Diels-Alder Reactions of 12-Hydroxy-9(10®20)-5aH-abeo-abieta-1(10),8(9),12(13)-triene-11,14-dione

Abstract
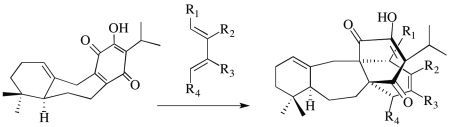
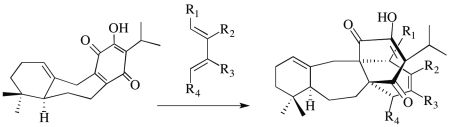
:
{kind=link}
{kind=link}
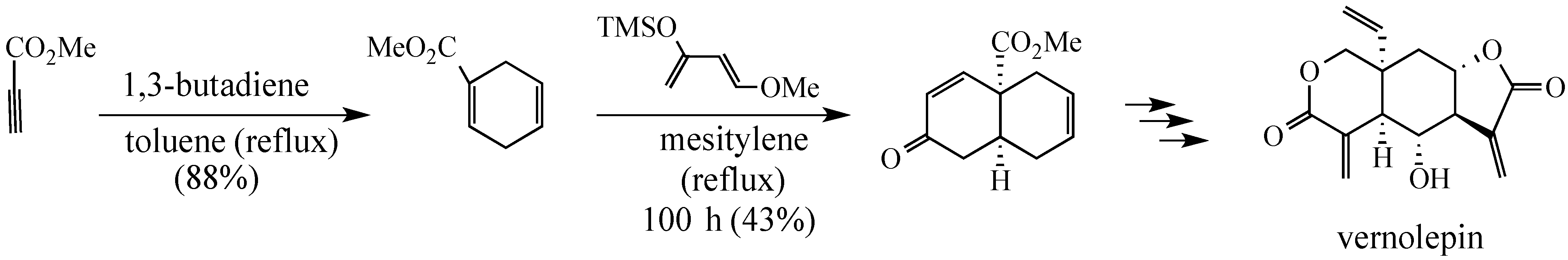{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
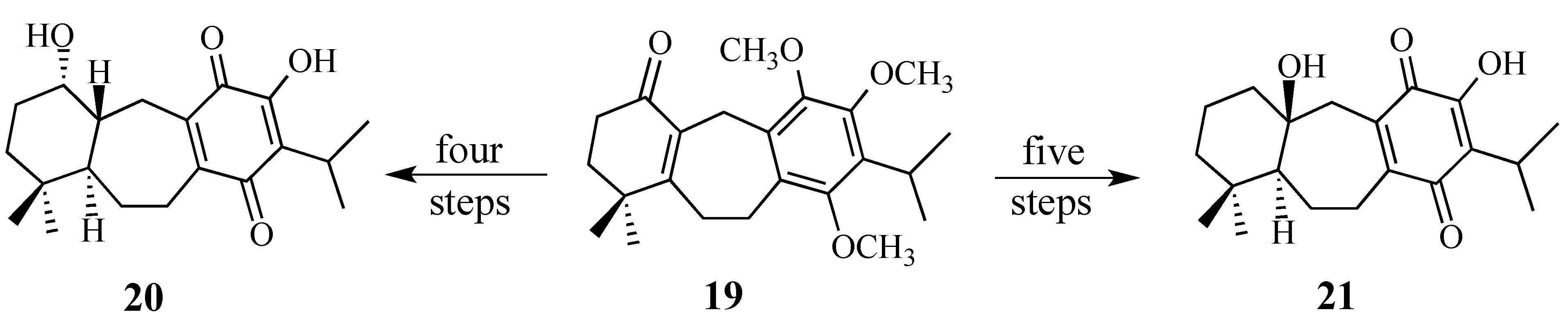
1. Introduction
In a Fall 1974 invited lecture before the Pittsburgh Section of the ACS, Professor Samuel Danishefsky stated, reporting on his group’s progress toward a synthesis of vernolepin, that: “A synthesis cannot be considered truly elegant if it does not contain at least one Diels-Alder reaction.”
