
Characterization of Global Transcriptome Using Illumina Paired-End Sequencing and Development of EST-SSR Markers in Two Species of Gynostemma (Cucurbitaceae)

Abstract
:1. Introduction
2. Results
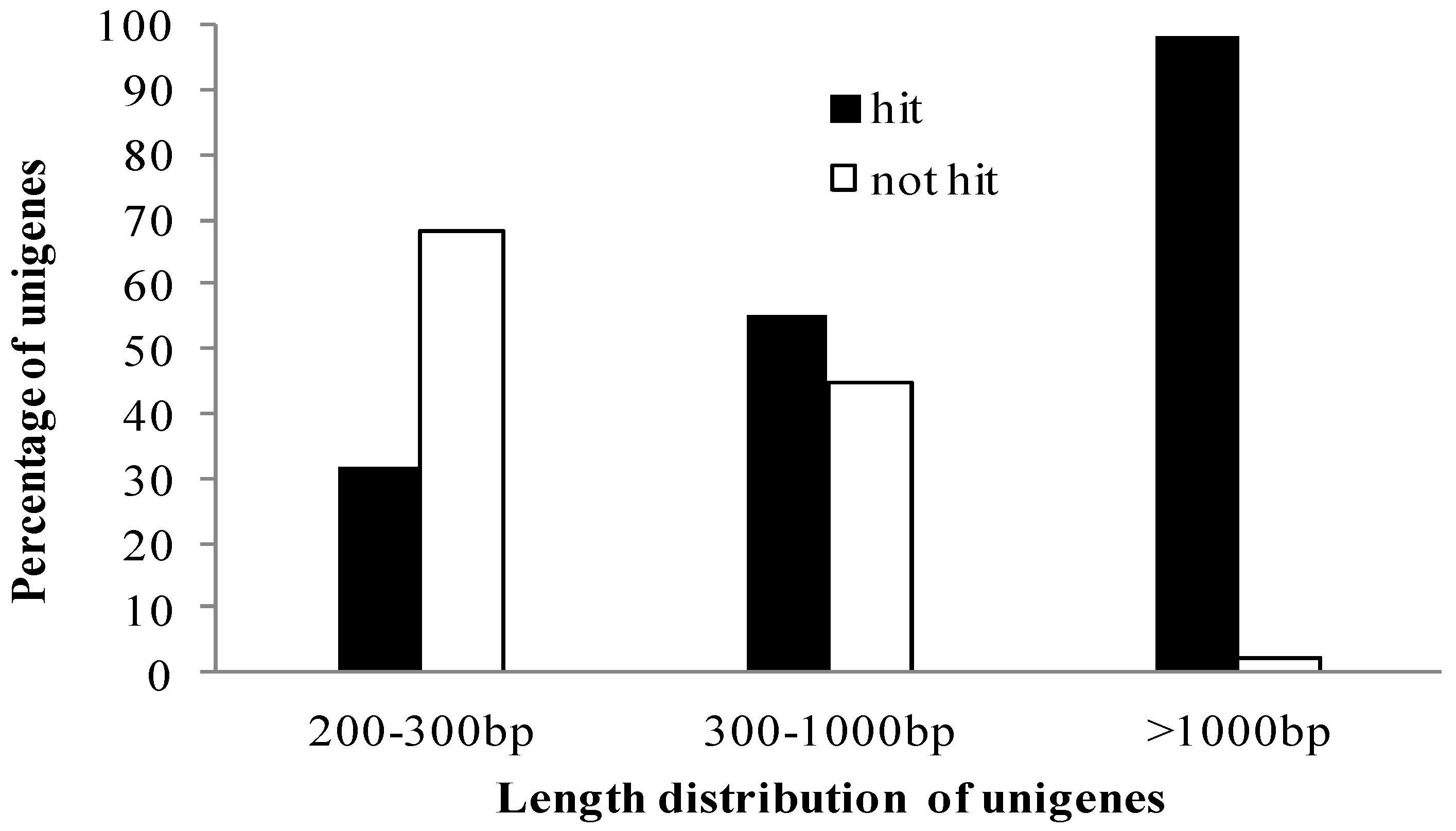
2.1. Assembly of Gynostemma Transcriptome Data from Illumina Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total Reads | Total Clean Nucleotides (Nt) | Q30 Percentage | GC Percentage | Total Number of Contigs | Total Length of Contigs (Nt) | N50 of Contigs | Mean (Nt) | |
|---|---|---|---|---|---|---|---|---|---|
| G. pentaphyllum | 43,175,448 | 4,360,277,191 | 80.16% | 43.55% | 1,488,035 | 110,745,998 | 71 | 74 | |
| G. cardiospermum | 52,782,146 | 5,330,256,148 | 82.69% | 44.00% | 1,911,378 | 136,726,651 | 66 | 71 | |
| Unigene Source | Total Number of Unigenes | Total Length of Unigenes (Nt) | Mean Length of Unigenes (Nt) | N50 of Unigenes |
|---|---|---|---|---|
| G. pentaphyllum | 40,257 | 35,161,843 | 873.43 | 1516 |
| G. cardiospermum | 44,000 | 37,234,004 | 846.23 | 1504 |
| All | 71,607 | 61,367,129 | 857.00 | 1535 |
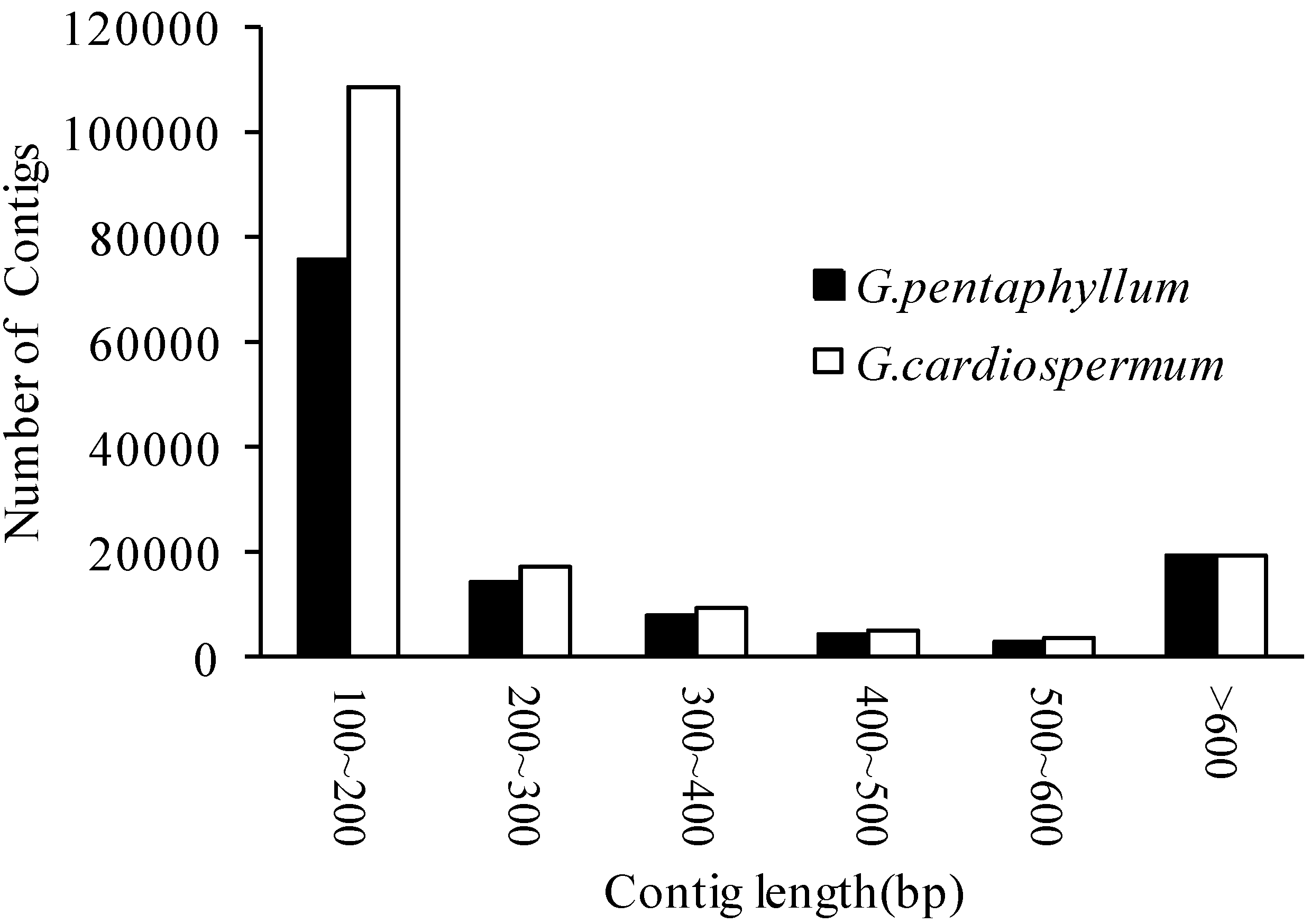
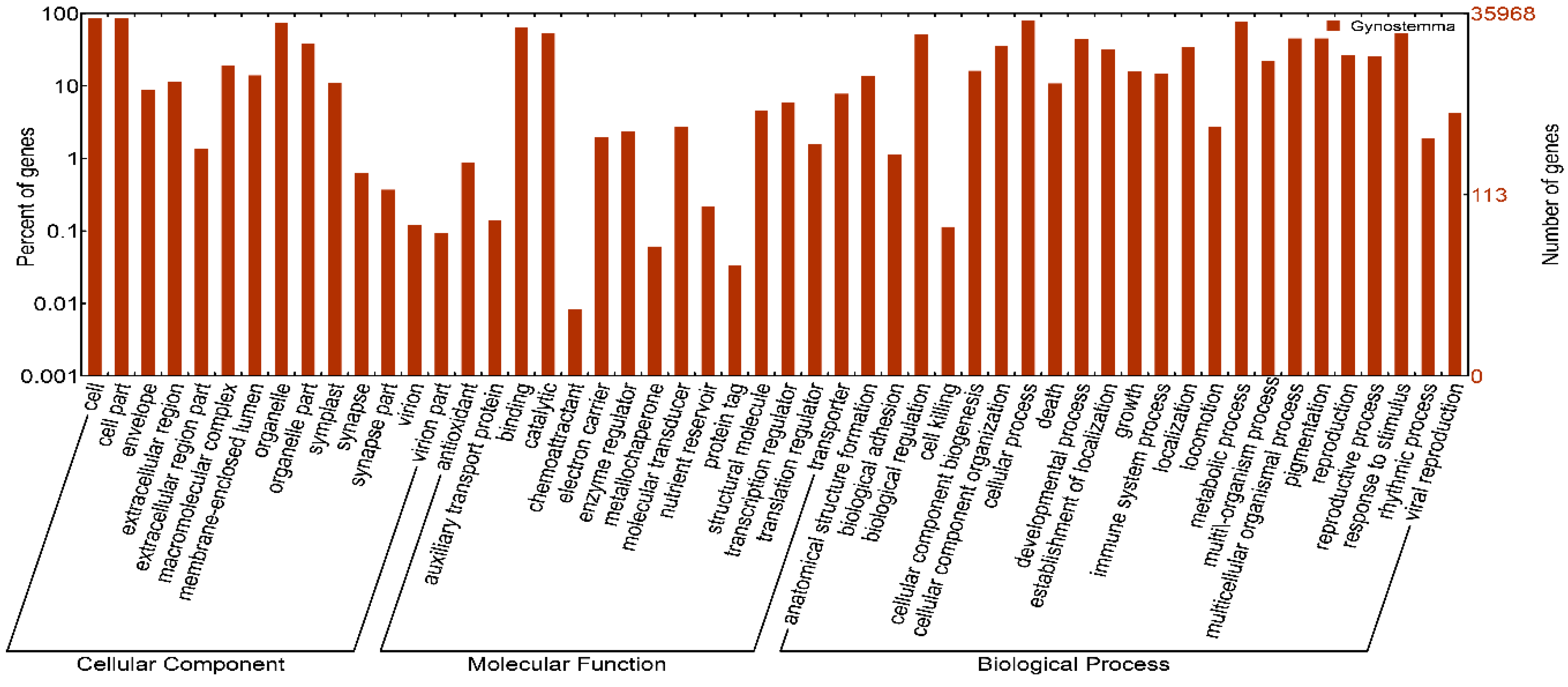
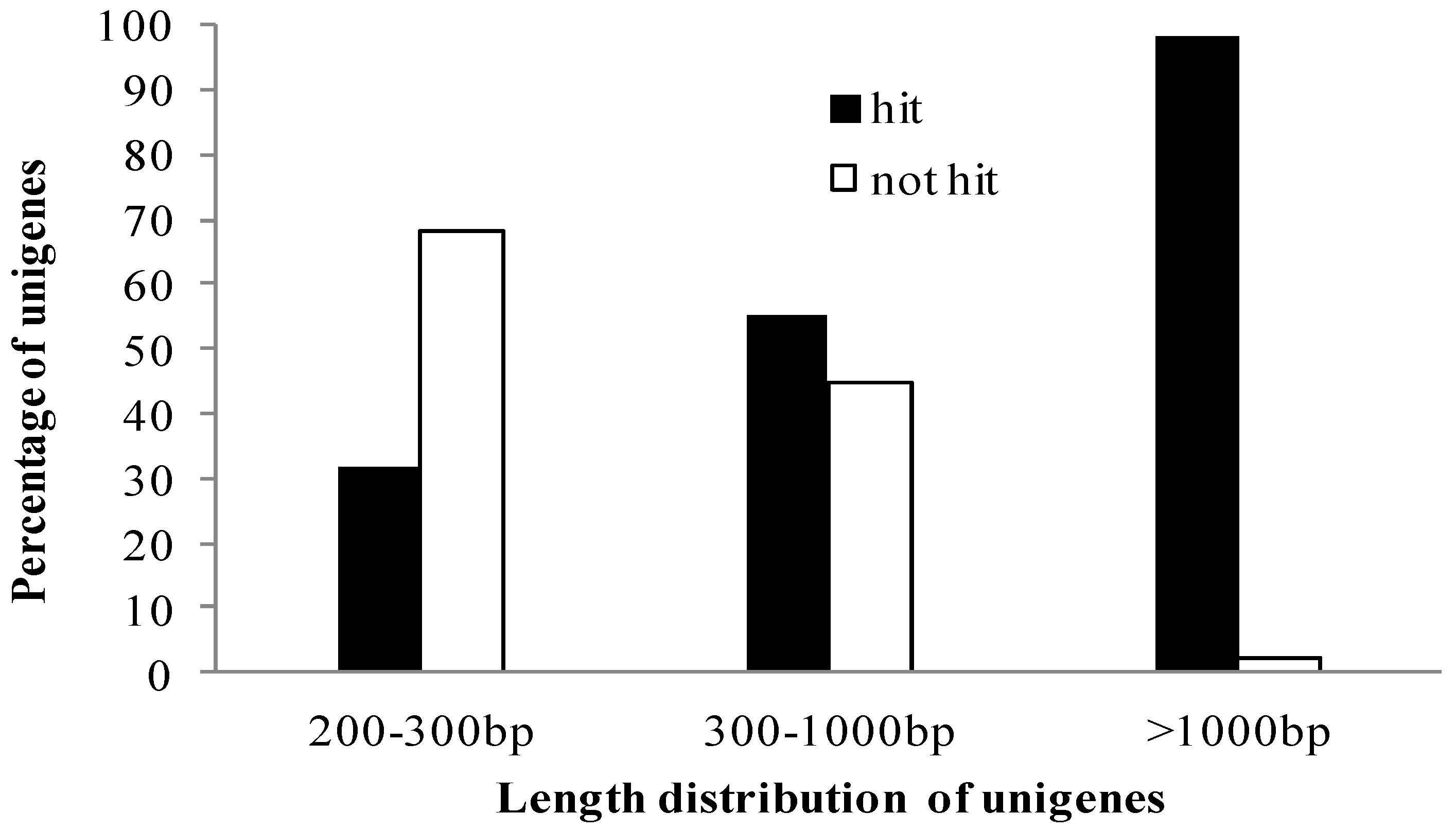
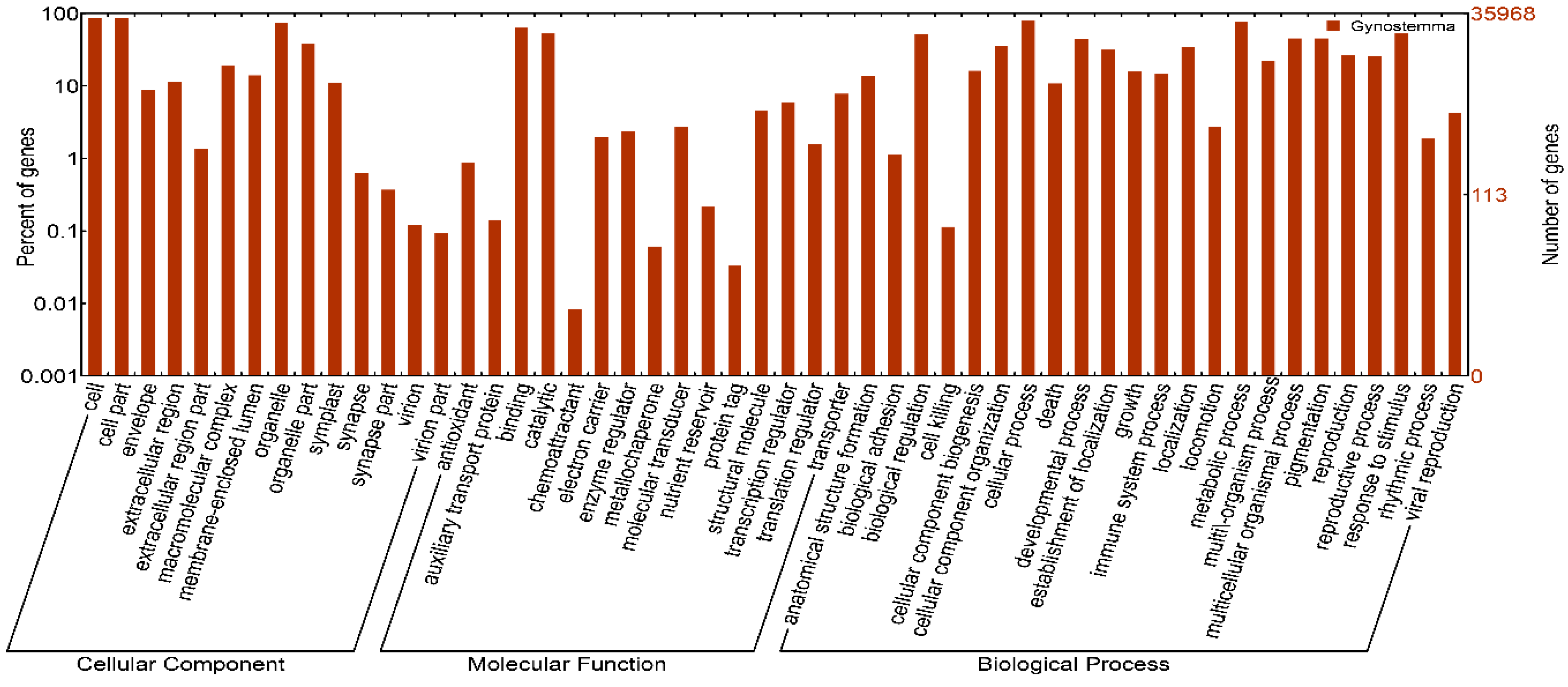
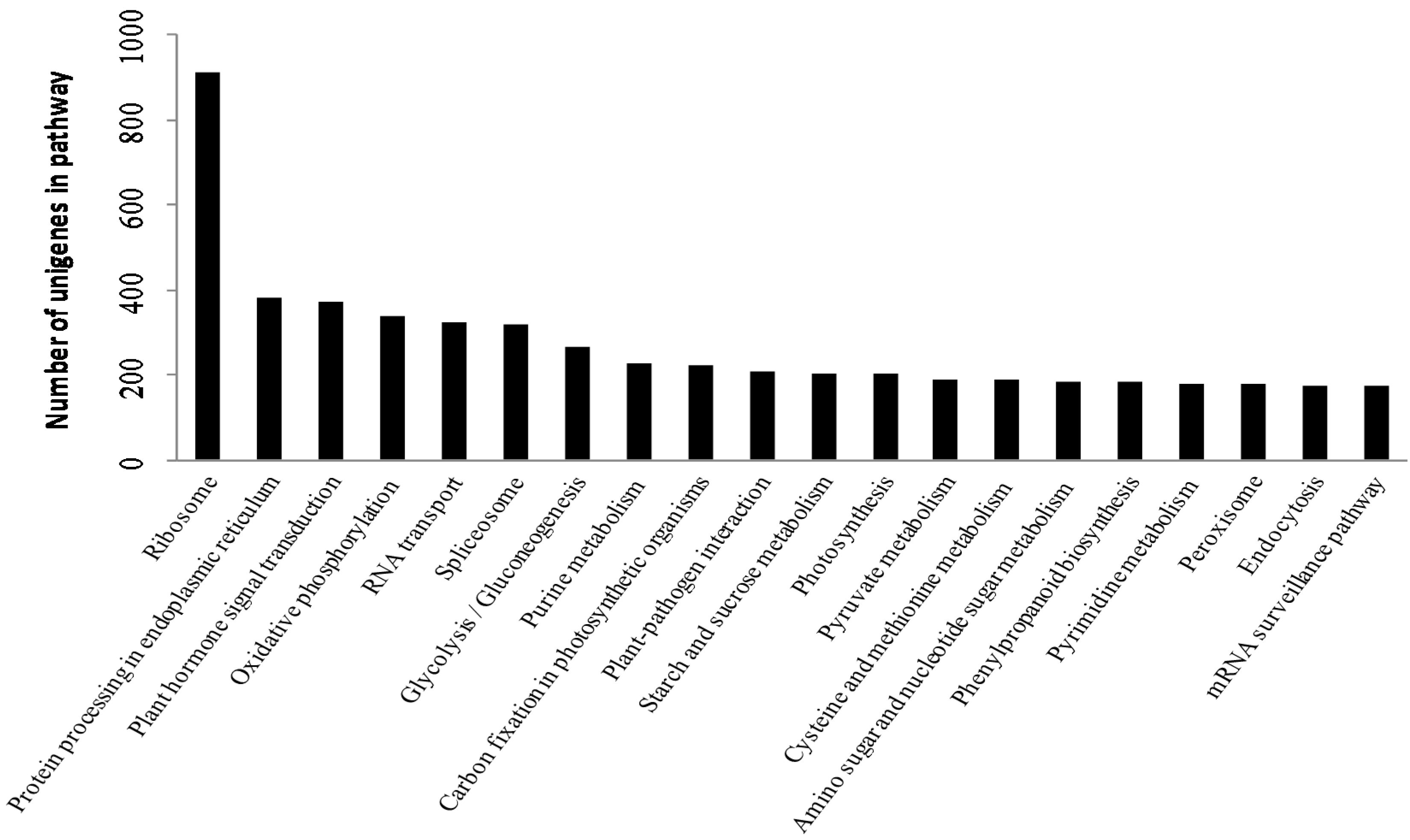
2.2. Functional Annotation and Classification

| Gene ID | Length | KO ID | Annotation |
|---|---|---|---|
| T3_Unigene_BMK.23540 | 2373 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| T3_Unigene_BMK.25990 | 2720 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| T3_Unigene_BMK.37882 | 301 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| T4_Unigene_BMK.23719 | 2489 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| T4_Unigene_BMK.30793 | 3057 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| T4_Unigene_BMK.33182 | 2483 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| CL10430Contig1 | 2754 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| T3_Unigene_BMK.15665 | 2671 | K01662 | 1-deoxyxylulose-5-phosphate synthase, (EC:2.2.1.7) |
| CL9143Contig1 | 1586 | K00919 | 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase, (EC:2.7.1.148) |
| T3_Unigene_BMK.22099 | 359 | K03526 | 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase, (EC:1.17.7.1) |
| T3_Unigene_BMK.28355 | 906 | K03527 | 4-hydroxy-3-methylbut-2-enyl diphosphate reductase, (EC:1.17.1.2) |
| CL12440Contig1 | 616 | K03527 | 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase, (EC:1.17.1.2) |
| T3_Unigene_BMK.9388 | 411 | K03527 | 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase, (EC:1.17.1.2) |
| CL12699Contig1 | 1973 | K01823 | isopentenyl-diphosphate delta-isomerase, (EC:5.3.3.2) |
| T4_Unigene_BMK.25472 | 575 | K01823 | isopentenyl-diphosphate delta-isomerase, (EC:5.3.3.2) |
| CL7433Contig1 | 750 | K01823 | isopentenyl-diphosphate delta-isomerase, (EC:5.3.3.2) |
| T3_Unigene_BMK.21000 | 1177 | K13789 | geranyl geranyl pyrophosphate synthase, (EC:2.5.1.29) |
| T4_Unigene_BMK.20525 | 1569 | K14066 | geranylgeranyl pyrophosphate synthase, (EC:2.5.1.30) |
| T3_Unigene_BMK.18996 | 1754 | K14066 | geranylgeranyl pyrophosphate synthase, (EC:2.5.1.30) |
| T3_Unigene_BMK.35842 | 305 | K00626 | acetyl-CoA acetyltransferase, mitochondrial, (EC:2.3.1.9) |
