The Effect of Mini-PEG-Based Spacer Length on Binding and Pharmacokinetic Properties of a 68Ga-Labeled NOTA-Conjugated Antagonistic Analog of Bombesin


Abstract
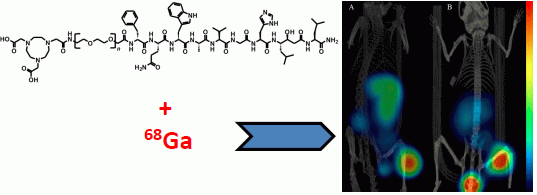
:
1. Introduction
2. Results and Discussion
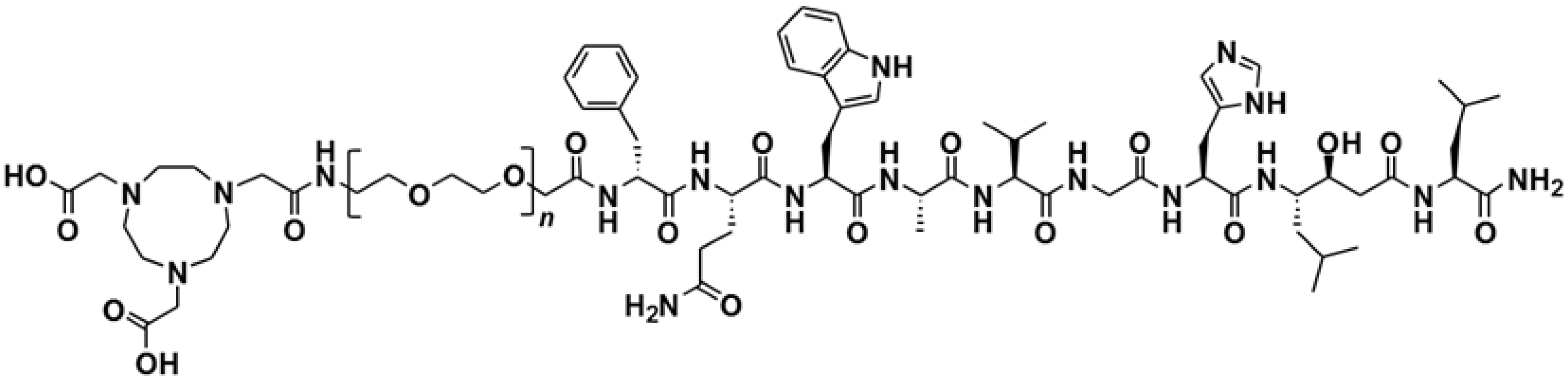
2.1. Peptide Synthesis

| PEG2 | PEG3 | PEG4 | PEG6 | |
|---|---|---|---|---|
| Synthetic yield (%) | 24 | 26 | 33 | 26 |
| m/z value (m/z theor. value) | 1543.88 (1542.82) | 1601.58 (1600.86) | 1646.06 (1644.89) | 1734.23 (1732.94) |
| Purity (%) | 98.3 | 99.3 | 99.8 | 99.5 |
2.2. Radiolabeling and Stability Test
| PEG2 | PEG3 | PEG4 | PEG6 | |
|---|---|---|---|---|
| Labeling yield for 68Ga, % | 99.4 ± 0.3 | 98 ± 1 | 99.2 ± 0.1 | 99.3 ± 0.1 |
| Stability under EDTA challenge, % | 99.3 ± 0.1 | 98.1 ± 0.2 | 98.5 ± 0.1 | 99.1 ± 0.2 |
| Labeling yield for 111In, % | 96 ± 2 | 97 ± 2 | 97 ± 2 | 97 ± 2 |
| Stability under EDTA challenge, % | 95.5 ± 0.3 | 92.5 ± 0.7 | 94.3 ± 0.6 | 94.6 ± 0.7 |
| LogD of 68Ga-NOTA-PEGn-RM26 | −2.27 ± 0.07 | −2.47 ± 0.06 | −2.49 ± 0.10 | −2.50 ± 0.09 |
2.3. In Vitro Studies
2.3.1. In Vitro Binding Specificity Assay
| PEG2 | PEG3 | PEG4 | PEG6 | |
|---|---|---|---|---|
| In vitro binding specificity test, % blocked uptake of 68Ga-labeled variants on PC-3 (BT474) cells in presence of excess of non-labeled tracer | 94.5 ± 0.1 | 98.0 ± 0.1 (95.3 ± 0.2) | 98.2 ± 0.1 | 95.5 ± 0.3 |
| In vitro binding specificity test, % blocked uptake of 111In-labeled variants on PC-3 (BT474) cells in the presence of excess of non-labeled tracer | 97.7 ± 0.2 | 94.0 ± 0.2 (95.0 ± 0.4) | 95.0 ± 0.9 | 94.8 ± 0.4 |
| Internalization rate by PC-3 (BT474) cells, % internalized radioactivity from total cell associated at 24 h continuous incubation | 33 ± 3 | 31 ± 2 (32 ± 2) | 30 ± 1 | 35 ± 2 |
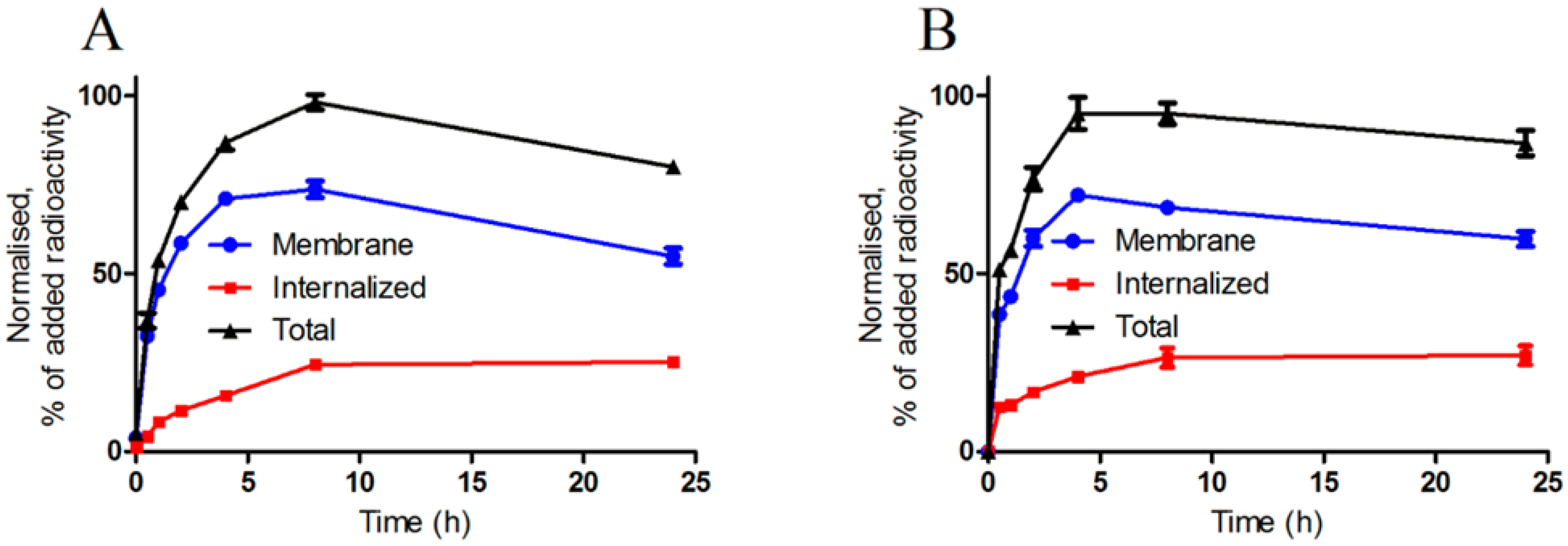
2.3.2. Cellular Uptake and Internalization
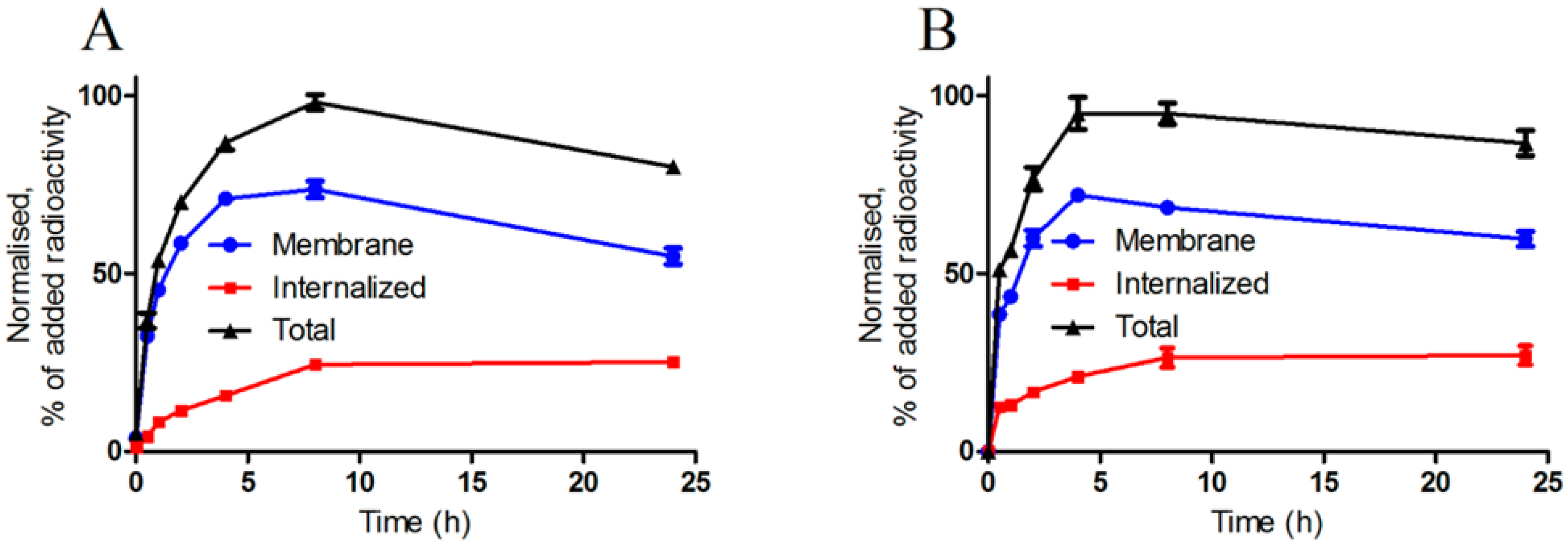
2.3.3. Real-Time Ligand Binding Kinetics: KD and Bmax Determination
| PEG2 | PEG3 | PEG4 | PEG6 | |
|---|---|---|---|---|
| Association rate, ka, M−1 s−1 | (2.9 ± 0.3) × 105 | (8 ± 4) × 105 | (5 ± 3) × 105 | (6 ± 5) × 105 |
