
Development of Advanced Macrosphelides: Potent Anticancer Agents

Abstract
:1. Introduction
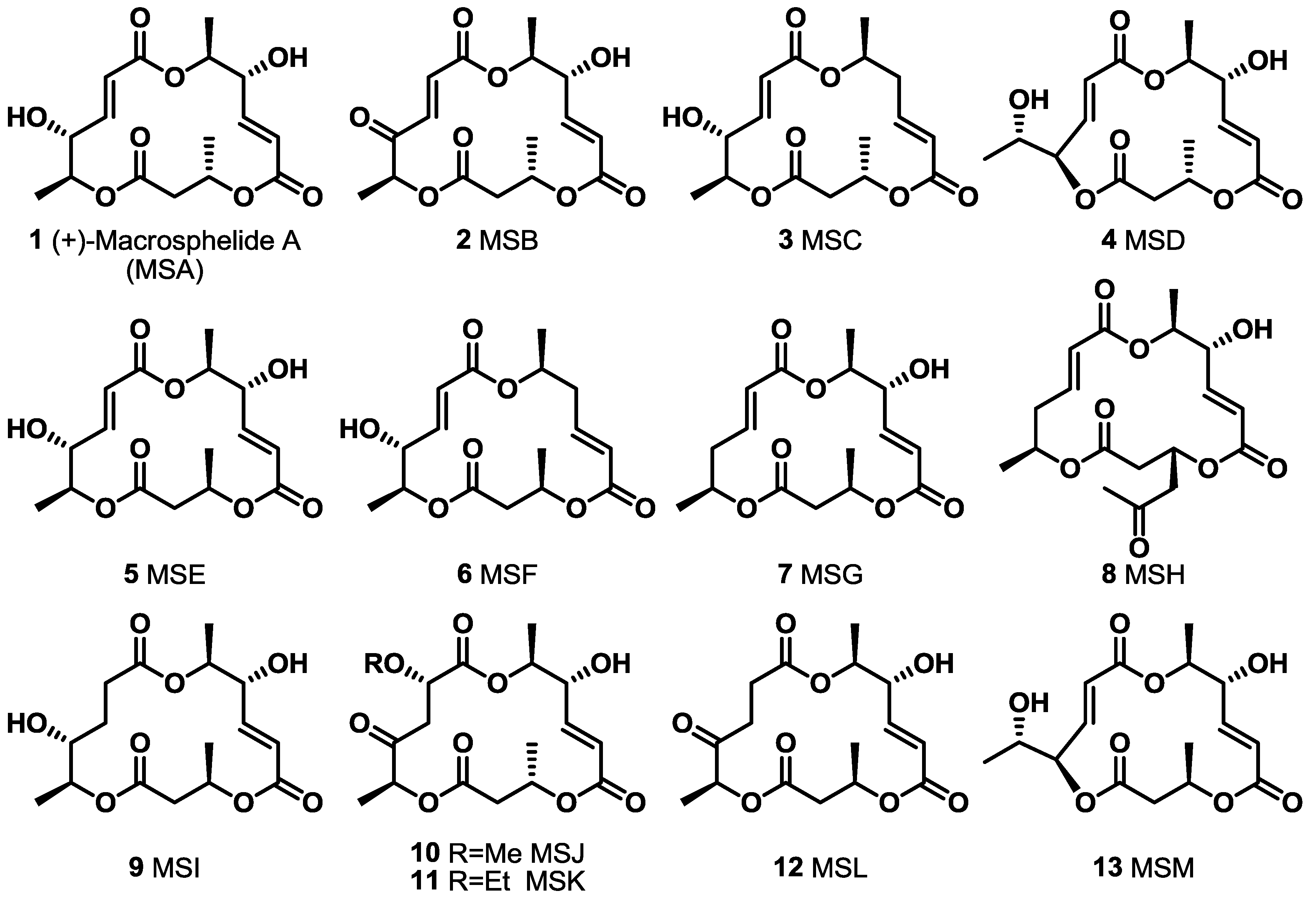
2. Results and Discussion
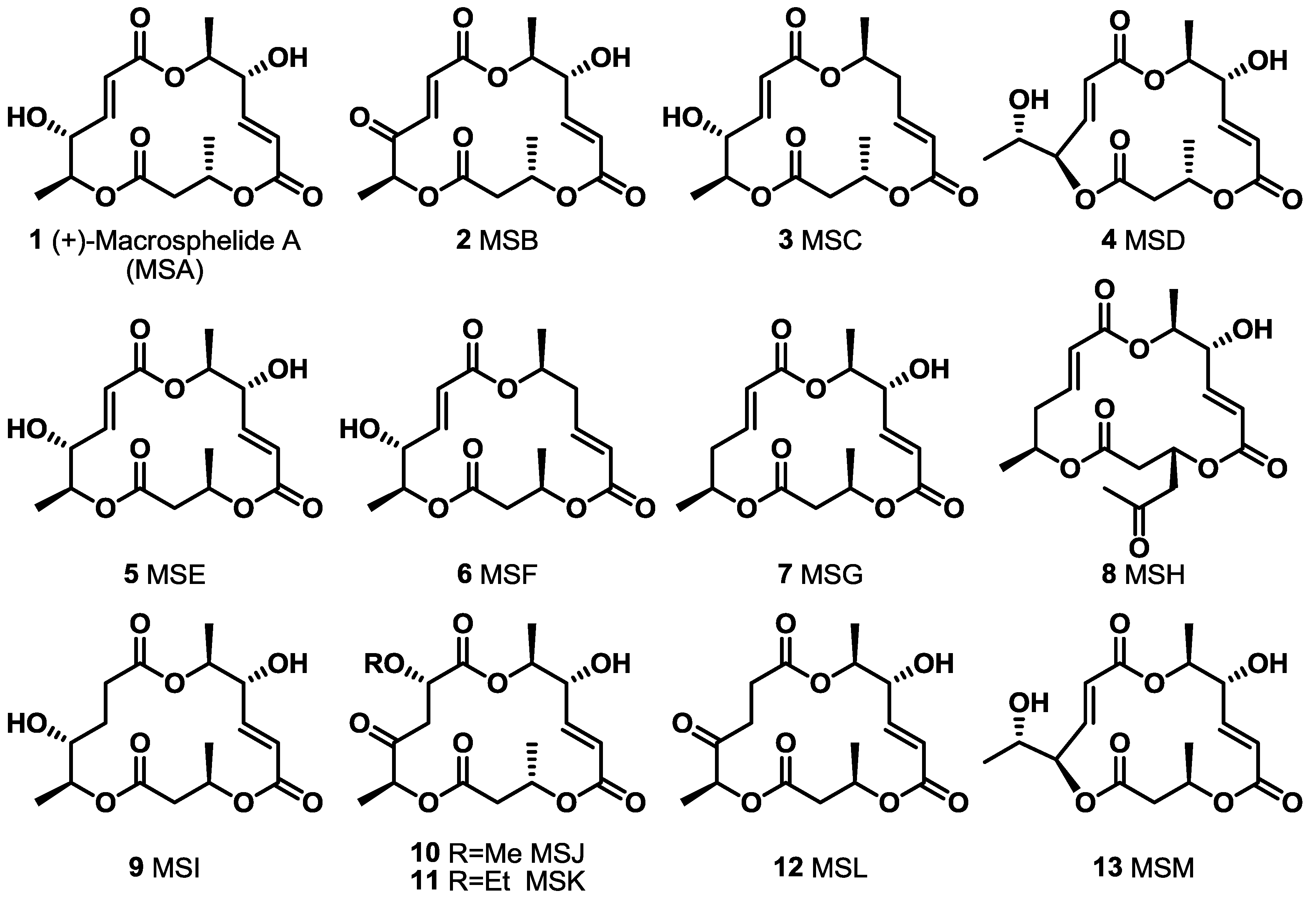
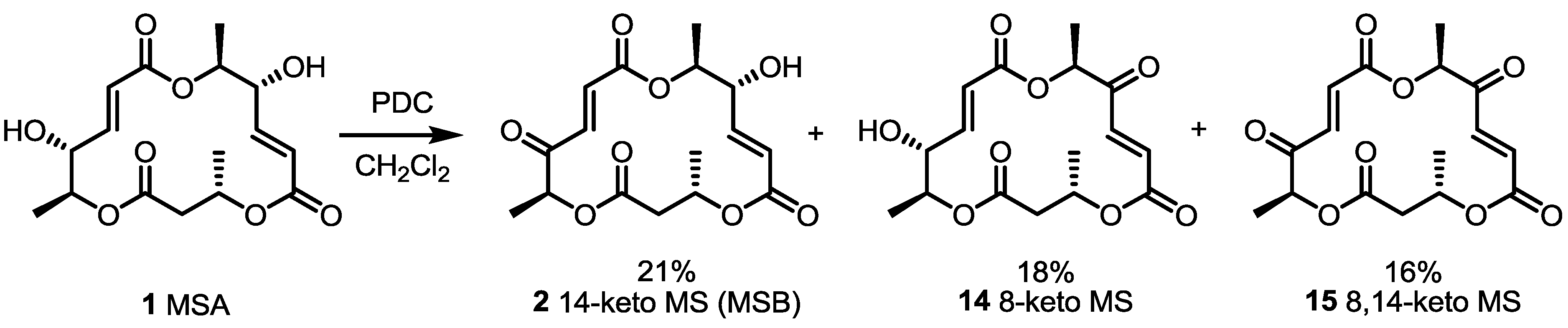
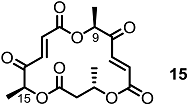
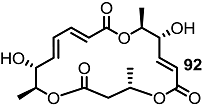
2.1. The First Unnatural Isomers of MS
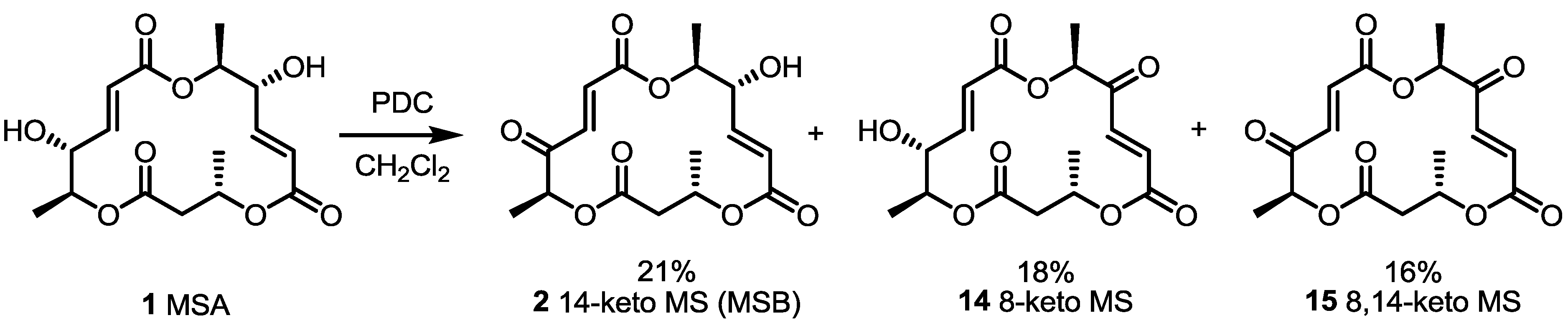
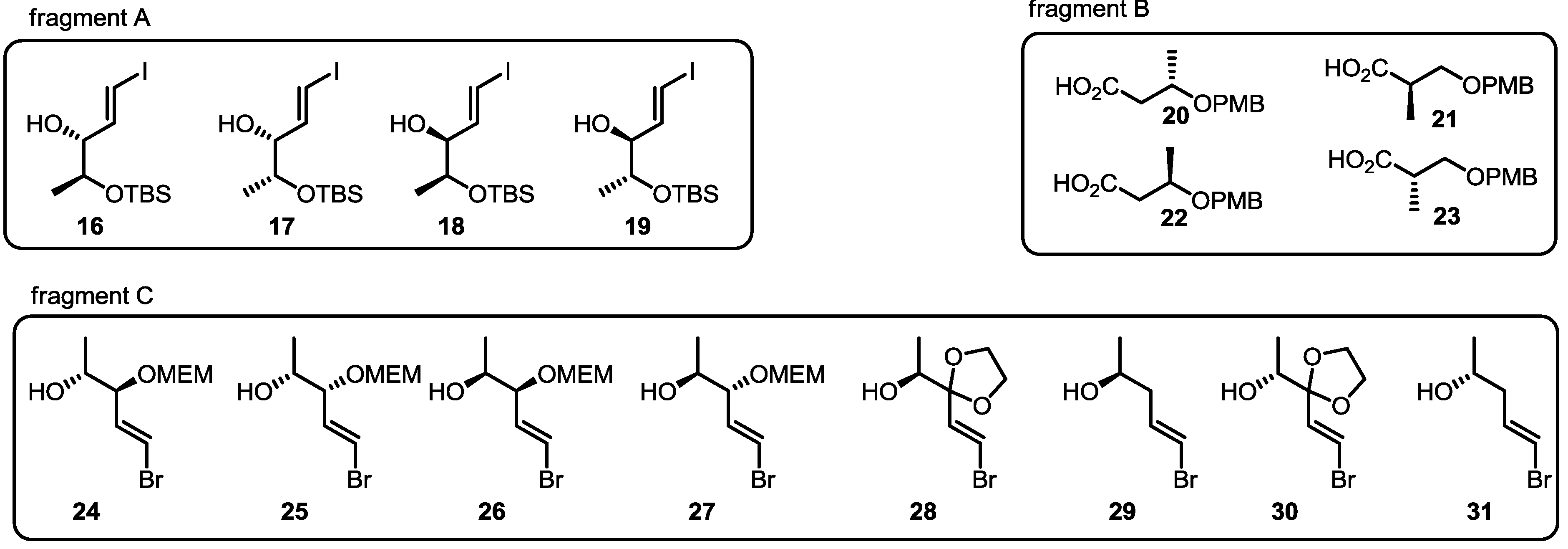
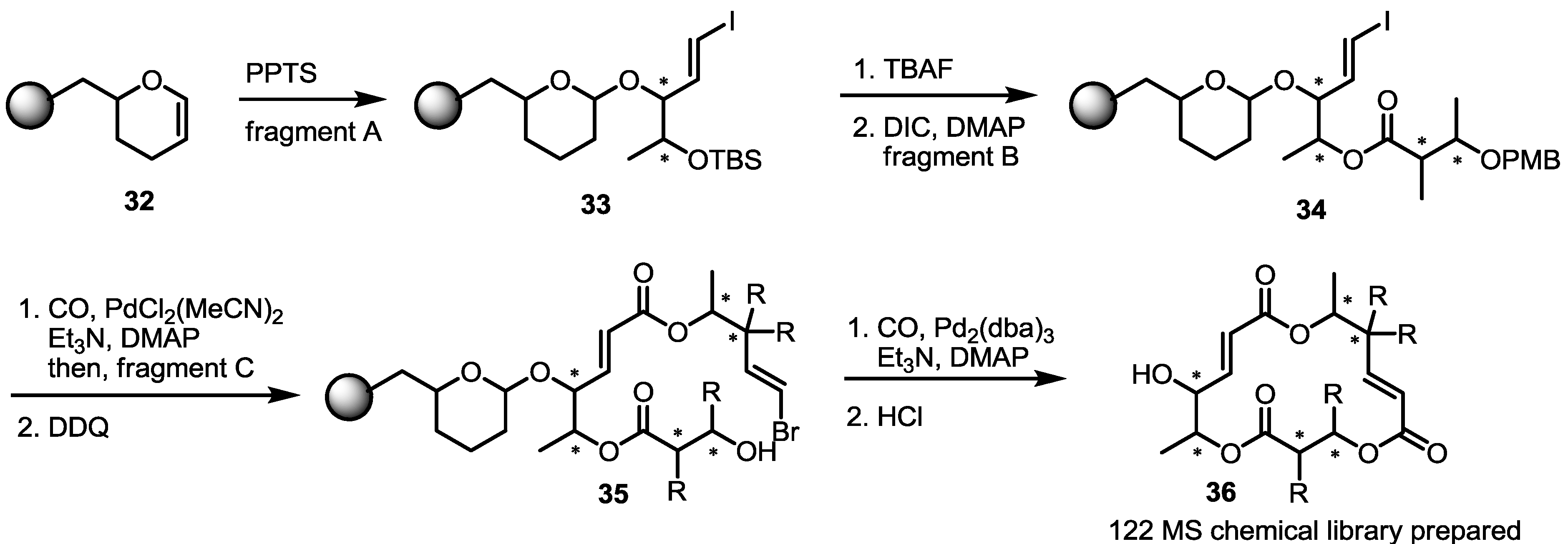
2.2. Combinatorial Chemistry of MS
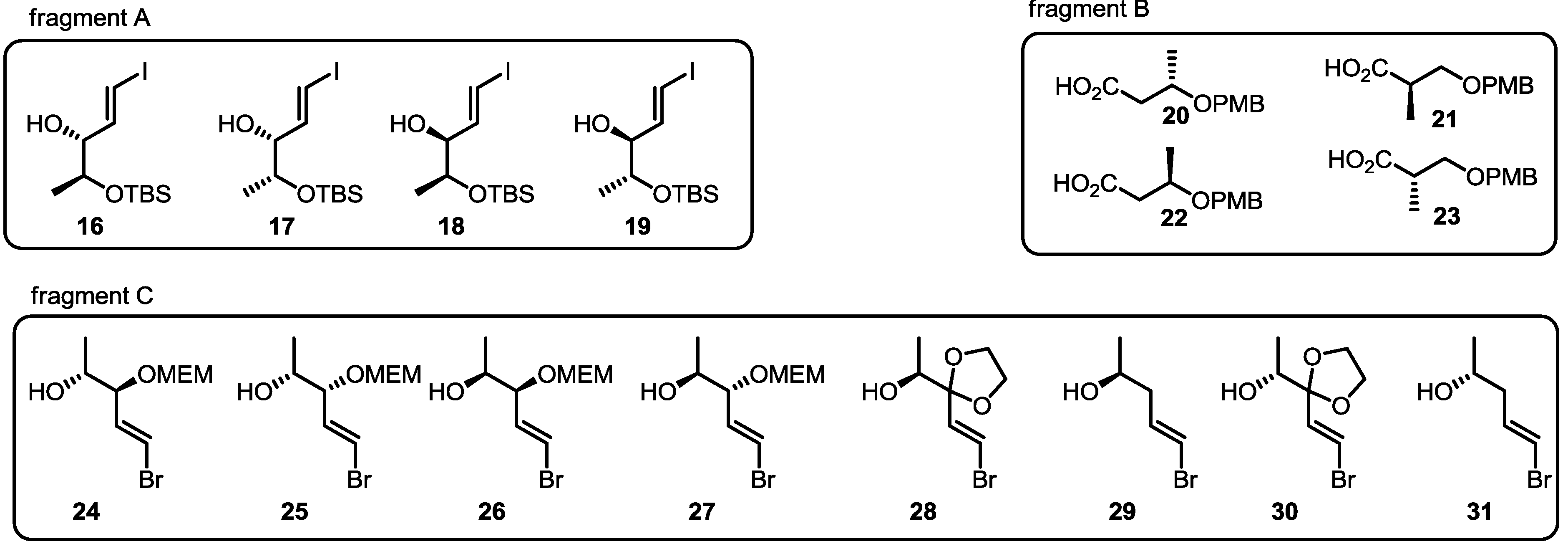
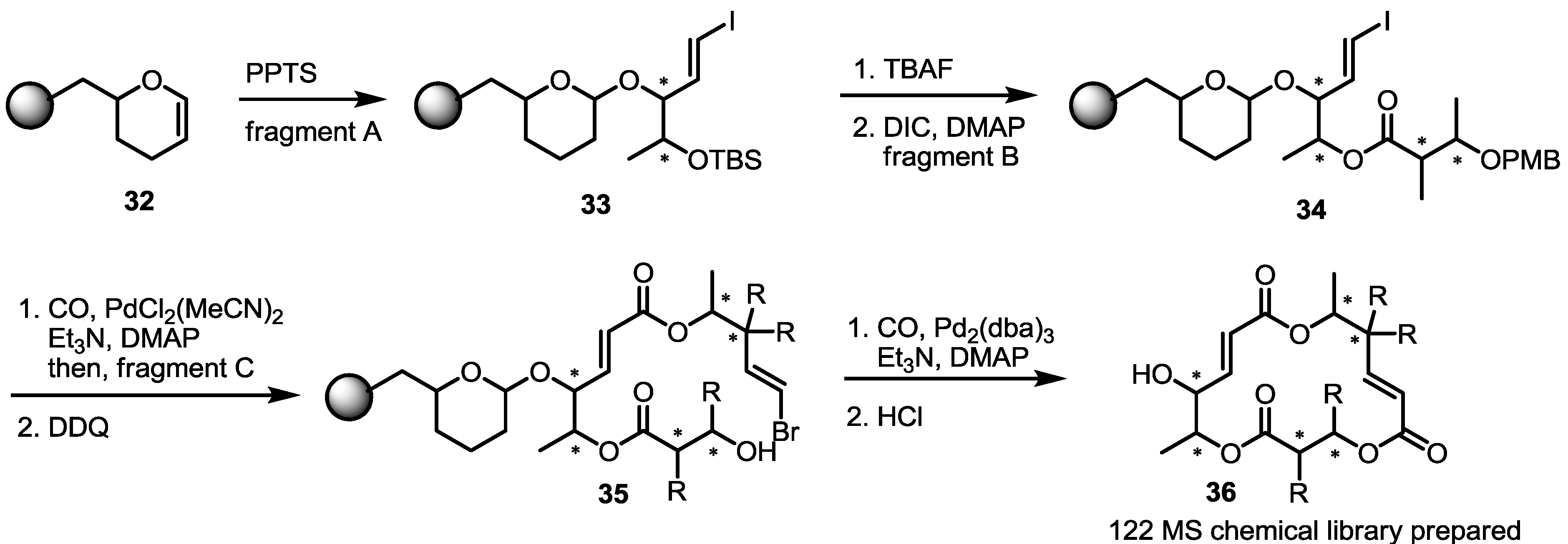
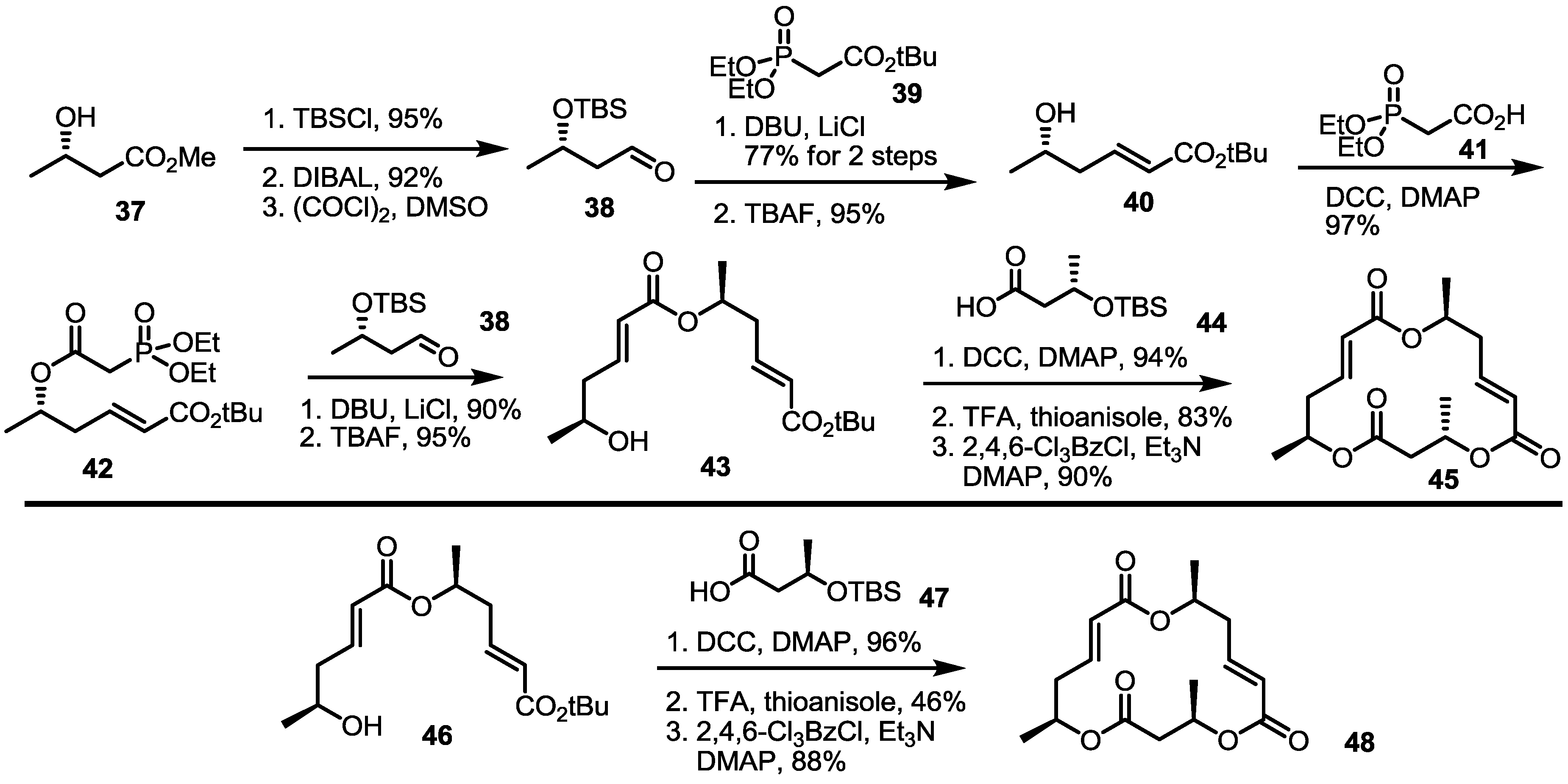
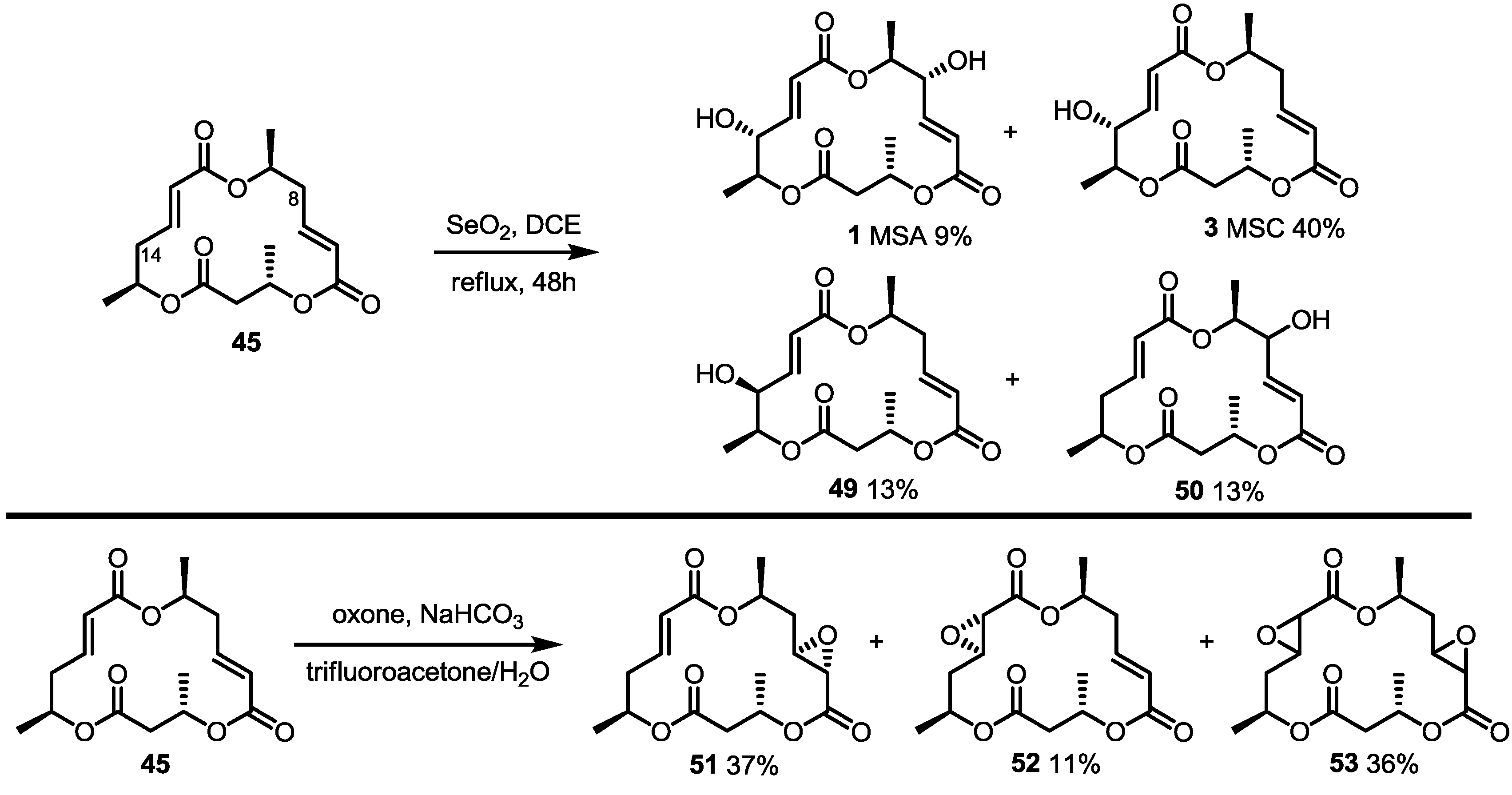
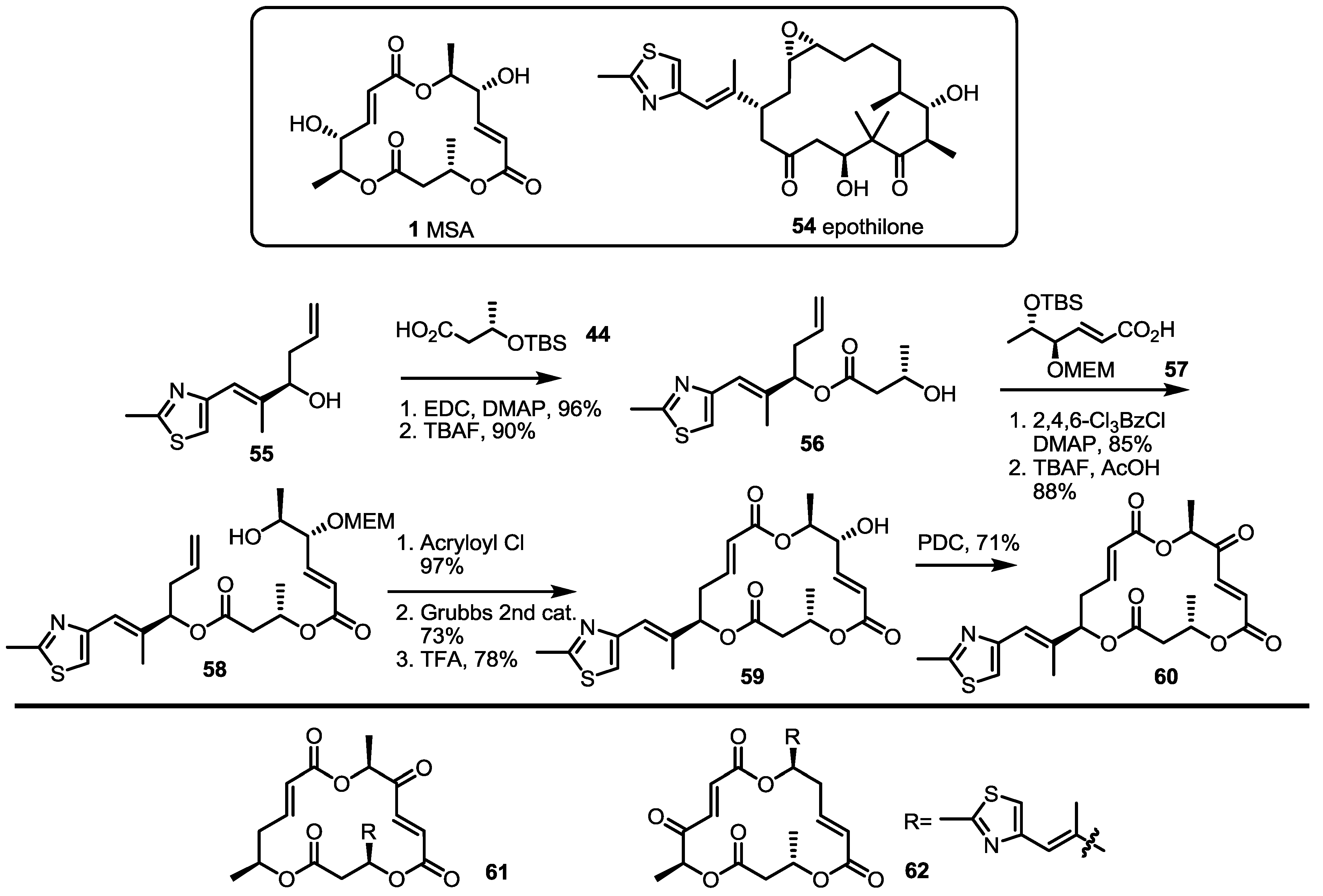
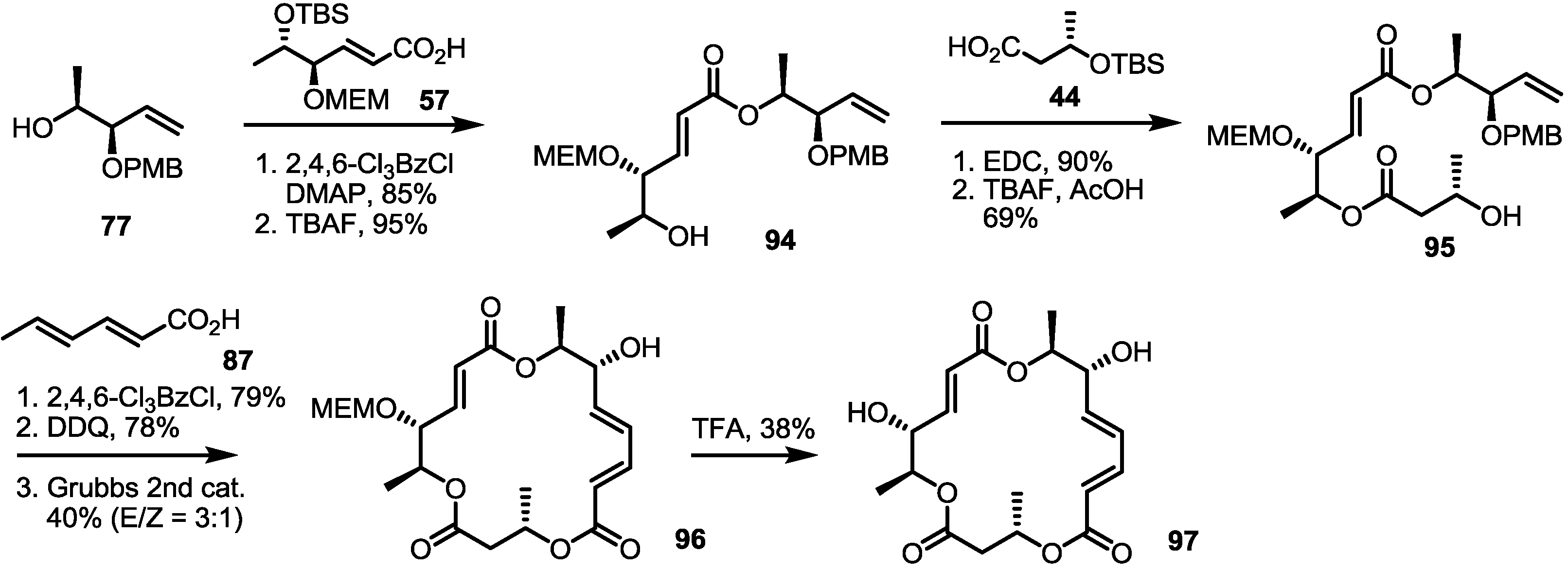
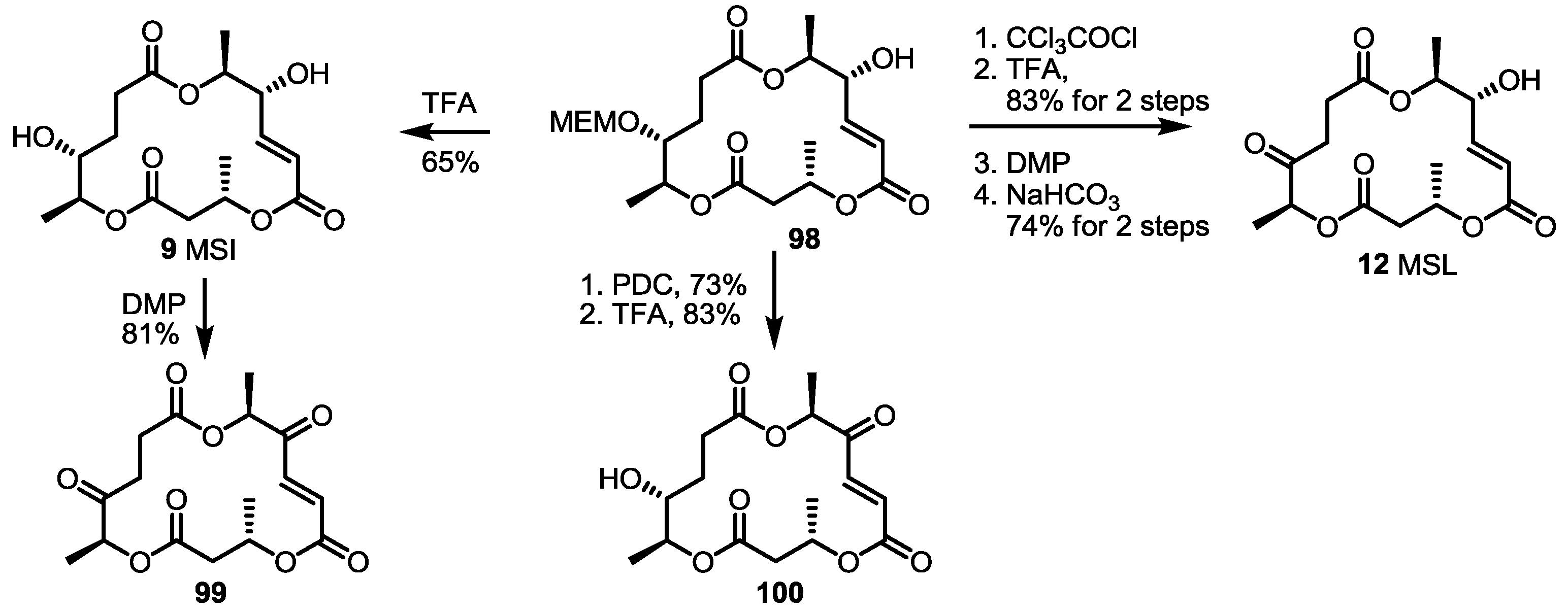
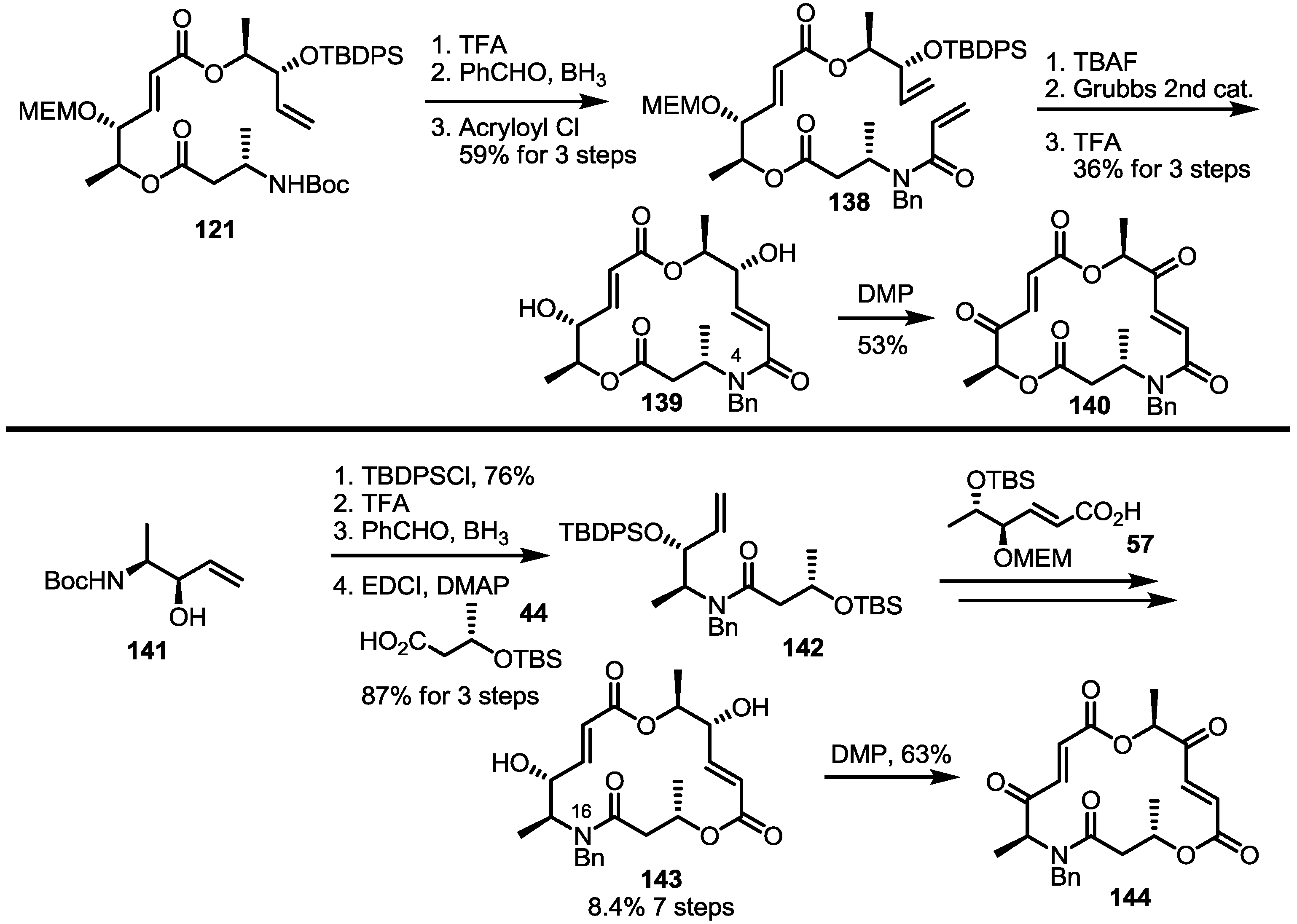
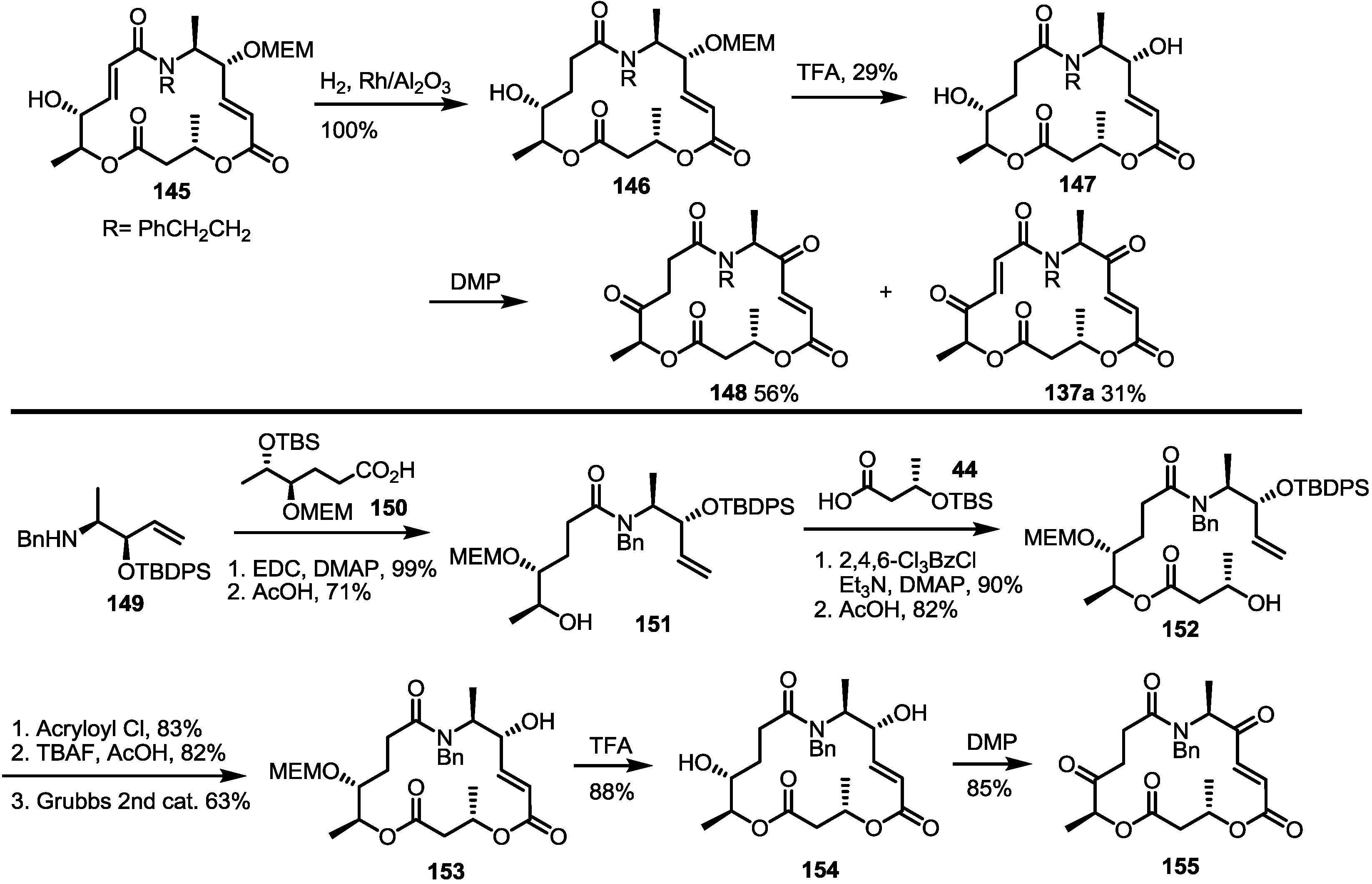
2.3. Synthesis of Core Structure and Its Derivatization
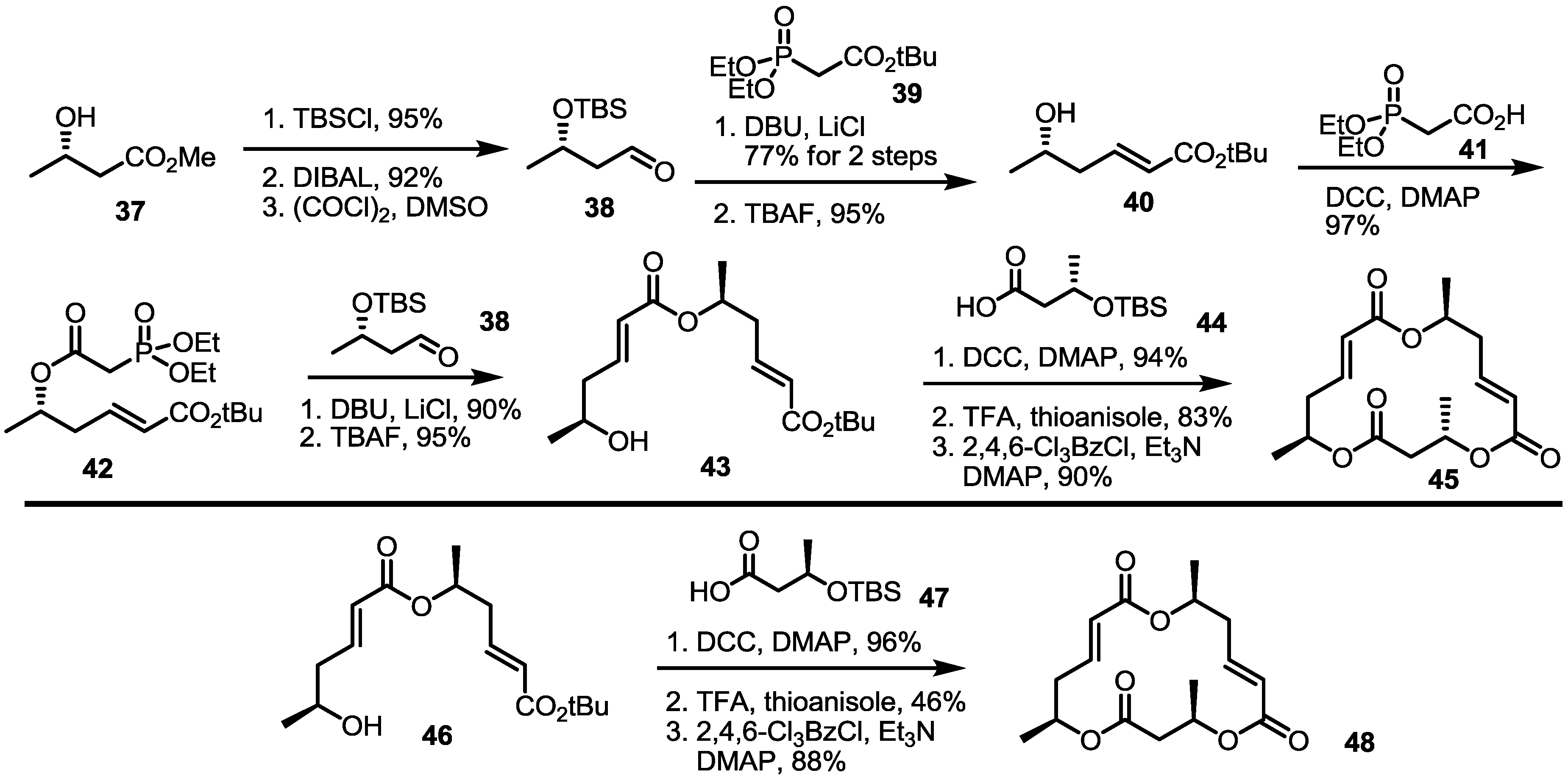
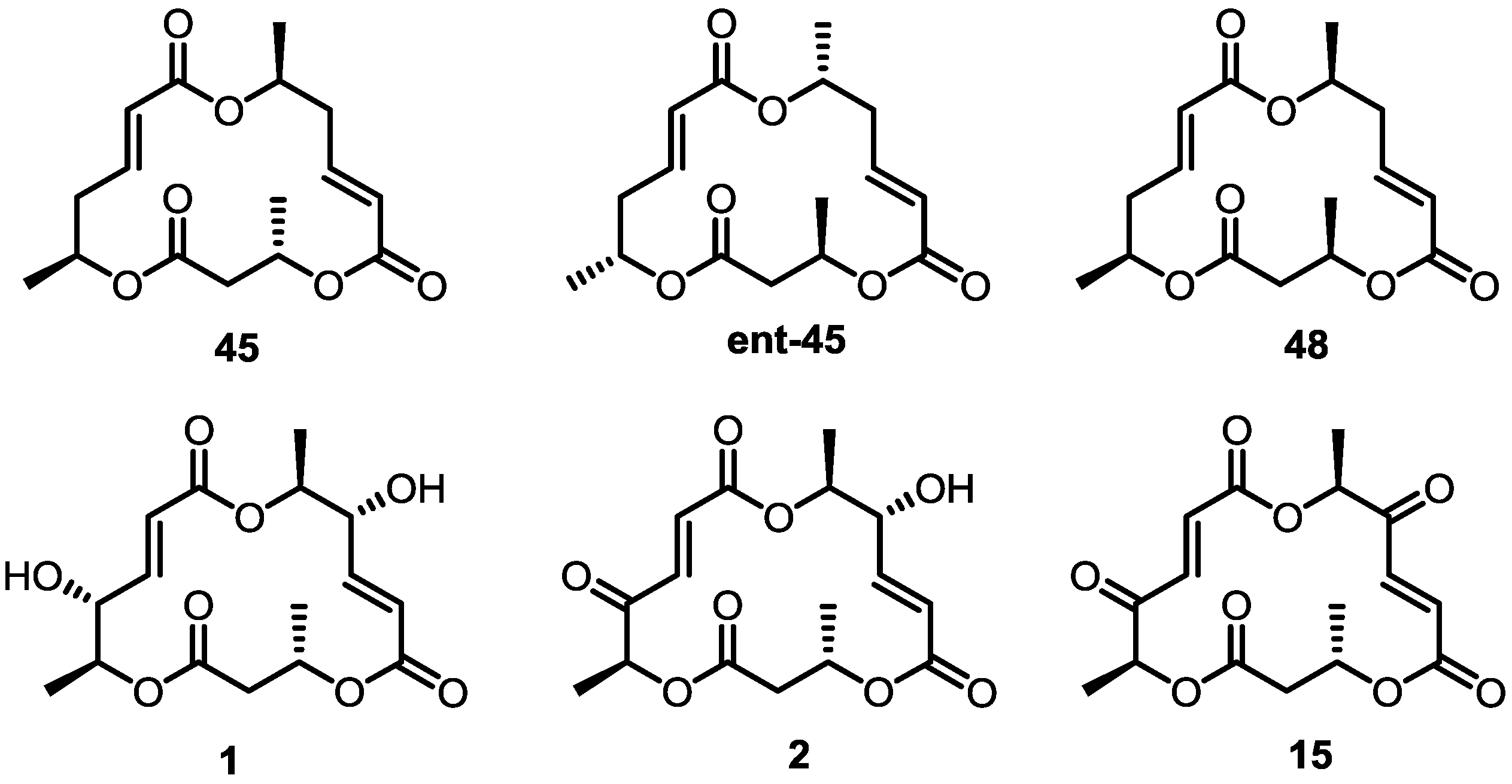
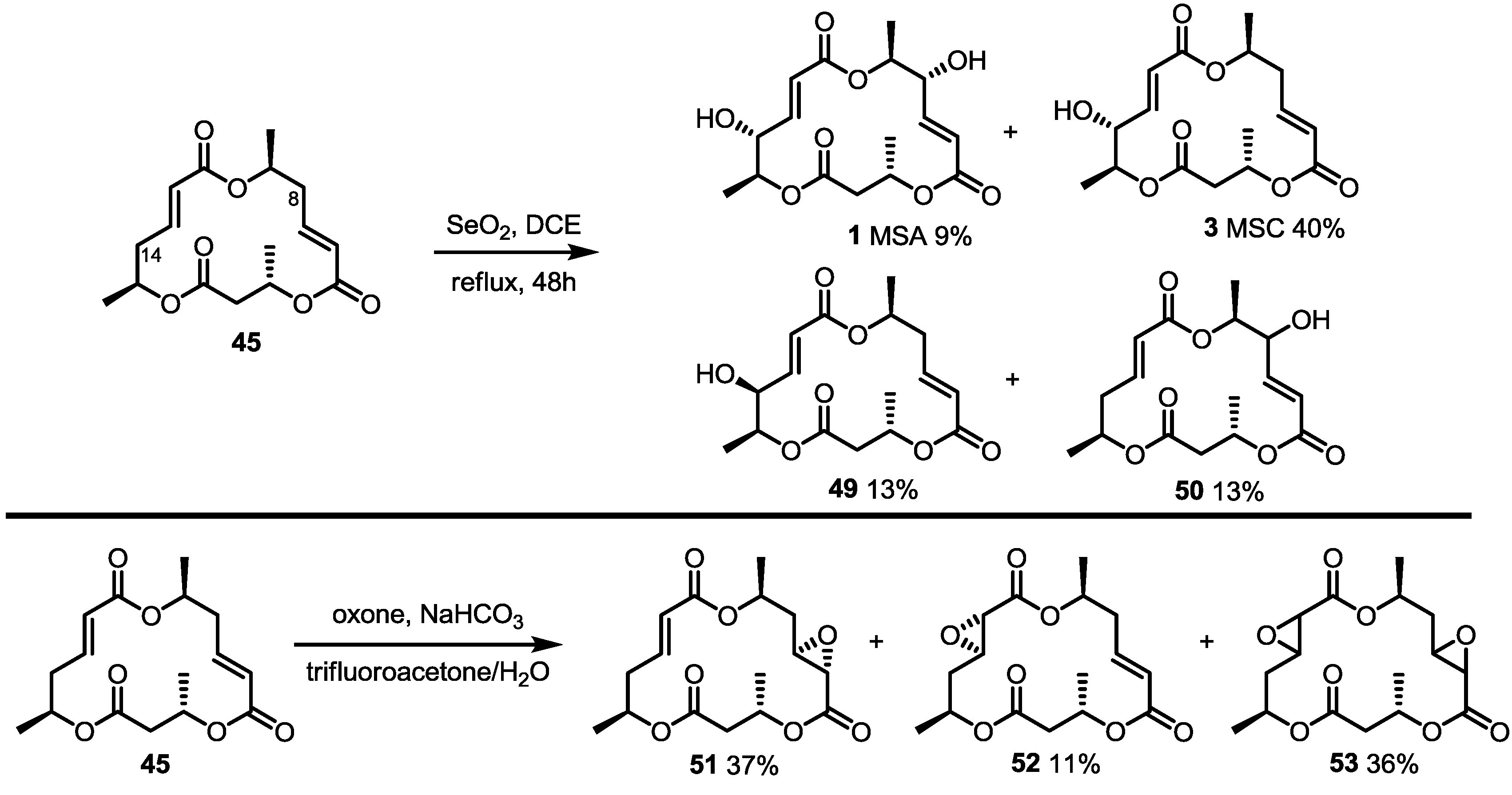
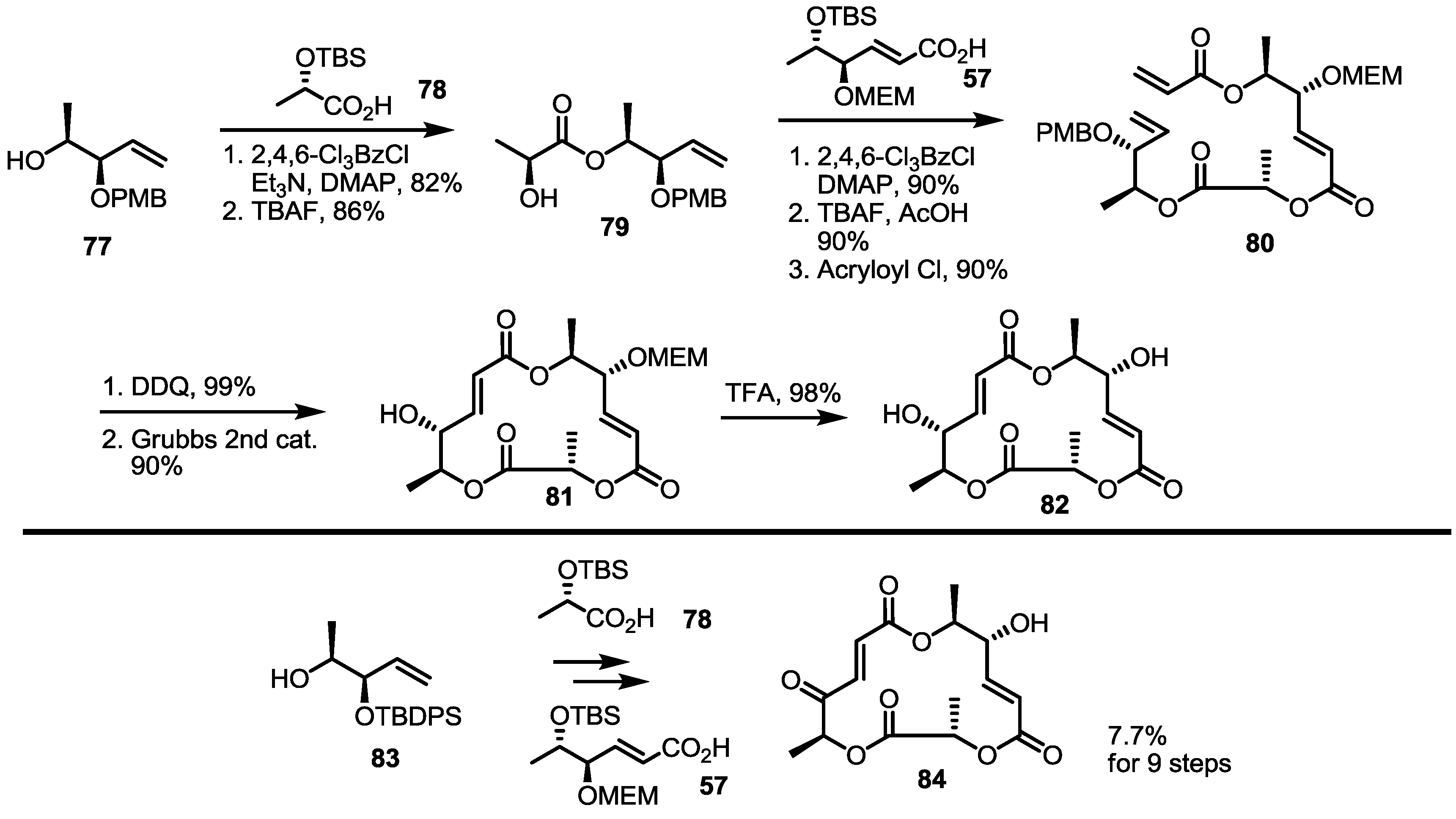
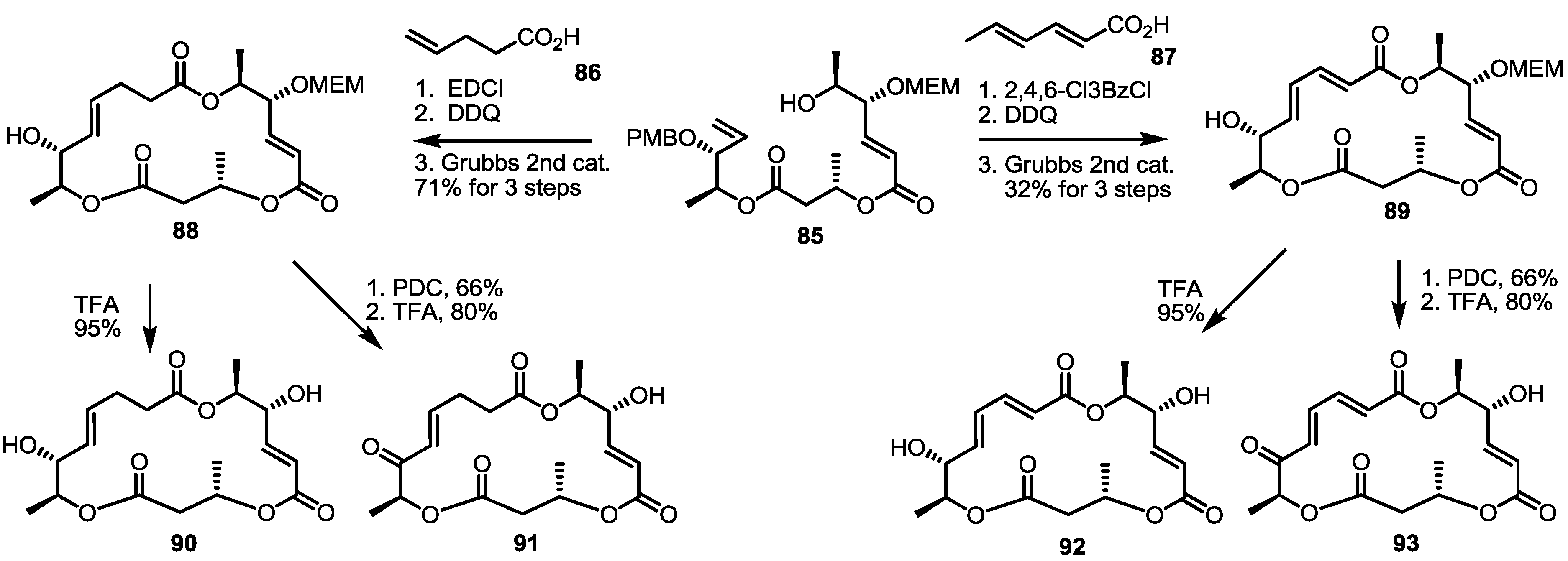
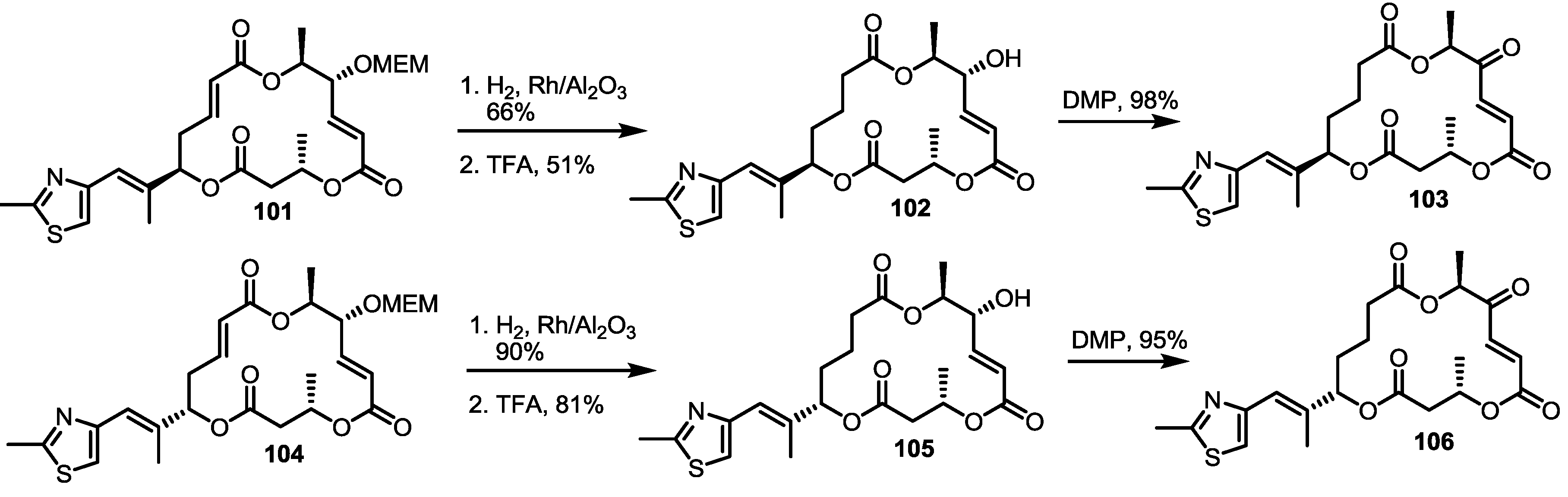
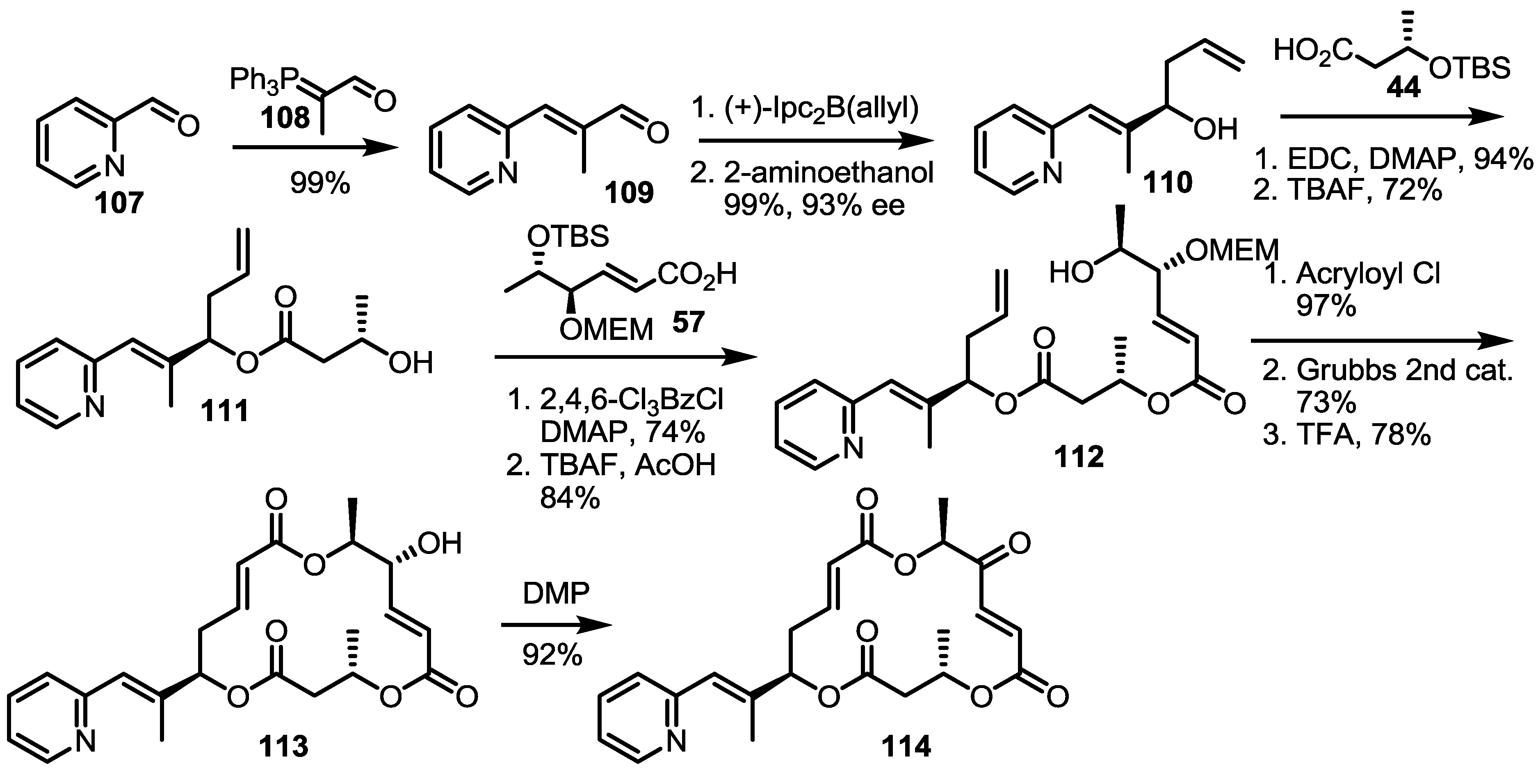
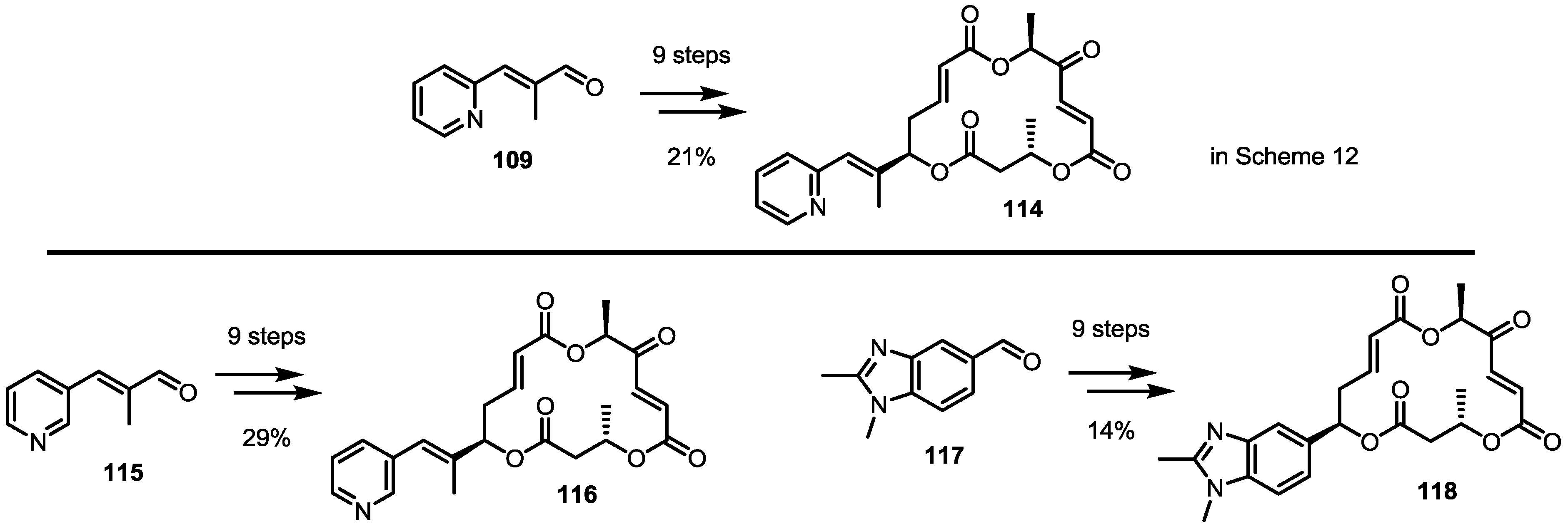
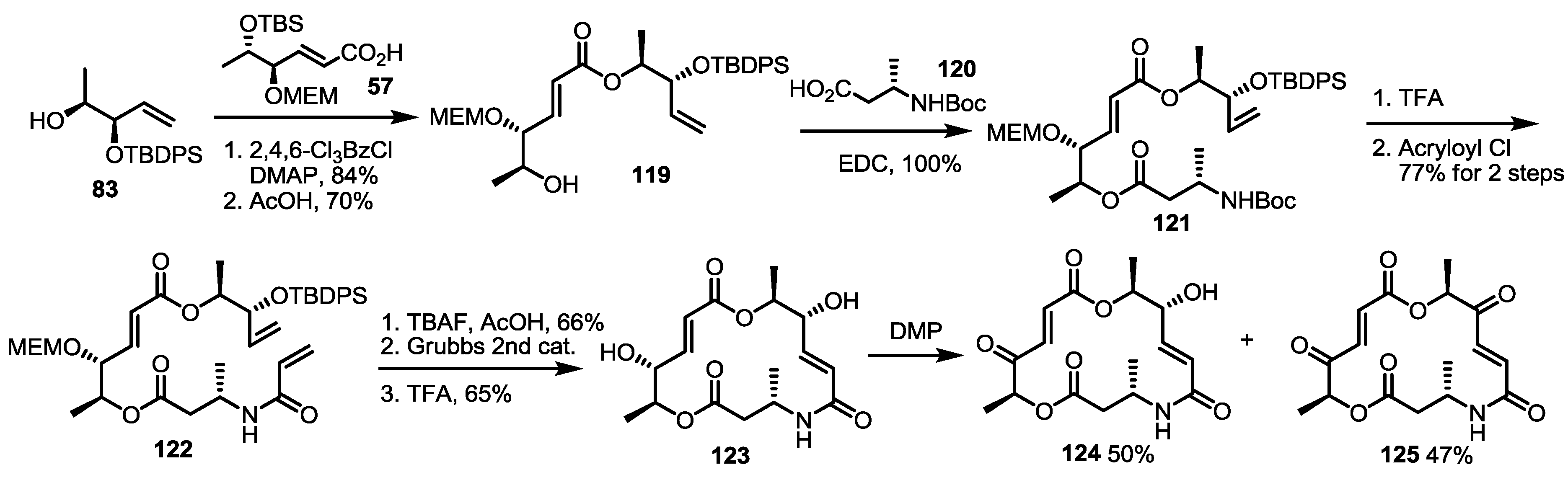
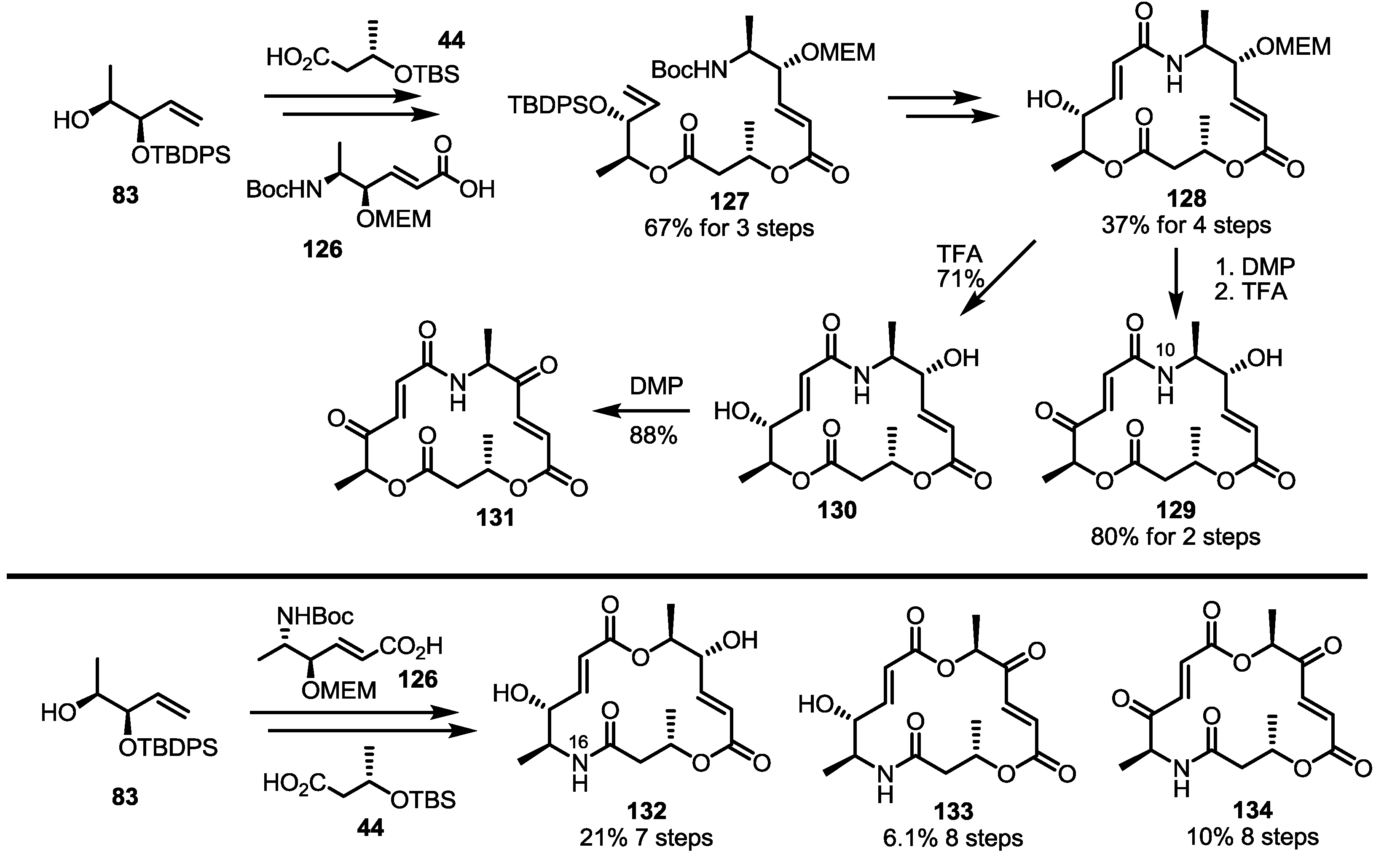
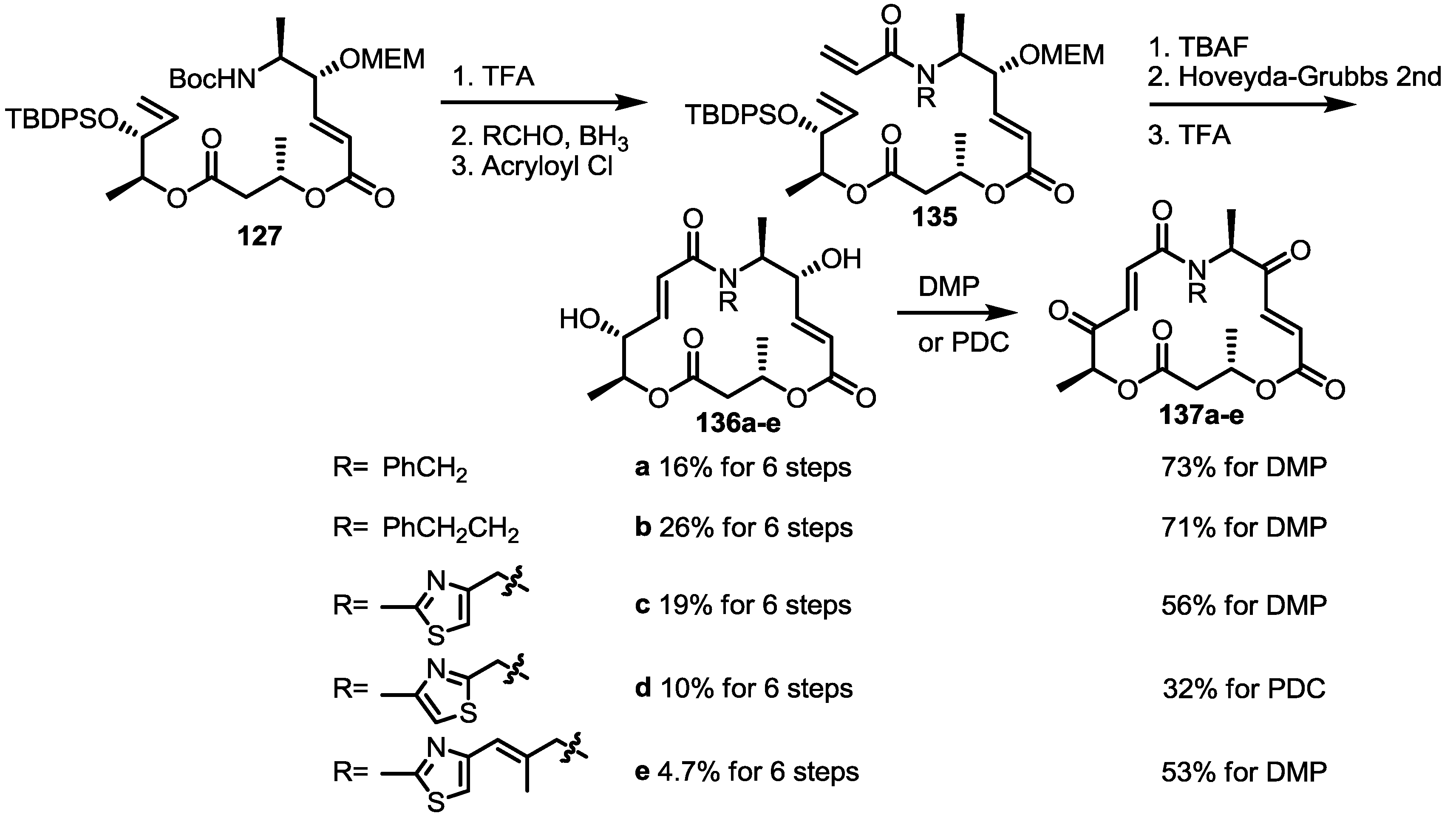
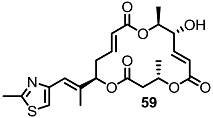
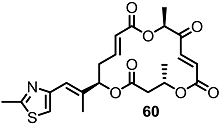
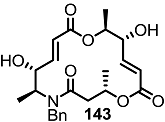
2.4. Modification of the MS Structure for Detailed SAR Studies
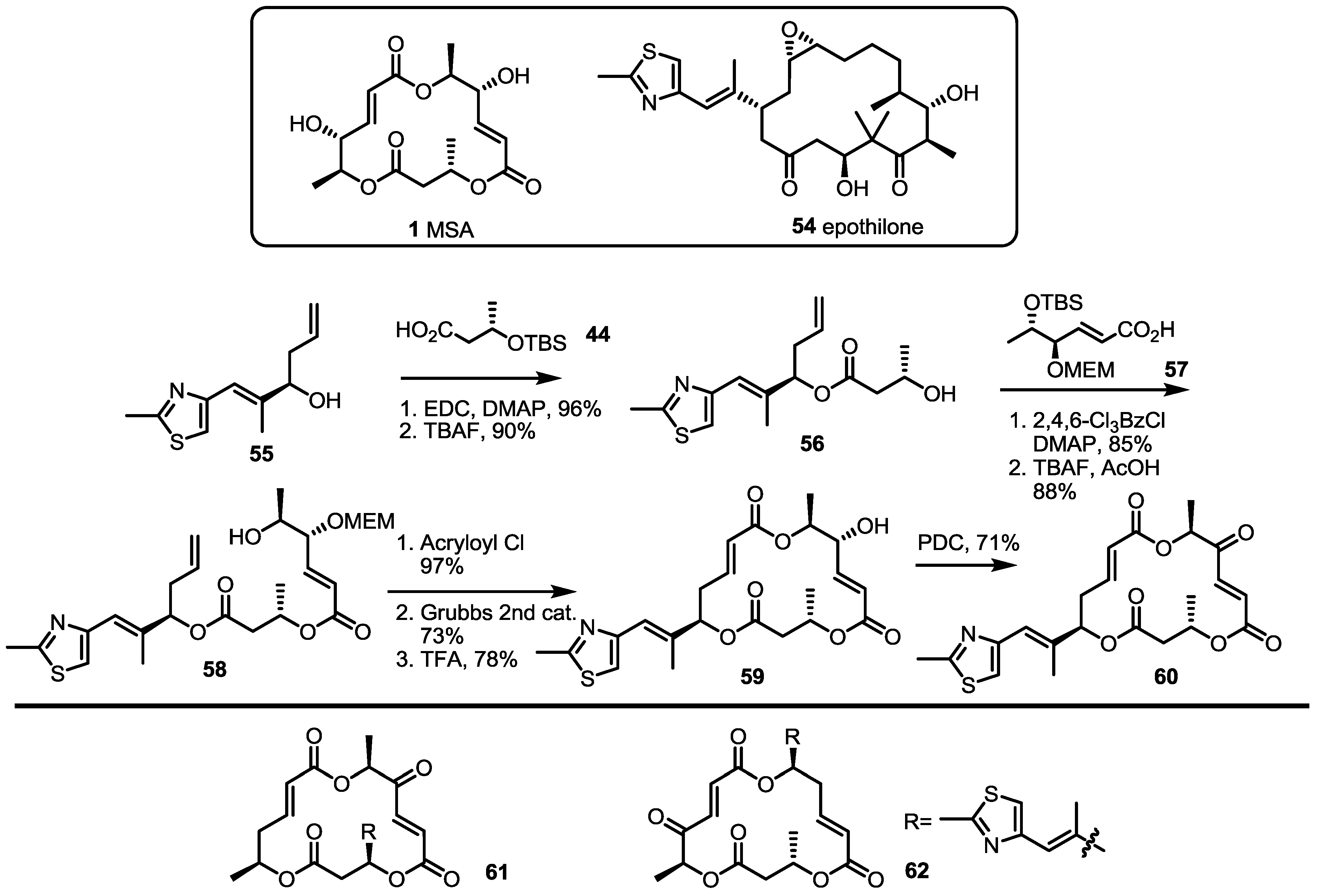
