
Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization

Abstract
:
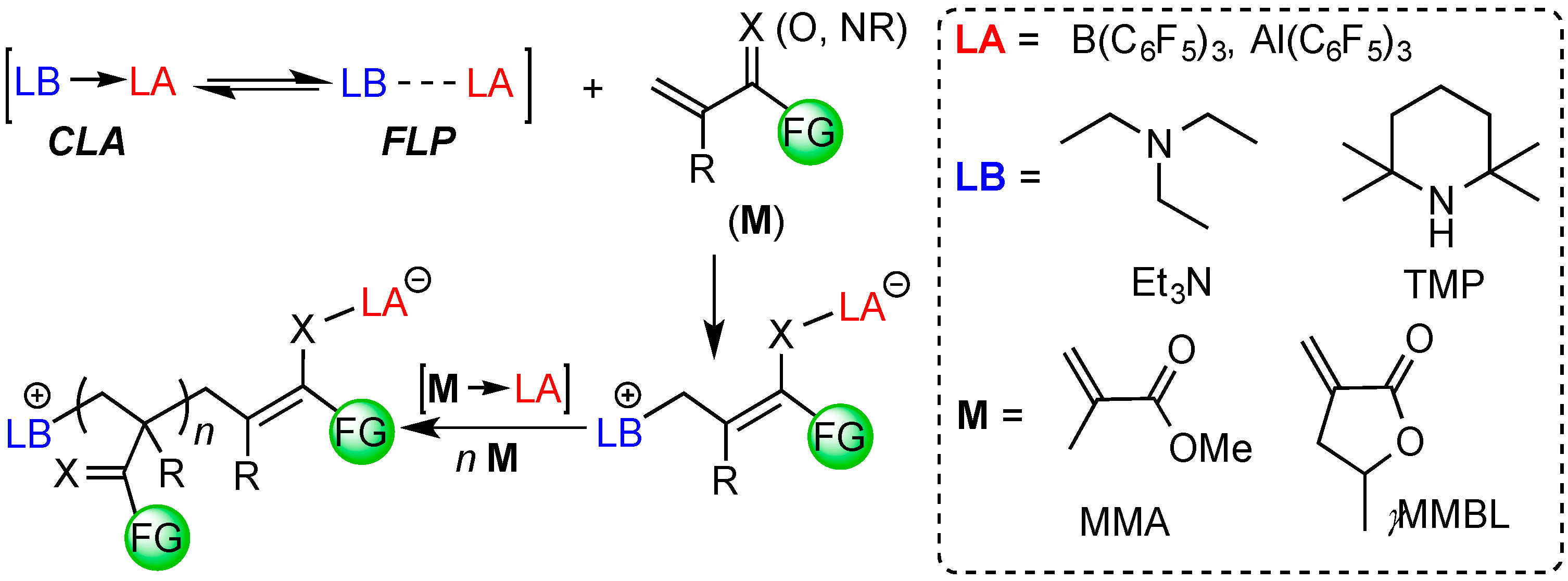
1. Introduction
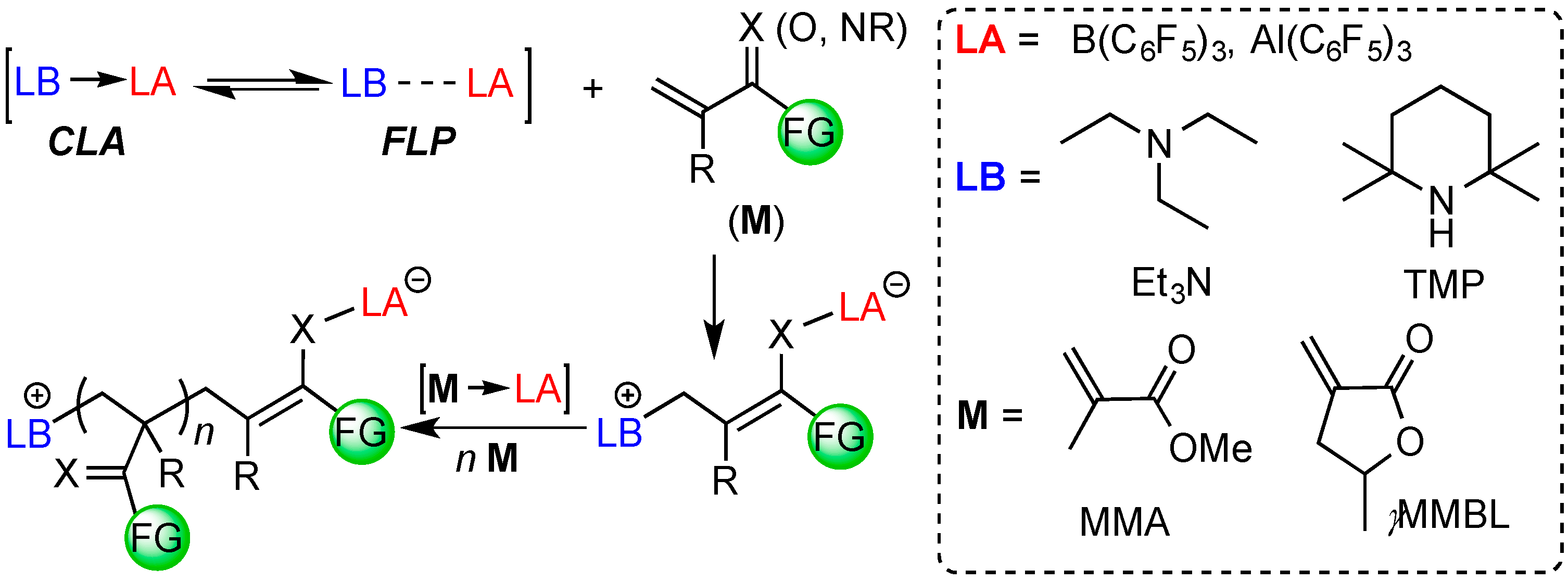
2. Results and Discussion
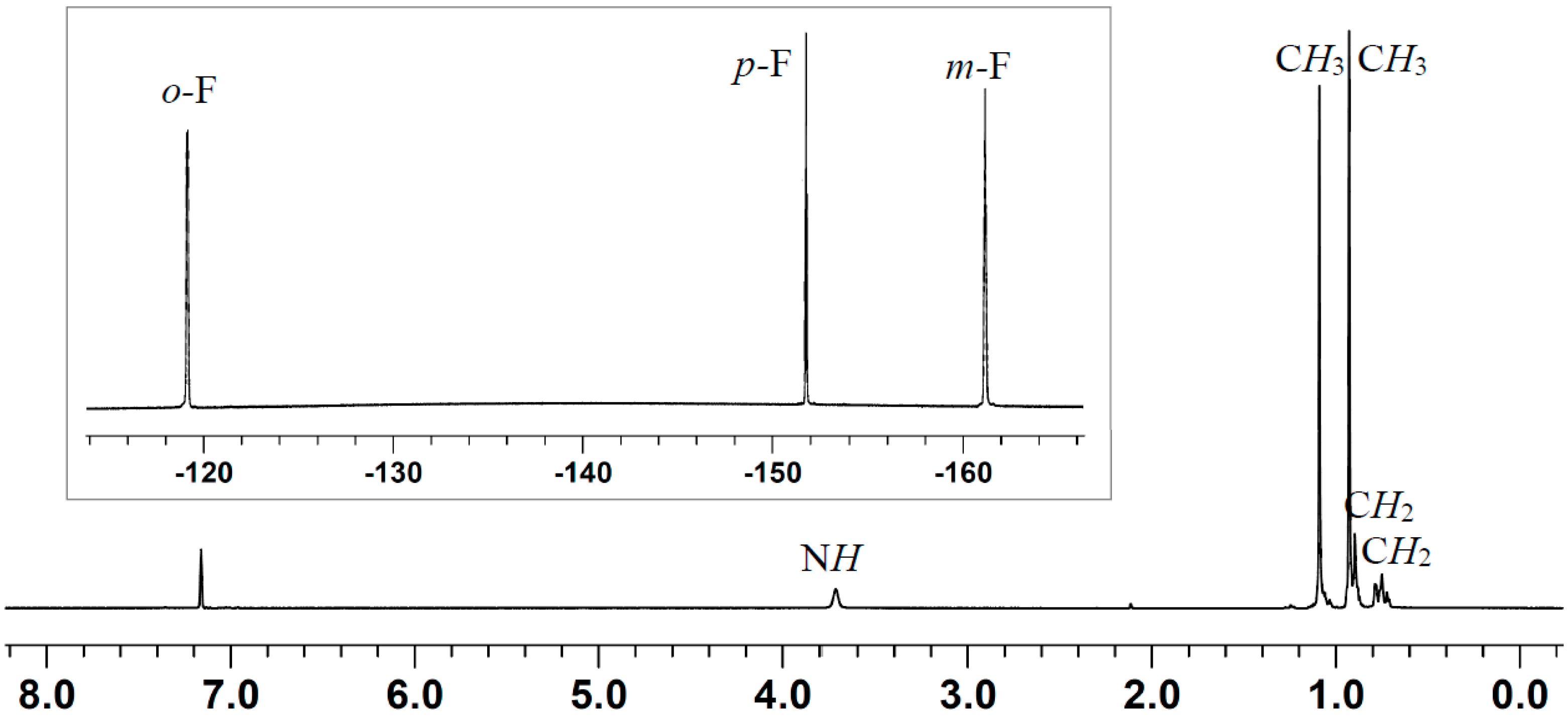
2.1. Reaction of Amines and E(C6F5)3 in the Absence of Monomer
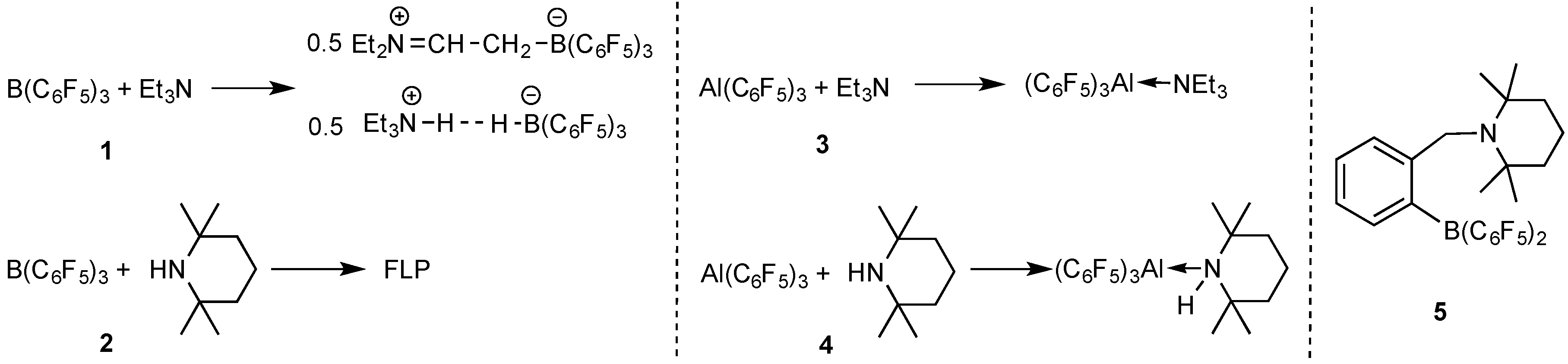
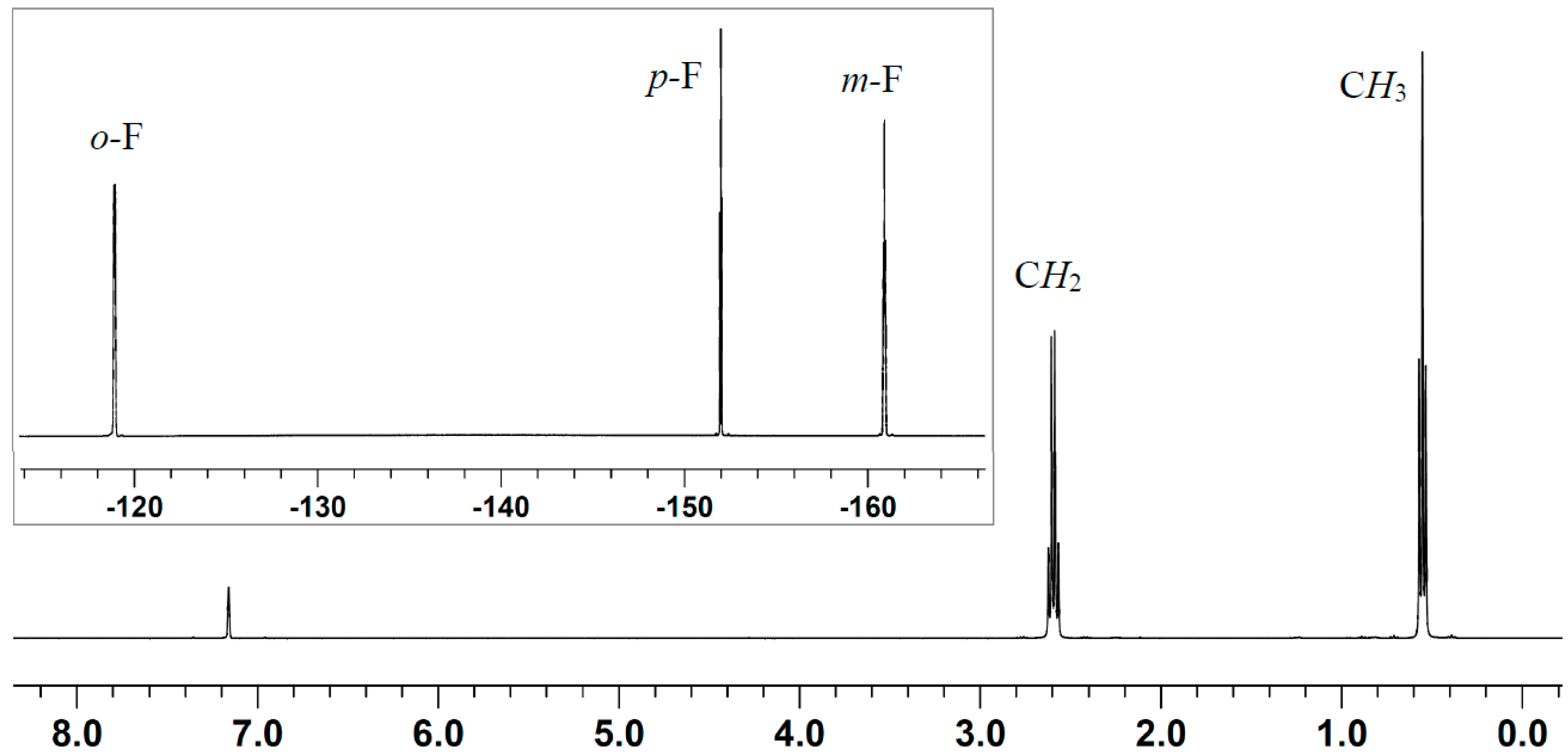
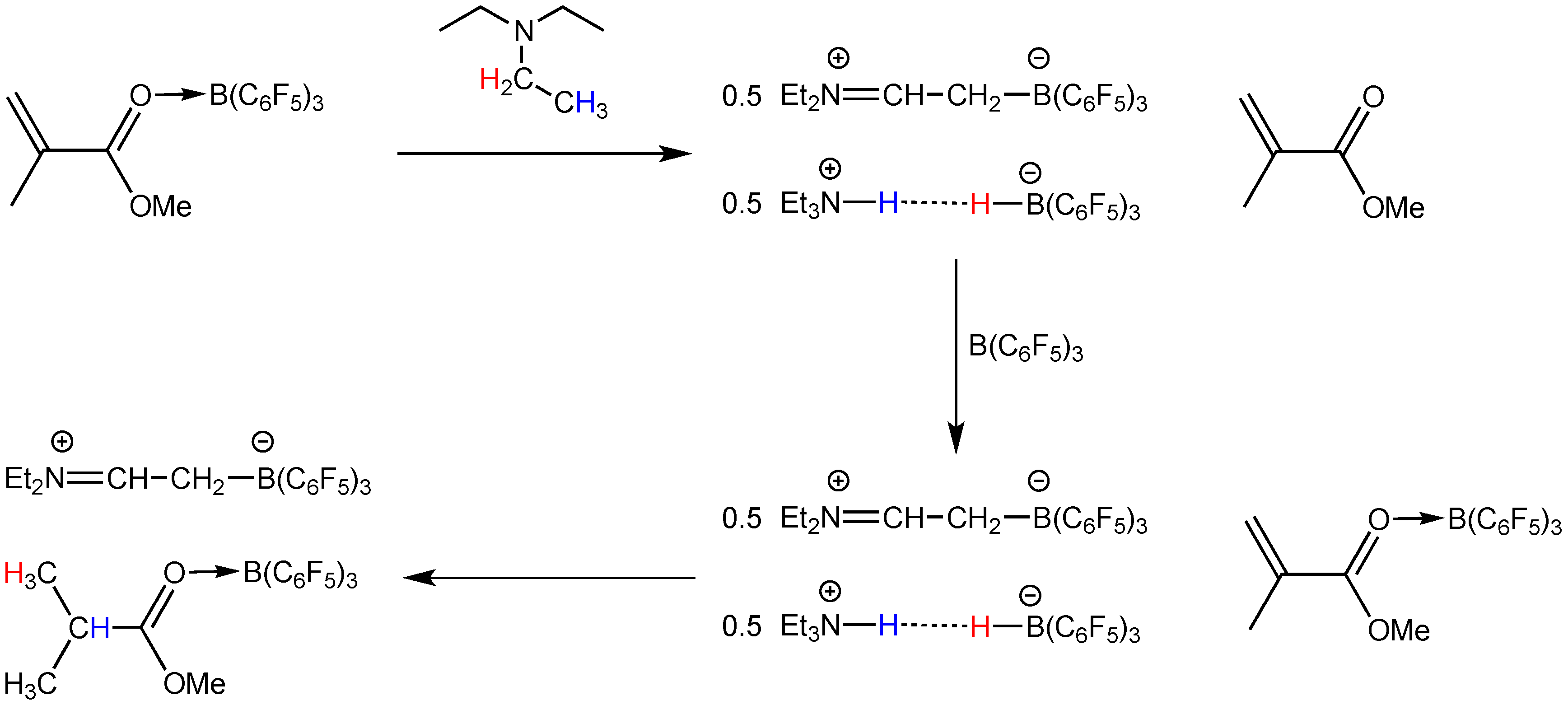
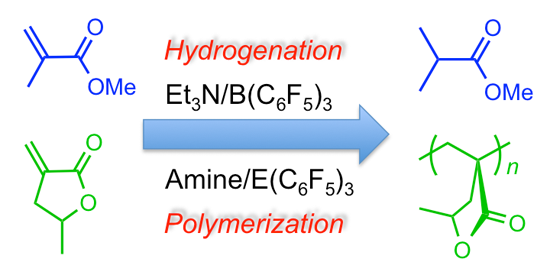
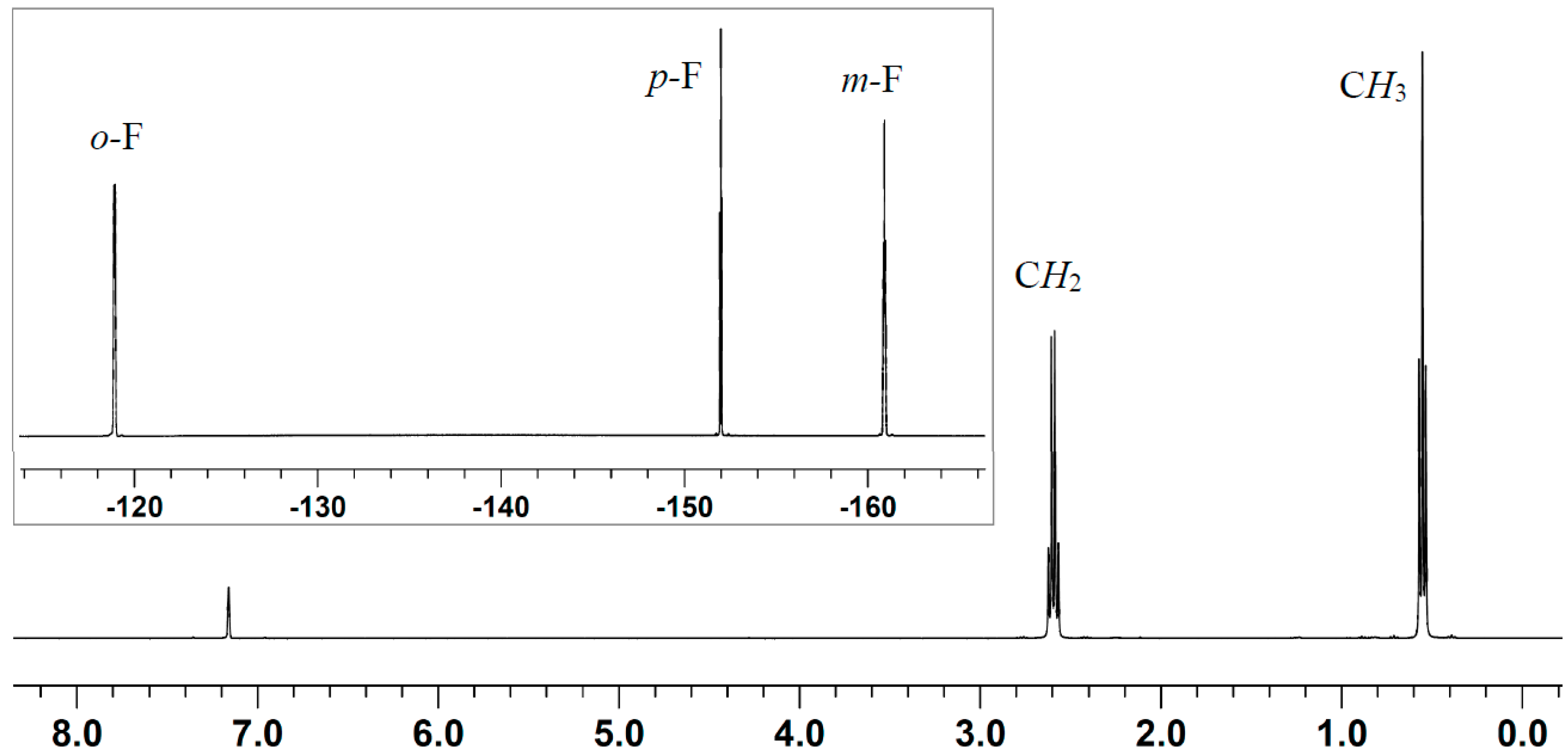
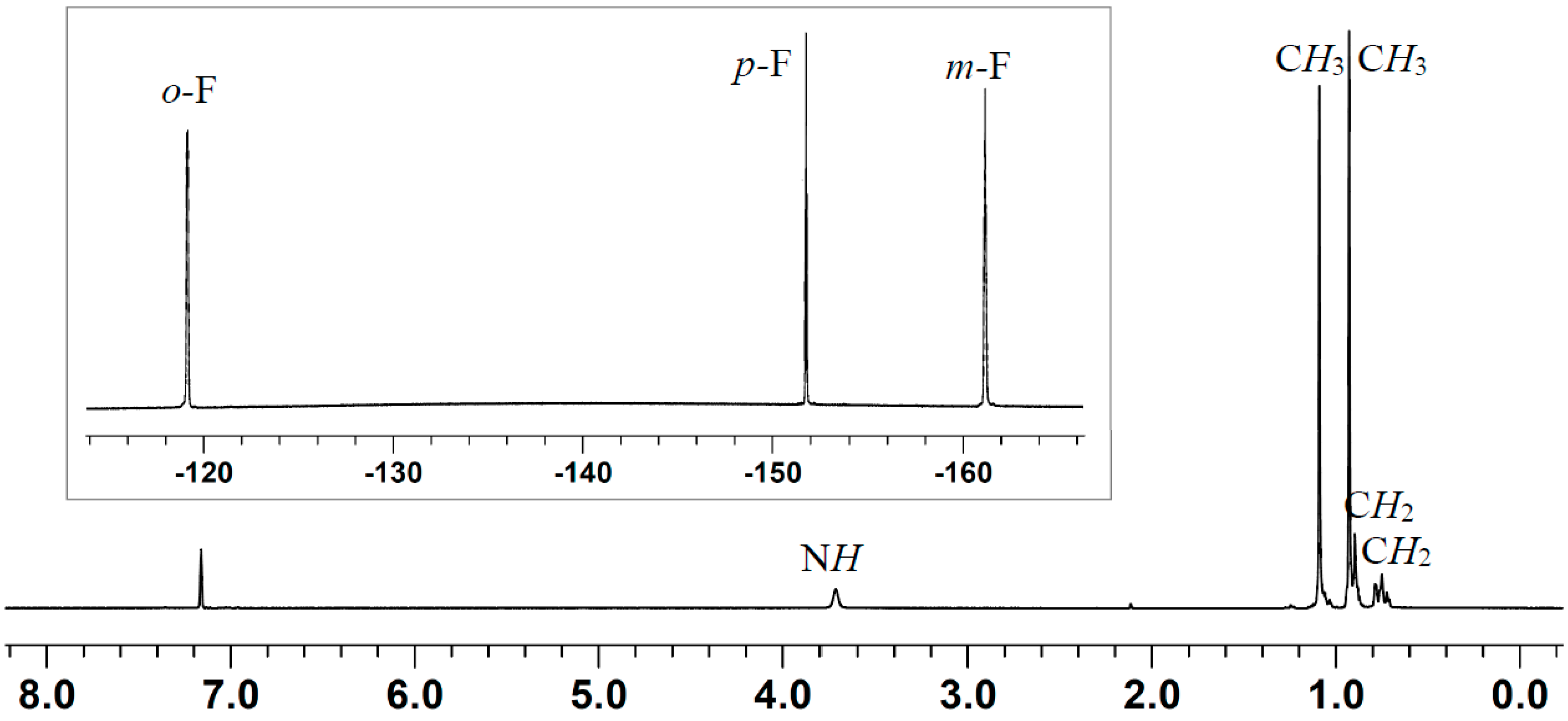