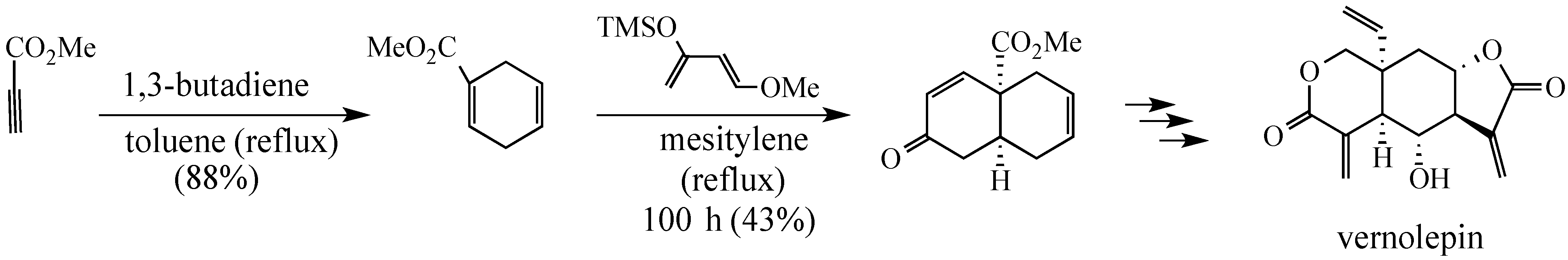
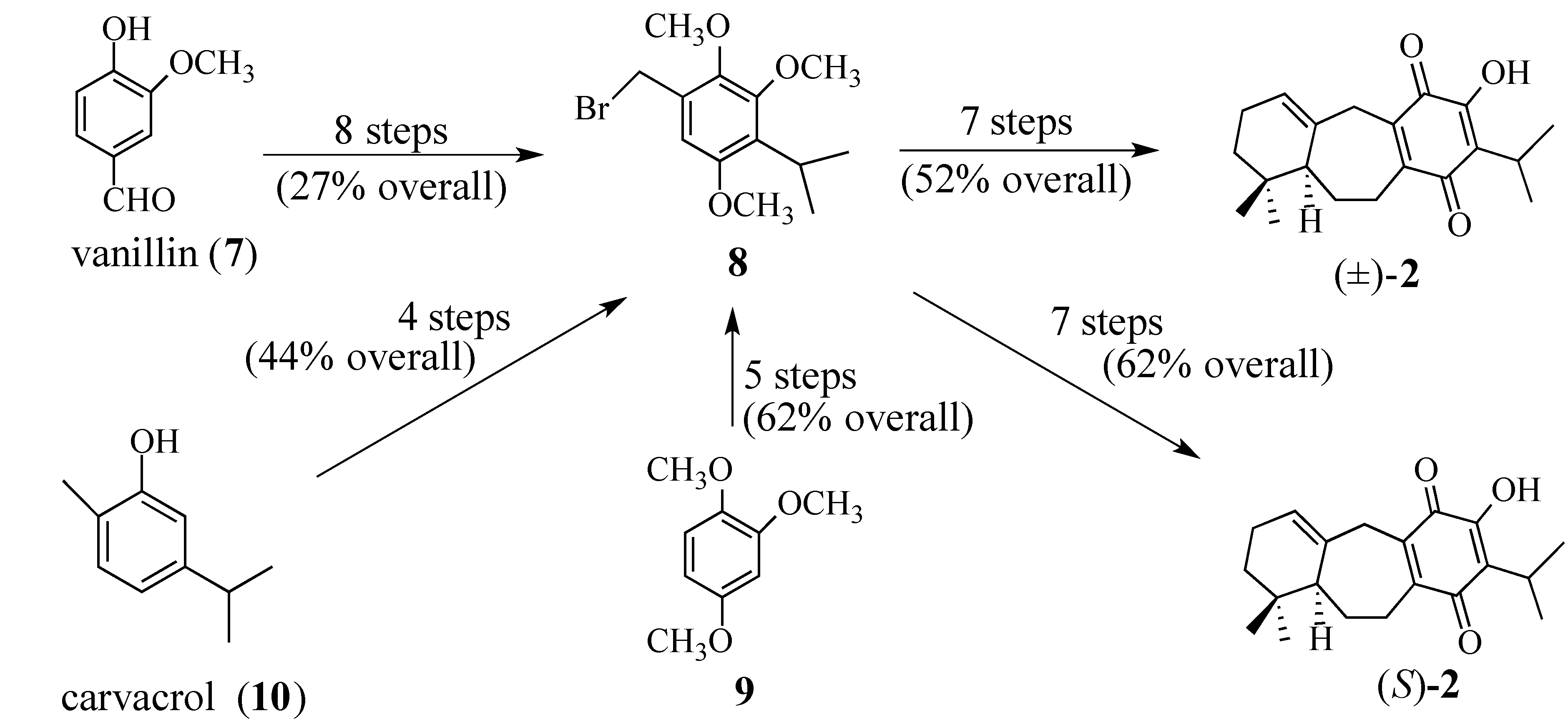
2. Results and Discussion
“Many syntheses only appear as terse communications in journals. Very rarely do chemists discuss the blind alleys and dead ends