| T3_Unigene_BMK.35857 | 278 | K00626 | acetyl-CoA acetyltransferase, mitochondrial, (EC:2.3.1.9) |
| T4_Unigene_BMK.23349 | 1785 | K00626 | acetyl-CoA C-acetyltransferase, (EC:2.3.1.9) |
| T3_Unigene_BMK.18569 | 1657 | K00626 | acetyl-CoA C-acetyltransferase, (EC:2.3.1.9) |
| CL11048Contig1 | 2014 | K01641 | hydroxymethylglutaryl-CoA synthase, (EC:2.3.3.10) |
| T3_Unigene_BMK.2712 | 475 | K00021 | hmg-CoA reductase, (EC:1.1.1.34) |
| CL14352Contig1 | 2368 | K00021 | hmg-CoA reductase, (EC:1.1.1.34) |
| T4_Unigene_BMK.28606 | 1468 | K00021 | hmg-CoA reductase, (EC:1.1.1.34) |
| T4_Unigene_BMK.28692 | 322 | K00021 | hmg-CoA reductase, (EC:1.1.1.34) |
| T3_Unigene_BMK.22340 | 296 | K00021 | hmg-CoA reductase, (EC:1.1.1.34) |
| T3_Unigene_BMK.3271 | 467 | K00869 | mevalonate kinase, (EC:2.7.1.36) |
| CL13115Contig1 | 526 | K00869 | mevalonate kinase, (EC:2.7.1.36) |
| T4_Unigene_BMK.23148 | 1576 | K00869 | mevalonate kinase, (EC:2.7.1.36) |
| T4_Unigene_BMK.26974 | 522 | K00869 | mevalonate kinase, (EC:2.7.1.36) |
| T3_Unigene_BMK.14474 | 1691 | K00869 | mevalonate kinase, (EC:2.7.1.36) |
| T3_Unigene_BMK.22024 | 1903 | K00938 | Phosphomevalonate kinase, (EC:2.7.4.2) |
| T4_Unigene_BMK.29327 | 2282 | K01597 | diphosphomevalonate decarboxylase, (EC:4.1.1.33) |
| T3_Unigene_BMK.15864 | 1831 | K01597 | diphosphomevalonate decarboxylase, (EC:4.1.1.33) |
| T3_Unigene_BMK.5322 | 215 | K11778 | undecaprenyl pyrophosphate synthetase, (EC:2.5.1.31) |
| T4_Unigene_BMK.33183 | 1237 | K11778 | undecaprenyl pyrophosphate synthetase, (EC:2.5.1.31) |
| T4_Unigene_BMK.21428 | 1774 | K00801 | farnesyl-diphosphate farnesyltransferase, (EC:2.5.1.21) |
| T3_Unigene_BMK.3061 | 271 | K00511 | Squalene monooxygenase, (EC:1.14.99.7) |
| CL1336Contig1 | 2048 | K00511 | Squalene monooxygenase, (EC:1.14.99.7) |
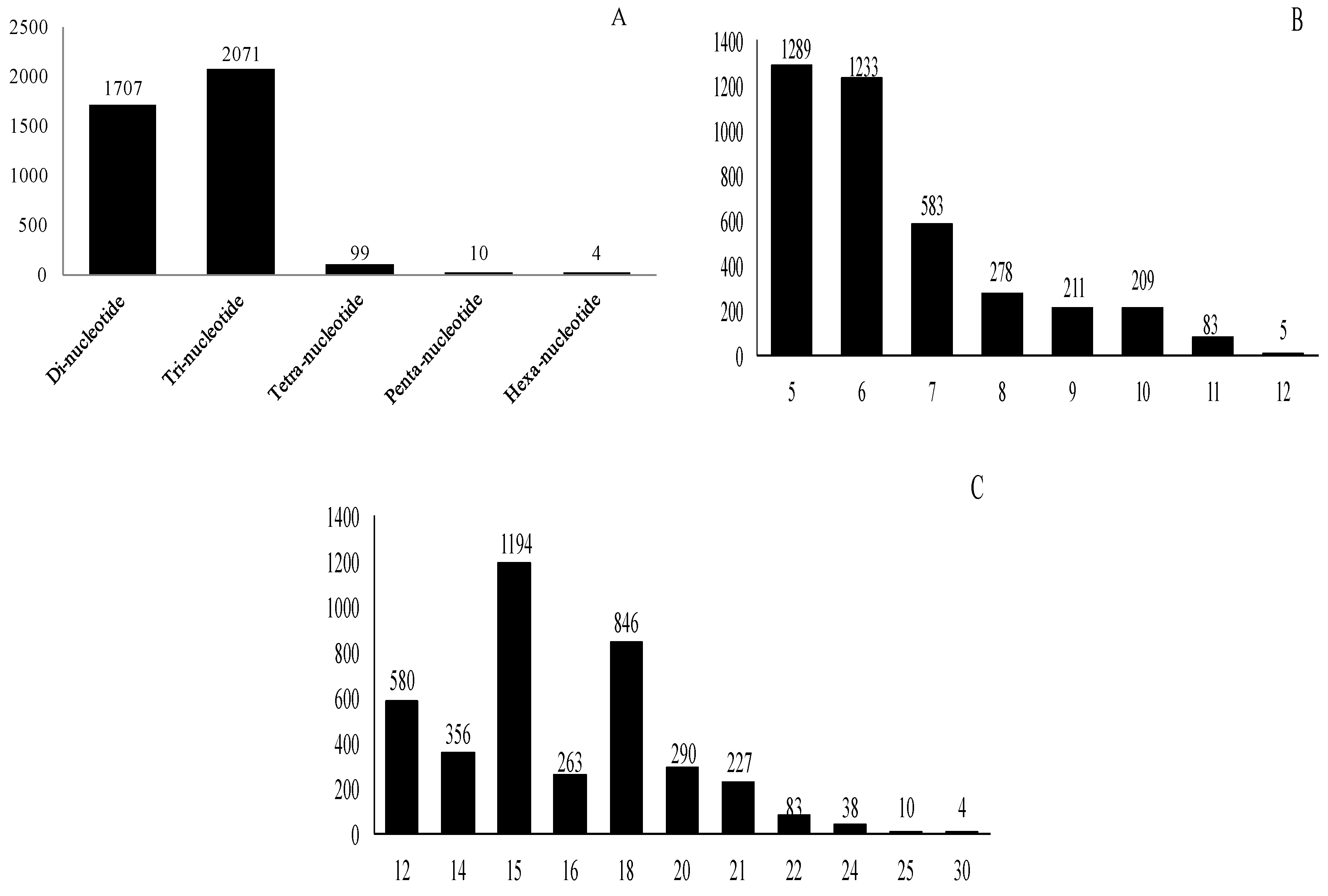
2.3. Frequency and Distribution of Different Types of SSR Markers

| Serial No. | Repeat Motif | Number of Reiterations of the Motif | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |||
| 1 | AG/CT | # | 456 | 285 | 217 | 189 | 192 | 81 | 3 | 1423 |
| 2 | AAG/CTT | 576 | 358 | 137 | 6 | 0 | 0 | 0 | 0 | 1077 |
| 3 | ATC/ATG | 159 | 74 | 25 | 0 | 0 | 0 | 0 | 0 | 258 |
| 4 | AT/AT | # | 95 | 54 | 36 | 18 | 14 | 2 | 1 | 220 |
| 5 | AAT/ATT | 97 | 50 | 19 | 2 | 0 | 0 | 0 | 0 | 168 |
| 6 | AGG/CCT | 90 | 41 | 12 | 1 | 0 | 0 | 0 | 0 | 144 |
| 7 | AGC/CTG | 83 | 24 | 9 | 1 | 0 | 0 | 0 | 0 | 117 |
| 8 | ACC/GGT | 52 | 32 | 6 | 1 | 0 | 0 | 0 | 0 | 91 |
| 9 | AAC/GTT | 49 | 24 | 14 | 2 | 0 | 0 | 0 | 0 | 89 |
| 13 | CCG/CGG | 49 | 17 | 3 | 0 | 0 | 0 | 0 | 0 | 69 |
| 10 | AC/GT | # | 28 | 17 | 10 | 4 | 3 | 0 | 1 | 63 |
| 11 | ACG/CGT | 24 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 34 |
| 12 | ACT/AGT | 15 | 5 | 2 | 2 | 0 | 0 | 0 | 0 | 24 |
| 14 | CG/CG | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 15 | Other motifs * | 95 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 113 |
| Total | 1289 | 1233 | 583 | 278 | 211 | 209 | 83 | 5 | 3891 | |
2.4. Development, Validation and Transferability of SSR Markers
| Primer Name | Base on Sequence ID | SSRs | EPS (bp) | OPS (bp) | Alleles | PIC |
|---|---|---|---|---|---|---|