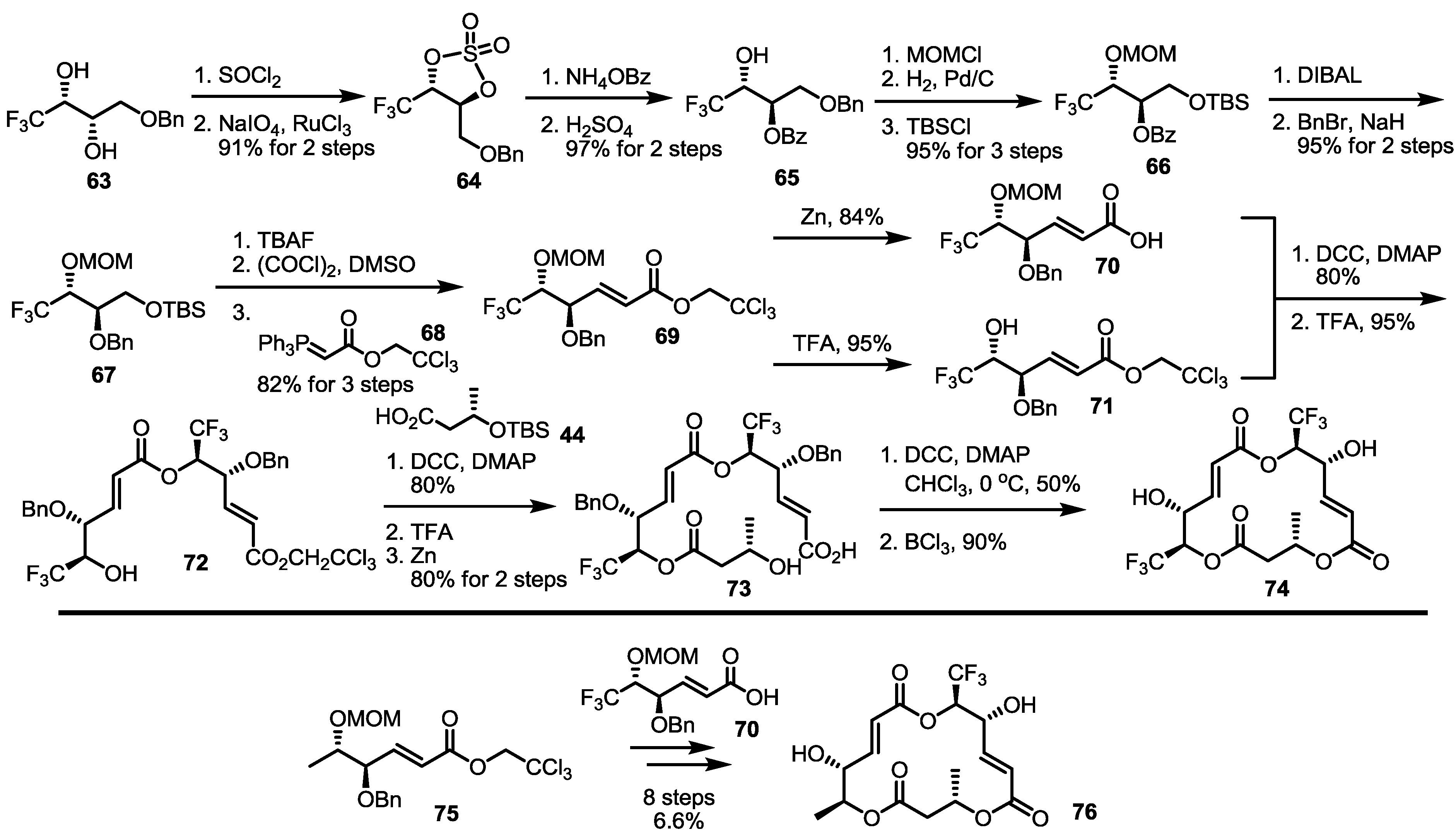
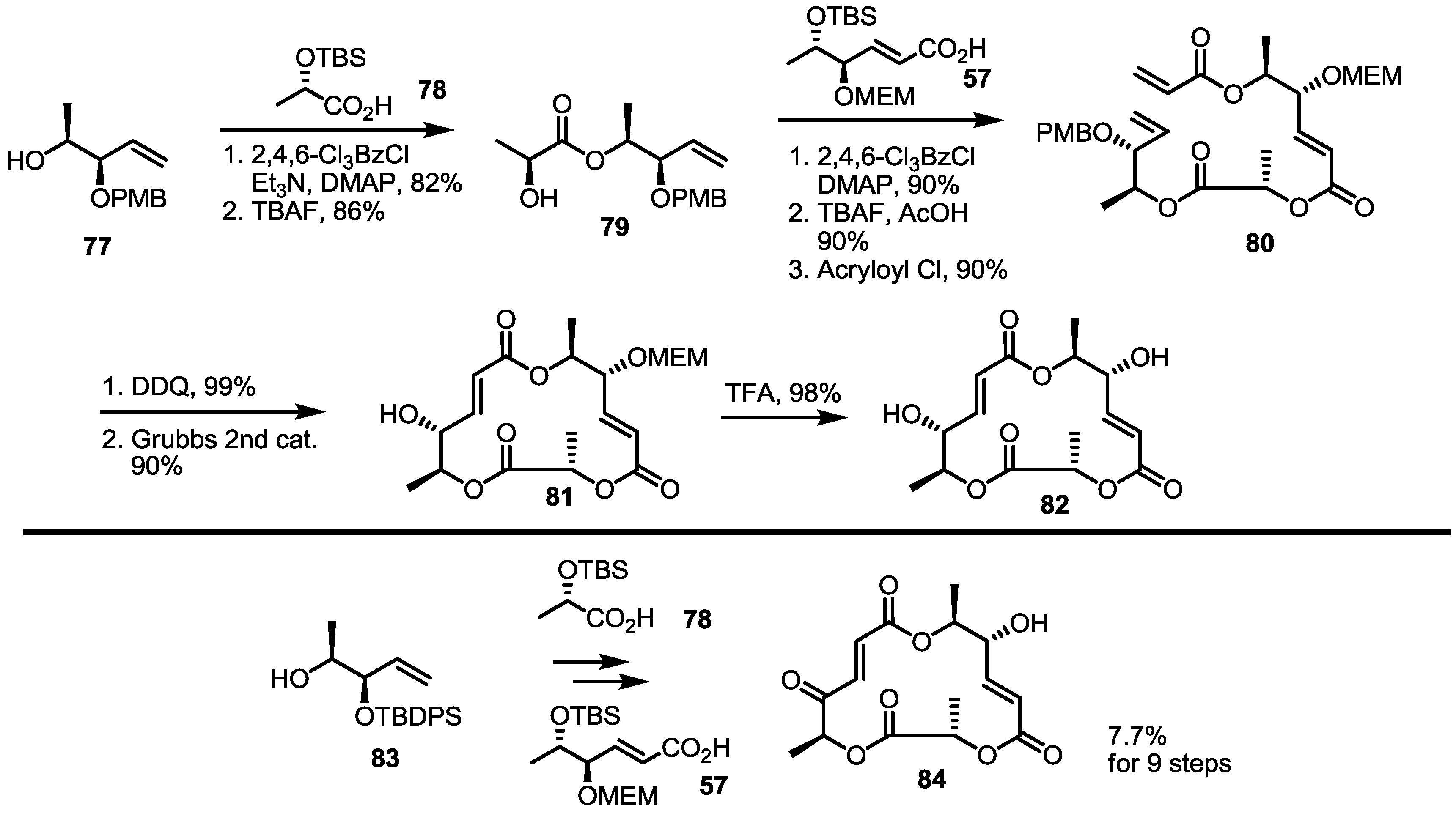
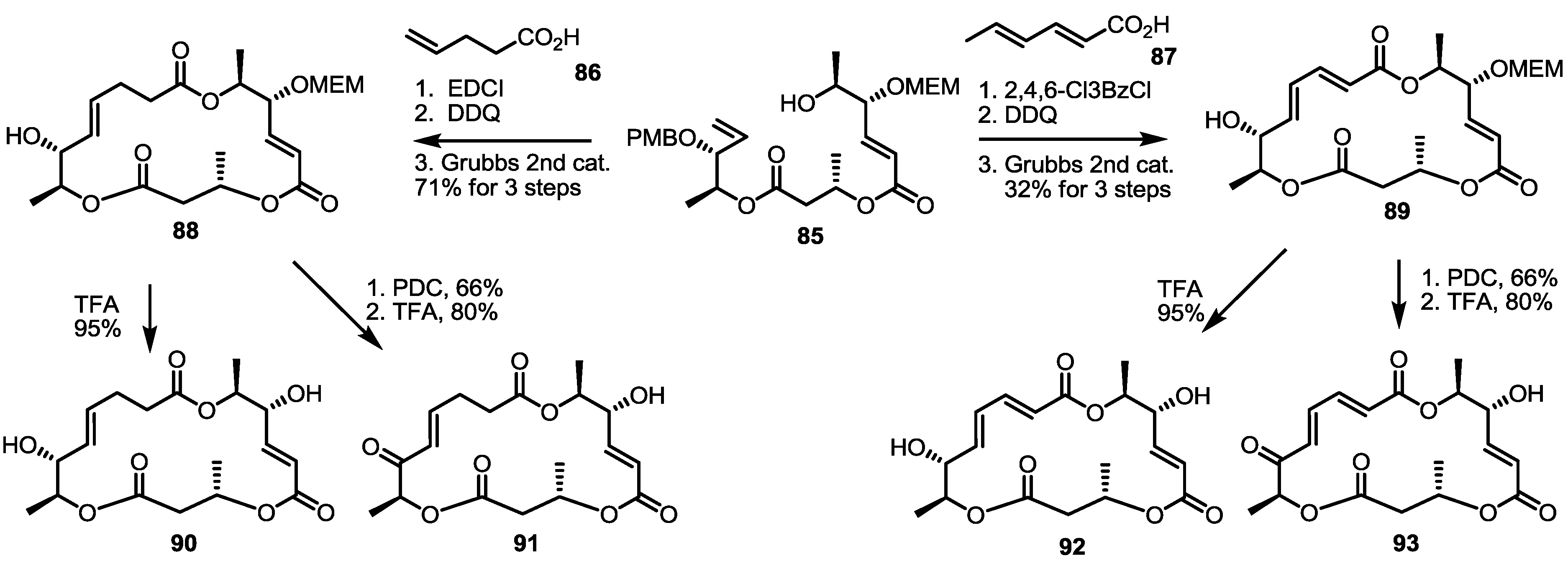
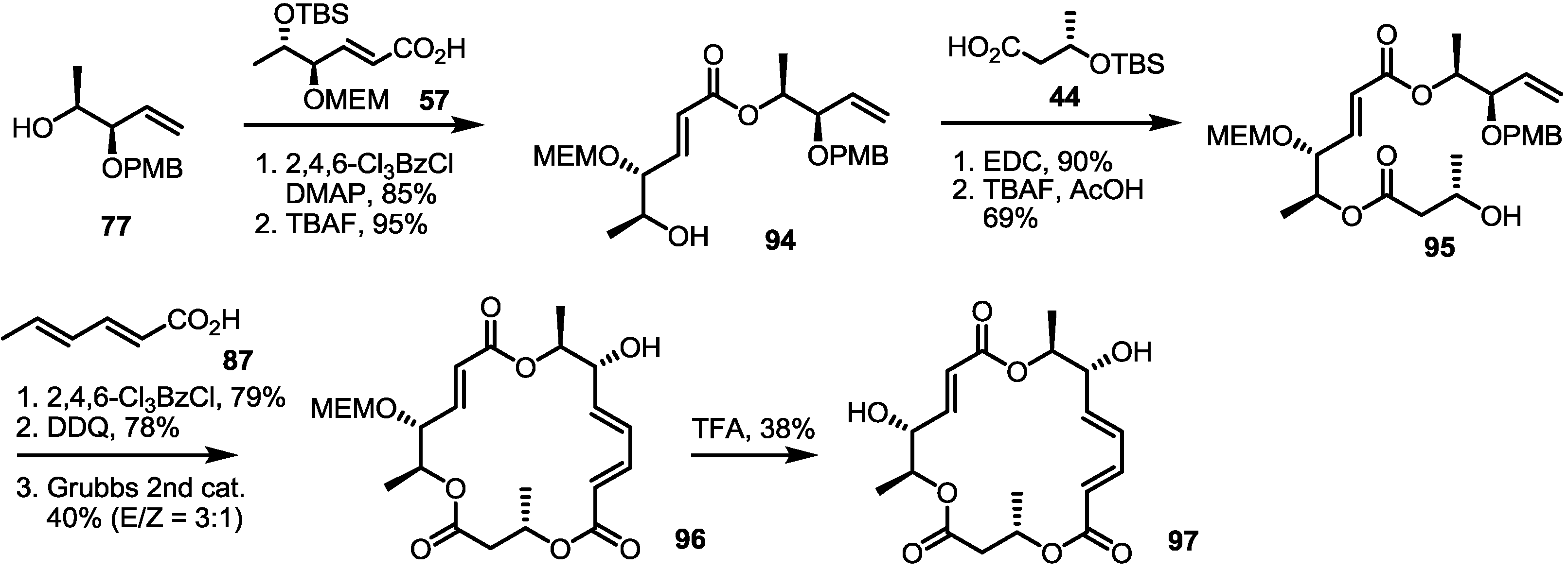
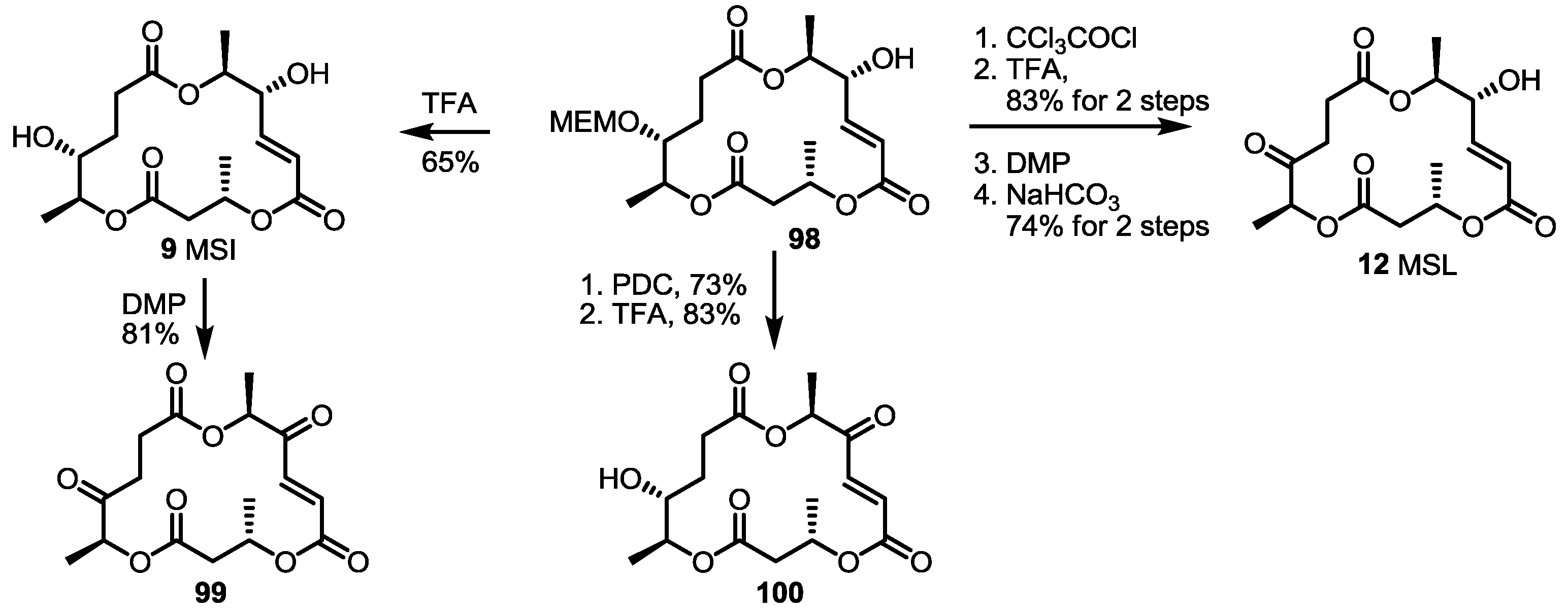
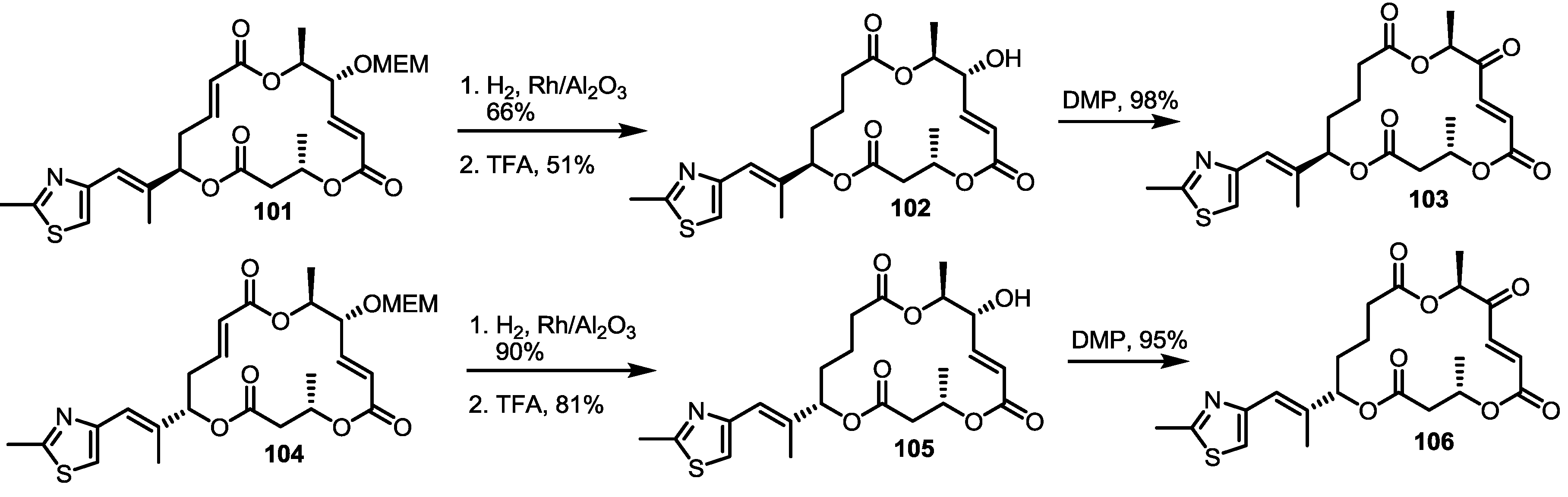
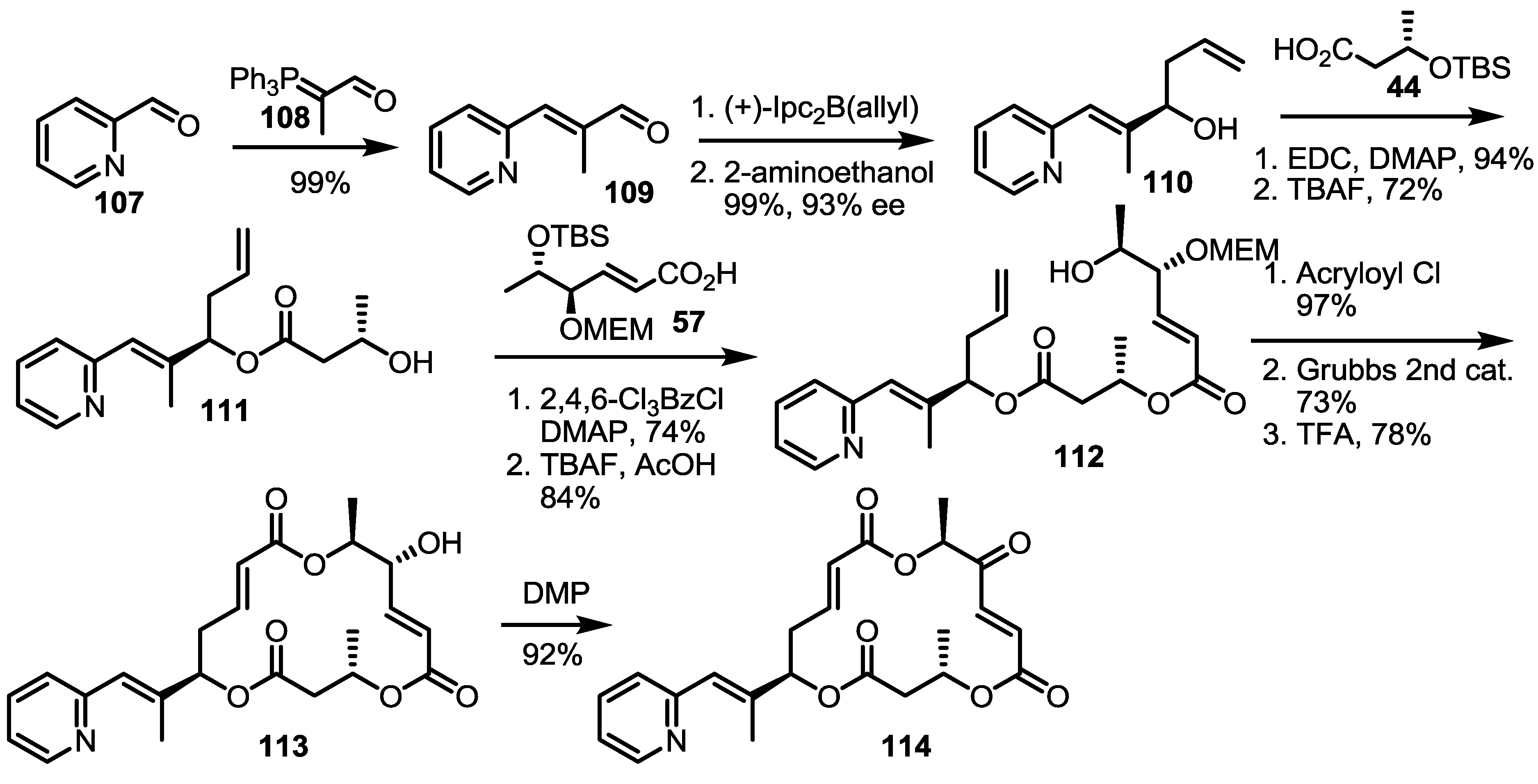
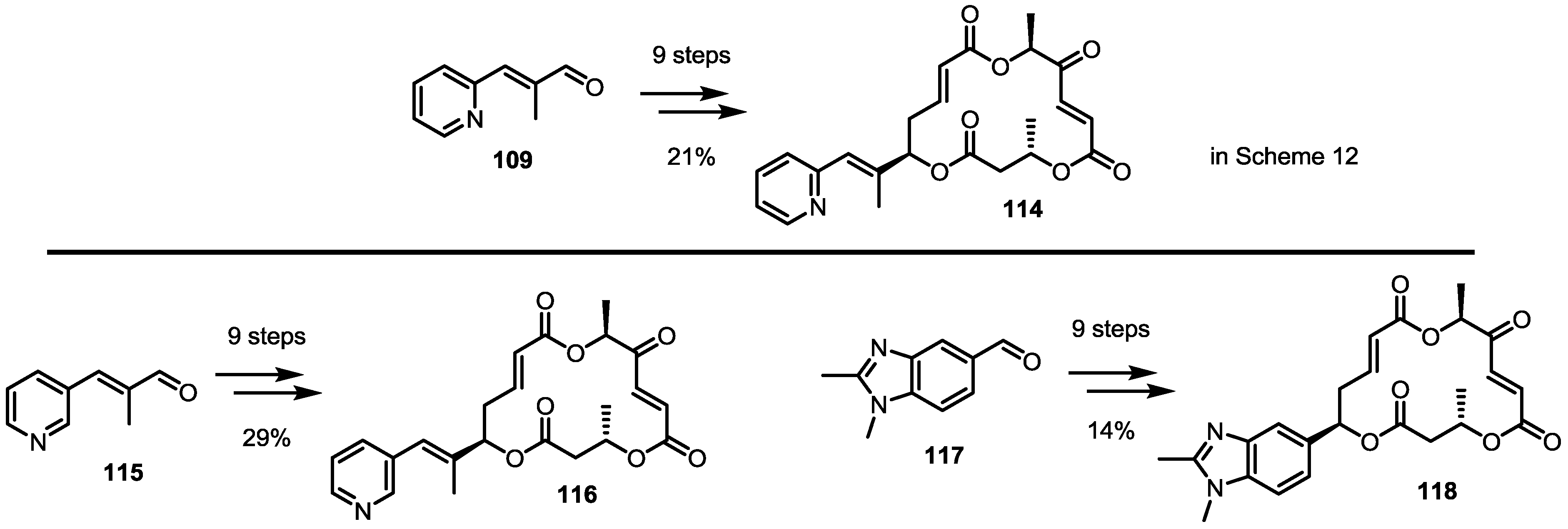
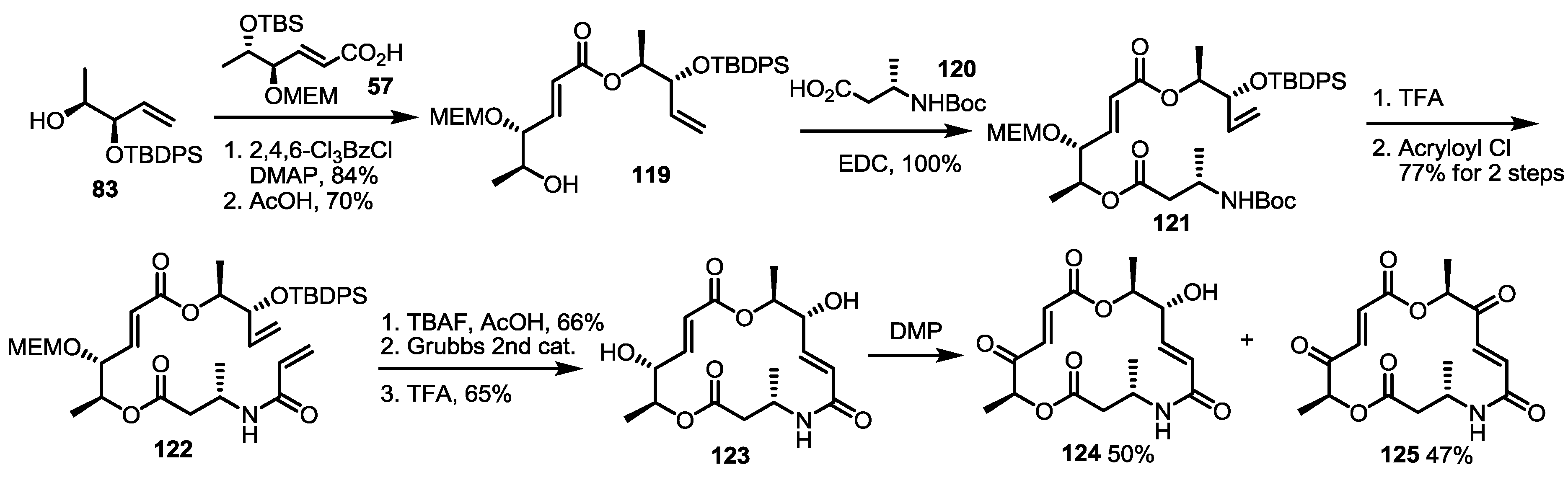

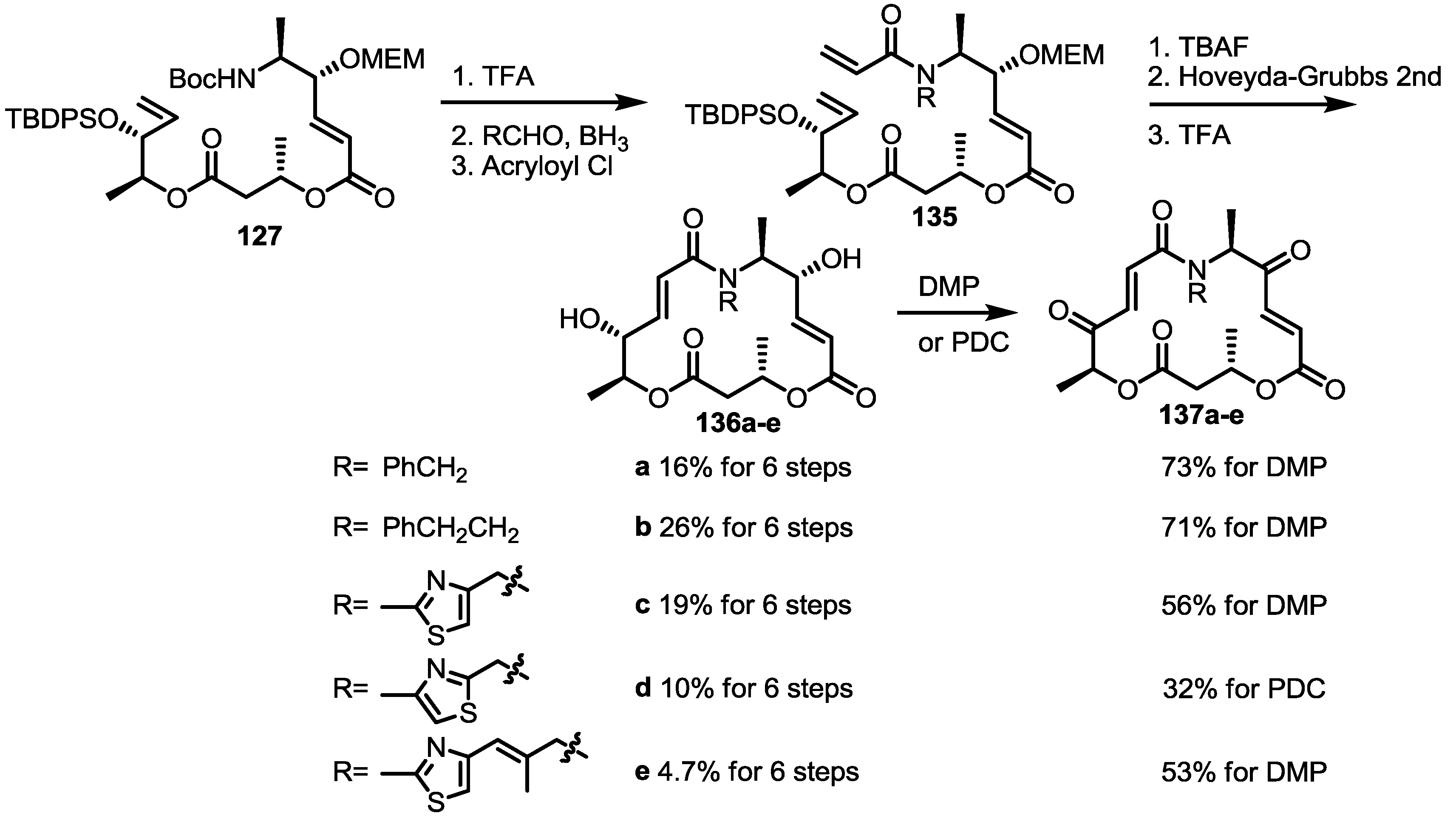
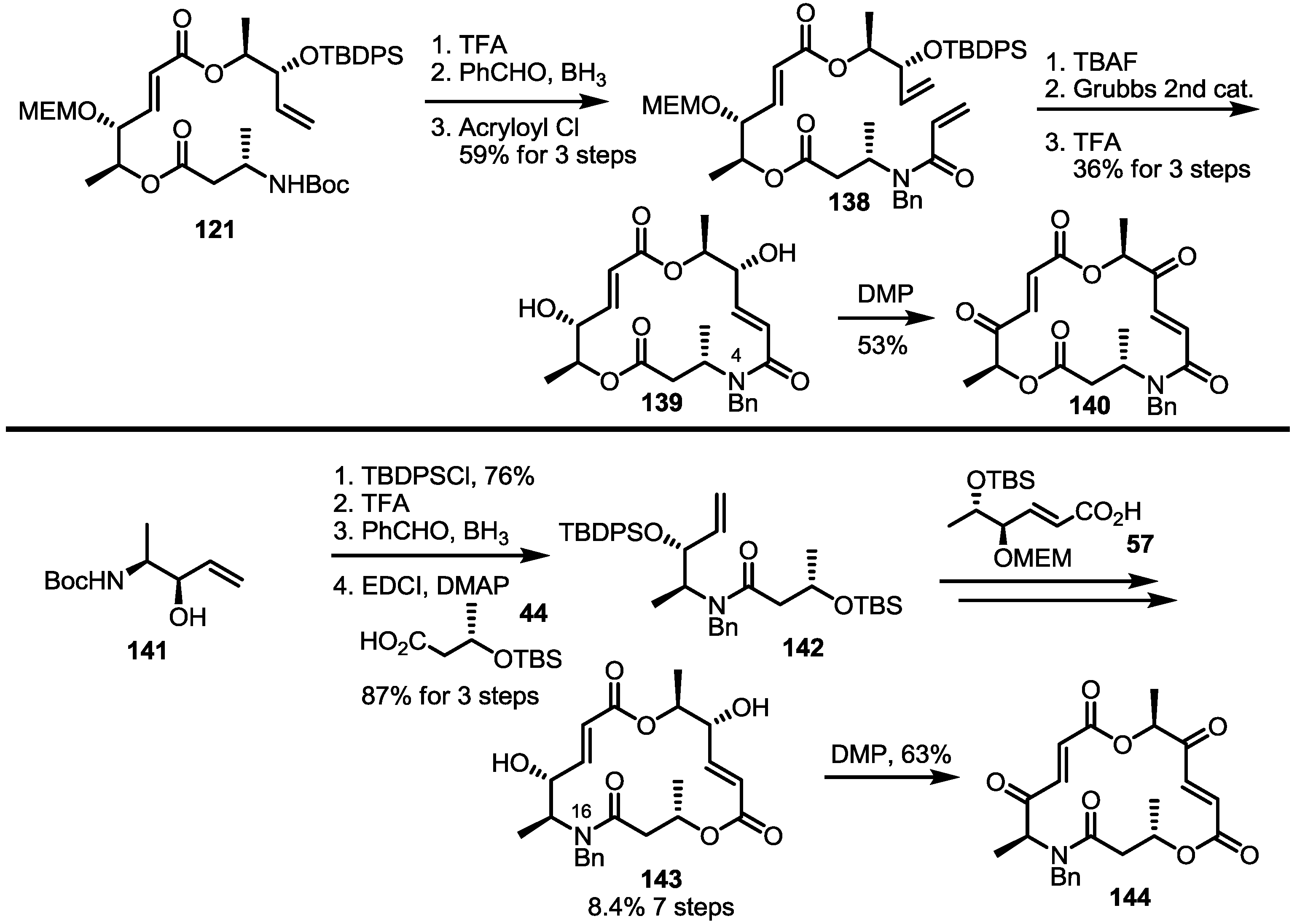
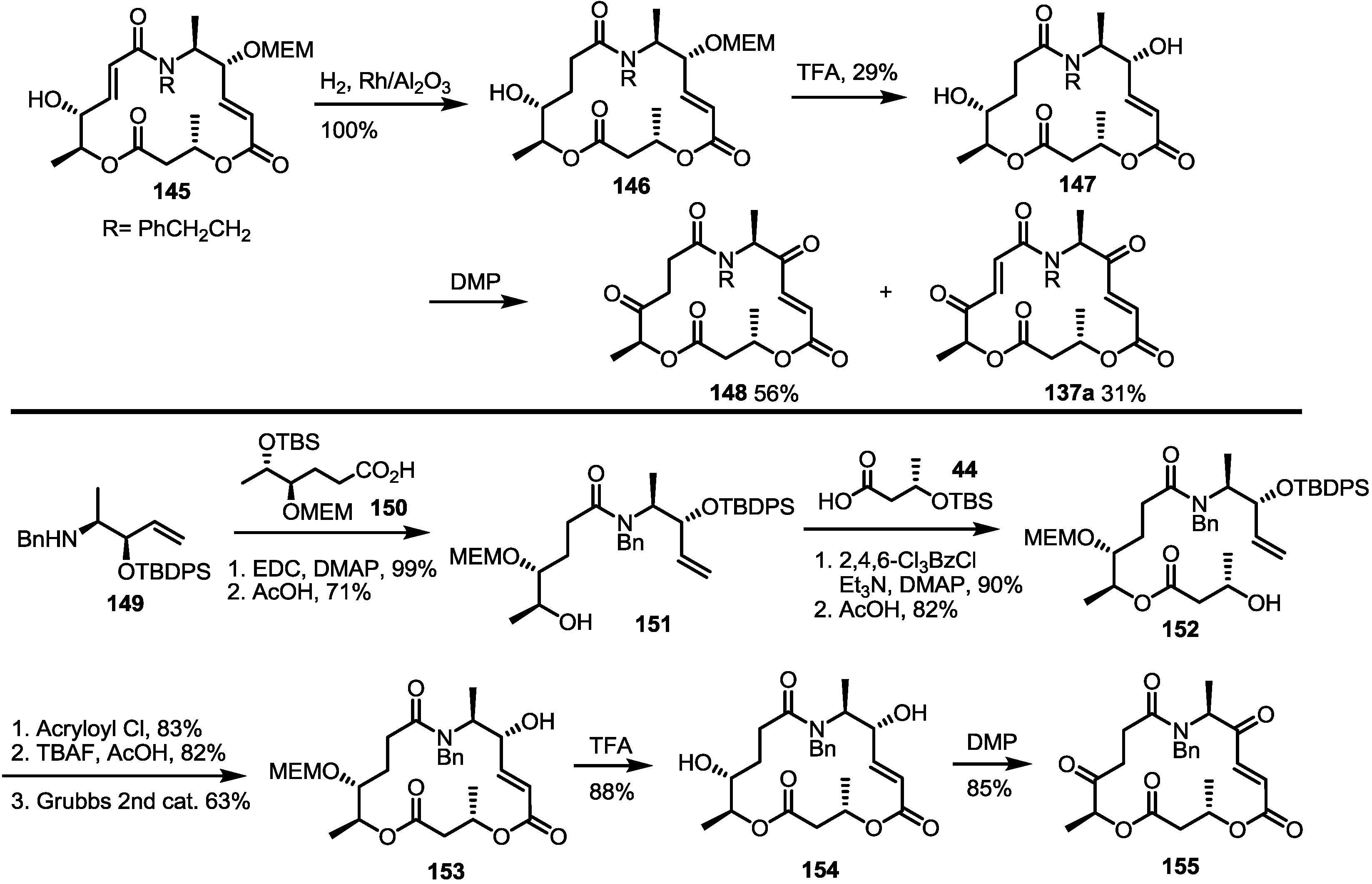
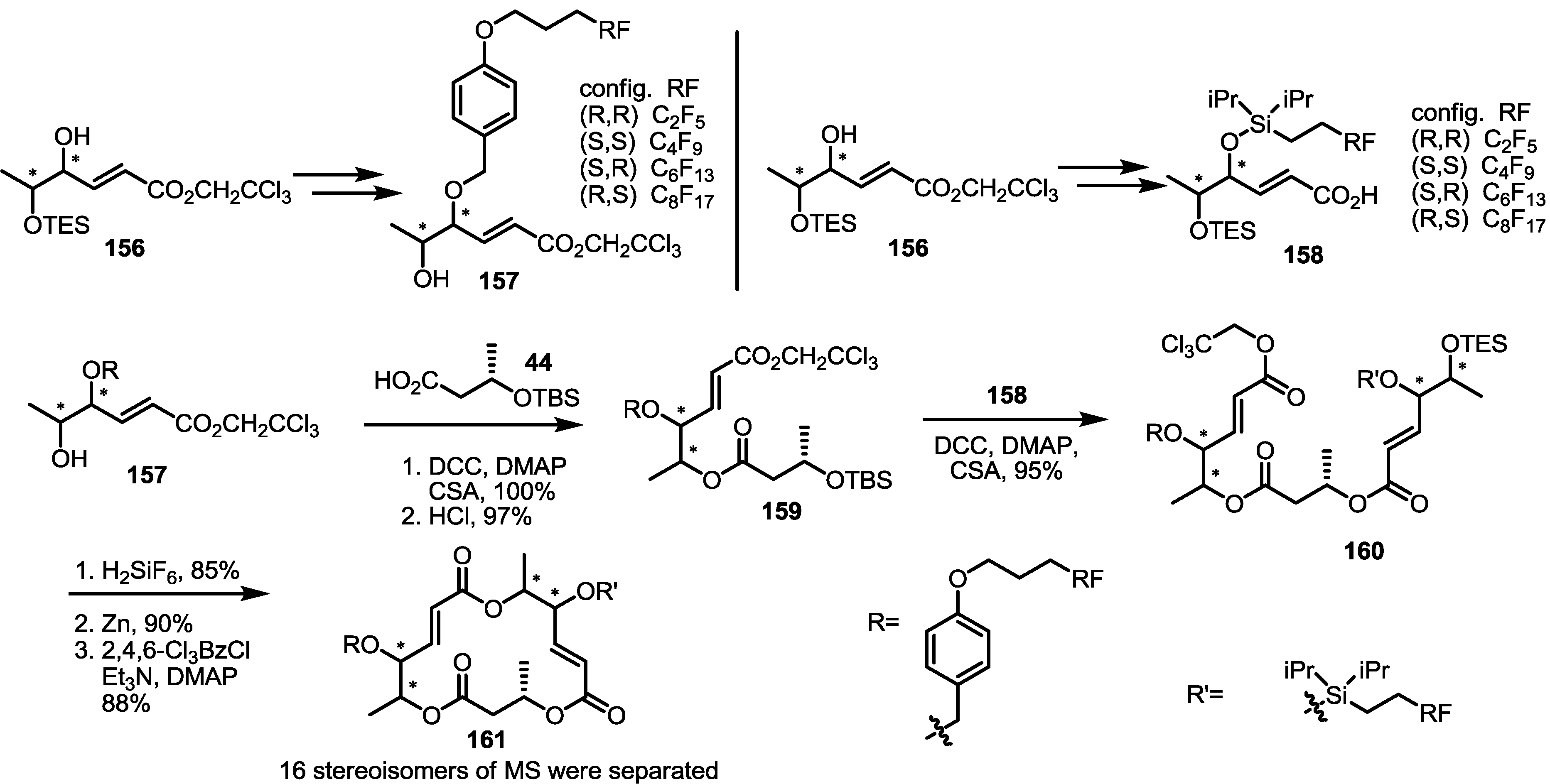
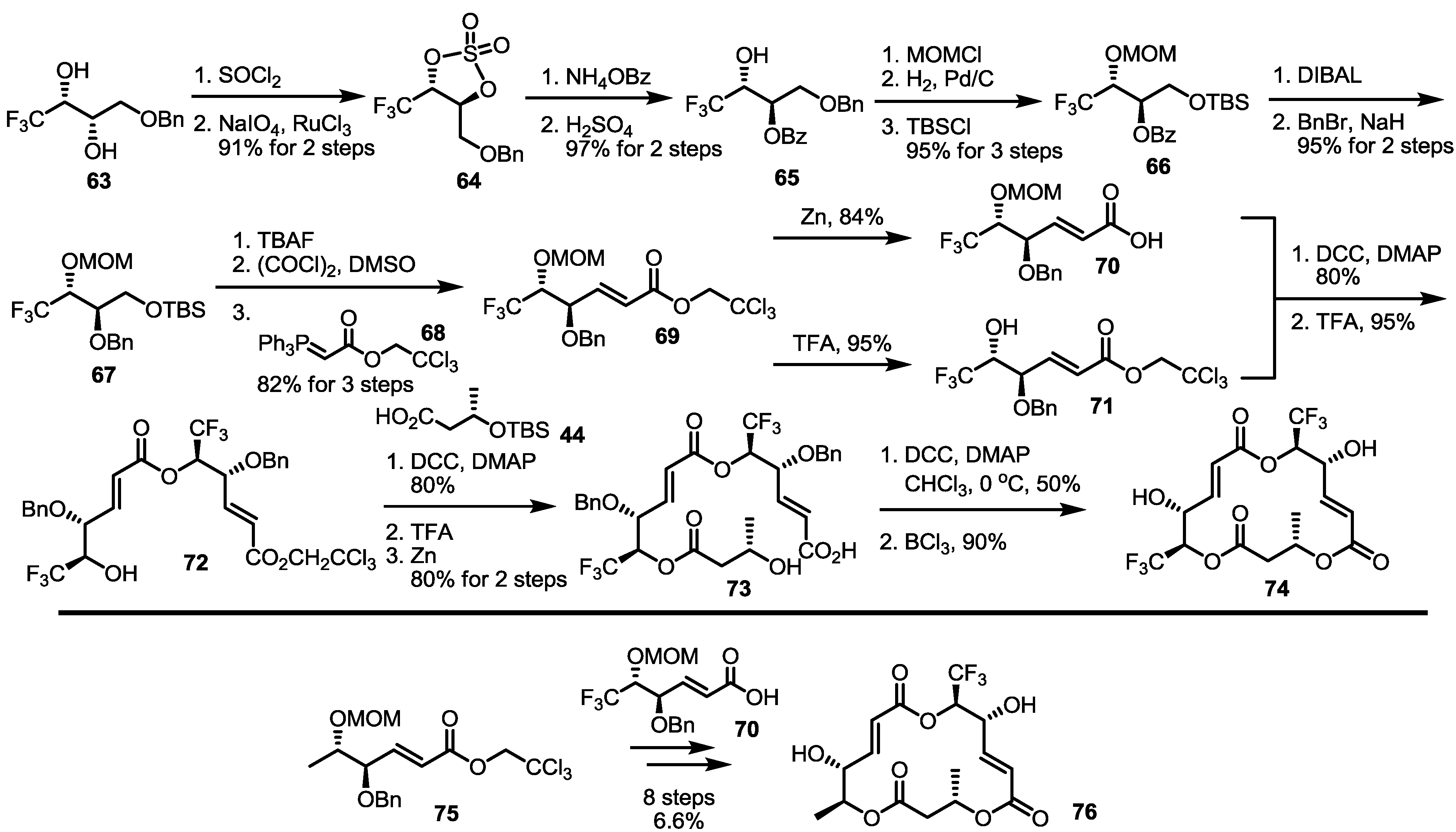
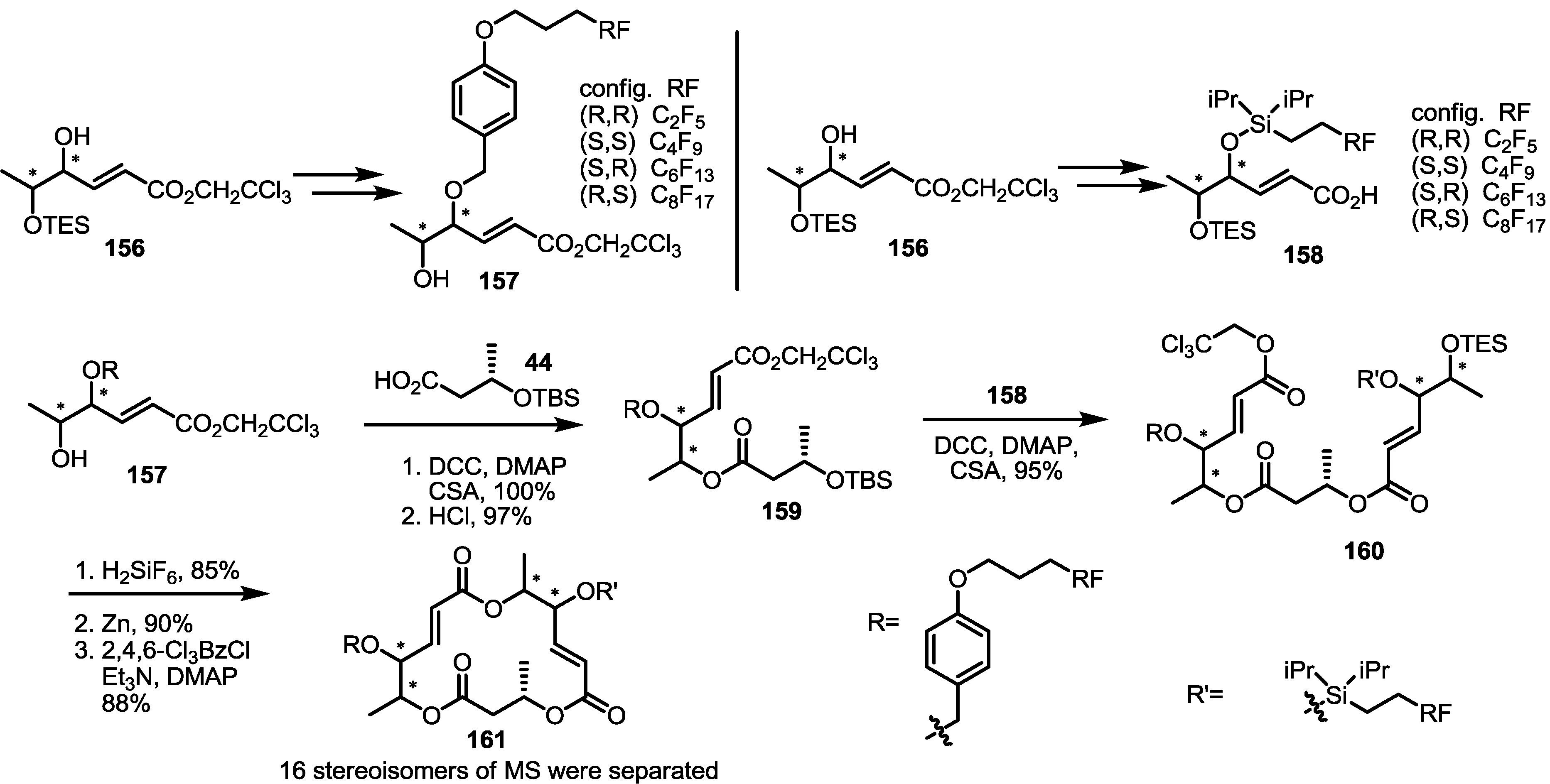
2.5. Fluorous Tagging Strategy for Diverse Synthesis of MS Stereoisomers
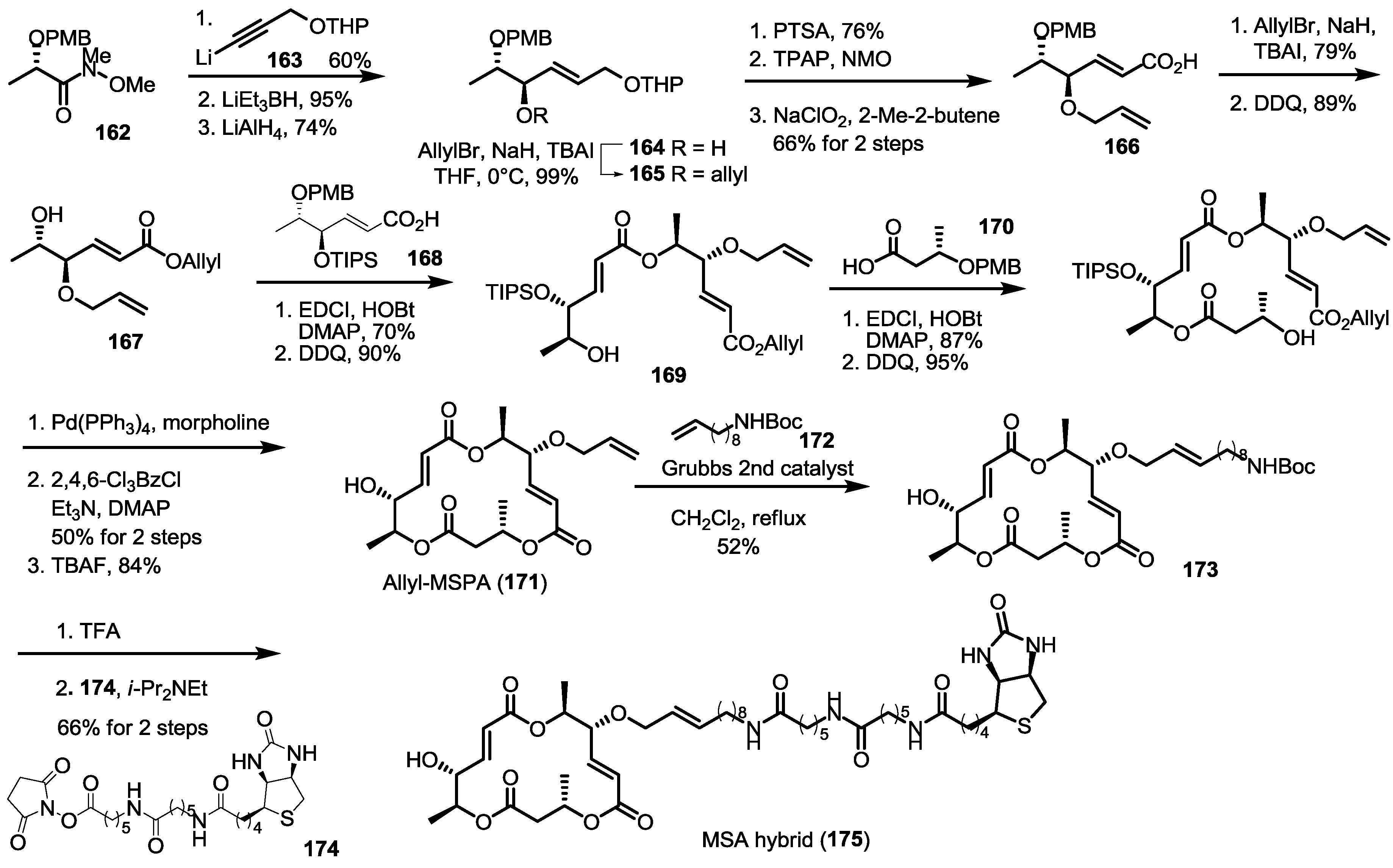
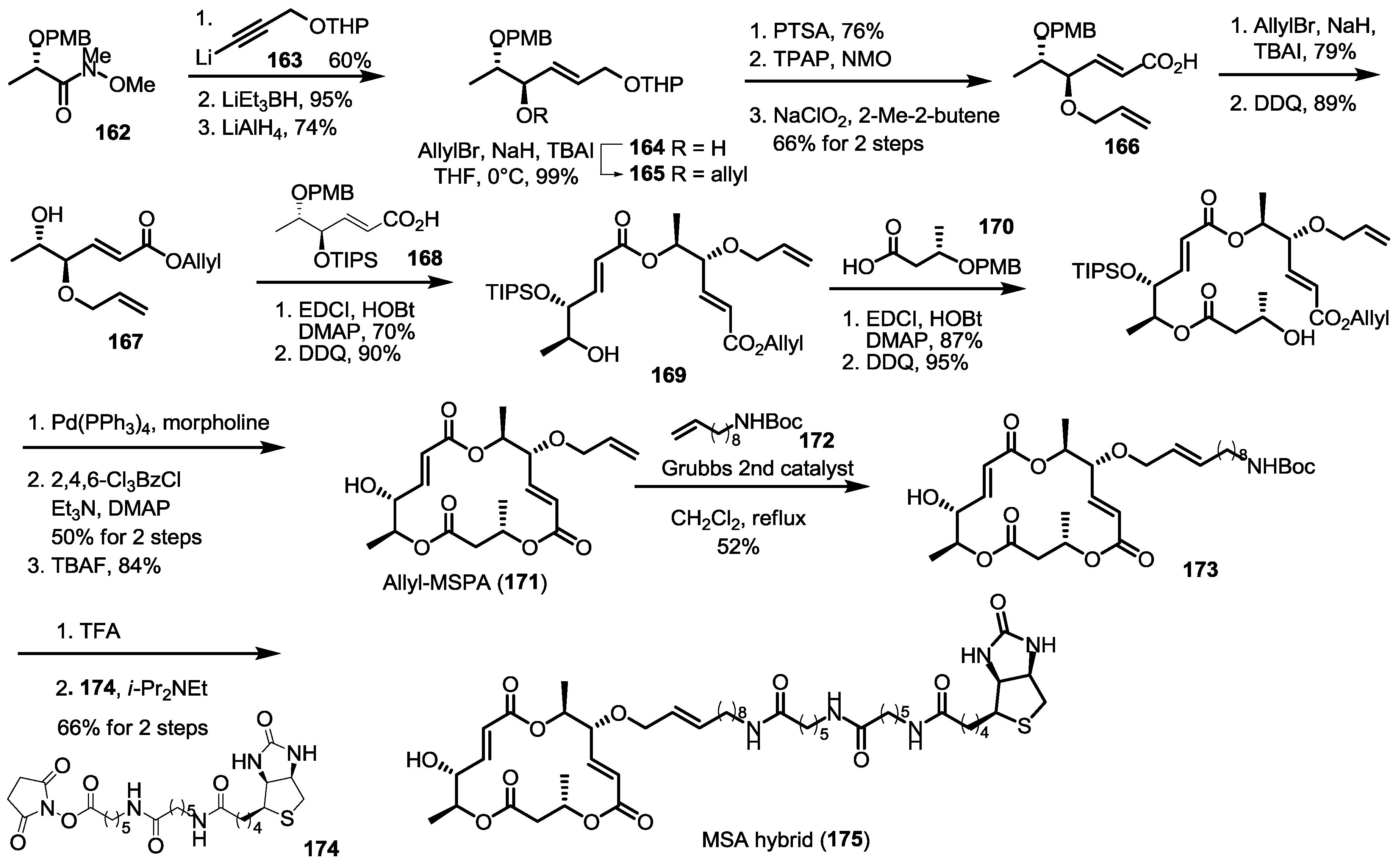
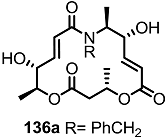
2.6. Preparation of MS-Biotin Hybrid
2.7. Biological Assay

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivative | Activity | Conc. | Derivative | Activity | Conc. |
|---|---|---|---|---|---|
 | <1% | 10 μM |  | 4%–5% | 1 μM |
 | <1% | 1 μM | 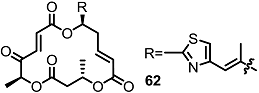 | <1% | 1 μM |
 | >10% | 1 μM |
| Derivative | Activity | Conc. | Derivative | Activity | Conc. |
|---|---|---|---|---|---|
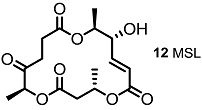
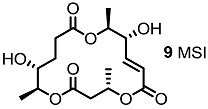 | <10% | 10 μM | 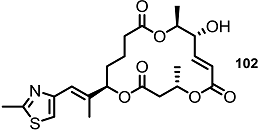 | <10% | 10 μM |
 | <10% | 10 μM | 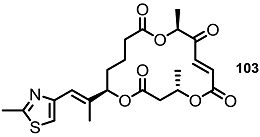 | 20% | 10 μM |
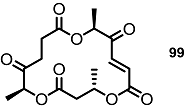 | >30% | 10 μM | 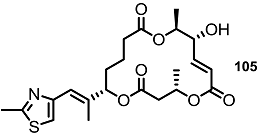 | <10% | 10 μM |
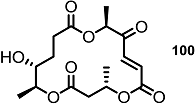 | 25% | 10 μM | 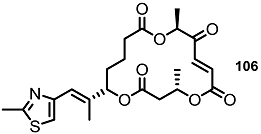 | 30% | 10 μM |