| Dissociation rate, kd, s−1 | (11.2 ± 0.0) × 10−6 | (2.7 ± 0.2) × 10−6 | (2.5 ± 0.0) × 10−6 | (7 ± 3) × 10−6 |
| KD, pM | 23 ± 13 | 5 ± 3 | 8 ± 5 | 18 ± 8 |
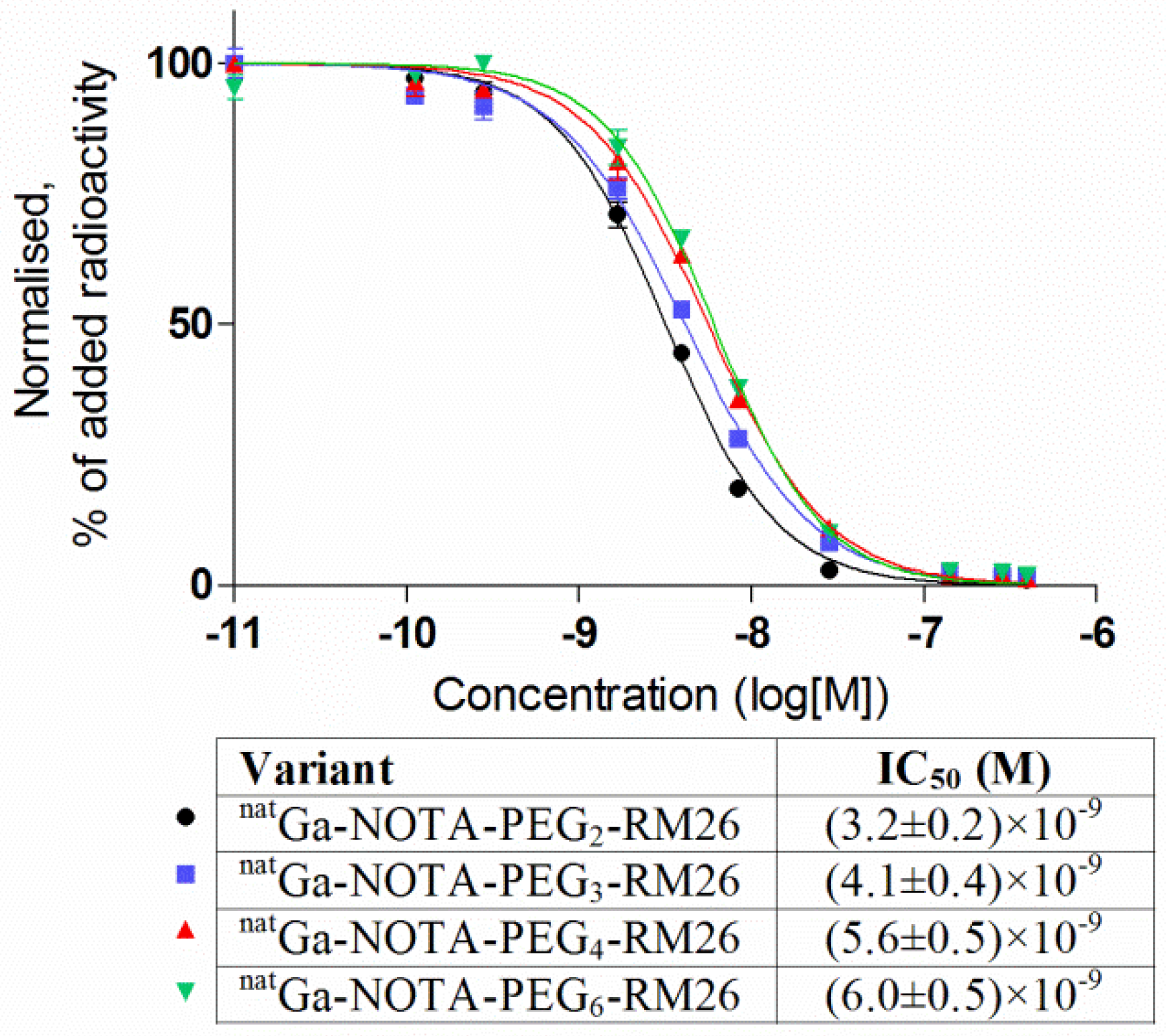
2.3.4. In Vitro Competitive Binding Assay: IC50 Determination
2.4. In Vivo Studies
2.4.1. Biodistribution in NMRI Mice
| PEG2 | PEG3 | PEG4 | PEG6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Organ | 1 h | 2 h | 1 h | 2 h | 1 h | 2 h | 1 h | 2 h | ||
| Blood | 0.4 ± 0.2 | 0.05 ± 0.01 | 0.19 ± 0.02 | 0.065 ± 0.002 | 0.25 ± 0.06 | 0.073 ± 0.007 | 0.27 ± 0.06 | 0.06 ± 0.03 | ||
| Lung | 0.3 ± 0.1 | 0.5 ± 0.4 | 0.20 ± 0.05 a | 0.17 ± 0.01 a | 0.32 ± 0.06 | 0.5 ± 0.1 | 0.5 ± 0.2 | 0.14 ± 0.08 | ||
| Liver | 0.9 ± 0.2 | 0.8 ± 0.2 | 0.6 ± 0.1 a,b | 0.7 ± 0.1 a,b | 1.1 ± 0.2 | 1.2 ± 0.2 | 1.06 ± 0.04 | 1.1 ± 0.1 | ||
| Spleen | 0.6 ± 0.2 | 0.46 ± 0.03 | 0.40 ± 0.04 b | 0.3 ± 0.1 a | 0.5 ± 0.2 | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.6 ± 0.2 | ||
| Pancreas | 7 ± 3 | 1.7 ± 0.6 | 6.6 ± 0.4 | 1.7 ± 0.2 | 6.1 ± 0.6 | 2.3 ± 0.7 | 6.2 ± 0.7 | 1.4 ± 0.3 | ||
| Stomach | 1.2 ± 0.3 | 0.63 ± 0.06 | 1.0 ± 0.2 | 0.60 ± 0.04 | 0.90 ± 0.08 | 0.7 ± 0.1 | 1.2 ± 0.2 | 0.57 ± 0.06 | ||
| Sm. intest. | 2.1 ± 0.8 | 0.8 ± 0.4 | 2.0 ± 0.3 | 0.9 ± 0.3 | 2.0 ± 0.3 | 1.1 ± 0.6 | 1.1 ± 0.6 | 0.51 ± 0.09 | ||
| Kidney | 3 ± 2 | 1.0 ± 0.4 | 1.6 ± 0.3 b | 1.0 ± 0.1 | 1.7 ± 0.6 | 1.3 ± 0.2 | 2.7 ± 0.5 | 1.08 ± 0.09 | ||
| Muscle | 0.10 ± 0.05 | 0.04 ± 0.02 | 0.06 ± 0.01 | 0.019 ± 0.002 | 0.09 ± 0.03 | 0.03 ± 0.02 | 0.08 ± 0.03 | 0.03 ± 0.02 | ||
| Bone | 0.18 ± 0.07 | 0.07 ± 0.02 | 0.12 ± 0.04 | 0.050 ± 0.003 | 0.12 ± 0.05 | 0.10 ± 0.04 | 0.13 ± 0.04 | 0.11 ± 0.06 | ||
| GI * | 4 ± 2 | 2.88 ± 0.06 | 3.7 ± 0.8 | 2.5 ± 0.7 | 3.5 ± 0.6 | 3.1 ± 0.9 | 2.8 ± 0.2 | 1.8 ± 0.5 | ||
| Carcass * | 4 ± 1 | 0.9 ± 0.4 | 2.1 ± 0.4 | 0.7 ± 0.1 | 2.3 ± 0.5 | 1.2 ± 0.3 | 3 ± 1 | 0.71 ± 0.05 | ||
2.4.2. Biodistribution and in vivo Binding Specificity Test in Female BALB/c nu/nu Mice Bearing PC-3 and BT-474 Xenografts
| PC-3 Cells | BT474 Cells | |||
|---|---|---|---|---|
| Organ | Non-Blocked | Blocked | Non-Blocked | Blocked |
| Blood | 0.11 ± 0.02 | 0.060 ± 0.010 * | 0.05 ± 0.02 | 0.17 ± 0.06 |
| Liver | 1.6 ± 0.1 | 1.6 ± 0.2 | 0.53 ± 0.06 | 0.81 ± 0.08 |
