
Synthesis, 18F-Radiolabelling and Biological Characterization of Novel Fluoroalkylated Triazine Derivatives for in Vivo Imaging of Phosphodiesterase 2A in Brain via Positron Emission Tomography

 ,
,  ,
,  , and
, and
Abstract
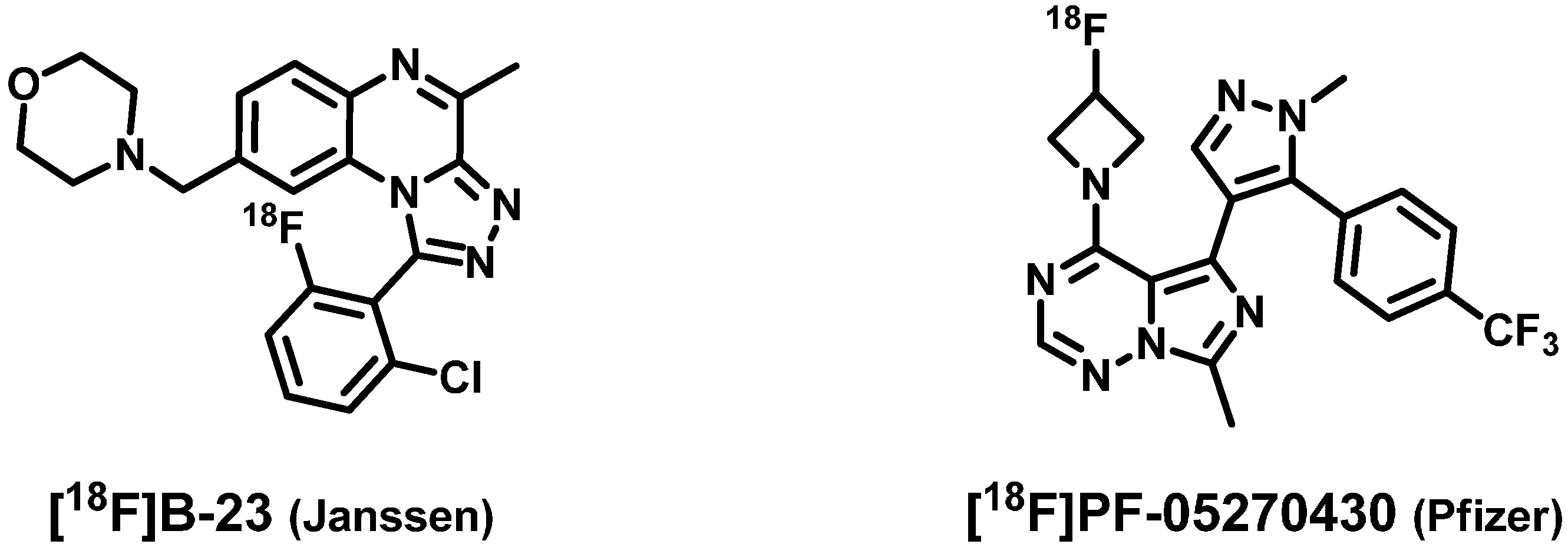
:1. Introduction
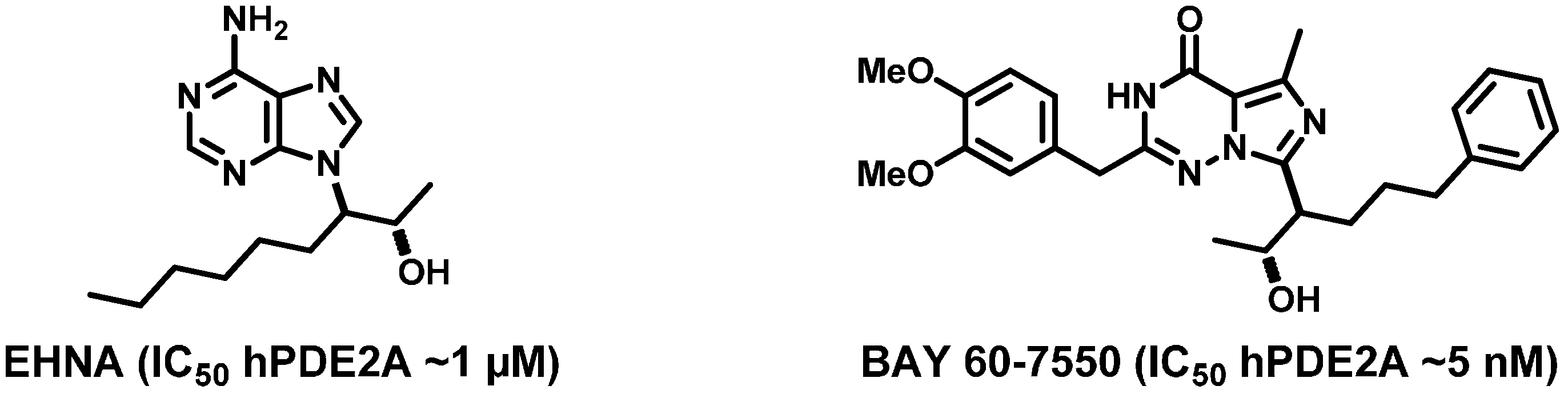
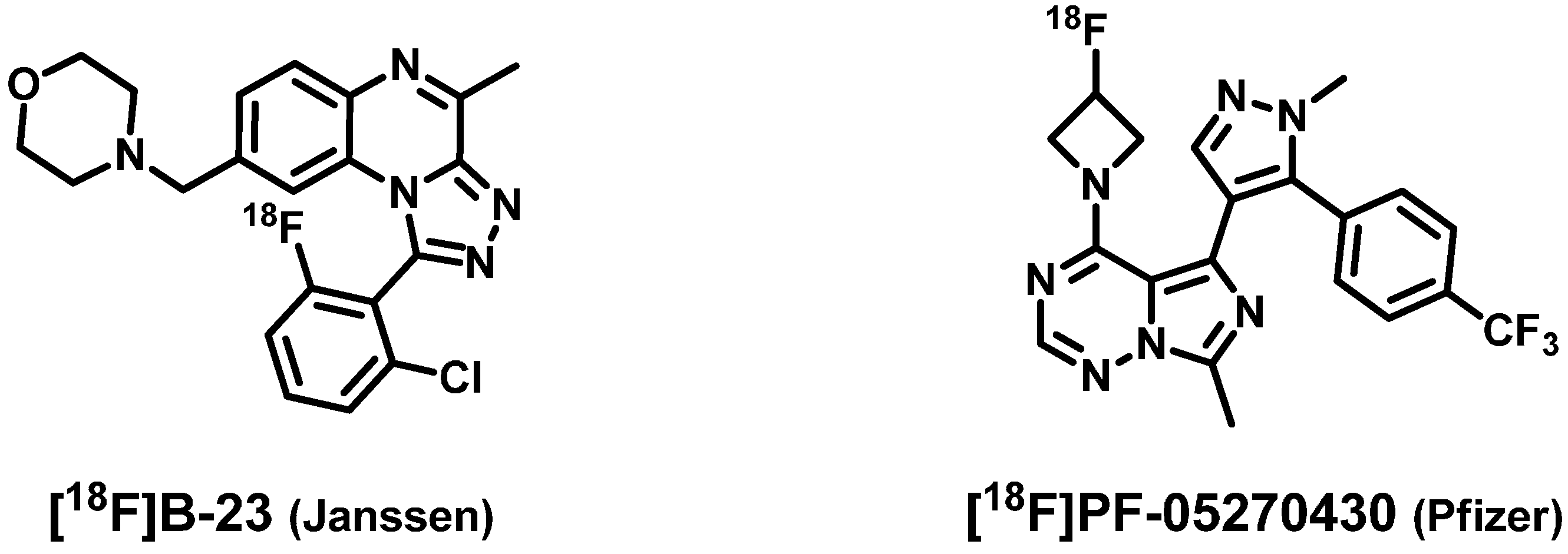
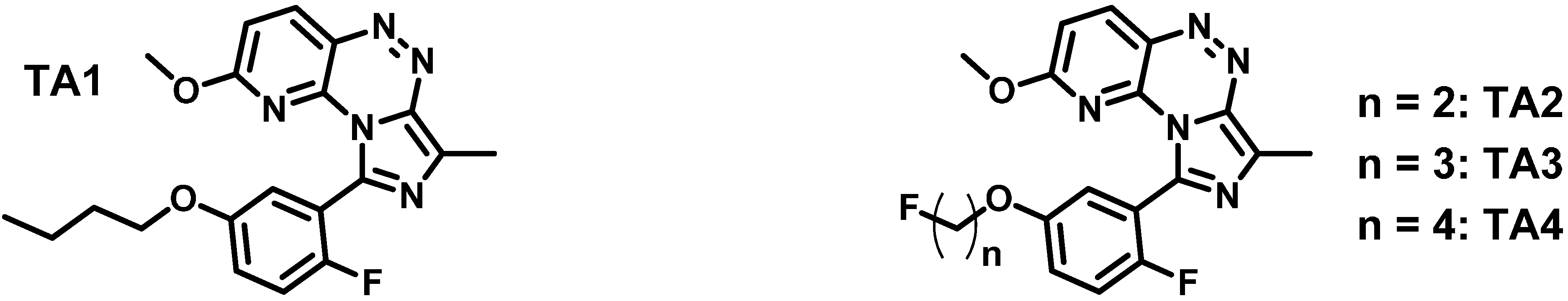
2. Results and Discussion
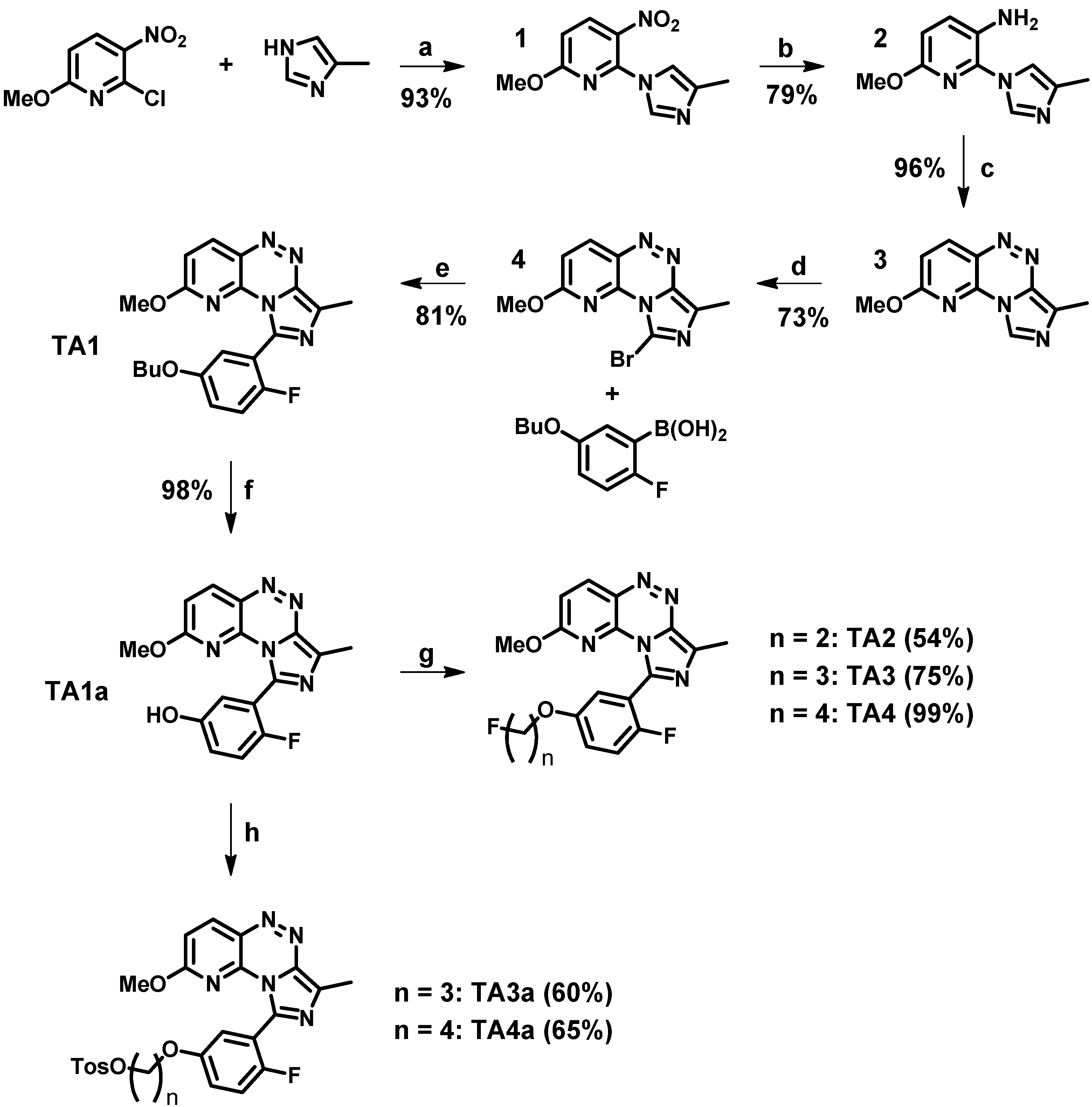
2.1. Synthesis and in Vitro Binding

{kind=link}
{kind=link}
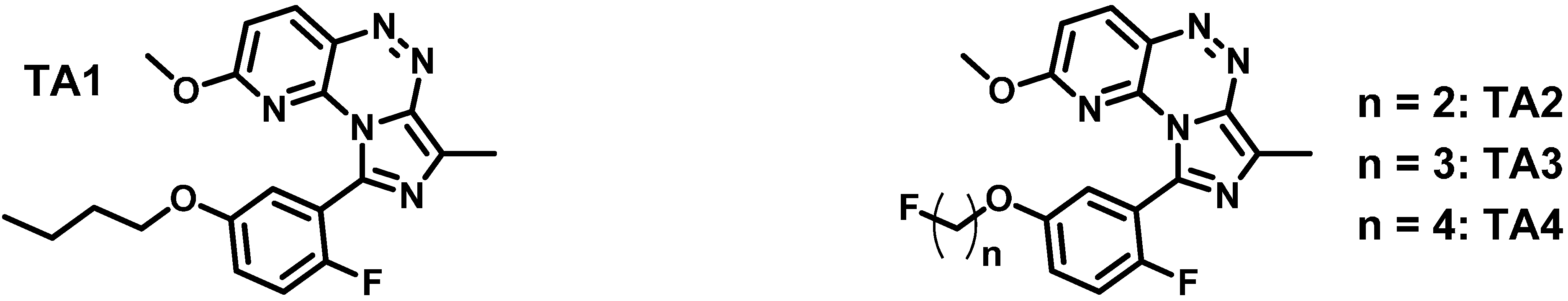{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
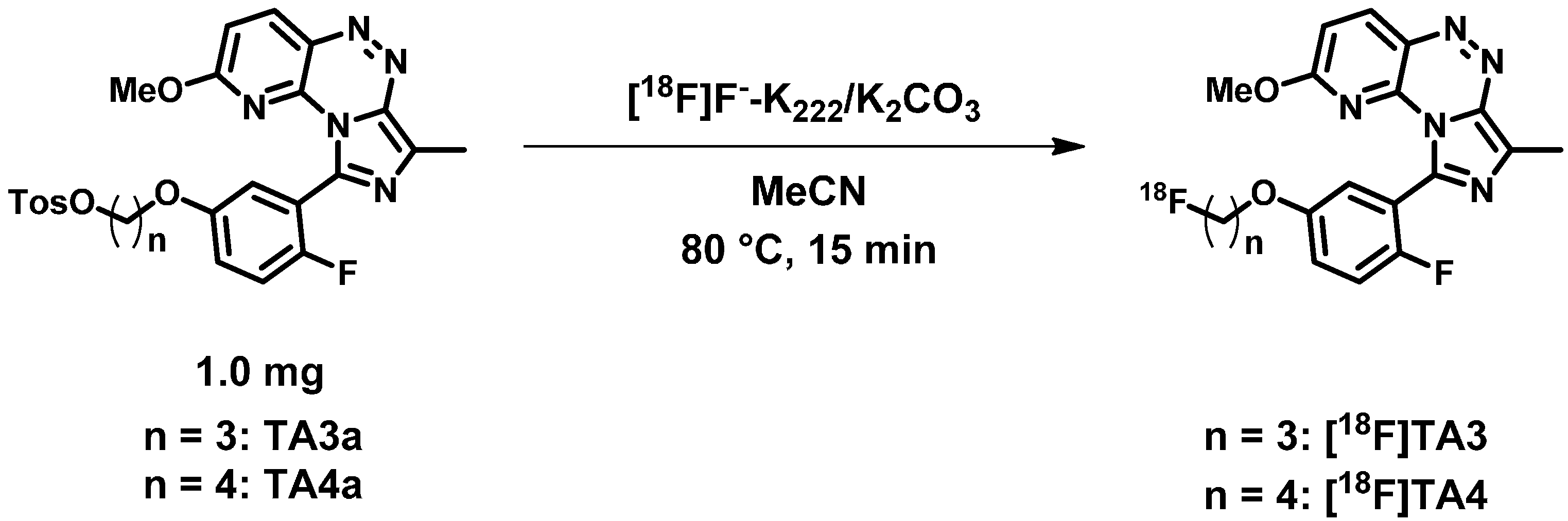{kind=link}
{kind=link}
| Ligand | IC50 hPDE2A | IC50 hPDE10A | Selectivity Ratio PDE10A/PDE2A |
|---|---|---|---|
| TA1 (lead) | 4.5 nM | 670 nM | 148.9 |
| TA2 (2-fluoroethyl) | 10.4 nM | 77 nM | 7.4 |
| TA3 (3-fluoropropyl) | 11.4 nM | 318 nM | 27.9 |
| TA4 (4-fluorobutyl) | 7.3 nM | 913 nM | 125.1 |
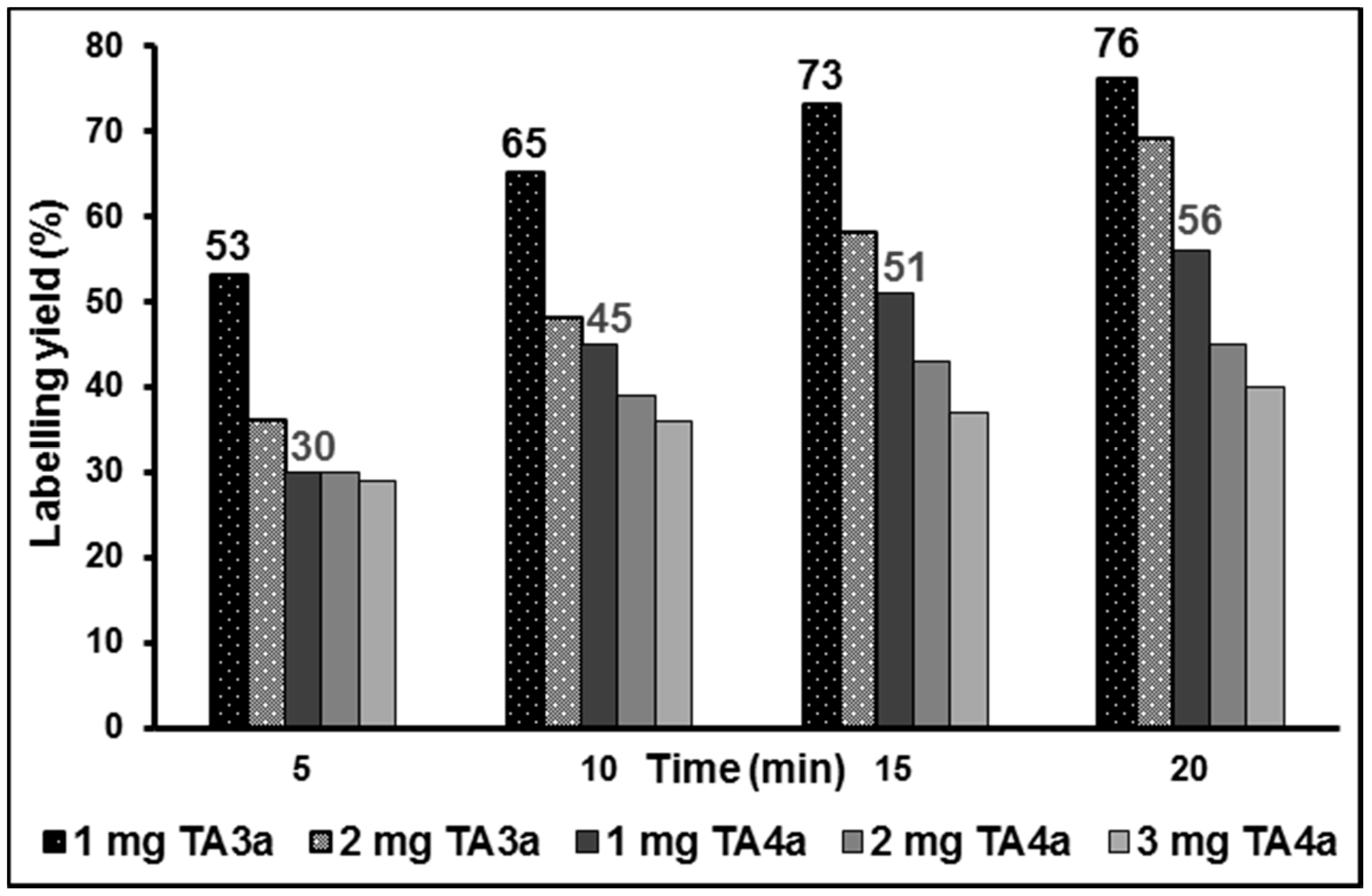
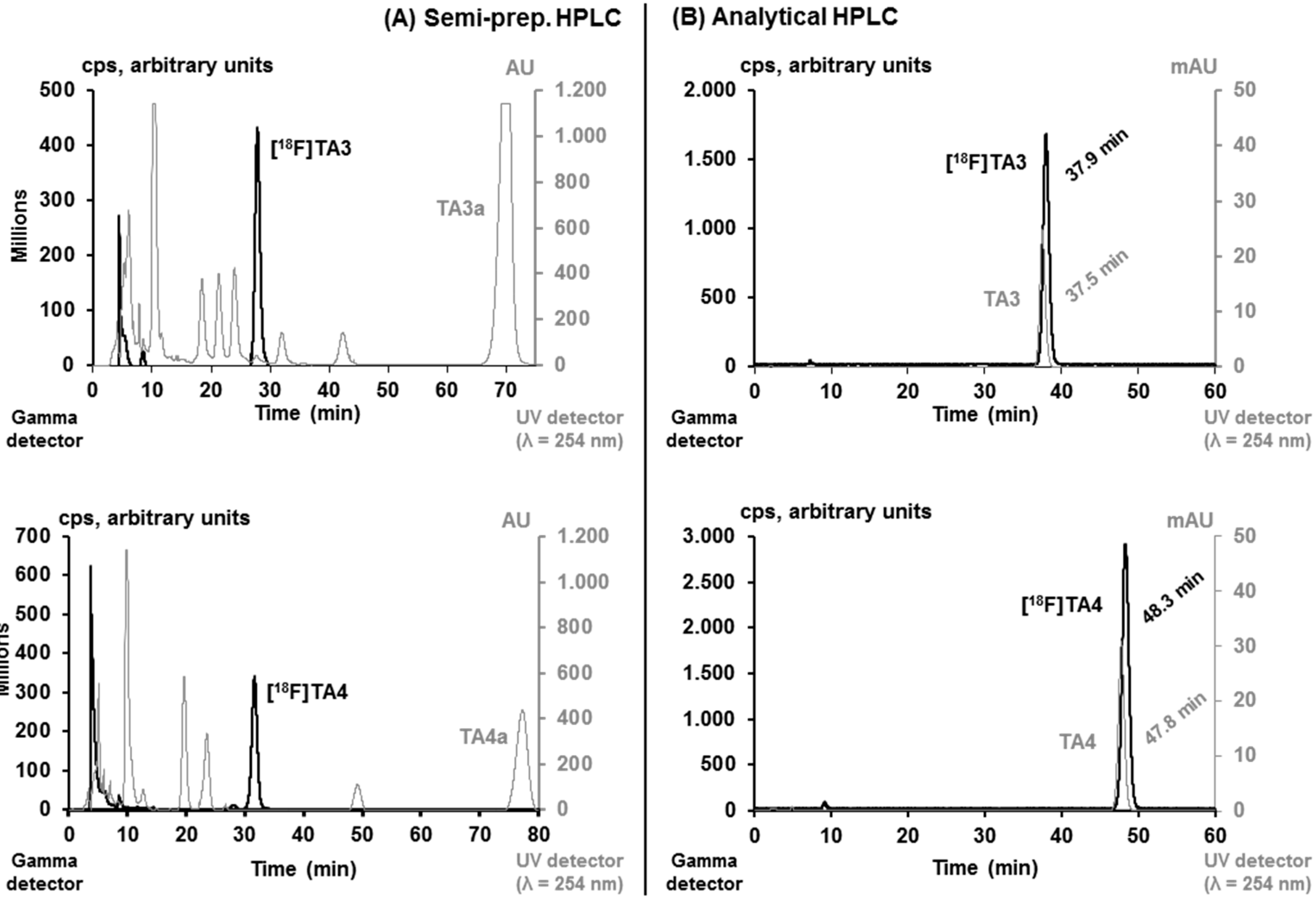
2.2. Radiochemistry, Lipophilicity and in Vitro Stability
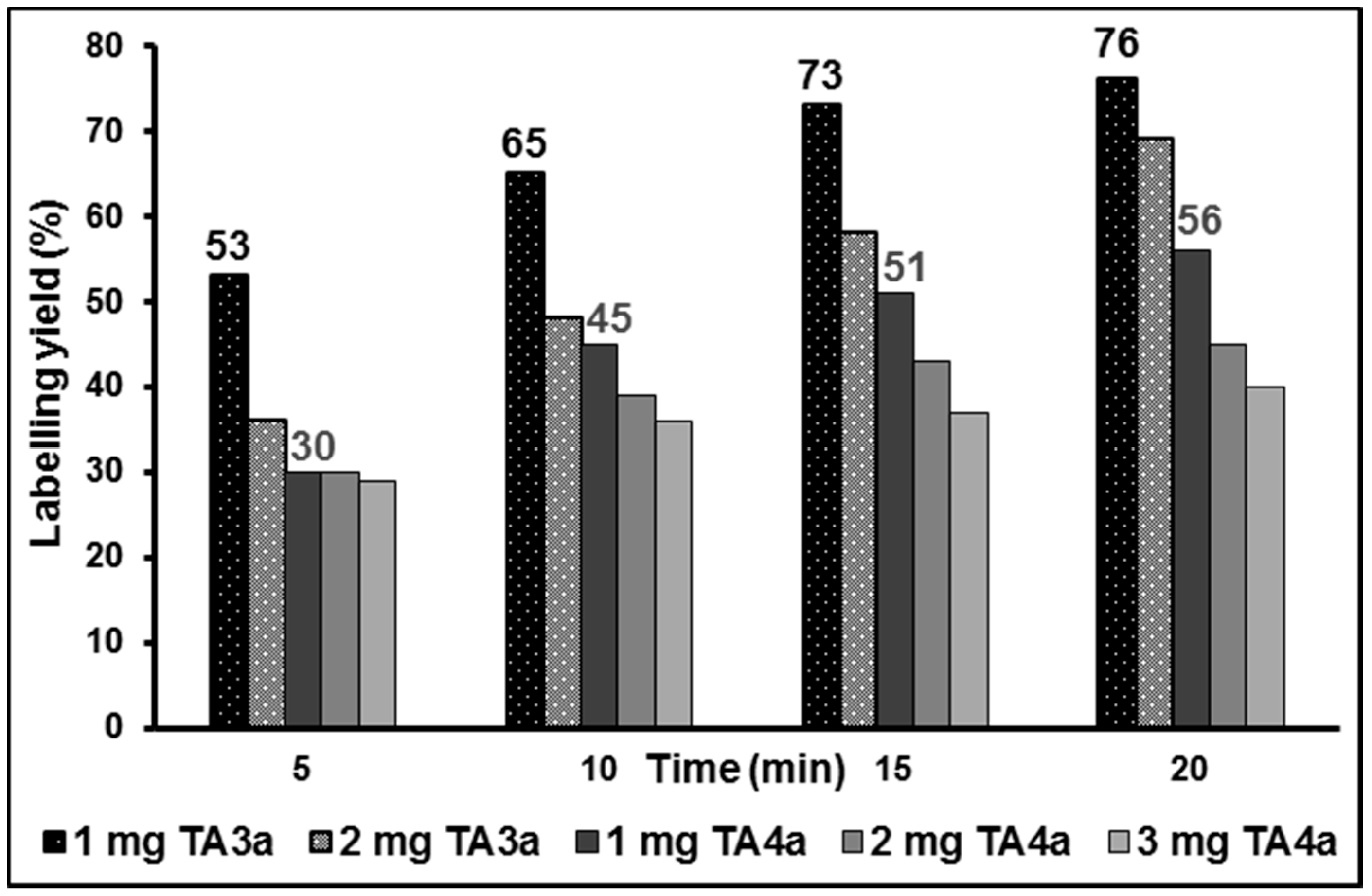
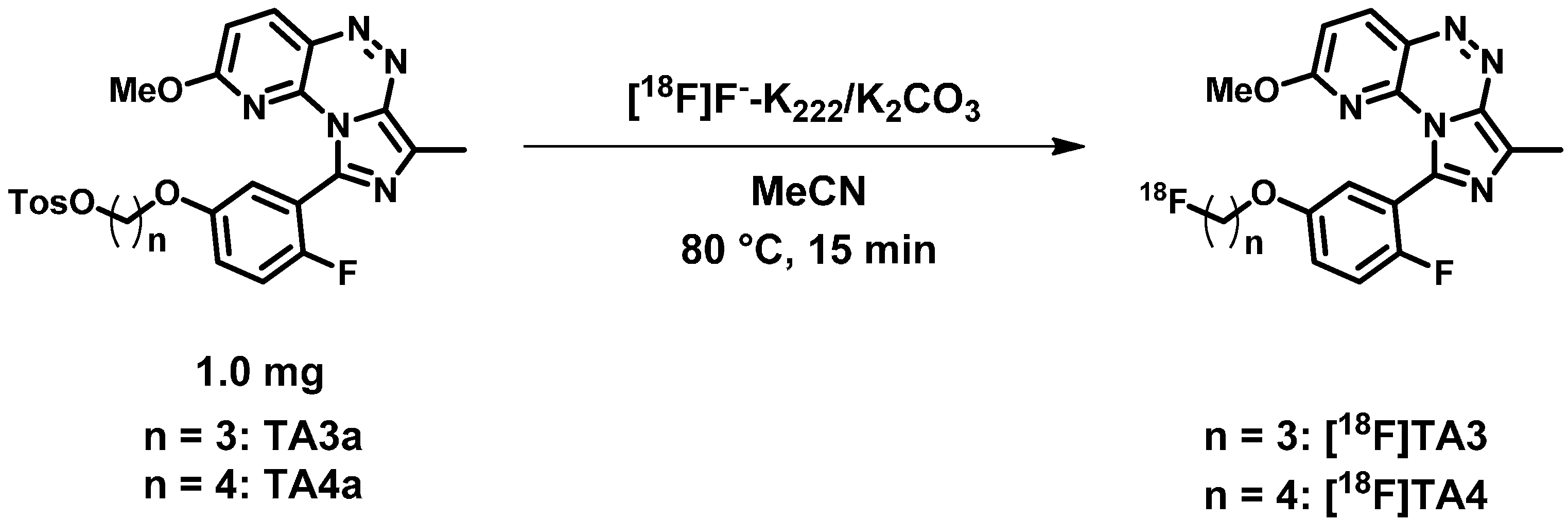
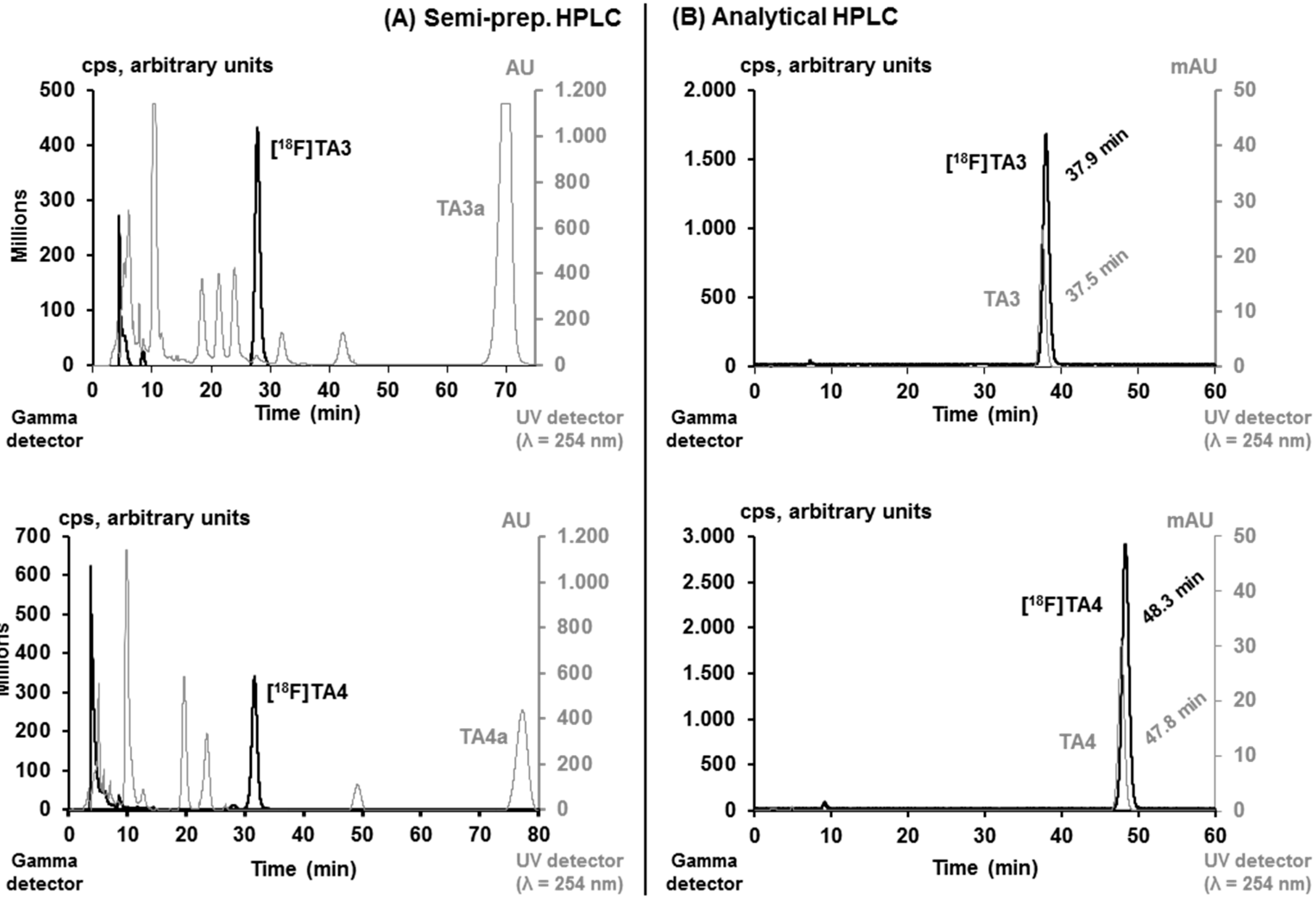
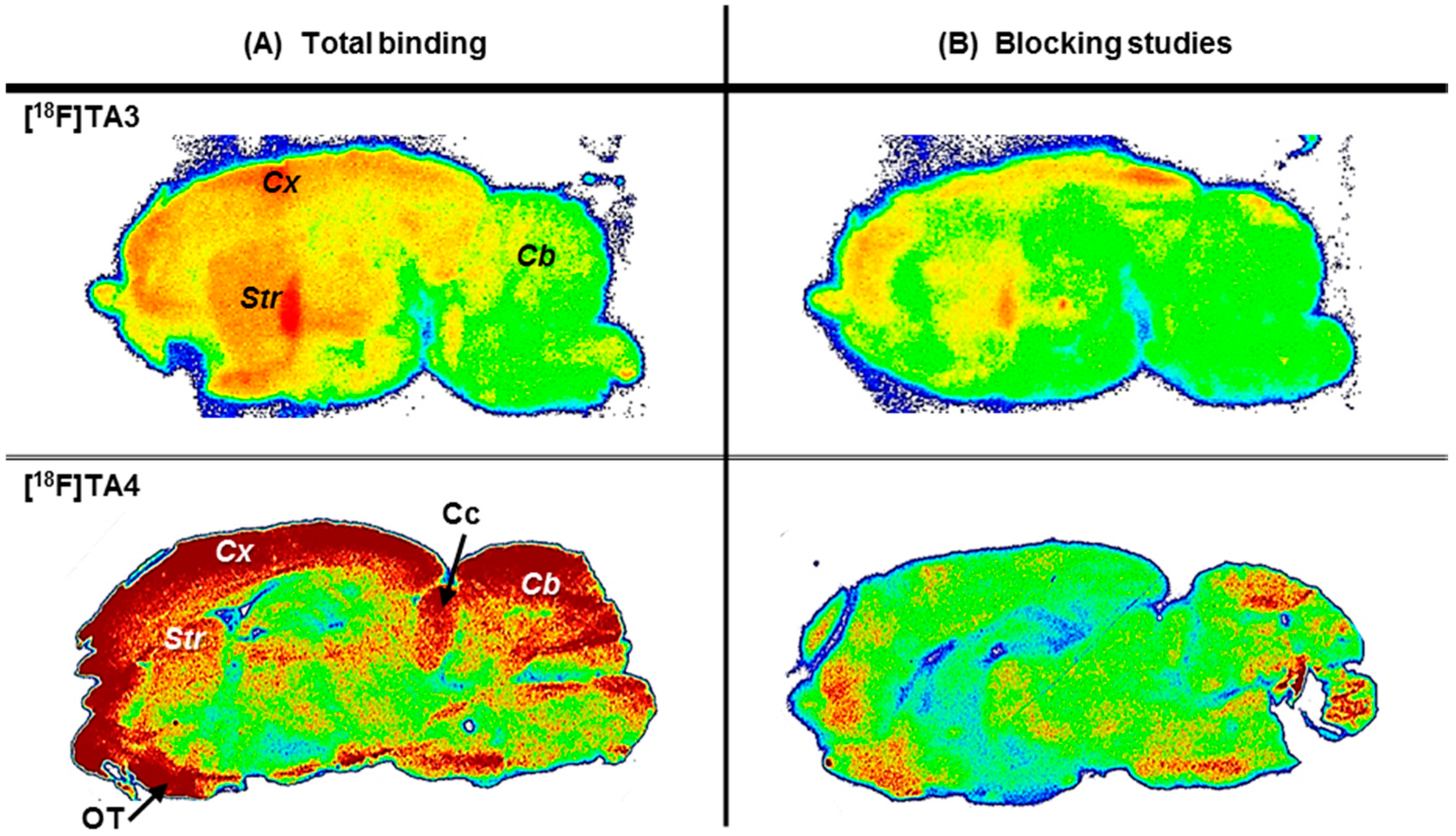
2.3. In Vitro Autoradiographic Studies in Rat Brain
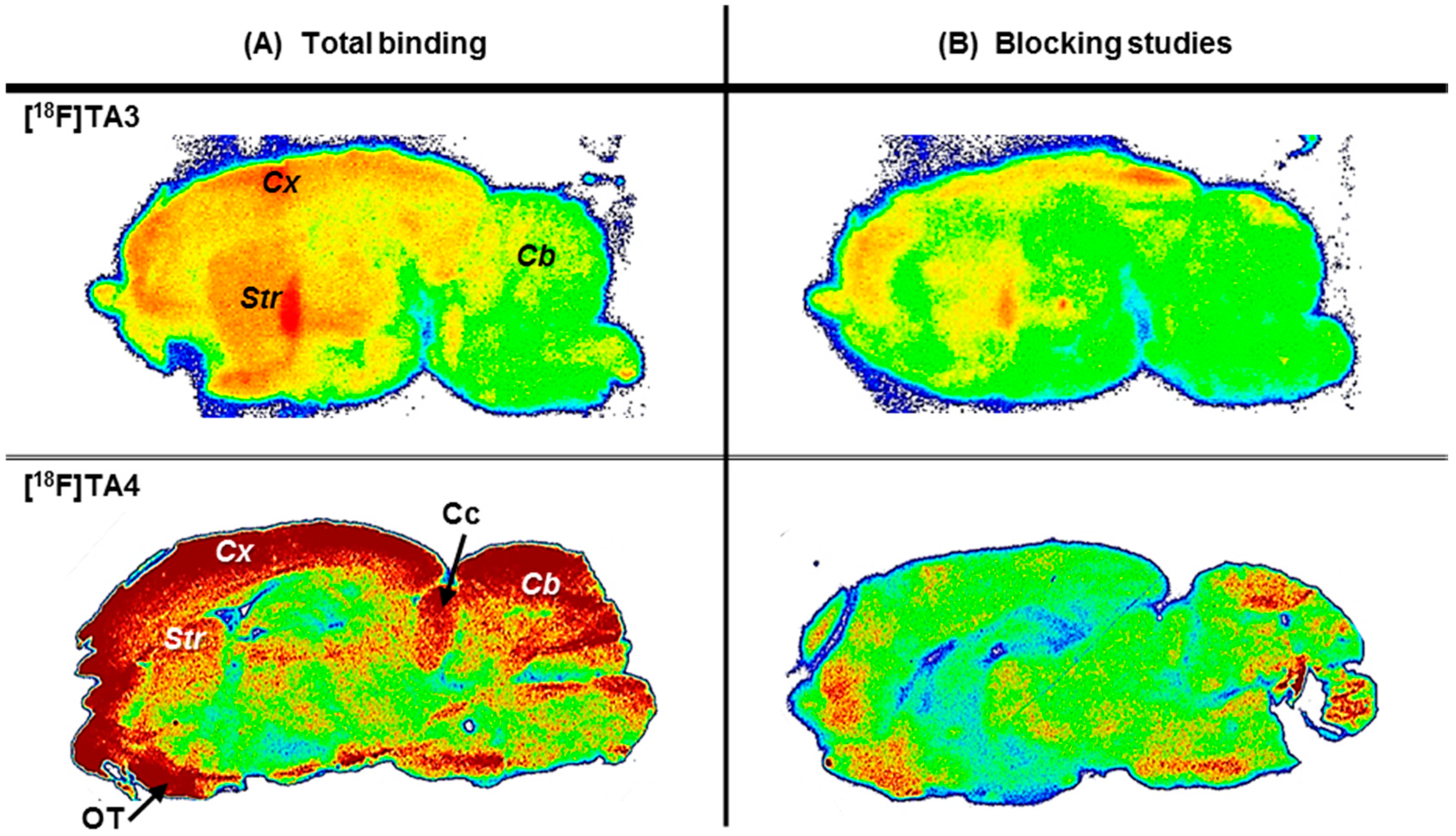
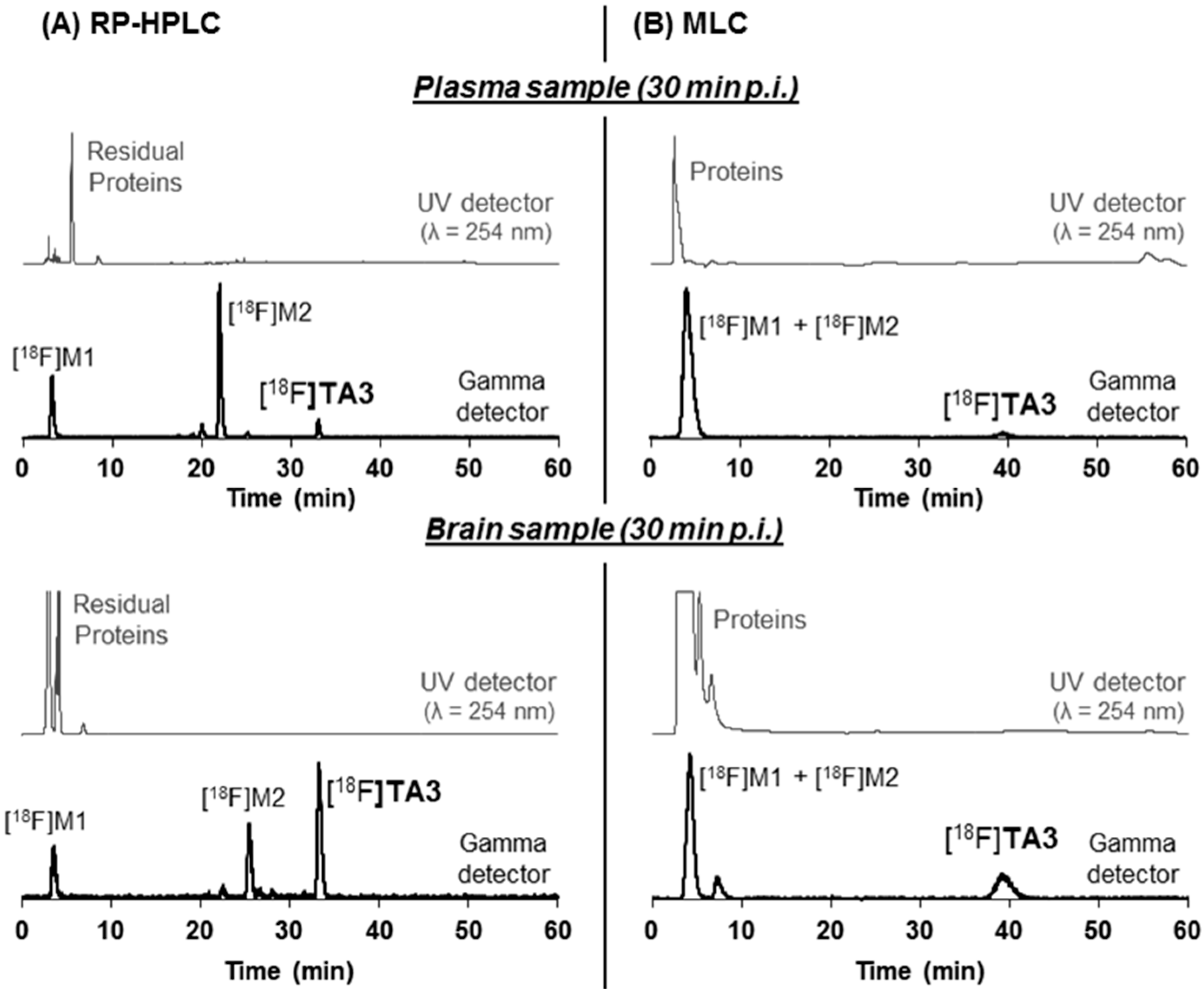
2.4. In Vivo Metabolism of [18F]TA3 and [18F]TA4 in Mice

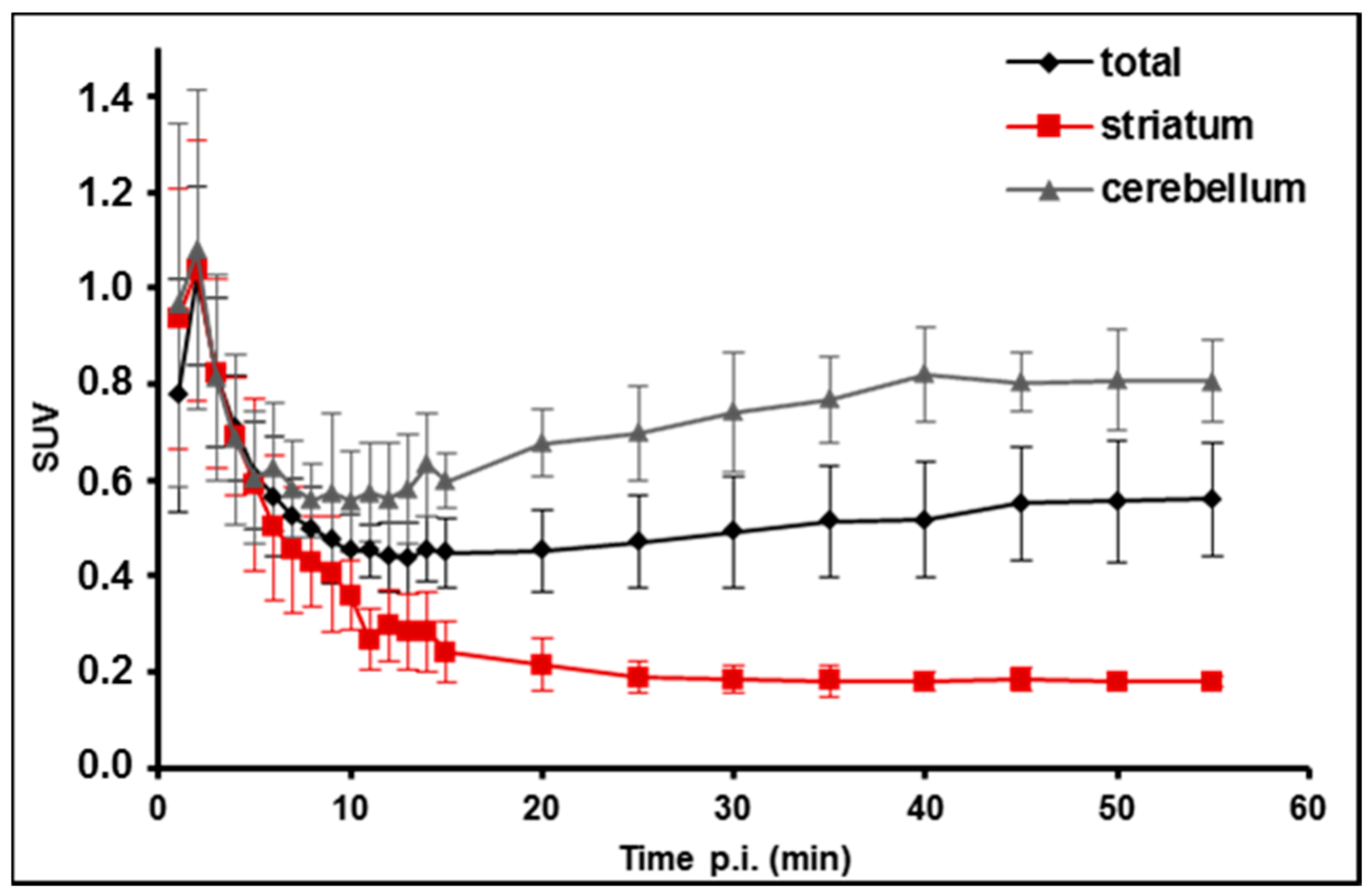
2.5. PET/MR Studies of [18F]TA3 in Mice
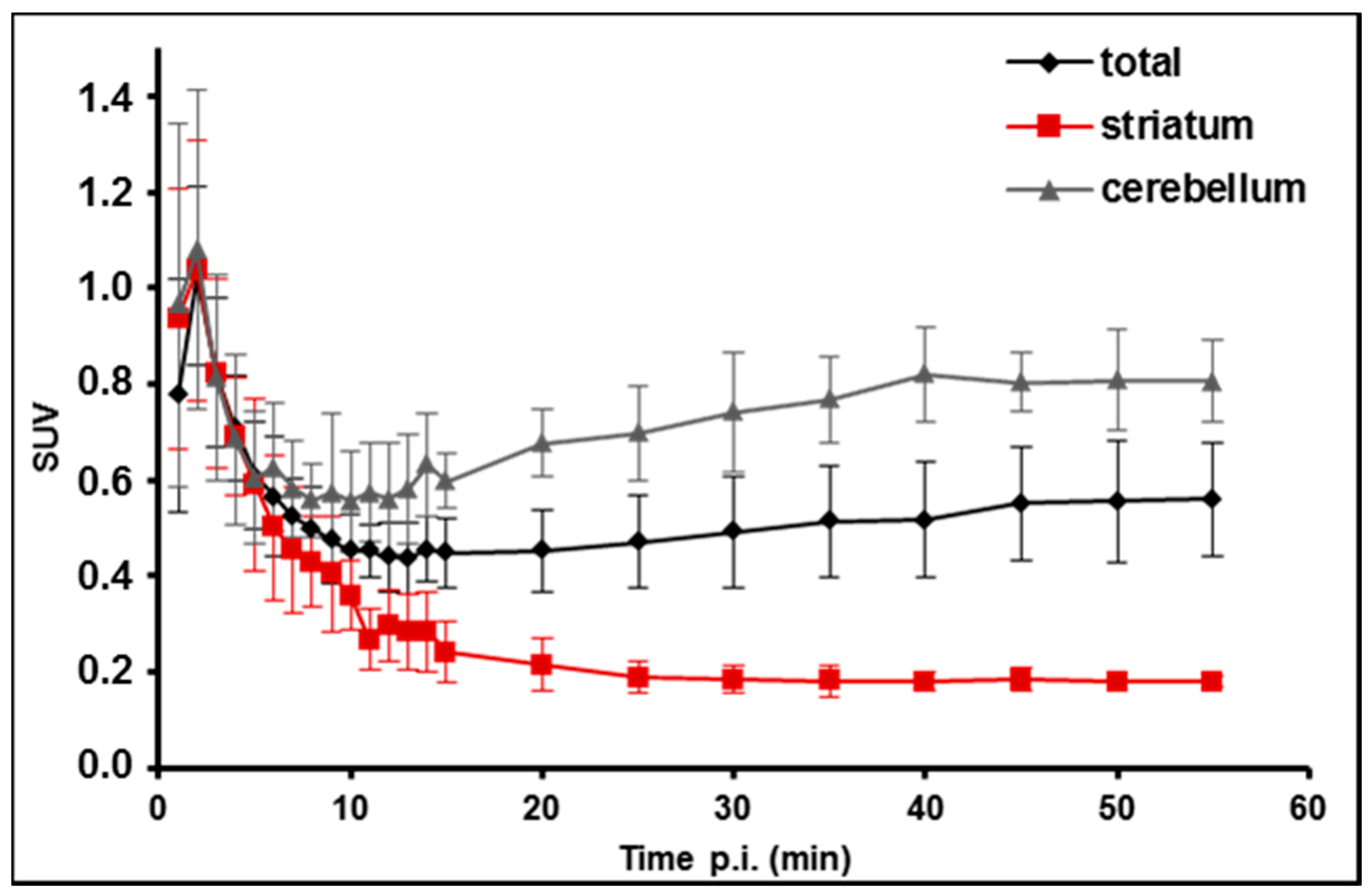
3. Experimental Section
3.1. General Information
3.2. Syntheses
3.2.1. General Procedure for the Preparation of Fluoroalkylated Triazine Derivatives TA2–4