| G-EST-SSR19 | CL10682Contig1 | (AT)6 | 149 | 149–161 | 5 | 0.67 |
| G-EST-SSR20 | CL10765Contig1 | (GAA)5 | 151 | 148–163 | 8 | 0.82 |
| G-EST-SSR40 | CL11560Contig1 | (TCT)5 | 151 | 151–160 | 4 | 0.68 |
| G-EST-SSR44 | CL13255Contig1 | (AG)6 | 164 | 162–196 | 11 | 0.87 |
| G-EST-SSR47 | CL13343Contig1 | (TCT)6 | 148 | 151–172 | 8 | 0.79 |
| G-EST-SSR57 | CL14153Contig1 | (TGA)5 | 149 | 152–170 | 6 | 0.74 |
| G-EST-SSR75 | CL400Contig1 | (TCA)6 | 172 | 172–202 | 7 | 0.80 |
| G-EST-SSR76 | CL435Contig1 | (TCT)5 | 131 | 122–137 | 6 | 0.76 |
| G-EST-SSR89 | CL1983Contig1 | (GAAA)5 | 146 | 146–166 | 6 | 0.79 |
| G-EST-SSR100 | CL2782Contig1 | (GAA)6 | 145 | 136–163 | 9 | 0.79 |
| G-EST-SSR118 | CL8580Contig1 | (CGG)5 | 141 | 138–153 | 6 | 0.62 |
| G-EST-SSR131 | CL9957Contig1 | (TCT)5 | 149 | 149–158 | 5 | 0.55 |
| G-EST-SSR140 | CL10320Contig1 | (CTA)6 | 158 | 158–170 | 6 | 0.73 |
| G-EST-SSR306 | CL11525Contig1 | (CT)7 | 169 | 169–189 | 6 | 0.73 |
| G-EST-SSR316 | CL12699Contig1 | (GAA)5 | 135 | 135–159 | 8 | 0.66 |
| Mean | - | - | - | 6.73 | 0.73 |
| No. | Species | Locality, Province | Latitude (N), longitude (E) | Characteristics |
|---|---|---|---|---|
| 1 | G. pentaphyllum | Ankang, Shaanxi | 32°25′N,109°04′E | wild |
| 2 | G. pentaphyllum | Ankang, Shaanxi | 32°25′N,109°04′E | Cultivar |
| 3 | G. pentaphyllum | Kunming, Yunnan | 25°14′N,102°49′E | Wild |
| 4 | G. pentaphyllum | Kunming, Yunnan | 25°14′N,102°49′E | Cultivar |
| 5 | G. pentaphyllum | Panzhihua, Sichuan | 26°36′N,101°43′E | Wild |
| 6 | G. pentaphyllum | Panzhihua, Sichuan | 26°36′N,101°43′E | Cultivar |
| 7 | G. burmanicum var. molle | Dehong, Yunnan | 24°48′N, 98°17′E | Wild |
| 8 | G. burmanicum | Menghai, Yunnan | 22°02′N, 100°22′E | Wild |
| 9 | G. burmanicum | Dehong, Yunnan | 24°36′N, 97°39′E | Wild |
| 10 | G.pentaphyllum var. dasycarpum | Mengla, Yunnan | 22°14′N, 101°15′E | Wild |
| 11 | G. pentaphyllum var. dasycarpum | Jinghong, Yunnan, | 22°01′N, 100°45′E | Wild |
| 12 | G. longipes | Xuancheng, Anhui | 30°55′N,118°46′E | Wild |
| 13 | G. longipes | Ankang, Shaanxi | 32°25′N,109°22′E | Wild |
| 14 | G. longipes | Enshi, Hubei | 30°03′N, 109°49′E | Wild |
| 15 | G. longipes | Zhaotong, Yunnan | 27°43′N,103°54′E | Wild |
| 16 | G. pubescens | Menghai, Yunnan | 21°56′N, 100°36′E | Wild |
| 17 | G. pubescens | Menglun, Yunnan | 21°56′N, 101°14′E | Wild |
| 18 | G. pubescens | Yingjiang, Yunnan | 24°42′N, 97°55′E | Wild |
| 19 | G. pubescens | Enshi, Hubei | 30°18′N, 109°31′E | Wild |
| 20 | G. laxum | Jiujiang, Jiangxi, | 29°17′N, 115°07′E | Wild |
| 21 | G. microspermum | Pu’er, Yunnan | 23°07′N, 100°22′E | Wild |
| 22 | G. laxiflorum | Xuancheng, Anhui | 30°41′N,118°39′E | Wild |
| 23 | G. yixingense | Tongling, Anhui | 30°57′N,117°48′E | Wild |
| 24 | G. caulopterum | Renhuai, Guizhou | 27°48′N, 106°26′E | Wild |
| 25 | G. cardiospermum | Ankang, Shaanxi | 32°13′N,109°01′E | Wild |
| 26 | G. cardiospermum | Ankang, Shaanxi | 32°13′N,109°01′E | Wild |
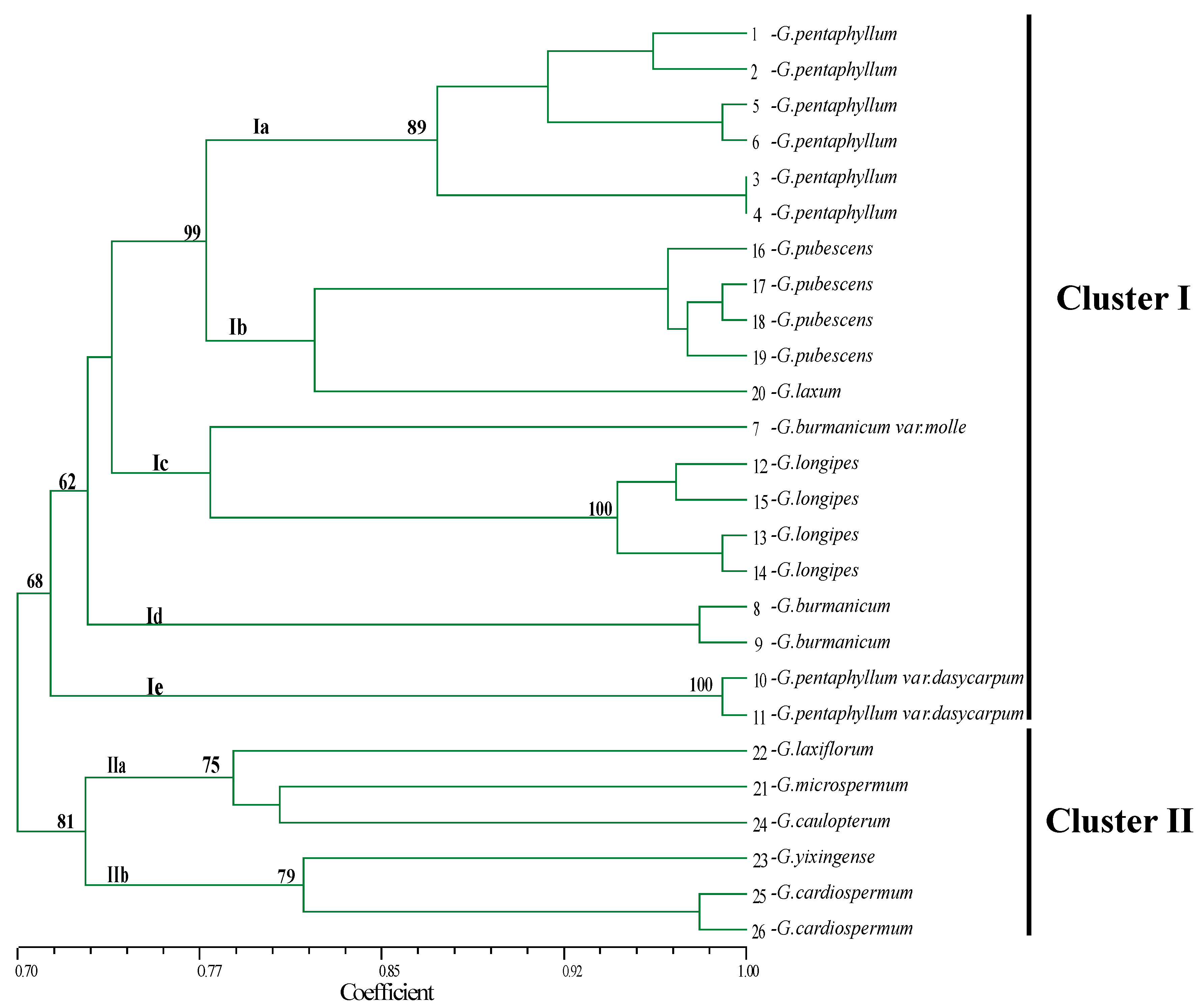
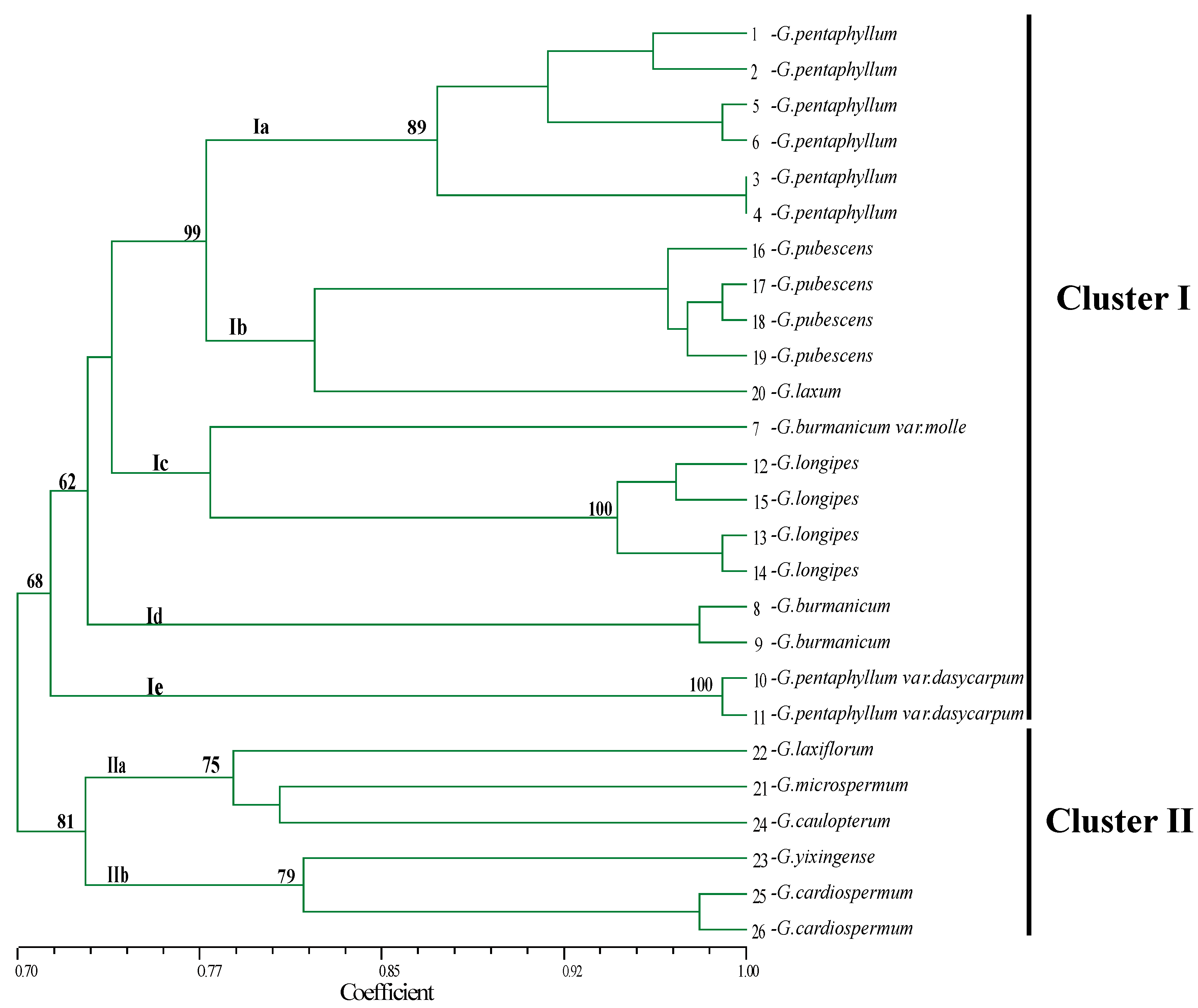
2.5. Genetic Diversity and Relatedness in the Genus Gynostemma
3. Discussion
3.1. Functional Annotation of Unigenes
3.2. SSR Marker Frequency and Distribution in Gynostemma Transcriptome