| Spleen | 1.4 ± 0.3 | 0.9 ± 0.3 | 0.3 ± 0.2 | 0.4 ± 0.1 |
| Pancreas | 3.9 ± 0.6 | 0.27 ± 0.04 * | 2.8 ± 0.6 | 0.3 ± 90.04 * |
| Small intestine | 1.2 ± 0.4 | 0.21 ± 0.08 * | 0.66 ± 0.08 | 0.48 ± 0.09 * |
| Kidney | 1.7 ± 0.2 | 3.3 ± 0.2 * | 1.65 ± 0.09 | 4.25 ± 0.05 * |
| Tumor | 4.6 ± 0.6 | 0.97 ± 0.04 * | 2.8 ± 0.4 | 0.4 ± 0.1 * |
| Muscle | 0.06 ± 0.02 | 0.030 ± 0.003 | 0.05 ± 0.02 | 0.13 ± 0.04 |
| Bone | 0.13 ± 0.03 | 0.13 ± 0.04 | 0.08 ± 0.03 | 0.14 ± 0.05 |
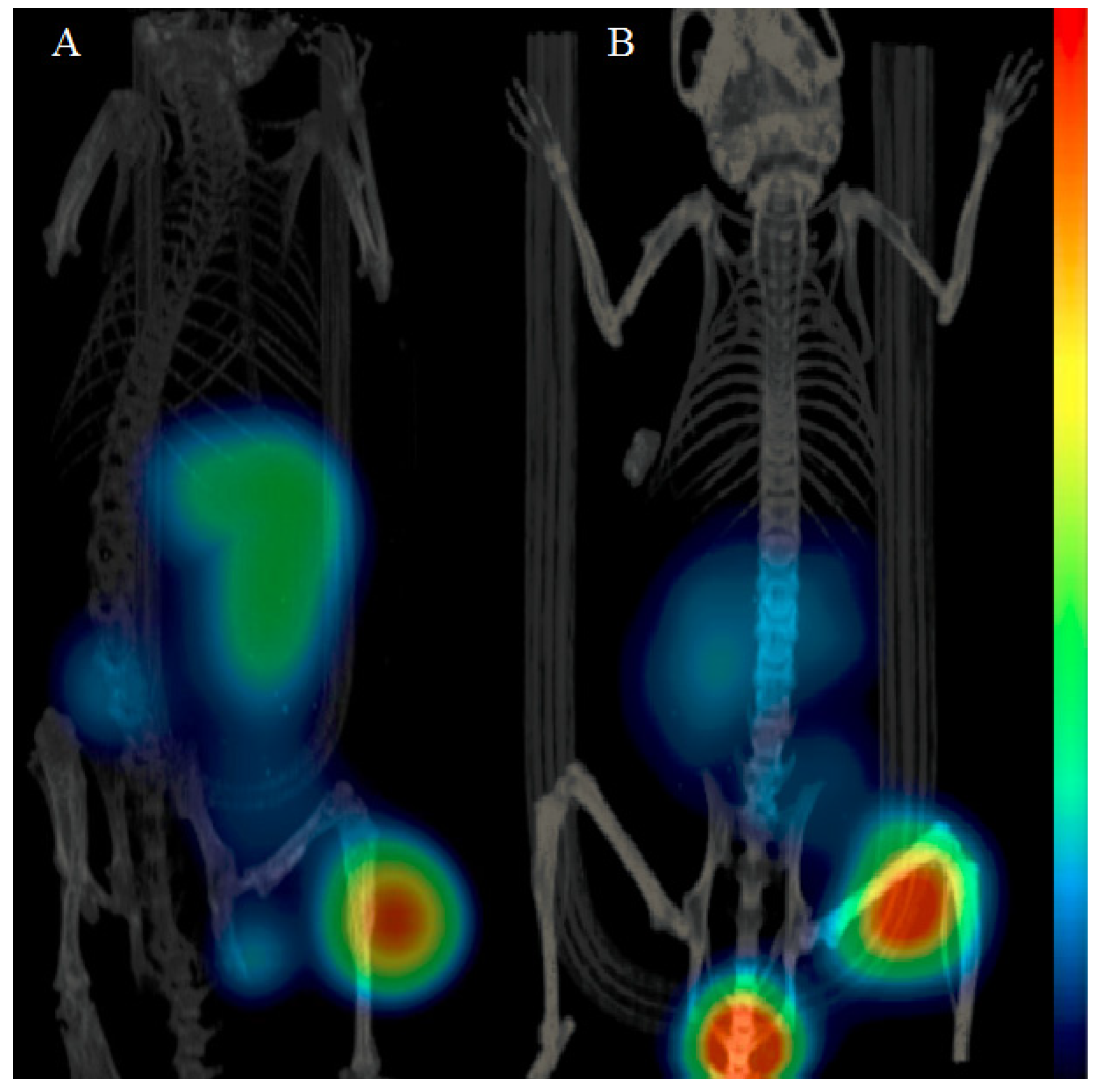
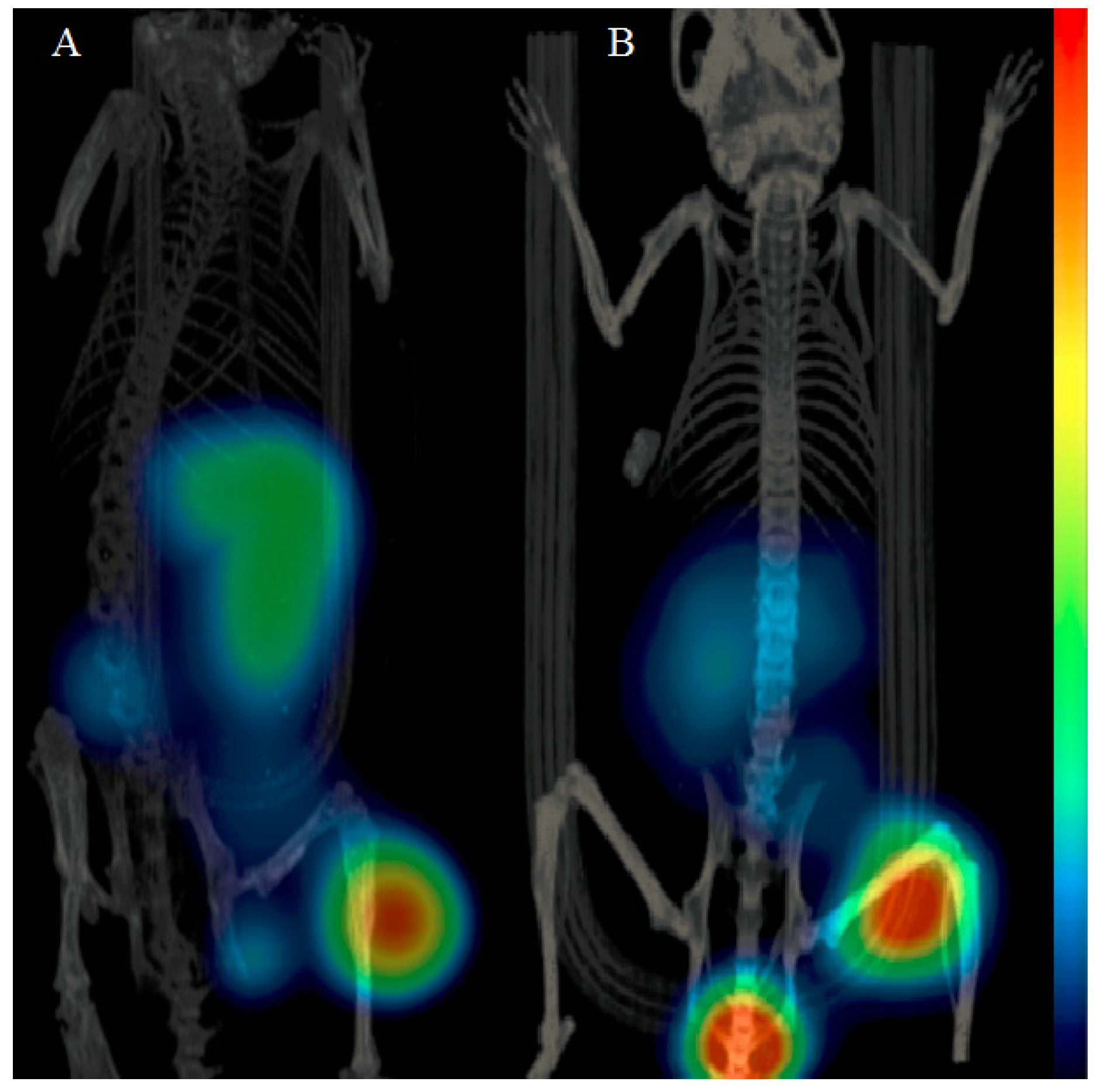
2.5. Imaging Studies
2.6. Discussion
3. Experimental Section
3.1. Peptide Synthesis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.2. Radiolabeling and Stability Test
3.3. In Vitro Studies
3.3.1. In Vitro Binding Specificity Assay
3.3.2. Real-Time Ligand Binding Kinetics: KD and Bmax Determination
3.3.3. In Vitro Competitive Binding Assay: IC50 Determination
3.3.4. Cellular Uptake and Internalization Assay
3.4. In Vivo Studies
3.4.1. Biodistribution in NMRI Mice
3.4.2. Biodistribution and in vivo Binding Specificity Test in BALB/c nu/nu Mice Bearing PC-3 Prostate Cancer and BT-474 Breast Cancer Xenografts
3.5. Imaging Studies
3.6. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ananias, H.J.; van den Heuvel, M.C.; Helfrich, W.; de Jong, I.J. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate 2009, 69, 1101–1108. [Google Scholar]
- Beer, M.; Montani, M.; Gerhardt, J.; Wild, P.J.; Hany, T.F.; Hermanns, T.; Müntener, M.; Kristiansen, G. Profiling gastrin-releasing peptide receptor in prostate tissues: Clinical implications and molecular correlates. Prostate 2012, 72, 318–325. [Google Scholar]
- Halmos, G.; Wittliff, J.L.; Schally, A.V. Characterization of bombesin/gastrin-releasing peptide receptors in human breast cancer and their relationship to steroid receptor expression. Cancer Res. 1995, 55, 280–287. [Google Scholar]
- Gugger, M.; Reubi, J.C. Gastrin-releasing peptide receptors in non-neoplastic and neoplastic human breast. Am. J. Pathol. 1999, 155, 2067–2076. [Google Scholar] [CrossRef]
- Chao, C.; Ives, K.; Hellmich, H.L.; Townsend, C.M., Jr.; Hellmich, M.R. Gastrin-releasing peptide receptor in breast cancer mediates cellular migration and interleukin-8 expression. J. Surg. Res. 2009, 156, 26–31. [Google Scholar]