3.2.2. General Procedure for the Preparation of alkyl bis(4-methylbenzenesulfonates)
3.2.3. General Procedure for the Preparation of Tosylate Precursors TA3a and TA4a
3.3. In Vitro PDE2A Affinity Assay
3.4. Radiochemistry
3.4.1. Manual Radiosyntheses of [18F]TA3 and [18F]TA4
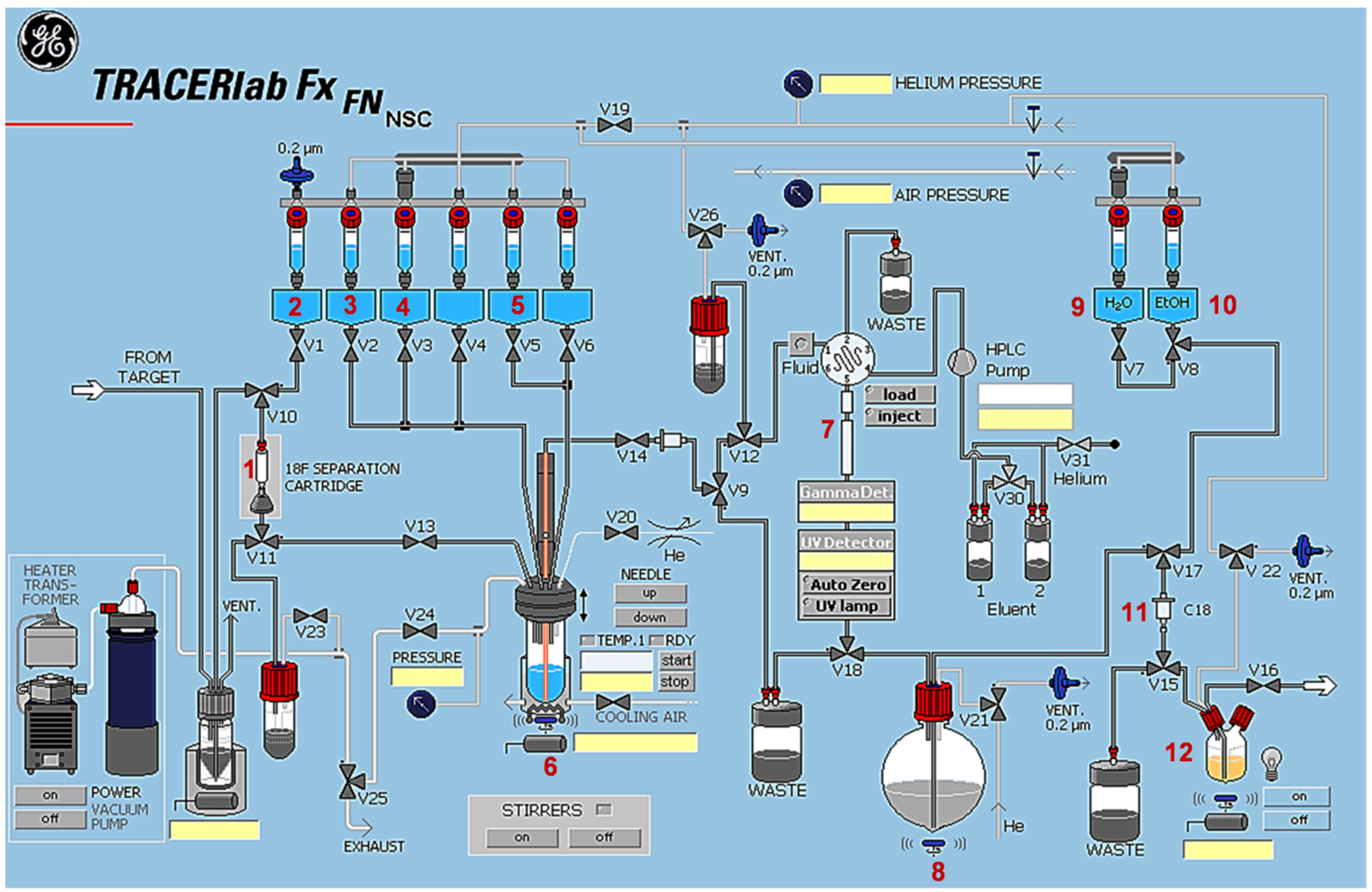
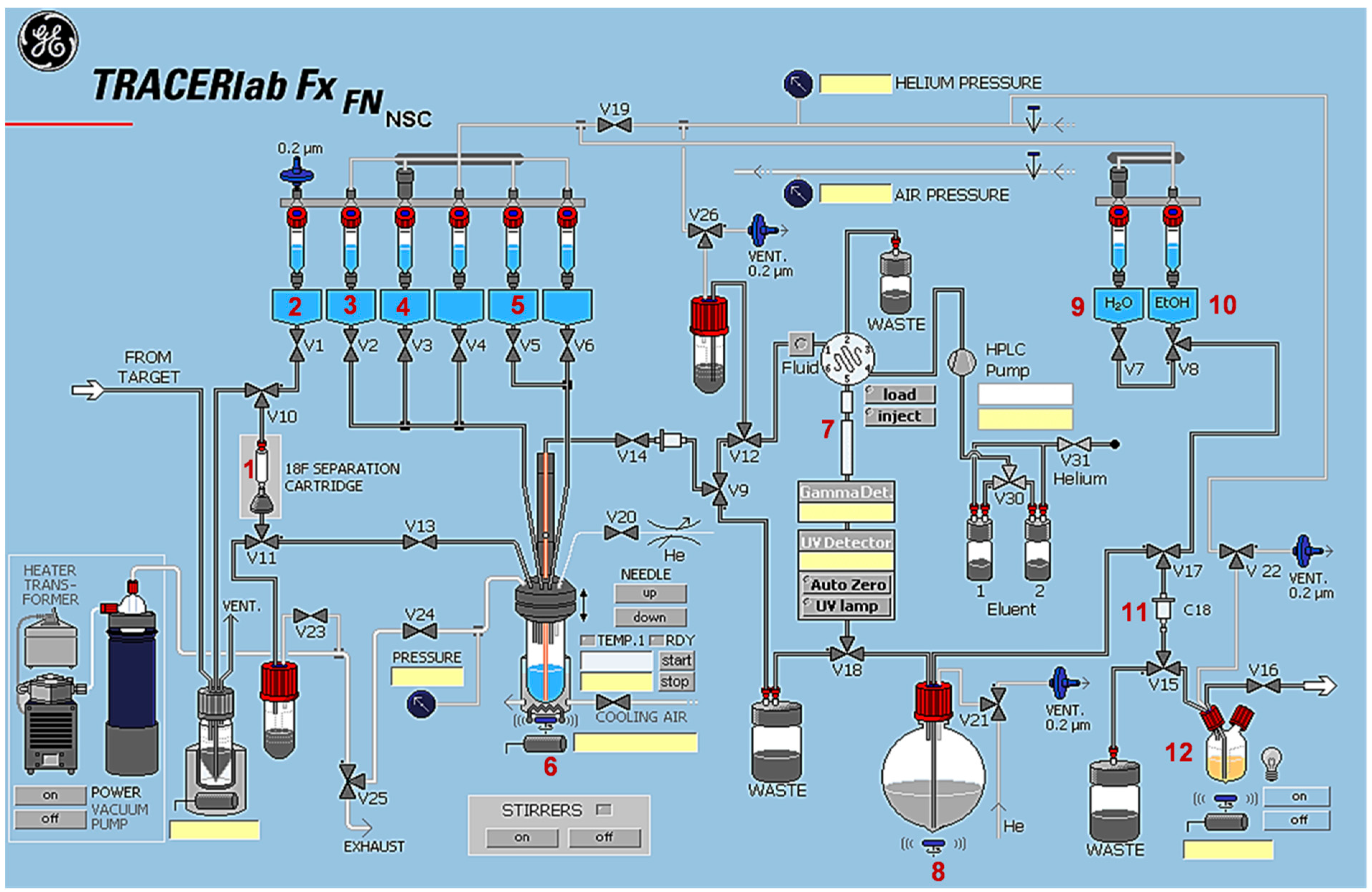
3.4.2. Automated Radiosynthesis of [18F]TA3
3.5. Determination of Lipophilicity (logD7.4) and in Vitro Stability
3.6. Animal Studies
3.6.1. In Vitro Autoradiographic Studies in Rat Brain
3.6.2. Small-Animal PET/MR Studies in Mice
3.6.3. In Vivo Metabolism Studies in Mice
3.7. Conventional Extraction Procedure
3.8. Micellar Chromatography (MLC)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kelly, M.P.; Adamowicz, W.; Bove, S.; Hartman, A.J.; Mariga, A.; Pathak, G.; Reinhart, V.; Romegialli, A.; Kleiman, R.J. Select 3′,5′-cyclic nucleotide phosphodiesterases exhibit altered expression in the aged rodent brain. Cell Signal. 2014, 26, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Breitenbucher, J.G. PDE2 inhibition: Potential for the treatment of cognitive disorders. Bioorg. Med. Chem. Lett. 2013, 23, 6522–6527. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.; Piazza, G.; Tinsley, H. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Zhang, Y.; Zhang, H.T.; Li, Y.F. Phosphodiesterase: An interface connecting cognitive deficits to neuropsychiatric and neurodegenerative diseases. Curr. Pharm. Des. 2015, 21, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Van Staveren, W.C.G.; Steinbusch, H.W.M.; Markerink-van Ittersum, M.; Repaske, D.R.; Goy, M.F.; Kotera, J.; Omori, K.; Beavo, J.A.; de Vente, J. MRNA expression patterns of the cGMP-hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J. Comp. Neurol. 