3.3. Transferability of SSR Markers and Genetic Relationships among Different Species of Gynostemma
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, Reverse Transcription and Sequencing
4.3. Transcript Assembly and Analysis
4.4. EST-SSR Detection and Pprimer Design
4.5. Plant DNA Extraction, PCR Conditions and Separation of SSR Markers
4.6. Genetic Analysis and Data Scoring
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gao, X.F.; Chen, S.K.; Gu, Z.J.; Zhao, J.Z. A chromosomal study on the genus Gynostemma (Cucurbitaceae). Acta Bot. Yunnanica 1995, 17, 312–316. [Google Scholar]
- Chen, S.K. A classificatory system and geographical distribution of the genus Gynostemma BL. (Cucurbitaceae). Acta Phytotaxon. Sin. 1995, 33, 403–410. [Google Scholar]
- Zhou, Z.T.; Wang, Y.; Zhou, Y.M.; Zhang, S.L. Effect of Gynostemma pentaphyllum mak on carcinomatous conversions of golden hamster cheek pouches induced by dimethylbenzanthracene: A histological study. Chin. Med. J. 1998, 111, 847–850. [Google Scholar] [PubMed]
- Lin, C.C.; Huang, P.C.; Lin, J.M. Antioxidant and hepatoprotective effects of Anoectochilus formosanus and Gynostemma pentaphyllum. Am. J. Chin. Med. 2000, 28, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Hu, L.H.; Pan, R.X. Novel dammarane-type glycosides from Gynostemma pentaphyllum. Chem. Pharm. Bull. 2004, 52, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Lin, R.; Hu, Z.H. Histochemical localization of Ginsenosides in Gynostemma Pentaphyllum. and the content changes of total gypenosides. Acta Biol. Exp. Sin. 2005, 38, 54–60. [Google Scholar]
- Liu, S.B.; Lin, R.; Hu, Z.H. Comparison of stem and leaf structures and total gypenosides among 5 species of Gynostemma. J. Fujian Agric. For. Univ. 2006, 35, 495–499. [Google Scholar]
- Yu, Y.F. A milestone of wild plant conservation in China. Plants 1999, 5, 3–11. [Google Scholar]
- Jiang, L.Y.; Qian, Z.Q.; Guo, Z.G.; Wang, C.; Zhao, G.F. Polyploid origins in Gynostemma pentaphyllum (Cucurbitaceae) inferred from multiple gene sequences. Mol. Phylogenet. Evol. 2009, 52, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Q.; Shen, Q.; Li, J.; Hu, L.H. Dammaranes from Gynostemma pentaphyllum and synthesis of their derivatives as inhibitors of protein tyrosine phosphatase 1B. Bioorg. Med. Chem. 2010, 18, 3934–3939. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Lin, C.L.; Chen, B.H. Preparative chromatography of flavonoids and saponins in Gynostemma pentaphyllum and their antiproliferation effect on hepatoma cell. Phytomedicine 2010, 18, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Razmovski-Naumovski, V.; Huang, T.H.W.; Tran, V.H.; Li, G.Q.; Duke, C.C.; Roufogalis, B.D. Chemistry and pharmacology of Gynostemma pentaphyllum. Phytochem. Rev. 2005, 4, 197–219. [Google Scholar] [CrossRef]