- Van de Wiele, C.; Dumont, F.; Broecke, R.V.; Oosterlinck, W.; Cocquyt, V.; Serreyn, R.; Peers, S.; Thornback, J.; Slegers, G.; Dierckx, R.A. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: A feasibility study. Eur. J. Nucl. Med. 2000, 27, 1694–1699. [Google Scholar] [CrossRef]
- Schwartsmann, G.; DiLeone, L.P.; Horowitz, M.; Schunemann, D.; Cancella, A.; Pereira, A.S.; Richter, M.; Souza, F.; da Rocha, A.B.; Souza, F.H.; et al. A phase I trial of the bombesin/gastrin-releasing peptide (BN/GRP) antagonist RC3095 in patients with advanced solid malignancies. Invest. New Drugs 2006, 24, 403–412. [Google Scholar] [CrossRef]
- Dijkgraaf, I.; Franssen, G.M.; McBride, W.J.; D’Souza, C.A.; Laverman, P.; Smith, C.J.; Goldenberg, D.M.; Oyen, W.J.; Boerman, O.C. PET of tumors expressing gastrin-releasing peptide receptor with an 18F-labeled bombesin analog. J. Nucl. Med. 2012, 53, 947–952. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, J.; Waldherr, C.; Hinni, K.; Waser, B.; Reubi, J.C.; Maecke, H.R. Synthesis and evaluation of bombesin derivatives on the basis of pan-bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for targeting bombesin receptor-expressing tumors. Cancer Res. 2004, 64, 6707–6715. [Google Scholar]
- Däpp, S.; Müller, C.; Garayoa, E.G.; Bläuenstein, P.; Maes, V.; Brans, L.; Tourwé, D.A.; Schibli, R. PEGylation, increasing specific activity and multiple dosing as strategies to improve the risk-benefit profile of targeted radionuclide therapy with 177Lu-DOTA-bombesin analogues. EJNMMI Res. 2012, 2, 24–36. [Google Scholar] [CrossRef]
- Wild, D.; Frischknecht, M.; Zhang, H.; Morgenstern, A.; Bruchertseifer, F.; Boisclair, J.; Provencher-Bolliger, A.; Reubi, J.C.; Maecke, H.R. Alpha- versus beta-particle radiopeptide therapy in a human prostate cancer model (213Bi-DOTA-PESIN and 213Bi-AMBA versus 177Lu-DOTA-PESIN). Cancer Res. 2011, 71, 1009–1018. [Google Scholar] [CrossRef]
- Garayoa, E.G.; Schweinsberg, C.; Maes, V.; Rüegg, D.; Blanc, A.; Bläuenstein, P.; Tourwé, D.A.; Beck-Sickinger, A.G.; Schubiger, P.A. New [99mTc]bombesin analogues with improved biodistribution for targeting gastrin releasing-peptide receptor-positive tumors. QJ Nucl. Med. Mol. Imaging 2007, 51, 42–50. [Google Scholar]