2003, 467, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, Y.; Ruan, L.; Wang, C.; Pan, J.; Klabnik, J.; Lueptow, L.; Zhang, H.T.; O’Donnell, J.M.; Xu, Y. The roles of phosphodiesterase 2 in the central nervous and peripheral systems. Curr. Pharm. Des. 2015, 21, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Van Staveren, W.C.G.; Markerink-van Ittersum, M.; Steinbusch, H.W.M.; de Vente, J. The effects of phosphodiesterase inhibition on cyclic GMP and cyclic AMP accumulation in the hippocampus of the rat. Brain Res. 2001, 888, 275–286. [Google Scholar] [CrossRef]
- Suvarna, N.U.; O’Donnell, J.M. Hydrolysis of N-methyl-d-aspartate receptor-stimulated cAMP and cGMP by PDE4 and PDE2 phosphodiesterases in primary neuronal cultures of rat cerebral cortex and hippocampus. J. Pharmacol. Exp. Ther. 2002, 302, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Blokland, A.; Schreiber, R.; Prickaerts, J. Improving memory: A role for phosphodiesterases. Curr. Pharm. Des. 2006, 12, 2511–2523. [Google Scholar] [CrossRef] [PubMed]
- Boess, F.G.; Hendrix, M.; van der Staay, F.J.; Erb, C.; Schreiber, R.; van Staveren, W.; de Vente, J.; Prickaerts, J.; Blokland, A.; Koenig, G. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 2004, 47, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Reneerkens, O.A.; Rutten, K.; Bollen, E.; Hage, T.; Blokland, A.; Steinbusch, H.W.; Prickaerts, J. Inhibition of phoshodiesterase type 2 or type 10 reverses object memory deficits induced by scopolamine or MK-801. Behav. Brain Res. 2013, 236, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Murata, T.; Shimizu, K.; Okumura, K.; Inui, M.; Tagawa, T. Characterization of phosphodiesterase 2A in human malignant melanoma PMP cells. Oncol. Rep. 2013, 29, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Drees, M.; Zimmermann, R.; Eisenbrand, G. 3′,5′-Cyclic nucleotide phosphodiesterase in tumor cells as potential target for tumor growth inhibition. Cancer Res. 1993, 53, 3058–3061. [Google Scholar] [PubMed]