- Xie, Z.; Liu, W.; Huang, H.Q.; Slavin, M.; Zhao, Y.; Whent, M.; Blackford, J.; Lutterodt, H.; Zhou, H.P.; Chen, P.; et al. Chemical composition of five commercial Gynostemma pentaphyllum samples and their radical scavenging, antiproliferative, and anti-inflammatory properties. J. Agric. Food Chem. 2010, 58, 11243–11249. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyam, S.; Mathiyalagan, R.; In, J.G.; Lee, B.; Lee, S.; Y., D.C. Transcriptome profiling and in silico analysis of Gynostemma pentaphyllum using a next generation sequencer. Plant. Cell. Rep. 2011, 30, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Zhao, Y.; Zhou, Y.; Wang, Y.G.; Wang, X.F.; Lu, F.; Song, Z.P. Microsatellite markers in the traditional Chinese medicinal herb Gynostemma pentaphyllum (Cucurbitaceae). Am. J. Bot. 2011, 98, e61–e63. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, H.; Qian, Z.Q.; Zhao, G.F. Genetic differentiation in endangered Gynostemma pentaphyllum (Thunb.) Makino based on ISSR polymorphism and its implications for conservation. Biochem. Syst. Ecol. 2008, 36, 699–705. [Google Scholar] [CrossRef]
- Zane, L.; Bargelloni, L.; Patarnello, T. Strategies for microsatellite isolation: A review. Mol. Ecol. 2002, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Squirrell, J.; Hollingsworth, P.M.; Woodhead, M.; Russell, J.; Lowe, A.J.; Gibby, M.; Powell, W. How much effort is required to isolate nuclear microsatellites from plants? Mol. Ecol. 2003, 12, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Bozhko, M.; Riegel, R.; Schubert, R.; Müller-Starck, G. A cyclophilin gene marker confirming geographical differentiation of Norway spruce populations and indicating viability response on excess soil-born salinity. Mol. Ecol. 2003, 12, 3147–3155. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Gilmore, S.; Keys, W.; Morgante, M.; Rafalski, A. Cross-species amplification of soybean (Glycine max) simple sequence repeats (SSRs) within the genus and other legume genera: Implications for the transferability of SSRs in plants. Mol. Biol. Evol. 1998, 15, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Eujayl, I.; Sledge, M.K.; Wang, L.; May, G.D.; Chekhovskiy, K.; Zwonitzer, J.C.; Mian, M.A. Medicago truncatula EST-SSRs reveal cross-species genetic markers for Medicago spp. Theor. Appl. Genet. 2004, 108, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Bernard, M.; Leroy, P.; Feuillet, C.; Sourdille, P. High transferability of bread wheat EST-derived SSRs to other cereals. Theor. Appl. Genet. 2005, 111, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Poncet, V.; Rondeau, M.; Tranchant, C.; Cayrel, A.; Hamon, S.; de Kochko, A.; Hamon, P. SSR mining in coffee tree EST databases: Potential use of EST-SSRs as markers for the Coffea genus. Mol. Genet. Genom. 2006, 276, 436–449. [Google Scholar] [CrossRef] [PubMed]
- Luro, F.L.; Costantino, G.; Terol, J.; Argout, X.; Allario, T.; Wincker, P.; Talon, M.; Ollitrault, P.; Morillon, R. Transferability of the EST-SSRs developed on Nules clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genom. 2008, 9, 287. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.F.; Pan, C.; Diao, Y.; You, Y.N.; Yang, C.Z.; Hu, Z.L. Development of microsatellite markers by transcriptome sequencing in two species of Amorphophallus (Araceae). BMC Genom. 2013, 14, 490. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Bouck, A.; Vision, T. The molecular ecologist’s guide to expressed sequence tags. Mol. Ecol. 2007, 16, 907–924. [Google Scholar] [CrossRef] [PubMed]