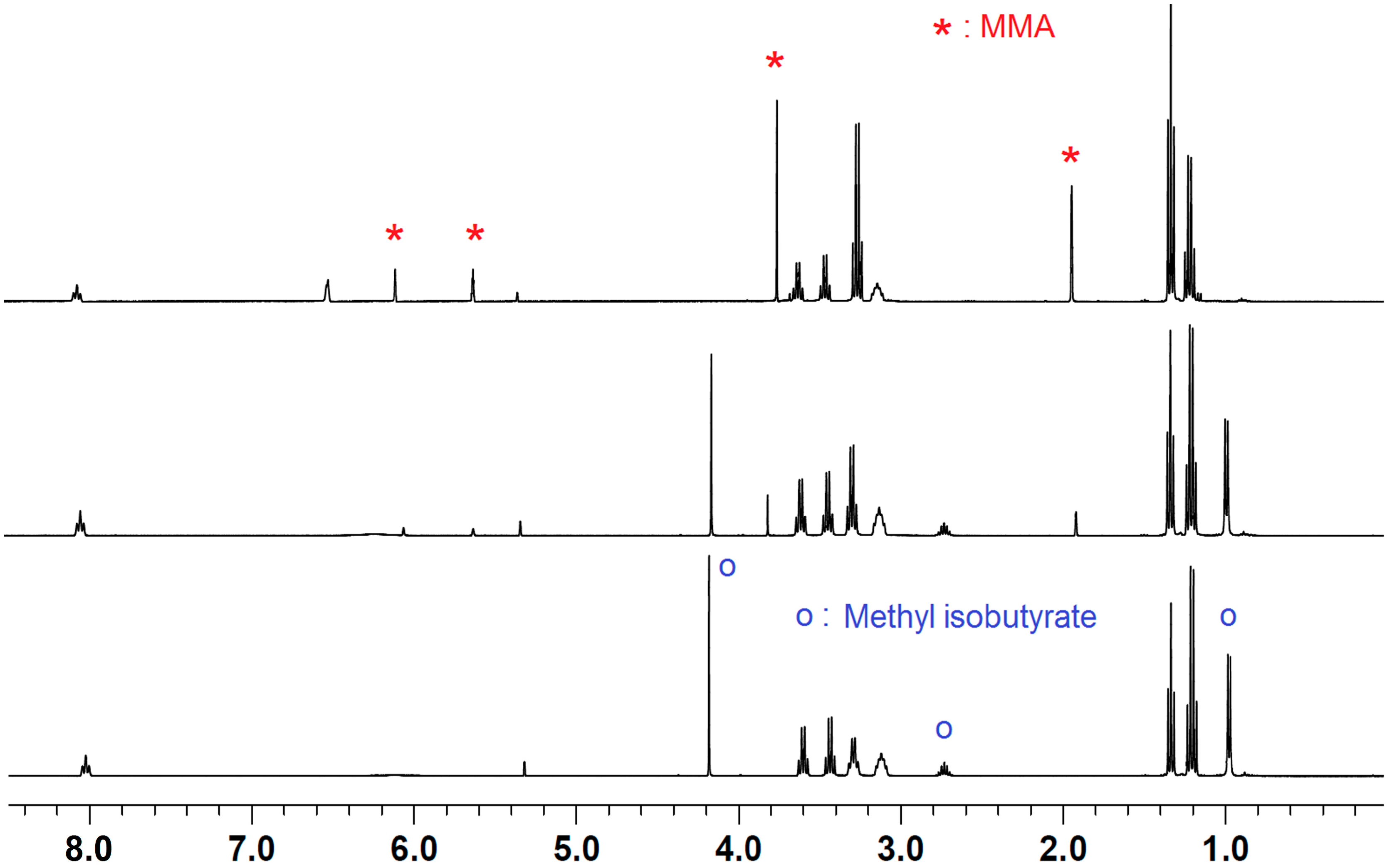
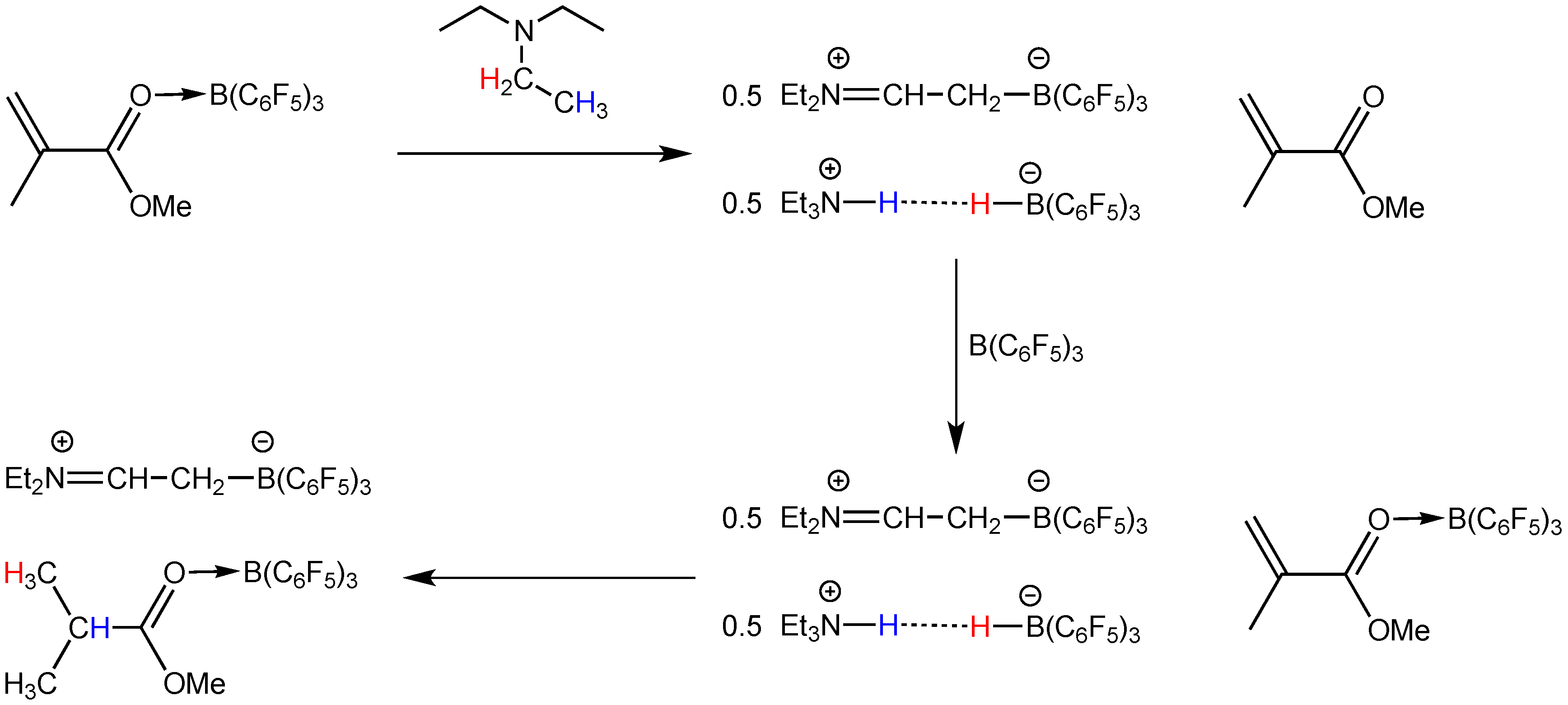
2.2. Stoichiometric Hydrogenation of MMA to Methyl Isobutyrate by Et3N/B(C6F5)3

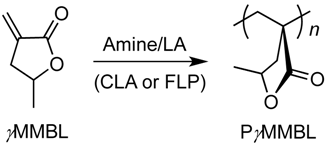
2.3. Characteristic of γMMBL Polymerization by Amine/E(C6F5)3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
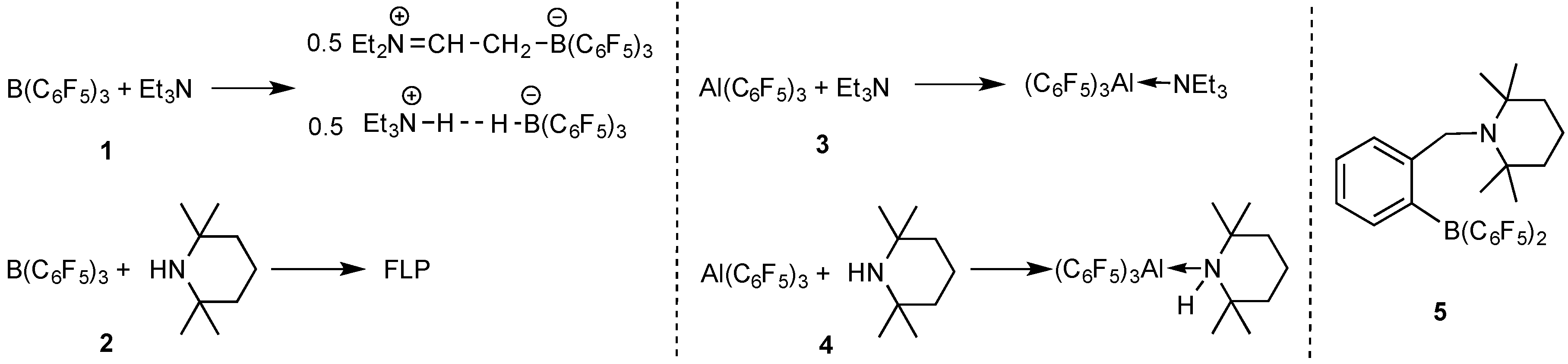{kind=link}
{kind=link}
| Entry | LP | [M]/[Base] | Time (min) | Conv. b (%) | TOF (h-1) | Mnc (kg∙mol−1) | Đ d (Mw/Mn) |
|---|---|---|---|---|---|---|---|
| 1 | B/Et3N (1) | 200 | 1 | 100 | 12,000 | 31.2 | 2.31 |