 | 10% | 10 μM | 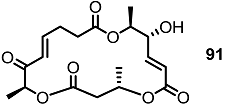 | 25% | 10 μM |
 | >10% | 10 μM |  | >10% | 10 μM |
| Derivative | Activity | Conc. | Derivative | Activity | Conc. |
|---|---|---|---|---|---|
 | <3% | 10 μM |  | <3% | 10 μM |
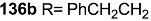 | <3% | 10 μM | 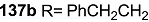 | 43% | 10 μM |
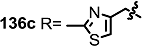 | <3% | 10 μM | 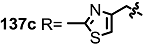 | 7% | 10 μM |
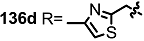 | <3% | 10 μM | 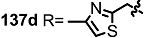 | 6% | 10 μM |
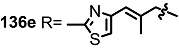 | <3% | 10 μM | 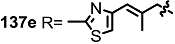 | 16% | 10 μM |
 | <3% | 10 μM | 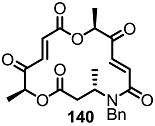 | <3% | 10 μM |
 | <3% | 10 μM | 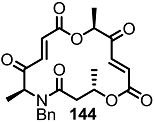 | 22% | 10 μM |
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2001, 75, 311–335. [Google Scholar]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar]
- Leslie, B.J.; Hergenrother, P.J. Identification of the cellular targets of bioactive small organic molecules using affinity reagents. Chem. Soc. Rev. 2008, 37, 1347–1360. [Google Scholar]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar]
- Teicher, B.A.; Andrews, P.A. Anticancer drug development guide; Humana Press: Totowa, NJ, USA, 2004; Volume 1. [Google Scholar]
- Kamb, A.; Wee, S.; Lengauer, C. Why is cancer drug discovery so difficult? Nat. Rev. Drug Discov. 2007, 6, 115–120. [Google Scholar]
- Hayashi, M.; Kim, Y.-P.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar]