that were encountered in a synthesis. This is unfortunate for the student who wants to learn about synthesis. I think many students have the mistaken impression that organic chemists conceive a brilliant ‘paper’ synthesis in 1 h and hand it over to their graduate students who see it through to completion without any problems or difficulties. However, in almost every synthesis there are problems to be overcome and obstacles to be surmounted.”[47]
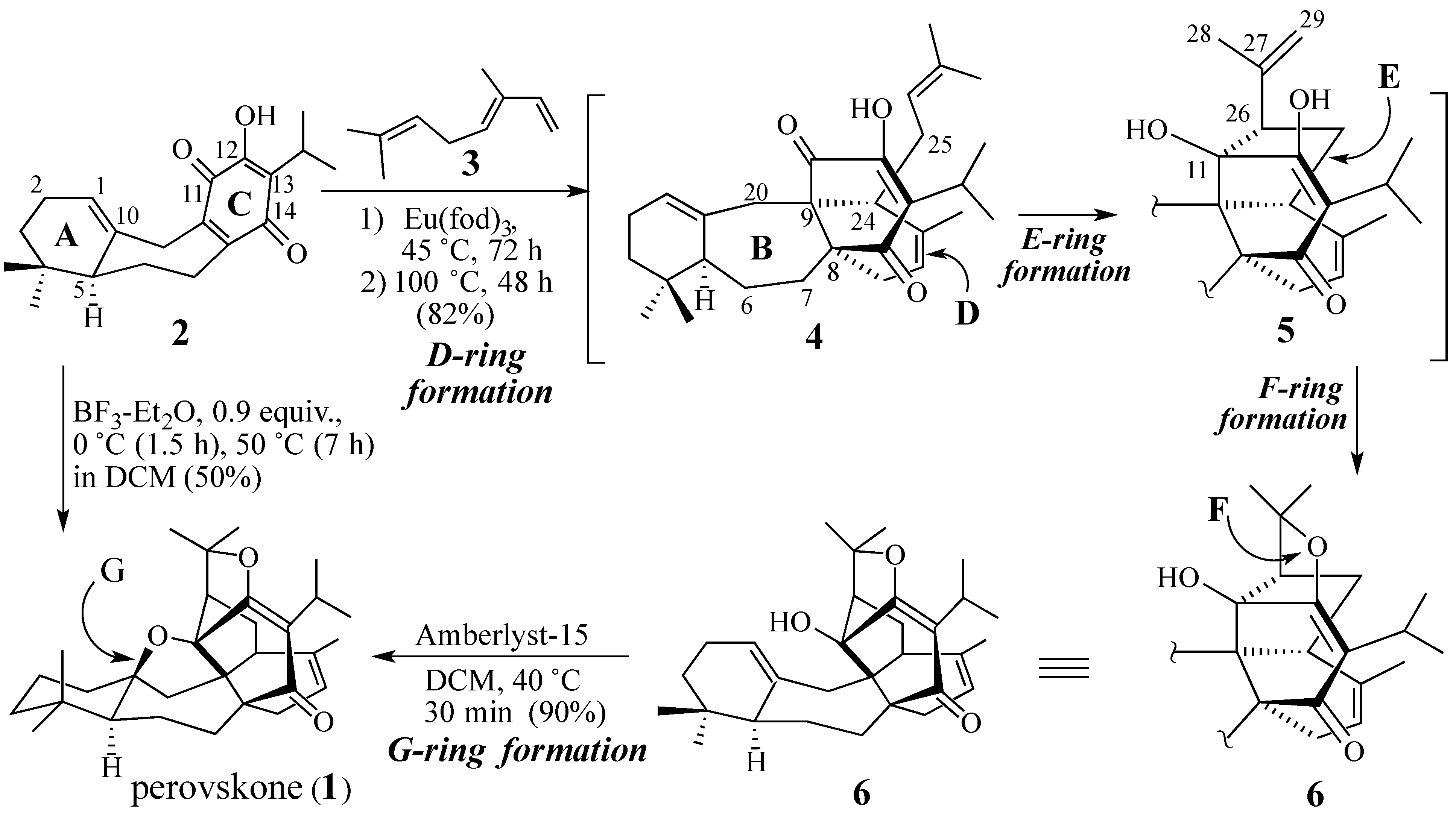
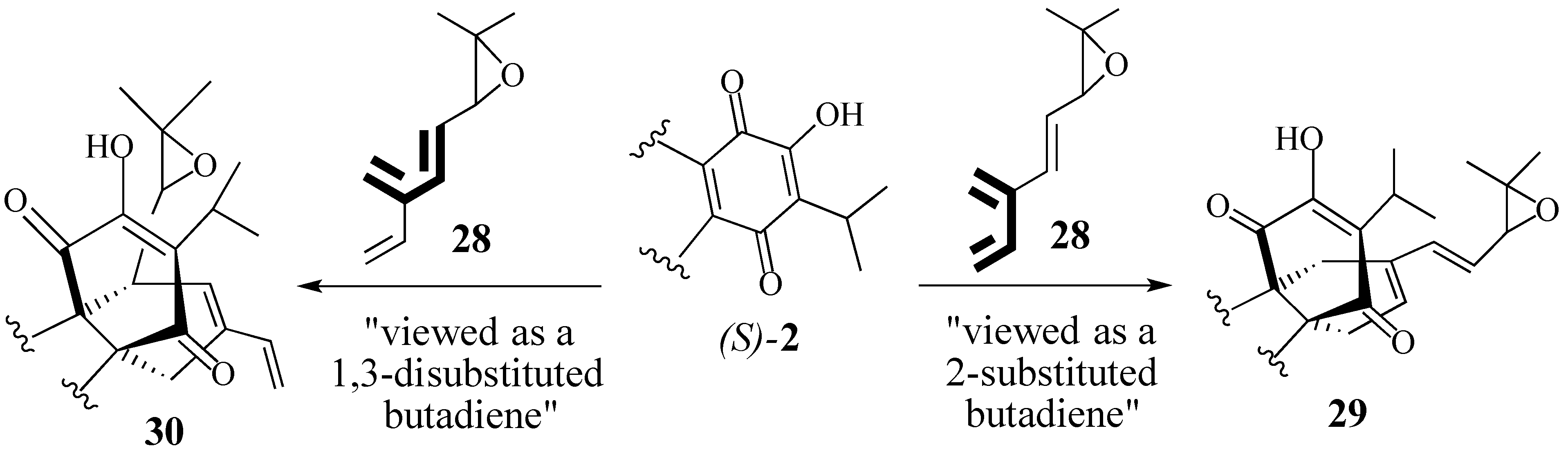
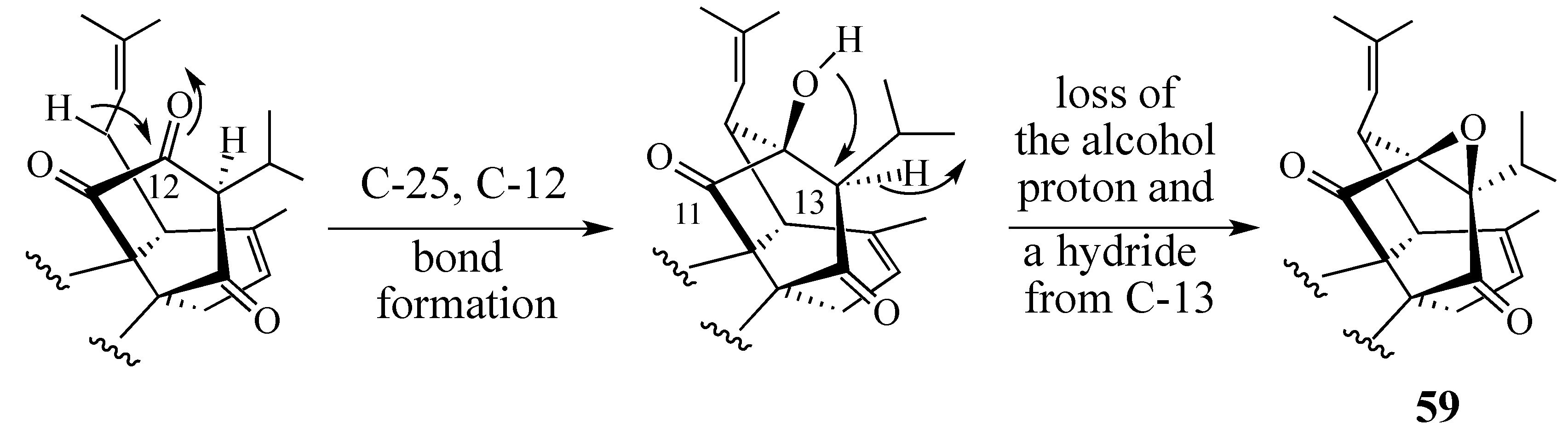
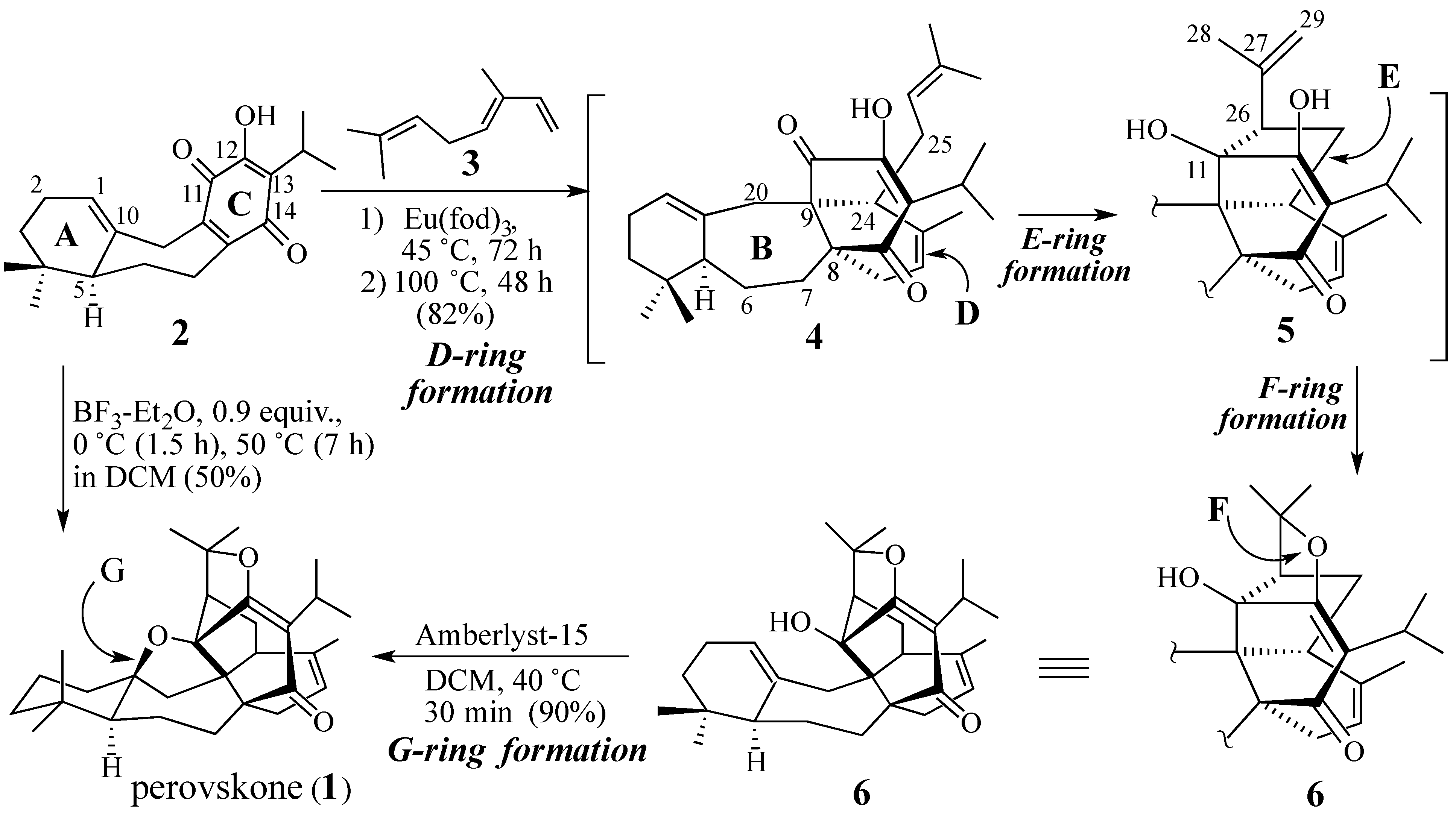
2.1. The Perovskone Diels-Alder Reaction
- Will the cycloaddition be facially selective?