- Abiraj, K.; Mansi, R.; Tamma, M.L.; Fani, M.; Forrer, F.; Nicolas, G.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 2011, 52, 1970–1978. [Google Scholar]
- Däpp, S.; Garayoa, E.G.; Maes, V.; Brans, L.; Tourwé, D.A.; Müller, C.; Schibli, R. PEGylation of (99m)Tc-labeled bombesin analogues improves their pharmacokinetic properties. Nucl. Med. Biol. 2011, 38, 997–1009. [Google Scholar]
- Biddlecombe, G.B.; Rogers, B.E.; de Visser, M.; Parry, J.J.; de Jong, M.; Erion, J.L.; Lewis, J.S. Molecular imaging of gastrin-releasing peptide receptor-positive tumors in mice using 64Cu- and 86Y-DOTA-(Pro1,Tyr4)-bombesin(1-14). Bioconjug. Chem. 2007, 18, 724–730. [Google Scholar] [CrossRef]
- Moody, T.W.; Crawley, J.N.; Jensen, R.T. Pharmacology and neurochemistry of bombesin-like peptides. Peptides 1982, 3, 559–563. [Google Scholar] [CrossRef]
- Breeman, W.A.; Hofland, L.J.; de Jong, M.; Bernard, B.F.; Srinivasan, A.; Kwekkeboom, D.J.; Visser, T.J.; Krenning, E.P. Evaluation of radiolabelled bombesin analogues for receptor-targeted scintigraphy and radiotherapy. Int. J. Cancer 1999, 81, 658–665. [Google Scholar]
- Hoffman, T.J.; Gali, H.; Smith, C.J.; Sieckman, G.L.; Hayes, D.L.; Owen, N.K.; Volkert, W.A. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J. Nucl. Med. 2003, 44, 823–831. [Google Scholar]
- Schweinsberg, C.; Maes, V.; Brans, L.; Bläuenstein, P.; Tourwé, D.A.; Schubiger, P.A.; Schibli, R.; Garayoa, E.G. Novel glycated [99mTc(CO)3]-labeled bombesin analogues for improved targeting of gastrin-releasing peptide receptor-positive tumors. Bioconjug. Chem. 2008, 19, 2432–2439. [Google Scholar] [CrossRef]
- Garayoa, G.E.; Schweinsberg, C.; Maes, V.; Brans, L.; Bläuenstein, P.; Tourwe, D.A.; Schibli, R.; Schubiger, P.A. Influence of the molecular charge on the biodistribution of bombesin analogues labeled with the [99mTc(CO)3]-core. Bioconjug. Chem. 2008, 19, 2409–2416. [Google Scholar] [CrossRef]
- Roberts, M.J.; Bentley, M.D.; Harris, J.M. Chemistry for peptide and protein PEGylation. Adv. Drug Deliv. Rev. 2002, 54, 459–476. [Google Scholar] [CrossRef]