| 2 | 800 | 60 | 100 | 800 | 53.1 | 2.47 | |
| 3 | Al/Et3N (3) | 200 | 0.5 | 100 | 24,000 | 19.2 | 2.02 |
| 4 | 800 | 30 | 100 | 1600 | 33.0 | 2.26 | |
| 5 | B/TMP (2) | 50 | 0.5 | 100 | 6000 | 67.5 d | 1.54 |
| 6 | 100 | 0.5 | 100 | 12,000 | 100 d | 1.64 | |
| 7 | 200 | 0.5 | 100 | 24,000 | 145 d | 1.78 | |
| 8 | 400 | 0.5 | 100 | 48,000 | 156 d | 1.92 | |
| 9 | 800 | 1 | 100 | 48,000 | 253 d | 1.64 | |
| 10 | 1600 | 3 | 100 | 32,000 | 255 d | 1.66 | |
| 11 | Al/TMP (4) | 50 | 0.5 | 100 | 6000 | 69.6 | 2.41 |
| 12 | 100 | 0.5 | 100 | 12,000 | 77.2 | 2.29 | |
| 13 | 200 | 0.5 | 100 | 24,000 | 113 | 2.16 | |
| 14 | 400 | 0.5 | 100 | 48,000 | 119 | 2.38 | |
| 15 | 800 | 0.5 | 100 | 96,000 | 129 | 2.21 | |
| 16 | 1600 | 3 | 100 | 32,000 | 138 | 2.16 | |
| 17 | B-TMP (5) | 200 | 1440 | 0 | - | - | - |
| 18 e | 5 + B | 200 | 1440 | 0 | - | - | - |
3. Experimental Section
3.1. Materials, Reagents, and Methods
3.2. Isolation of Adduct Et3N·Al(C6F5)3
3.3. Isolation of Adduct TMP·Al(C6F5)3
3.4. Stoichiometric Hydrogenation of MMA by Et3N/2B(C6F5)3 to Methyl Isobutyrate
3.5. Typical Procedure for the NMR Scale Reactions (TMP/B(C6F5)3/MMA as an Example)
3.6. General Polymerization Procedures
3.7. Polymer Characterizations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stephan, D.W. Frustrated Lewis pairs: From concept to catalysis. Acc. Chem. Res. 2015, 48, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry of carbon, nitrogen and sulfur oxides. Chem. Sci. 2014, 5, 2625–2641. [Google Scholar] [CrossRef]
- Erker, G.; Stephan, D.W. (Eds.) Frustrated Lewis Pairs I & II; Springer-Verlag: Berlin, Germany, 2013.