- Abusnina, A.; Keravis, T.; Zhou, Q.; Justiniano, H.; Lobstein, A.; Lugnier, C. Tumour growth inhibition and anti-angiogenic effects using curcumin correspond to combined PDE2 and PDE4 inhibition. Thromb. Haemost. 2015, 113, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.J.; Lou, Y.R.; Peng, Q.Y.; Li, T.; Lu, Y.P. PDE2 is a novel target for attenuating tumor formation in a mouse model of UVB-induced skin carcinogenesis. PLoS ONE 2014, 9, e109862. [Google Scholar] [CrossRef] [PubMed]
- Podzuweit, T.; Nennstiel, P.; Müller, A. Isozyme selective inhibition of cGMP-stimulated cyclic nucleotide phosphodiesterases by erythro-9-(2-hydroxy-3-nonyl)adenine. Cell Signal. 1995, 7, 733–738. [Google Scholar] [CrossRef]
- Biagi, G.; Giorgi, I.; Livi, O.; Pacchini, F.; Rum, P.; Scartoni, V.; Costa, B.; Mazzoni, M.R.; Giusti, L. Erythro- and threo-2-hydroxynonyl substituted 2-phenyladenines and 2-phenyl-8-azaadenines: Ligands for A1 adenosine receptors and adenosine deaminase. Farmaco 2002, 57, 221–233. [Google Scholar] [CrossRef]
- Boess, F.G.; Grosser, R.; Hendrix, M.; Koenig, G.; Niewoehner, U.; Schauss, D.; Schlemmer, K.H.; Schreiber, R.; van der Staay, F.J. Novel Substituted Imidazotriazines as PDE II Inhibitors. Patent WO 2002/050078 A1, 27 June 2002. [Google Scholar]
- Masood, A.; Huang, Y.; Hajjhussein, H.; Xiao, L.; Li, H.; Wang, W.; Hamza, A.; Zhan, C.G.; O’Donnell, J.M. Anxiolytic effects of phosphodiesterase-2 inhibitors associated with increased cGMP signaling. J. Pharmacol. Exp. Ther. 2009, 331, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Pan, J.; Chen, L.; Zhang, C.; Sun, J.; Li, J.; Nguyen, L.; Nair, N.; Zhang, H.; O’Donnell, J.M. Phosphodiesterase-2 inhibitor reverses corticosterone-induced neurotoxicity and related behavioural changes via cGMP/PKG dependent pathway. Int. J. Neuropsychopharmacol. 