- Emrich, S.J.; Barbazuk, W.B.; Li, L.; Schnable, P.S. Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 2007, 17, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Wheat, C.W.; Fescemyer, H.W.; Frilander, M.J.; Crawford, D.L.; Hanski, I.; Marden, J.H. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol. Ecol. 2008, 17, 1636–1647. [Google Scholar] [CrossRef] [PubMed]
- Barbazuk, W.B.; Emrich, S.J.; Chen, H.D.; Li, L.; Schnable, P.S. SNP discovery via 454 transcriptome sequencing. Plant. J. 2007, 51, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J., Jr.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Namroud, M.C.; Beaulieu, J.; Juge, N.; Laroche, J.; Bousquet, J. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol. Ecol. 2008, 17, 3599–3613. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.H.; Xiao, G.; Guo, J.; Fei, Z.J.; Xu, Y.Q.; Roe, B.A.; Wang, Y. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genom. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, Y.; Wu, Q.; Luo, H.M.; Sun, Y.Z.; Song, J.Y.; Lui, E.M.; Chen, S.L. De novo sequencing and analysis of the American ginseng root transcriptome using a GS FLX Titanium platform to discover putative genes involved in ginsenoside biosynthesis. BMC Genom. 2010, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.M.; Sun, C.; Sun, Y.Z.; Wu, Q.; Li, Y.; Song, J.Y.; Niu, Y.Y.; Cheng, X.L.; Xu, H.X.; Li, C.Y.; et al. Analysis of the transcriptome of Panax notoginseng root uncovers putative triterpene saponin-biosynthetic genes and genetic markers. BMC Genom. 2011, 12, S5. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.L.; Qi, X.Q.; Wang, L.H.; Zhang, Y.X.; Hua, W.; Li, D.H.; Lv, H.X.; Zhang, X.R. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Fang, B.P.; Chen, J.Y.; Zhang, X.J.; Luo, Z.X.; Huang, L.F.; Chen, X.L.; Li, Y.J. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genom. 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Li, D.J.; Deng, Z.; Qin, B.; Liu, X.H.; Men, Z.H. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Luikart, G.; England, P.R.; Tallmon, D.; Jordan, S.; Taberlet, P. The power and promise of population genomics: From genotyping to genome typing. Nat. Rev. Genet. 2003, 4, 981–994. [Google Scholar] [CrossRef] [PubMed]
- Kumpatla, S.P.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar, S.; Priya, P.; Garg, R.; Parida, S.K.; Tyagi, A.K.; Jain, M. Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant. Biotechnol. J. 2012, 10, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.N.; Zhang, Y.Q.; Jin, M.D.; Li, H.K.; Song, Z.P.; Wang, Y.G.; Chen, J.K. Characterization and high cross-species transferability of microsatellite markers from the floral transcriptome of Aspidistra saxicola (Asparagaceae). Mol. Ecol. Resour. 2014, 14, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.; Niu, Z.X.; Datla, R.; Duguid, S. Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor. Appl. Genet. 2009, 119, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Thiel, T.; Stein, N.; Langridge, P.; Graner, A. In silico analysis on frequency and distribution of microsatellites in ESTs of some cereal species. Cell. Mol. Biol. Lett. 2002, 7, 537–546. [Google Scholar] [PubMed]
- Kaur, S.; Pembleton, L.W.; Cogan, N.O.; Savin, K.W.; Leonforte, T.; Paull, J.; Materne, M.; Forster, J.W. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genom. 2012, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [PubMed]