- Akita, H.; Nakamura, H.; Ono, M. Total synthesis of (+)-macrosphelides a, c, e, f, and g based on enzymatic function. Chirality 2003, 15, 352–359. [Google Scholar]
- Matsuya, Y.; Nemoto, H. Recent advances in macrosphelide synthesis. Heterocycles 2005, 65, 1741–1749. [Google Scholar]
- Sunazuka, T.; Hirose., T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Omura, S.; Sprengeler, P.A.; Smith, A.B., III. Relative and Absolute Stereochemistries and Total Synthesis of (+)-Macrosphelides A and B, Potent, Orally Bioavailable Inhibitors of Cell-Cell Adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar]
- Omura, S.; Hayashi, M.; Tomoda, H. Recent progress of the research on novel microbial metabolites. Pure Appl. Chem. 1999, 71, 1673–1681. [Google Scholar]
- Fukami, A.; Iijima, K.; Hayashi, M.; Komiyama, K.; Omura, S. Macrosphelide B Suppressed Metastasis through Inhibition of Adhesion of sLex/E-Selectin Molecules. Biochem. Biophy. Res. Commun. 2002, 291, 1065–1070. [Google Scholar]
- Tomprefa, N.; McQuilken, M.; Hill, R.; Whipps, J. Antimicrobial activity of Coniothyrium minitans and its macrolide antibiotic macrosphelide A. J. Appl. Microbiol. 2009, 106, 2048–2056. [Google Scholar]
- Omura, S.; Komiyama, K. Immunosuppressant macrosphelide B. WO0147516 A1 5 July 2001. [Google Scholar]
- Paek, S.-M. Synthetic Advance in Macrosphelides: Natural Anticancer Agents. Molecules 2014, 19, 15982–16000. [Google Scholar]
- Matsuya, Y.; Nemoto, H. Artificial macrosphelides as a novel apoptosis-inducing compound. Heterocycles 2010, 81, 57–66. [Google Scholar]