2013, 16, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Domek-Łopacińska, K.; Strosznajder, J.B. The effect of selective inhibition of cyclic GMP hydrolyzing phosphodiesterases 2 and 5 on learning and memory processes and nitric oxide synthase activity in brain during aging. Brain Res. 2008, 1216, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Rutten, K.; Prickaerts, J.; Hendrix, M.; van der Staay, F.J.; Şik, A.; Blokland, A. Time-dependent involvement of cAMP and cGMP in consolidation of object memory: Studies using selective phosphodiesterase type 2, 4 and 5 inhibitors. Eur. J. Pharmacol. 2007, 558, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Abarghaz, M.; Biondi, S.; Duranton, J.; Limanton, E.; Mondadori, C.; Wagner, P. Benzo[1,4]diazepin-2-one Derivatives as Phosphodiesterase PDE2 Inhibitors, Preparation and Therapeutic Use Thereof. Patent WO 2005/063723 A1, 14 July 2005. [Google Scholar]
- Dost, R.; Egerland, U.; Grunwald, C.; Höfgen, N.; Langen, B.; Lankau, H.J.; Stange, H. (1,2,4)Triazolo[4,3-a]quinoxaline Derivatives as Inhibitors of Phosphodiesterases. Patent WO 2012/104293 A1, 09 August 2012. [Google Scholar]
- Andres, J.I.; Buijnsters, P.; de Angelis, M.; Langlois, X.; Rombouts, F.; Trabanco, A.A.; Vanhoof, G. Discovery of a new series of [1,2,4]triazolo[4,3-a]quinoxalines as dual phosphodiesterase 2/phosphodiesterase 10 (PDE2/PDE10) inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Helal, C.J.; Chappie, T.A.; Humphrey, J.M.; Verhoest, P.R.; Yang, E. Imidazo[5,1-f][1,2,4]triazines for the Treatment of Neurological Disorders. U.S. Patent WO 2012/114222 A1, 23 August 2012. [Google Scholar]