- Han, Z.G.; Guo, W.Z.; Song, X.L.; Zhang, T.Z. Genetic mapping of EST-derived microsatellites from the diploid Gossypium arboreum in allotetraploid cotton. Mol. Genet. Genom. 2004, 272, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Hisano, H.; Sato, S.; Isobe, S.; Sasamoto, S.; Wada, T.; Matsuno, A.; Fujishiro, T.; Yamada, M.; Nakayama, S.; Nakamura, Y.; Watanabe, S.; Harada, K.; Tabata, S. Characterization of the soybean genome using EST-derived microsatellite markers. DNA Res. 2007, 14, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Cogan, N.O.; Pembleton, L.W.; Shinozuka, M.; Savin, K.W.; Materne, M.; Forster, J.W. Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genom. 2011, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- La Rota, M.; Kantety, R.V.; Yu, J.K.; Sorrells, M.E. Nonrandom distribution and frequencies of genomic and EST-derived microsatellite markers in rice, wheat, and barley. BMC Genom. 2005, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.X.; Zhou, P.; Choi, Y.A.; Huang, S.; Gmitter, F.G., Jr. Mining and characterizing microsatellites from citrus ESTs. Theor. Appl. Genet. 2006, 112, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar] [PubMed]
- Parida, S.K.; Kumar, K.A. R.; Dalal, V.; Singh, N.K.; Mohapatra, T. Unigene derived microsatellite markers for the cereal genomes. Theor. Appl. Genet. 2006, 112, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.K.; Dalal, V.; Singh, A.K.; Singh, N.K.; Mohapatra, T. Genic non-coding microsatellites in the rice genome: Characterization, marker design and use in assessing genetic and evolutionary relationships among domesticated groups. BMC Genom. 2009, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K.; et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea [Cajanus cajan (L.) Millspaugh]. BMC Plant. Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Rungis, D.; Berube, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Le Provost, G.; Leger, V.; Klopp, C.; Noirot, C.; Frigerio, J.M.; Salin, F.; Salse, J.; Abrouk, M.; Murat, F.; et al. Bioinformatic analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species: Oak. BMC Genom. 2010, 11, 650. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Zaki, N.; Ting, N.-C.; Rosli, R.; Tan, S.-G.; Low, E.-T.; Ithnin, M.; Cheah, S.-C. Exploiting an oil palm EST database for the development of gene-derived SSR markers and their exploitation for assessment of genetic diversity. Biologia 2008, 63, 227–235. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, H.F.; Wu, W.; Yu, H.; Ge, X.J. Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonii). BMC Genom. 2013, 14, 329. [Google Scholar] [CrossRef] [PubMed]
- Torales, S.L.; Rivarola, M.; Pomponio, M.F.; Fernandez, P.; Acuna, C.V.; Marchelli, P.; Gonzalez, S.; Azpilicueta, M.M.; Hopp, H.E.; Gallo, L.A.; et al. Transcriptome survey of Patagonian southern beech Nothofagus nervosa (= N. Alpina.): Assembly, annotation and molecular marker discovery. BMC Genom. 2012, 13, 291. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.C.; Mian, M.R.; Eujayl, I.; Zwonitzer, J.C.; Wang, L.; May, G.D. Tall fescue EST-SSR markers with transferability across several grass species. Theor. Appl. Genet. 2004, 109, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.G.; Ishii, T.; Temnykh, S.; Chen, X.; Lipovich, L.; McCOUCH, S.R.; Park, W.D.; Ayres, N.; Cartinhour, S. Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 2000, 100, 713–722. [Google Scholar] [CrossRef]