- Will the diene component be stable under the Diels-Alder reaction conditions employed?
- Will the Diels-Alder adduct be stable under the Diels-Alder reaction conditions used?
- Will the cycloaddition be regiospecific?
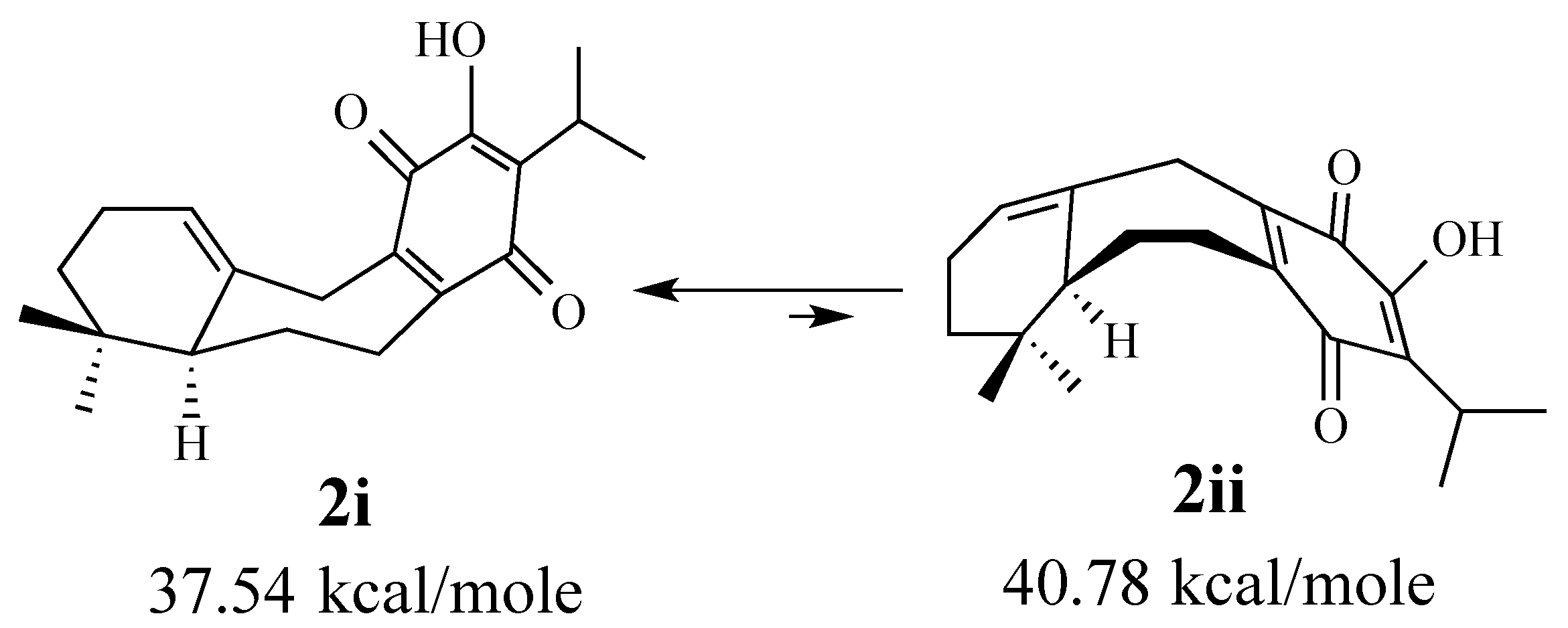
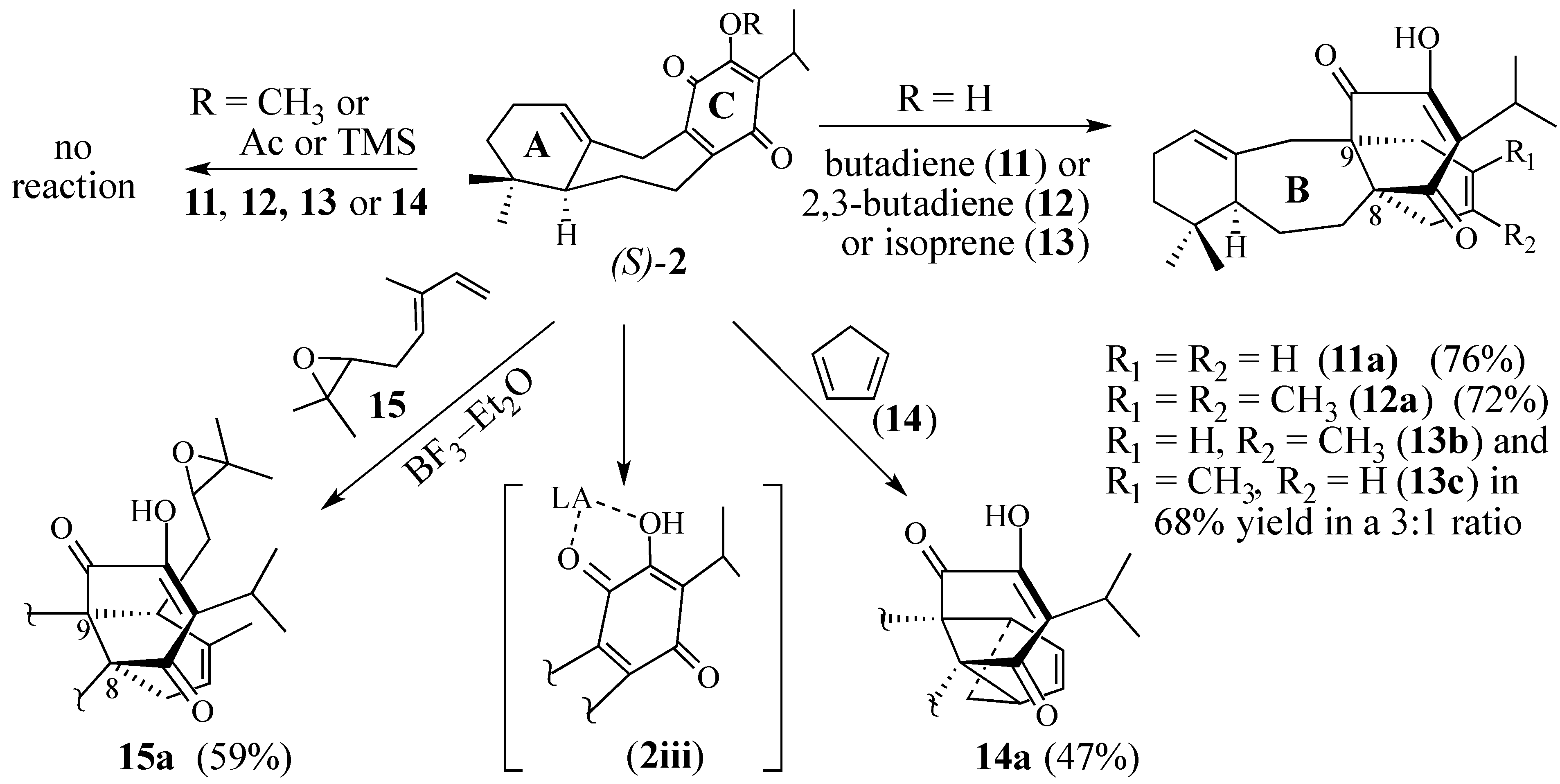