- Schmidt, B.; Weinbrenner, S.; Flockerzi, D.; Kuelzer, R.; Tenor, H.; Kley, H.P. Triazolophthalazines. Patent WO 2006/024640 A2, 09 March 2006. [Google Scholar]
- Flockerzi, D.; Kley, H.P.; Kuelzer, R.; Schmidt, B.; Tenor, H.; Weinbrenner, S. Triazolophthalazines as PDE2-inhibitors. Patent WO 2006/072612 A2, 13 July 2006. [Google Scholar]
- De Leon, P.; Egbertson, M.; Hills, I.D.; Johnson, A.W.; Machacek, M. Quinolinone PDE2 Inhibitors. U.S. Patent WO 2011/011312 A1, 27 January 2011. [Google Scholar]
- Andres, J.I.; Rombouts, F.J.R.; Trabanco, A.A.; Vanhoof, G.C.P.; de Angelis, M.; Buijnsters, P.J.J.A.; Guillemont, J.E.G.; Bormans, G.M.R.; Celen, S.J.L. 1-Aryl-4-methyl-[1,2,4]triazolo[4,3-a]quinoxaline Derivatives. Patent WO 2013/000924 A1, 03 January 2013. [Google Scholar]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Stange, H.; Langen, B.; Egerland, U.; Hoefgen, N.; Priebs, M.; Malamas, M.S.; Erdel, J.J.; Ni, Y. Triazine Derivatives as Inhibitors of Phosphodiesterases. Patent WO 2010/054253 A1, 14 May 2010. [Google Scholar]
- Brust, P.; van den Hoff, J.; Steinbach, J. Development of 18F-labeled radiotracers for neuroreceptor imaging with positron emission tomography. Neurosci. Bull. 2014, 30, 777–811. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Ni, Y.; Erdei, J.; Stange, H.; Schindler, R.; Lankau, H.J.; Grunwald, C.; Fan, K.Y.; Parris, K.; Langen, B.; et al. Highly potent, selective, and orally active phosphodiesterase 10A inhibitors. J. Med. Chem. 2011, 54, 7621–7638. [Google Scholar] [CrossRef] [PubMed]