- Eujayl, I.; Sorrells, M.; Baum, M.; Wolters, P.; Powell, W. Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor. Appl. Genet. 2002, 104, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.R.; Burke, J.M. EST-SSRs as a resource for population genetic analyses. Heredity 2007, 99, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Barbara, T.; Palma-Silva, C.; Paggi, G.M.; Bered, F.; Fay, M.F.; Lexer, C. Cross-species transfer of nuclear microsatellite markers: Potential and limitations. Mol. Ecol. 2007, 16, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Q.J.; Li, Z.H.; Zhao, G.F. Genetic diversity and population structure of Gynostemma pentaphyllun. Chin. Tradit. Herb. Drugs 2015, 46, 1958–1965. [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinform. 2008, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Porebski, S.; Bailey, L.G.; Baum, B.R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 1997, 15, 8–15. [Google Scholar] [CrossRef]
- Creste, S.; Neto, A.T.; Figueira, A. Detection of single sequence repeat polymorphisms in denaturing polyacrylamide sequencing gels by silver staining. Plant Mol. Biol. Rep. 2001, 19, 299–306. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314. [Google Scholar] [PubMed]
- Rolf, J. Numerical Taxonomy and Multivariate Analysis System, Version 2.11; T Exeter Software: Setauket, NY, USA, 2000. [Google Scholar]
- Pavlicek, A.; Hrda, S.; Flegr, J. Free-Tree-freeware program for construction of phylogenetic trees on the basis of distance data and bootstrap/jackknife analysis of the tree robustness. Application in the RAPD analysis of genus Frenkelia. Folia Biol. 1999, 97–99. [Google Scholar]
- Sample Availability: All samples are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.-M.; Zhou, T.; Li, Z.-H.; Zhao, G.-F. Characterization of Global Transcriptome Using Illumina Paired-End Sequencing and Development of EST-SSR Markers in Two Species of Gynostemma (Cucurbitaceae). Molecules 2015, 20, 21214-21231. https://doi.org/10.3390/molecules201219758
Zhao Y-M, Zhou T, Li Z-H, Zhao G-F. Characterization of Global Transcriptome Using Illumina Paired-End Sequencing and Development of EST-SSR Markers in Two Species of Gynostemma (Cucurbitaceae). Molecules. 2015; 20(12):21214-21231. https://doi.org/10.3390/molecules201219758
Chicago/Turabian StyleZhao, Yue-Mei, Tao Zhou, Zhong-Hu Li, and Gui-Fang Zhao. 2015. "Characterization of Global Transcriptome Using Illumina Paired-End Sequencing and Development of EST-SSR Markers in Two Species of Gynostemma (Cucurbitaceae)" Molecules 20, no. 12: 21214-21231. https://doi.org/10.3390/molecules201219758
APA StyleZhao, Y.-M., Zhou, T., Li, Z.-H., & Zhao, G.-F. (2015). Characterization of Global Transcriptome Using Illumina Paired-End Sequencing and Development of EST-SSR Markers in Two Species of Gynostemma (Cucurbitaceae). Molecules, 20(12), 21214-21231. https://doi.org/10.3390/molecules201219758