- Sunazuka, T.; Hirose, T.; Chikaraishi, N.; Harigaya, Y.; Hayashi, M.; Komiyama, K.; Sprengeler, P.A.; Smith, A.B., III; Ōmura, S. Absolute stereochemistries and total synthesis of (+)/(−)-macrosphelides, potent, orally bioavailable inhibitors of cell–cell adhesion. Tetrahedron 2005, 61, 3789–3803. [Google Scholar]
- Takahashi, T.; Kusaka, S.-i.; Doi, T.; Takayuki, T.; Sunazuka, T.; Omura, S. A combinatorial synthesis of a macrosphelide library utilizing a palladium-catalyzed carbonylation on a polymer support. Angew. Chem. Int. Ed. 2003, 42, 5230–5234. [Google Scholar]
- Kusaka, S.-I.; Dohi, S.; Doi, T.; Takahashi, T. Total synthesis of macrosphelide A by way of palladium-catalyzed carbonylative esterification. Tetrahedron Lett. 2003, 44, 8857–8859. [Google Scholar]
- Ono, M.; Nakamura, H.; Arakawa, S.; Honda, N.; Akita, H. Formal total synthesis of (+)-macrosphelide A based on regioselective hydrolysis using lipase. Chem. Pharm. Bull. 2002, 50, 692–696. [Google Scholar]
- Ishihara, K.; Kawaguchi, T.; Matsuya, Y.; Sakurai, H.; Saiki, I.; Nemoto, H. Synthesis and biological evaluation of macrosphelide cores. Eur. J. Org. Chem. 2004, 3973–3978. [Google Scholar]
- Ahmed, K.; Zhao, Q.-L.; Matsuya, Y.; Yu, D.-Y.; Feril, L.B., Jr.; Nemoto, H.; Kondo, T. Rapid and transient intracellular oxidative stress due to novel macrosphelides trigger apoptosis via Fas/caspase-8-dependent pathway in human lymphoma U937 cells. Chem.-Biol. Interact. 2007, 170, 86–99. [Google Scholar]
- Ahmed, K.; Zhao, Q.-L.; Matsuya, Y.; Yu, D.-Y.; Salunga, T.; Nemoto, H.; Kondo, T. Enhancement of macrosphelide-induced apoptosis by mild hyperthermia. Int. J. Hyperthermia 2007, 23, 353–361. [Google Scholar]