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar]
- Zhang, H.; Schuhmacher, J.; Waser, B.; Wild, D.; Eisenhut, M.; Reubi, J.; Maecke, H. DOTA-PESIN, a DOTA-conjugated bombesin derivative designed for imaging and targeting radionuclide treatment of bombesin receptor-positive tumours. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1198–1208. [Google Scholar] [CrossRef]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef]
- Wu, Z.; Li, Z-B.; Cai, W.; He, L.; Chin, F.T.; Li, F.; Chen, X. 18F-labeled mini-PEG spacered RGD dimer (18F-FPRGD2): Synthesis and microPET imaging of αvβ3 integrin expression. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1823–1831. [Google Scholar] [CrossRef]
- Fang, W.; He, J.; Kim, Y.S.; Zhou, Y.; Liu, S. Evaluation of 99mTc-labeled cyclic RGD peptide with a PEG4 linker for thrombosis imaging: Comparison with DMP444. Bioconjug. Chem. 2011, 22, 1715–1722. [Google Scholar] [CrossRef]
- Veronese, F.M. Peptide and protein PEGylation: A review of problems and solutions. Biomaterials 2001, 22, 405–417. [Google Scholar]
- Monfardini, C.; Schiavon, O.; Caliceti, P.; Morpurgo, M.; Harris, J.M.; Veronese, F.M. A branched monomethoxypoly(ethylene glycol) for protein modification. Bioconjug. Chem. 1995, 6, 62–69. [Google Scholar]
- Pradhananga, S.; Wilkinson, I.; Ross, R.J. Pegvisomant: Structure and function. J. Mol. Endocrinol. 2002, 29, 11–14. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Tsunoda, S.; Kamada, H.; Kihira, T.; Nakagawa, S.; Kaneda, Y.; Kanamori, T.; Mayumi, T. Molecular design of hybrid tumour necrosis factor-alpha. II: The molecular size of polyethylene glycol-modified tumour necrosis factor-alpha affects its anti-tumour potency. Br. J. Cancer 1996, 74, 1090–1095. [Google Scholar] [CrossRef]
- Varasteh, Z.; Velikyan, I.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Sandström, M.; Selvaraju, R.K.; Malmberg, J.; Tolmachev, V.; Orlova, A. Synthesis and characterization of a high-affinity NOTA-conjugated bombesin antagonist for GRPR-targeted tumor imaging. Bioconjug. Chem. 2013, 24, 1144–1153. [Google Scholar] [CrossRef]