- Stephan, D.W. “Frustrated Lewis pair” hydrogenations. Org. Biomol. Chem. 2012, 10, 5740–5746. [Google Scholar] [CrossRef] [PubMed]
- Erker, G. Frustrated Lewis pairs: Some recent developments. Pure Appl. Chem. 2012, 84, 2203–2217. [Google Scholar] [CrossRef]
- Erker, G. Organometallic frustrated Lewis pair chemistry. Dalton Trans. 2011, 40, 7475–7483. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis pairs: Metal-free hydrogen activation and more. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis pairs: A new strategy to small molecule activation and hydro-genation catalysis. Dalton Trans. 2009, 17, 3129–3136. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. “Frustrated Lewis pairs”: A concept for new reactivity and catalysis. Org. Biomol. Chem. 2008, 6, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, E.J.; Oganesyan, V.S.; Hughes, D.L.; Ashley, A.E.; Wildgoose, G.G. An electrochemical study of frustrated Lewis pairs: A metal-free route to hydrogen oxidation. J. Am. Chem. Soc. 2014, 136, 6031–6036. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Elmer, L.M.; Rosorius, C.; Daniliuc, C.G.; Grimme, S.; Kehr, G.; Erker, G. Facile carbon monoxide reduction at intramolecular frustrated phosphane/borane Lewis pair templates. Angew. Chem. Int. Ed. 2013, 52, 2243–2246. [Google Scholar] [CrossRef] [PubMed]
- Dobrovetsky, R.; Stephan, D.W. Stoichiometric metal-free reduction of CO in syn-gas. J. Am. Chem. Soc. 2013, 135, 4974–4977. [Google Scholar] [CrossRef] [PubMed]
- Appelt, C.; Slootweg, J.C.; Lammertsma, K.; Uhl, W. Reaction of a P/Al-based frustrated Lewis pair with ammonia, borane, and amine-boranes: Adduct formation and catalytic dehydrogenation. Angew. Chem. Int. Ed. 2013, 52, 4256–4259. [Google Scholar] [CrossRef] [PubMed]
- Bertini, F.; Lyaskoyskyy, V.; Timmer, B.J.J.; de Kanter, F.J.J.; Lutz, M.; Ehlers, A.W.; Slootweg, J.C.; Lammertsma, K. Preorganized frustrated Lewis pairs. J. Am. Chem. Soc. 2012, 134, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Reissmann, M.; Schafer, A.; Saak, W.; Haase, D.; Muller, T. A new synthesis of triarylsilylium ions and their application in dihydrogen activation. Angew. Chem. Int. Ed. 2011, 50, 12636–12638. [Google Scholar] [CrossRef] [PubMed]
- Marwitz, A.J.V.; Dutton, J.L.; Mercier, L.G.; Piers, W.E. Dihydrogen activation with tBu3P/B(C6F5)3: A chemically competent indirect mechanism via in situ-generated p-tBu2P-C6F4-B(C6F5)2. J. Am. Chem. Soc. 2011, 133, 10026–10029. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.P.; Cheng, Z.H.; Chen, Z.X.; Weng, L.H.; Li, Z.H.; Wang, H.D. Heterolytic cleavage of dihydrogen by “frustrated Lewis pairs” comprising bis(2,4,6-tris(trifluoromethyl)phenyl)borane and amines: Stepwise versus concerted mechanism. Angew. Chem. Int. Ed. 2011, 50, 12227–12231. [Google Scholar] [CrossRef] [PubMed]
- Ekkert, O.; Kehr, G.; Frohlich, R.; Erker, G. P-C bond activation chemistry: Evidence for 1,1-carboboration reactions proceeding with phosphorus-carbon bond cleavage. J. Am. Chem. Soc. 2011, 133, 4610–4616. [Google Scholar] [CrossRef] [PubMed]
- Appelt, C.; Westenberg, H.; Bertini, F.; Ehlers, A.W.; Slootweg, J.C.; Lammertsma, K.; Uhl, W. Geminal phosphorus/aluminum-based frustrated Lewis pairs: C-H versus C equivalent to C activation and CO2 fixation. Angew. Chem. Int. Ed. 2011, 50, 3925–3928. [Google Scholar]
- Ines, B.; Holle, S.; Goddard, R.; Alcarazo, M. Heterolytic S-S bond cleavage by a purely carbogenic frustrated Lewis pair. Angew. Chem. Int. Ed. 2010, 49, 8389–8391. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Kruse, H.; Goerigk, L.; Erker, G. The mechanism of dihydrogen activation by frustrated Lewis pairs revisited. Angew. Chem. Int. Ed. 2010, 49, 1402–1405. [Google Scholar] [CrossRef] [PubMed]
- Momming, C.M.; Otten, E.; Kehr, G.; Frohlich, R.; Grimme, S.; Stephan, D.W.; Erker, G. Reversible metal-free carbon dioxide binding by frustrated Lewis pairs. Angew. Chem. Int. Ed. 2009, 48, 6643–6646. [Google Scholar] [CrossRef] [PubMed]
- Holschumacher, D.; Bannenberg, T.; Hrib, C.G.; Jones, P.G.; Tamm, M. Heterolytic dihydrogen activation by a frustrated carbene-borane Lewis pair. Angew. Chem. Int. Ed. 2008, 47, 7428–7432. [Google Scholar]
- Chase, P.A.; Stephan, D.W. Hydrogen and amine activation by a frustrated Lewis pair of a bulky N-heterocyclic carbene and B(C6F5)3. Angew. Chem. Int. Ed. 2008, 47, 7433–7437. [Google Scholar] [CrossRef] [PubMed]
- Sumerin, V.; Schulz, F.; Nieger, M.; Leskelä, M.; Repo, T.; Rieger, B. Facile heterolytic H2 activation by amines and B(C6F5)3. Angew. Chem. Int. Ed. 2008, 47, 6001–6003. [Google Scholar] [CrossRef] [PubMed]
- Hounjet, L.J.; Bannwarth, C.; Garon, C.N.; Caputo, C.B.; Grimme, S.; Stephan, D.W. Combinations of ethers and B(C6F5)3 function as hydrogenation catalysts. Angew. Chem. Int. Ed. 2013, 52, 7492–7495. [Google Scholar] [CrossRef] [PubMed]
- Greb, L.; Daniliuc, C.G.; Bergander, K.; Paradies, J. Functional-group tolerance in frustrated Lewis pairs: Hydrogenation of nitroolefins and acrylates. Angew. Chem. Int. Ed. 2013, 52, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Chernichenko, K.; Madarasz, A.; Papai, I.; Nieger, M.; Leskela, M.; Repo, T. A frustrated-Lewis-pair approach to catalytic reduction of alkynes to cis-alkenes. Nat. Chem. 2013, 5, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.M.; Labinger, J.A.; Bercaw, J.E. Homogeneous CO hydrogenation: Dihydrogen activation involves a frustrated Lewis pair instead of a platinum complex. J. Am. Chem. Soc. 2010, 132, 3301–3303. [Google Scholar] [CrossRef] [PubMed]
- Eros, G.; Mehdi, H.; Papai, I.; Rokob, T.A.; Kiraly, P.; Tarkanyi, G.; Soos, T. Expanding the scope of metal-free catalytic hydrogenation through frustrated Lewis pair design. Angew. Chem. Int. Ed. 2010, 49, 6559–6563. [Google Scholar] [CrossRef] [PubMed]
- Axenov, K.V.; Kehr, G.; Frohlich, R.; Erker, G. Catalytic hydrogenation of sensitive organometallic compounds by antagonistic N/B Lewis pair catalyst systems. J. Am. Chem. Soc. 2009, 131, 3454–3455. [Google Scholar] [CrossRef] [PubMed]