- Matsuya, Y.; Kawaguchi, T.; Nemoto, H. Synthesis of various macrosphelides by oxidative derivatization of the macrosphelide core. Heterocycles 2003, 61, 39–43. [Google Scholar]
- Matsuya, Y.; Kawaguchi, T.; Ishihara, K.; Ahmed, K.; Zhao, Q.-L.; Kondo, T.; Nemoto, H. Synthesis of Macrosphelides with a Thiazole Side Chain: New Antitumor Candidates Having Apoptosis-Inducing Property. Org. Lett. 2006, 8, 4609–4612. [Google Scholar]
- Höfle, G.; Bedorf, N.; Steinmetz, H.; Schomburg, D.; Gerth, K.; Reichenbach, H. Epothilone A and B-Novel 16-Membered Macrolides with Cytotoxic Activity: Isolation, Crystal Structure, and Conformation in Solution. Angew. Chem. Int. Ed. 1996, 35, 1567–1569. [Google Scholar]
- Meng, D.; Bertinato, P.; Balog, A.; Su, D.-S.; Kamenecka, T.; Sorensen, E.J.; Danishefsky, S.J. Total syntheses of epothilones A and B. J. Am. Chem. Soc. 1997, 119, 10073–10092. [Google Scholar]
- Matsuya, Y.; Kawaguchi, T.; Nemoto, H. New strategy for the total synthesis of macrosphelides A and B based on ring-closing metathesis. Org. Lett. 2003, 5, 2939–2941. [Google Scholar]
- Kawaguchi, T.; Funamori, N.; Matsuya, Y.; Nemoto, H. Total Synthesis of Macrosphelides A, B, and E: First Application of Ring-Closing Metathesis for Macrosphelide Synthesis. J. Org. Chem. 2004, 69, 505–509. [Google Scholar]
- Ahmed, K.; Matsuya, Y.; Nemoto, H.; Zaidi, S.F.H.; Sugiyama, T.; Yoshihisa, Y.; Shimizu, T.; Kondo, T. Mechanism of apoptosis induced by a newly synthesized derivative of macrosphelides with a thiazole side chain. Chem.-Biol. Interact. 2009, 177, 218–216. [Google Scholar]