- Varasteh, Z.; Aberg, O.; Velikyan, I.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Antoni, G.; Sandström, M.; Tolmachev, V.; Orlova, A. In vitro and in vivo evaluation of a (18)F-labeled high affinity NOTA conjugated bombesin antagonist as a PET ligand for GRPR-targeted tumor imaging. PLoS One 2013, 8, e81932. [Google Scholar]
- Dimitrakopoulou-Strauss, A.; Hohenberger, P.; Haberkorn, U.; Mäcke, H.R.; Eisenhut, M.; Strauss, L.G. 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: Comparison with 18F-FDG. J. Nucl. Med. 2007, 48, 1245–1250. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Phonteyne, P.; Pauwels, P.; Goethals, I.; van den Broecke, R.; Cocquyt, V.; Dierckx, R.A. Gastrin-releasing peptide receptor imaging in human breast carcinoma versus immunohistochemistry. J. Nucl. Med. 2008, 49, 260–264. [Google Scholar] [CrossRef]
- Nock, B.A.; Nikolopoulou, A.; Galanis, A.; Cordopatis, P.; Waser, B.; Reubi, J.C.; Maina, T. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: A preclinical study. J. Med. Chem. 2005, 48, 100–110. [Google Scholar]
- Kunstler, J.U.; Veerendra, B.; Figueroa, S.D.; Sieckman, G.L.; Rold, T.L.; Hoffman, T.J.; Smith, C.J.; Pietzsch, H.J. Organometallic 99m Tc(III) ‘4 + 1’ bombesin(7-14) conjugates: Synthesis, radiolabeling, and in vitro/in vivo studies. Bioconjug. Chem. 2007, 18, 1651–1661. [Google Scholar] [CrossRef]
- Prasanphanich, A.F.; Nanda, P.K.; Rold, T.L.; Ma, L.; Lewis, M.R.; Garrison, J.C.; Hoffman, T.J.; Sieckman, G.L.; Figueroa, S.D.; Smith, C.J. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc. Natl. Acad. Sci. USA 2007, 104, 12462–12467. [Google Scholar] [CrossRef]
- Garayoa, E.G.; Ruegg, D.; Blauenstein, P.; Zwimpfer, M.; Khan, I.U.; Maes, V.; Blanc, A.; Beck-Sickinger, A.G.; Tourwe, D.A.; Schubiger, P.A. Chemical and biological characterization of new Re(CO)3/[99mTc](CO)3bombesin analogues. Nucl. Med. Biol. 2007, 34, 17–28. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A. Influence of labelling methods on biodistribution and imaging properties of radiolabelled peptides for visualisation of molecular therapeutic targets. Curr. Med. Chem. 2010, 17, 2636–2655. [Google Scholar] [CrossRef]
- Humle, E.C.; Trevetchick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef]