- Ashley, A.E.; Thompson, A.L.; O’Hare, D. Non-metal-mediated homogeneous hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2009, 48, 9839–9843. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Kehr, G.; Daniliuc, C.G.; Erker, G. Internal adduct formation of active intramolecular C-4-bridged frustrated phosphane/borane Lewis pairs. J. Am. Chem. Soc. 2014, 136, 3293–3303. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Ciancaleoni, G.; Zuccaccia, C.; Macchioni, A. Probing the association of frustrated phosphine-borane Lewis pairs in solution by NMR spectroscopy. J. Am. Chem. Soc. 2014, 136, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Henthorn, J.T.; Agapie, T. Dioxygen reactivity with a ferrocene-Lewis acid pairing: Reduction to a boron peroxide in the presence of tris(pentafluorophenyl)borane. Angew. Chem. Int. Ed. 2014, 53, 12893–12896. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Kehr, G.; Wiegand, T.; Eckert, H.; Schwickert, C.; Pottgen, R.; Cardenas, A.J.P.; Warren, T.H.; Frohlich, R.; Daniliuc, C.G.; et al. Noninteracting, vicinal frustrated P/B-Lewis pair at the norbornane framework: Synthesis, characterization, and reactions. J. Am. Chem. Soc. 2013, 135, 8882–8895. [Google Scholar] [CrossRef] [PubMed]
- Menard, G.; Hatnean, J.A.; Cowley, H.J.; Lough, A.J.; Rawson, J.M.; Stephan, D.W. C-H bond activation by radical ion pairs derived from R3P/Al(C6F5)3 frustrated Lewis pairs and N2O. J. Am. Chem. Soc. 2013, 135, 6446–6449. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.X.; Stephan, D.W. Olefin-borane “van der Waals complexes”: Intermediates in frustrated Lewis pair addition reactions. J. Am. Chem. Soc. 2011, 133, 12448–12450. [Google Scholar] [CrossRef] [PubMed]
- Menard, G.; Stephan, D.W. Stoichiometric reduction of CO2 to CO by aluminum-based frustrated Lewis pairs. Angew. Chem. Int. Ed. 2011, 50, 8396–8399. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.M.; Haddow, M.F.; Wass, D.F. Frustrated Lewis pairs beyond the main group: Cationic zirconocene-phosphinoaryloxide complexes and their application in catalytic dehydrogenation of amine boranes. J. Am. Chem. Soc. 2011, 133, 8826–8829. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.J.P.; Culotta, B.J.; Warren, T.H.; Grimme, S.; Stute, A.; Frohlich, R.; Kehr, G.; Erker, G. Capture of NO by a frustrated Lewis pair: A new type of persistent N-oxyl radical. Angew. Chem. Int. Ed. 2011, 50, 7567–7571. [Google Scholar] [CrossRef] [PubMed]
- Momming, C.M.; Kehr, G.; Wibbeling, B.; Frohlich, R.; Schirmer, B.; Grimme, S.; Erker, G. Formation of cyclic allenes and cumulenes by cooperative addition of frustrated Lewis pairs to conjugated enynes and diynes. Angew. Chem. Int. Ed. 2010, 49, 2414–2417. [Google Scholar] [CrossRef] [PubMed]
- Menard, G.; Stephan, D.W. Room temperature reduction of CO2 to methanol by Al-based frustrated Lewis pairs and ammonia borane. J. Am. Chem. Soc. 2010, 132, 1796–1797. [Google Scholar] [CrossRef] [PubMed]
- Berkefeld, A.; Piers, W.E.; Parvez, M. Tandem frustrated Lewis pair/tris(pentafluorophenyl) borane-catalyzed deoxygenative hydrosilylation of carbon dioxide. J. Am. Chem. Soc. 2010, 132, 10660–10661. [Google Scholar] [CrossRef] [PubMed]
- Alcarazo, M.; Gomez, C.; Holle, S.; Goddard, R. Exploring the reactivity of carbon(0)/borane-based frustrated Lewis pairs. Angew. Chem. Int. Ed. 2010, 49, 5788–5791. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, M.; Borre, K.; Axenov, K.; Kótai, B.; Nieger, M.; Leskelä, M.; Pápai, I.; Repo, T. Chiral molecular tweezers: Synthesis and reactivity in asymmetric hydrogenation. J. Am. Chem. Soc. 2015, 137, 4038–4041. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.M.; Du, H.F. A highly enantioselective hydrogenation of silyl enol ethers catalyzed by chiral frustrated Lewis pairs. J. Am. Chem. Soc. 2014, 136, 12261–12264. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lalancette, R.A.; Jäkle, F. Chiral organoborane Lewis pairs derived from pyridylferrocene. Chem. Eur. J. 2014, 20, 9120–9129. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.B.; Du, H.F. Chiral dienes as “ligands” for borane-catalyzed metal-free asymmetric hydrogenation of imines. J. Am. Chem. Soc. 2013, 135, 6810–6813. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lalancette, R.A.; Jäkle, F. Synthesis and Lewis acid properties of a ferrocene-based planar-chiral borenium cation. Chem. Commun. 2013, 49, 4893–4895. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, G.; Chen, D.J.; Pan, F.F.; Klankermayer, J. Asymmetric hydrogenation of imines with a recyclable chiral frustrated Lewis pair catalyst. Dalton Trans. 2012, 41, 9026–9028. [Google Scholar] [CrossRef] [PubMed]
- Sumerin, V.; Chernichenko, K.; Nieger, M.; Leskela, M.; Rieger, B.; Repo, T. Highly active metal-free catalysts for hydrogenation of unsaturated nitrogen-containing compounds. Adv. Synth. Catal. 2011, 353, 2093–2110. [Google Scholar] [CrossRef]
- Chen, J.; Venkatasubbaiah, K.; Pakkirisamy, T.; Doshi, A.; Yusupov, A.; Patel, Y.; Lalancette, R.A.; Jäkle, F. Planar chiral organoborane Lewis acids derived from naphthylferrocene. Chem. Eur. J. 2010, 16, 8861–8867. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Wang, Y.T.; Klankermayer, J. Enantioselective hydrogenation with chiral frustrated Lewis pairs. Angew. Chem. Int. Ed. 2010, 49, 9475–9478. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.J.; Klankermayer, J. Metal-free catalytic hydrogenation of imines with tris(perfluorophenyl)borane. Chem. Commun. 2008, 18, 2130–2131. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.X. Polymerization by classical and frustrated Lewis pairs. Top. Curr. Chem. 2013, 334, 239–260. [Google Scholar] [PubMed]
- Chen, J.; Chen, E.Y.X. Lewis pair polymerization of acrylic monomers by N-heterocyclic carbenes and B(C6F5)3. Isr. J. Chem. 2015, 55, 216–225. [Google Scholar] [CrossRef]
- Zhang, Y.T.; Miyake, G.M.; John, M.G.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. Lewis pair polymerization by classical and frustrated Lewis pairs: Acid, base and monomer scope and polymerization mechanism. Dalton Trans. 2012, 41, 9119–9134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.T.; Miyake, G.M.; Chen, E.Y.X. Alane-based classical and frustrated Lewis pairs in polymer synthesis: Rapid polymerization of MMA and naturally renewable methylene butyrolactones into high-molecular-weight polymers. Angew. Chem. Int. Ed. 2010, 49, 10158–10162. [Google Scholar] [CrossRef] [PubMed]