- Zhang, H.; Wang, X.-N.; Lin, L.-P.; Ding, J.; Yue, J.-M. Indole alkaloids from three species of the Ervatamia genus: E. officinalis, E. divaricata, and E. divaricata Gouyahua. J. Nat. Prod. 2007, 70, 54–59. [Google Scholar]
- Wang, B.-L.; Jiang, Z.-X.; You, Z.-W.; Qing, F.-L. Total synthesis of trifluoromethylated analogs of macrosphelide A. Tetrahedron 2007, 63, 12671–12680. [Google Scholar]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar]
- Jiang, Z.-X.; Qin, Y.-Y.; Qing, F.-L. Qing Asymmetric Synthesis of Both Enantiomers of anti-4,4,4-Trifluorothreonine and 2-Amino-4,4,4-trifluorobutanoic Acid. J. Org. Chem. 2003, 68, 7544–7547. [Google Scholar]
- Matsuya, Y.; Matsushita, T.; Sakamoto, K.; Nemoto, H. Total synthesis of 2-nor-macrosphelides A and B. Heterocycles 2009, 77, 483–492. [Google Scholar]
- Matsuya, Y.; Kobayashi, Y.; Kawaguchi, T.; Hori, A.; Watanabe, Y.; Ishihara, K.; Ahmed, K.; Wei, Z.-L.; Yu, D.-Y.; Zhao, Q.-L.; et al. Design, Synthesis, and Biological Evaluation of Artificial Macrosphelides in the Search for New Apoptosis-Inducing Agents. Chem. Eur. J. 2009, 15, 5799–5819. [Google Scholar]
- Matsuya, Y.; Hori, A.; Kawamura, T.; Emam, H.F.; Ahmed, K.; Yu, D.-Y.; Kondo, T.; Toyooka, N.; Nemoto, H. Synthesis of macrosphelides containing a heterocyclic side chain as a novel apoptosis inducer. Heterocycles 2010, 80, 579–591. [Google Scholar]
- Sugimoto, K.; Kobayashi, Y.; Hori, A.; Kondo, T.; Toyooka, N.; Nemoto, H.; Matsuya, Y. Syntheses of aza-analogues of macrosphelides via RCM strategy and their biological evaluation. Tetrahedron 2011, 67, 7681–7685. [Google Scholar]