- Xu, B.; Varasteh, Z.; Orlova, A.; Andersson, K.; Larhammar, D.; Björkelund, H. Detecting interactions with GPCR in real-time on living cells to understand receptor dynamics. Biochem. Biophys. Res. Commun. 2013, 441, 820–824. [Google Scholar] [CrossRef]
- Wållberg, H.; Orlova, A. Slow internalization of anti-HER2 synthetic Affibody monomer 111In-DOTA-ZHER2:342-pep2: Implications for development of labeled tracers. Cancer Biother. Radiopharm. 2008, 23, 435–442. [Google Scholar]
- Sample Availability: Samples of the compounds NOTA-PEGn-RM26 (n = 2, 3, 4 and 6) are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varasteh, Z.; Rosenström, U.; Velikyan, I.; Mitran, B.; Altai, M.; Honarvar, H.; Rosestedt, M.; Lindeberg, G.; Sörensen, J.; Larhed, M.; et al. The Effect of Mini-PEG-Based Spacer Length on Binding and Pharmacokinetic Properties of a 68Ga-Labeled NOTA-Conjugated Antagonistic Analog of Bombesin. Molecules 2014, 19, 10455-10472. https://doi.org/10.3390/molecules190710455
Varasteh Z, Rosenström U, Velikyan I, Mitran B, Altai M, Honarvar H, Rosestedt M, Lindeberg G, Sörensen J, Larhed M, et al. The Effect of Mini-PEG-Based Spacer Length on Binding and Pharmacokinetic Properties of a 68Ga-Labeled NOTA-Conjugated Antagonistic Analog of Bombesin. Molecules. 2014; 19(7):10455-10472. https://doi.org/10.3390/molecules190710455
Chicago/Turabian StyleVarasteh, Zohreh, Ulrika Rosenström, Irina Velikyan, Bogdan Mitran, Mohamed Altai, Hadis Honarvar, Maria Rosestedt, Gunnar Lindeberg, Jens Sörensen, Mats Larhed, and et al. 2014. "The Effect of Mini-PEG-Based Spacer Length on Binding and Pharmacokinetic Properties of a 68Ga-Labeled NOTA-Conjugated Antagonistic Analog of Bombesin" Molecules 19, no. 7: 10455-10472. https://doi.org/10.3390/molecules190710455
APA StyleVarasteh, Z., Rosenström, U., Velikyan, I., Mitran, B., Altai, M., Honarvar, H., Rosestedt, M., Lindeberg, G., Sörensen, J., Larhed, M., Tolmachev, V., & Orlova, A. (2014). The Effect of Mini-PEG-Based Spacer Length on Binding and Pharmacokinetic Properties of a 68Ga-Labeled NOTA-Conjugated Antagonistic Analog of Bombesin. Molecules, 19(7), 10455-10472. https://doi.org/10.3390/molecules190710455