- He, J.H.; Zhang, Y.T.; Falivene, L.; Caporaso, L.; Cavallo, L.; Chen, E.Y.X. Chain propagation and termination mechanisms for polymerization of conjugated polar alkenes by [Al]-Based frustrated Lewis pairs. Macromolecules 2014, 47, 7765–7774. [Google Scholar] [CrossRef]
- He, J.H.; Zhang, Y.T.; Chen, E.Y.X. Synthesis of pyridine- and 2-oxazoline-functionalized vinyl polymers by alane-based frustrated Lewis pairs. Synlett 2014, 25, 1534–1538. [Google Scholar]
- Piedra-Arroni, E.; Ladaviere, C.; Amgoune, A.; Bourissou, D. Ring-opening polymerization with Zn(C6F5)2-based Lewis pairs: Original and efficient approach to cyclic polyesters. J. Am. Chem. Soc. 2013, 135, 13306–13309. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Stute, A.; Cardenas, A.J.P.; Culotta, B.J.; Hepperle, J.A.M.; Warren, T.H.; Schirmer, B.; Grimme, S.; Studer, A.; Daniliuc, C.G.; et al. N,N-Addition of frustrated Lewis pairs to nitric oxide: An easy entry to a unique family of aminoxyl radicals. J. Am. Chem. Soc. 2012, 134, 10156–10168. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.Q.; Chen, E.Y.X. Probing site cooperativity of frustrated phosphine/borane Lewis pairs by a polymerization study. J. Am. Chem. Soc. 2014, 136, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.B.; Ren, W.M.; Liu, S.J.; Xu, T.Q.; Wang, Y.B.; Lu, X.B. Controlled divinyl monomer polymerization mediated by Lewis pairs: A powerful synthetic strategy for functional polymers. ACS Macro Lett. 2014, 3, 896–899. [Google Scholar] [CrossRef]
- Eisenberger, P.; Bailey, A.M.; Crudden, C.M. Taking the F out of FLP: Simple Lewis acid-base pairs for mild reductions with neutral boranes via borenium ion catalysis. J. Am. Chem. Soc. 2012, 134, 17384–17387. [Google Scholar] [CrossRef] [PubMed]
- Sumerin, V.; Schulz, F.; Atsumi, M.; Wang, C.; Nieger, M.; Leskela, M.; Repo, T.; Pyykko, P.; Rieger, B. Molecular tweezers for hydrogen: Synthesis, characterization, and reactivity. J. Am. Chem. Soc. 2008, 130, 14117–14119. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Sumerin, V.; Heikkinen, S.; Pedersen, B.; Wang, C.; Atsumi, M.; Leskela, M.; Repo, T.; Pyykko, P.; Petry, W.; et al. Molecular hydrogen tweezers: Structure and mechanisms by neutron diffraction, NMR, and deuterium labeling studies in solid and solution. J. Am. Chem. Soc. 2011, 133, 20245–20257. [Google Scholar] [CrossRef] [PubMed]
- Mountford, A.J.; Lancaster, S.J.; Coles, S.J.; Horton, P.N.; Hughes, D.L.; Hursthouse, M.B.; Light, M.E. Intra- and intermolecular N-H···F-C hydrogen-bonding interactions in amine adducts of tris(pentafluorophenyl)borane and -alane. Inorg. Chem. 2005, 44, 5921–5933. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Zhu, H.P.; Chen, E.Y.X. Remarkable Lewis acid effects on polymerization of functionalized alkenes by metallocene and lithium ester enolates. J. Organomet. Chem. 2007, 692, 4535–4544. [Google Scholar] [CrossRef]
- Rodriguez-Delgado, A.; Chen, E.Y.X. Single-site anionic polymerization. Monomeric ester enolaluminate propagator synthesis, molecular structure, and polymerization mechanism. J. Am. Chem. Soc. 2005, 127, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.X.; Kruper, W.J.; Roof, G.; Wilson, D.R. “Double activation” of constrained geometry and ansa-metallocene group 4 metal dialkyls: Synthesis, structure, and olefin polymerization study of mono-and dicationic aluminate complexes. J. Am. Chem. Soc. 2001, 123, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Bolig, A.D.; Chen, E.Y.X. Reversal of polymerization stereoregulation in anionic polymerization of MMA by chiral metallocene and non-metallocene initiators: A new reaction pathway for metallocene-initiated MMA polymerization. J. Am. Chem. Soc. 2001, 123, 7943–7944. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, E.Y.X. Elusive silane alane complex [Si-H···Al]: Isolation, characterization, and multifaceted frustrated-Lewis-pair-type catalysis. Angew. Chem. Int. Ed. 2015. [Google Scholar] [CrossRef]
- Di Saverio, A.; Focante, F.; Camurati, I.; Resconi, L.; Beringhelli, T.; D’Alfonso, G.; Donghi, D.; Maggioni, D.; Mercandelli, P.; Sironi, A. Oxygen-bridged borate anions from tris(pentafluorophenyl)borane: Synthesis, NMR characterization, and reactivity. Inorg. Chem. 2005, 44, 5030–5041. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.D.; Long, T.E.; Mcgrath, J.E. Preparation of high-purity, anionic-polymerization grade alkyl methacrylate monomers. Polym. Bull. 1986, 15, 127–134. [Google Scholar] [CrossRef]
- Feng, S.G.; Roof, G.R.; Chen, E.Y.X. Tantalum (V)-based metallocene, half-metallocene, and non-metallocene complexes as ethylene-1-octene copolymerization and methyl methacrylate polymerization catalysts. Organometallics 2002, 21, 832–839. [Google Scholar] [CrossRef]
- Lee, C.H.; Lee, S.J.; Park, J.W.; Kim, K.H.; Lee, B.Y.; Oh, J.S. Preparation of Al(C6F5)3 and its use for the modification of methylalumoxane. J. Mol. Catal. A Chem. 1998, 132, 231–239. [Google Scholar] [CrossRef]
- Biagini, P.; Lugli, G.; Abis, L.; Andreussi, P. Organometallic derivatives of group IIIA and process for their preparation. U.S. Patent 5602269, 11 February 1997. [Google Scholar]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Chen, E.X.-Y. Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization. Molecules 2015, 20, 9575-9590. https://doi.org/10.3390/molecules20069575
Chen J, Chen EX-Y. Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization. Molecules. 2015; 20(6):9575-9590. https://doi.org/10.3390/molecules20069575
Chicago/Turabian StyleChen, Jiawei, and Eugene X.-Y. Chen. 2015. "Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization" Molecules 20, no. 6: 9575-9590. https://doi.org/10.3390/molecules20069575
APA StyleChen, J., & Chen, E. X.-Y. (2015). Reactivity of Amine/E(C6F5)3 (E = B, Al) Lewis Pairs toward Linear and Cyclic Acrylic Monomers: Hydrogenation vs. Polymerization. Molecules, 20(6), 9575-9590. https://doi.org/10.3390/molecules20069575