- Rae, A.; Aliev, A.E.; Anderson, J.E.; Castro, J.; Ker, J.; Parsons, S.; Stchedroff, M.; Thomas, S.; Tabor, A. Preparation and structure of homochiral tetrahydro-2 H-1, 3-oxazines. J. Chem. Soc. Perkin Trans. 1 1999, 1933–1942. [Google Scholar]
- Sugimoto, K.; Kobayashi, Y.; Kondo, T.; Toyooka, N.; Nemoto, H.; Matsuya, Y. Dihydroazamacrosphelides: Synthesis and apoptosis inducing activities. Heterocycles 2011, 83, 2823–2835. [Google Scholar]
- Curran, D.P.; Sinha, M.K.; Zhang, K.; Sabatini, J.J.; Cho, D.-H. Binary fluorous tagging enables the synthesis and separation of a 16-stereoisomer library of macrosphelides. Nat. Chem. 2012, 4, 124–129. [Google Scholar]
- Paek, S.-M.; Seo, S.-Y.; Kim, S.-H.; Jung, J.-W.; Lee, Y.-S.; Jung, J.-K.; Suh, Y.-G. Concise Syntheses of (+)-Macrosphelide A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar]
- Yun, H.; Sim, J.; An, H.; Lee, J.; Lee, H.S.; Shin, Y.K.; Paek, S.-M.; Suh, Y.-G. Design and synthesis of a macrosphelide A-biotin chimera. Org. Biomol. Chem. 2014, 12, 7127–7135. [Google Scholar]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paek, S.-M. Development of Advanced Macrosphelides: Potent Anticancer Agents. Molecules 2015, 20, 4430-4449. https://doi.org/10.3390/molecules20034430
Paek S-M. Development of Advanced Macrosphelides: Potent Anticancer Agents. Molecules. 2015; 20(3):4430-4449. https://doi.org/10.3390/molecules20034430
Chicago/Turabian StylePaek, Seung-Mann. 2015. "Development of Advanced Macrosphelides: Potent Anticancer Agents" Molecules 20, no. 3: 4430-4449. https://doi.org/10.3390/molecules20034430
APA StylePaek, S.-M. (2015). Development of Advanced Macrosphelides: Potent Anticancer Agents. Molecules, 20(3), 4430-4449. https://doi.org/10.3390/molecules20034430