carew, J.S.; Espitia, C.M.; Esquivel, J.A.; Mahalingam, D.; Kelly, K.R.; Reddy, G.; Giles, F.J.; Nawrocki, S.T. Lucanthone is a novel inhibitor of autophagy that induces cathepsin d-mediated apoptosis. J. Biol. Chem. 2011, 286, 6602–6613. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, A.S.; Miller, J.; Arrasate, M.; Wong, J.S.; Pleiss, M.A.; Finkbeiner, S. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc. Natl. Acad. Sci. USA 2010, 107, 16982–16987. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-Methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta. Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Fujiwara, K.; Bogler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, K.; Oral, O.; Gozuacik, D. Chapter 13—Induction of Autophagic Cell Death by Anticancer Agents. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2014; pp. 179–202. [Google Scholar]
- Li, X.; Fan, Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the beclin 1/hVps34 complex. Cancer Res. 2010, 70, 5942–5952. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Yang, P.M.; Shun, C.T.; Wu, M.S.; Weng, J.R.; Chen, C.C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Paiva, A.M.; Pinto, M.M.; Sousa, E. A century of thioxanthones: Through synthesis and biological applications. Curr. Med. Chem. 2013, 20, 2438–2457. [Google Scholar] [CrossRef] [PubMed]
- Naidu, M.D.; Agarwal, R.; Pena, L.A.; Cunha, L.; Mezei, M.; Shen, M.; Wilson, D.M.; Liu, Y.; Sanchez, Z.; Chaudhary, P.; et al. Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (APE1) by direct protein binding. PLoS ONE 2011, 6, e23679. [Google Scholar] [CrossRef] [PubMed]
- Safety and Efficacy Study of Lucanthone When Used in Combination With Temozolomide(TMZ) and Radiation to Treat Glioblastoma Multiforme(GBM). Available online: https://clinicaltrials.gov/ct2/show/NCT01587144 (accessed on 7 October 2016).
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [PubMed]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 294, F777–F787. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, D.; Wang, W.; Piao, S.; Zhou, J.; Saiyin, W.; Zheng, C.; Sun, H.; Li, Y. Inhibition of autophagy by 3-MA enhances IL-24-induced apoptosis in human oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 97. [Google Scholar] [CrossRef] [PubMed]
- Seca, H.; Lima, R.T.; Lopes-Rodrigues, V.; Guimarães, J.E.; Almeida, G.M.; Vasconcelos, M.H. Targeting miR-21 induces autophagy and chemosensitivity of leukemia cells. Curr. Drug Targets 2013, 14, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Y.; Yang, R.; Wang, H.J.; Huang, H.; Wu, D.; Tashiro, S.; Onodera, S.; Ikejima, T. Mechanism of autophagy induction and role of autophagy in antagonizing mitomycin C-induced cell apoptosis in silibinin treated human melanoma A375-S2 cells. Eur. J. Pharmacol. 2011, 659, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cheng, Y.; Zhang, B.; Bian, H.J.; Bao, J.K. Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett. 2009, 275, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. BioTechniques 1993, 14, 1010–1013. [Google Scholar] [PubMed]
- Zhang, L. A descent modified Polak-Ribière-Polyak conjugate gradient method and its global convergence. IMA J. Numer. Anal. 2006, 26, 11. [Google Scholar] [CrossRef]