- Funke, U.; Deuther-Conrad, W.; Schwan, G.; Maisonial, A.; Scheunemann, M.; Fischer, S.; Hiller, A.; Briel, D.; Brust, P. Radiosynthesis and radiotracer properties of a 7-(2-[18F]fluoroethoxy)-6-methoxypyrrolidinylquinazoline for imaging of phosphodiesterase 10A with PET. Pharmaceuticals 2012, 5, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, Q.; Dai, D.; Huang, Q. X-ray crystal structure of phosphodiesterase 2 in complex with a highly selective, nanomolar inhibitor reveals a binding-induced pocket important for selectivity. J. Am. Chem. Soc. 2013, 135, 11708–11711. [Google Scholar] [CrossRef] [PubMed]
- Blom, E.; Karimi, F.; Eriksson, O.; Hall, H.; Långström, B. Synthesis and in vitro evaluation of 18F-β-carboline alkaloids as PET ligands. J. Label. Compd. Rad. 2008, 51, 277–282. [Google Scholar]
- Nakao, R.; Schou, M.; Halldin, C. Direct plasma metabolite analysis of positron emission tomography radioligands by micellar liquid chromatography with radiometric detection. Anal. Chem. 2012, 84, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Rambla-Alegre, M. Basic principles of MLC. Chromatogr. Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET imaging of the dopamine transporter with 18F-FECNT: A polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Evens, N.; Vandeputte, C.; Muccioli, G.G.; Lambert, D.M.; Baekelandt, V.; Verbruggen, A.M.; Debyser, Z.; van Laere, K.; Bormans, G.M. Synthesis, in vitro and in vivo evaluation of fluorine-18 labelled FE-GW405833 as a PET tracer for type 2 cannabinoid receptor imaging. Bioorg. Med. Chem. 2011, 19, 4499–4505. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.H.; Chan, H.K.; Miller, J.D.; Jayne, C.L.; Eichhorn, D.M. Synthesis, modification, and characterization of a family of homologues of exo-calix[4]arene: Exo-[n.m.n.m]metacyclophanes, n,m ≥ 3. J. Org. Chem. 2000, 65, 5185–5196. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds TA1–4 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schröder, S.; Wenzel, B.; Deuther-Conrad, W.; Teodoro, R.; Egerland, U.; Kranz, M.; Scheunemann, M.; Höfgen, N.; Steinbach, J.; Brust, P. Synthesis, 18F-Radiolabelling and Biological Characterization of Novel Fluoroalkylated Triazine Derivatives for in Vivo Imaging of Phosphodiesterase 2A in Brain via Positron Emission Tomography. Molecules 2015, 20, 9591-9615. https://doi.org/10.3390/molecules20069591
Schröder S, Wenzel B, Deuther-Conrad W, Teodoro R, Egerland U, Kranz M, Scheunemann M, Höfgen N, Steinbach J, Brust P. Synthesis, 18F-Radiolabelling and Biological Characterization of Novel Fluoroalkylated Triazine Derivatives for in Vivo Imaging of Phosphodiesterase 2A in Brain via Positron Emission Tomography. Molecules. 2015; 20(6):9591-9615. https://doi.org/10.3390/molecules20069591
Chicago/Turabian StyleSchröder, Susann, Barbara Wenzel, Winnie Deuther-Conrad, Rodrigo Teodoro, Ute Egerland, Mathias Kranz, Matthias Scheunemann, Norbert Höfgen, Jörg Steinbach, and Peter Brust. 2015. "Synthesis, 18F-Radiolabelling and Biological Characterization of Novel Fluoroalkylated Triazine Derivatives for in Vivo Imaging of Phosphodiesterase 2A in Brain via Positron Emission Tomography" Molecules 20, no. 6: 9591-9615. https://doi.org/10.3390/molecules20069591
APA StyleSchröder, S., Wenzel, B., Deuther-Conrad, W., Teodoro, R., Egerland, U., Kranz, M., Scheunemann, M., Höfgen, N., Steinbach, J., & Brust, P. (2015). Synthesis, 18F-Radiolabelling and Biological Characterization of Novel Fluoroalkylated Triazine Derivatives for in Vivo Imaging of Phosphodiesterase 2A in Brain via Positron Emission Tomography. Molecules, 20(6), 9591-9615. https://doi.org/10.3390/molecules20069591