- Abreu, R.M.V.; Ferreira, I.C.F.R.; Calhelha, R.C.; Lima, R.T.; Vasconcelos, M.H.; Adega, F.; Chaves, R.; Queiroz, M.J.R.P. Anti-hepatocellular carcinoma activity using human HepG2 cells and hepatotoxicity of 6-substituted methyl 3-aminothieno[3,2-bpyridine-2-carboxylate derivatives: In vitro evaluation, cell cycle analysis and QSAR studies. Eur. J. Med. Chem. 2011, 46, 5800–5806. [Google Scholar] [PubMed]
- Lima, R.T.; Martins, L.M.; Guimaraes, J.E.; Sambade, C.; Vasconcelos, M.H. Specific downregulation of bcl-2 and xIAP by RNAi enhances the effects of chemotherapeutic agents in MCF-7 human breast cancer cells. Cancer Gene Ther. 2004, 11, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.J.; Calhelha, R.C.; Vale-Silva, L.A.; Pinto, E.; Lima, R.T.; Vasconcelos, M.H. Efficient synthesis of 6-(hetero)arylthieno[3,2-b]pyridines by Suzuki-Miyaura coupling. Evaluation of growth inhibition on human tumor cell lines, SARs and effects on the cell cycle. Eur. J. Med. Chem. 2010, 45, 5628–5634. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.J.; Calhelha, R.C.; Vale-Silva, L.A.; Pinto, E.; Almeida, G.M.; Vasconcelos, M.H. Synthesis and evaluation of tumor cell growth inhibition of methyl 3-amino-6-[(hetero)arylethynyl]thieno[3,2-b]pyridine-2-carboxylates. Structure-activity relationships, effects on the cell cycle and apoptosis. Eur. J. Med. Chem. 2011, 46, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Barron, G.A.; Grabowska, J.A.; Bermano, G.; Kaur, S.; Roy, N.; Vasconcelos, M.H.; Lin, P.K. Cytotoxicity and cell death mechanisms induced by a novel bisnaphthalimidopropyl derivative against the NCI-H460 non-small lung cancer cell line. Anti-Cancer Agents Med. Chem. 2013, 13, 414–421. [Google Scholar]
- Preto, A.; Goncalves, J.; Rebocho, A.P.; Figueiredo, J.; Meireles, A.M.; Rocha, A.S.; Vasconcelos, H.M.; Seca, H.; Seruca, R.; Soares, P.; et al. Proliferation and survival molecules implicated in the inhibition of BRAF pathway in thyroid cancer cells harbouring different genetic mutations. BMC Cancer 2009, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Birame, B.M.; Jigui, W.; Fuxian, Y.; Shuang, W.; Li, Y.D.; Jiazeng, S.; Zhili, L.; Bao, Y.; Weiquan, L. Co-expression of apoptin (VP3) and antibacterial peptide cecropin B mutant (ABPS1) genes induce higher rate of apoptosis in HepG2 and A375 cell lines. Afr. J. Biotechnol. 2012, 11, 8. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compound TXA1 are available from the authors.




© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, R.T.; Sousa, D.; Paiva, A.M.; Palmeira, A.; Barbosa, J.; Pedro, M.; Pinto, M.M.; Sousa, E.; Vasconcelos, M.H. Modulation of Autophagy by a Thioxanthone Decreases the Viability of Melanoma Cells. Molecules 2016, 21, 1343. https://doi.org/10.3390/molecules21101343
Lima RT, Sousa D, Paiva AM, Palmeira A, Barbosa J, Pedro M, Pinto MM, Sousa E, Vasconcelos MH. Modulation of Autophagy by a Thioxanthone Decreases the Viability of Melanoma Cells. Molecules. 2016; 21(10):1343. https://doi.org/10.3390/molecules21101343
Chicago/Turabian StyleLima, Raquel T., Diana Sousa, Ana M. Paiva, Andreia Palmeira, João Barbosa, Madalena Pedro, Madalena M. Pinto, Emília Sousa, and M. Helena Vasconcelos. 2016. "Modulation of Autophagy by a Thioxanthone Decreases the Viability of Melanoma Cells" Molecules 21, no. 10: 1343. https://doi.org/10.3390/molecules21101343
APA StyleLima, R. T., Sousa, D., Paiva, A. M., Palmeira, A., Barbosa, J., Pedro, M., Pinto, M. M., Sousa, E., & Vasconcelos, M. H. (2016). Modulation of Autophagy by a Thioxanthone Decreases the Viability of Melanoma Cells. Molecules, 21(10), 1343. https://doi.org/10.3390/molecules21101343