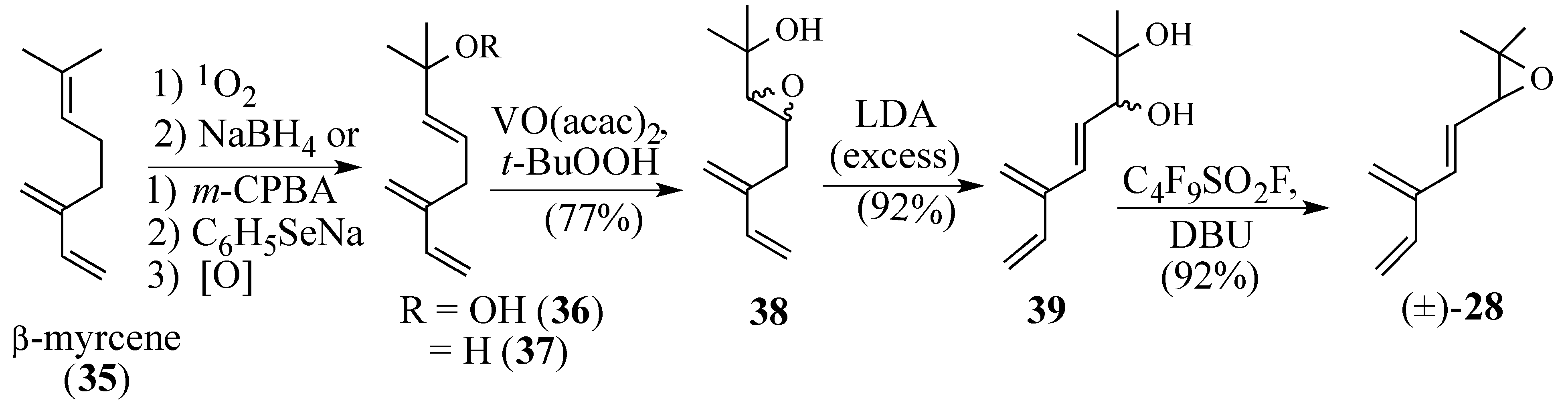
2.1.1. Facial Selectivity of Quinone 2
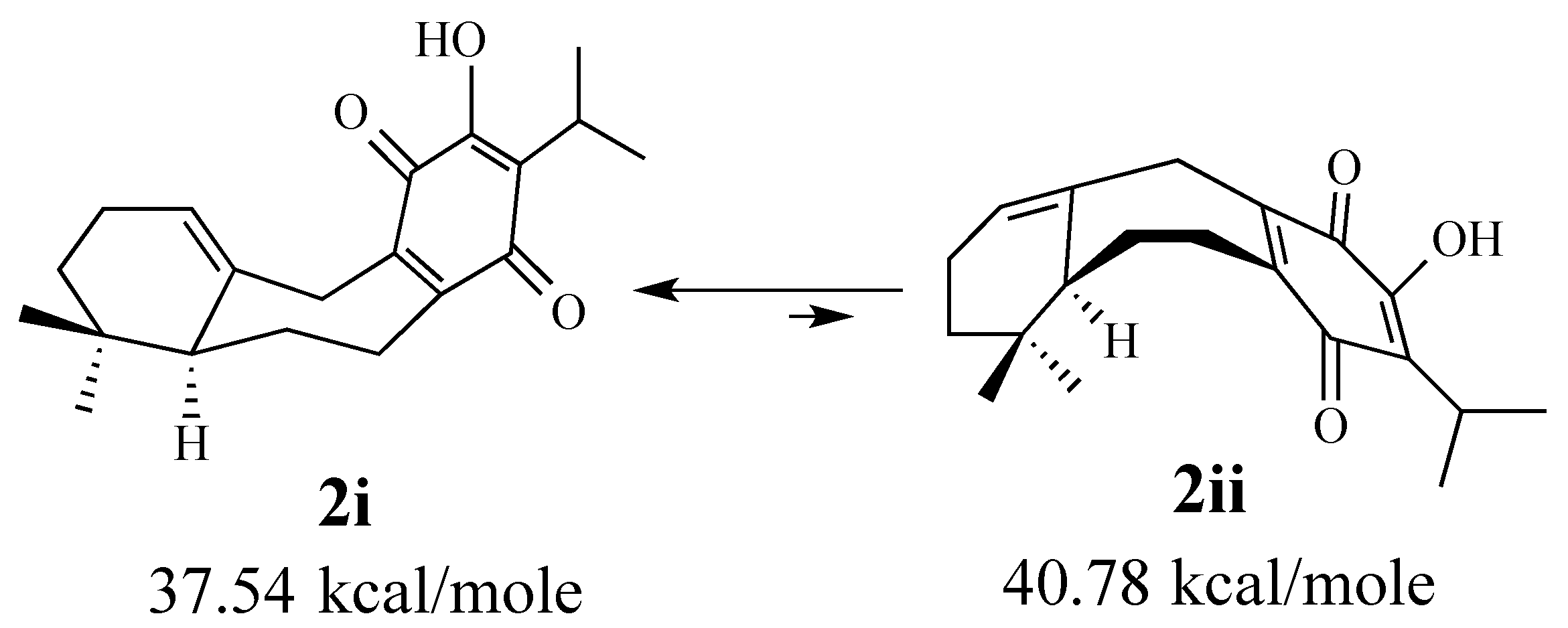
2.1.2. Other Dienes Studied
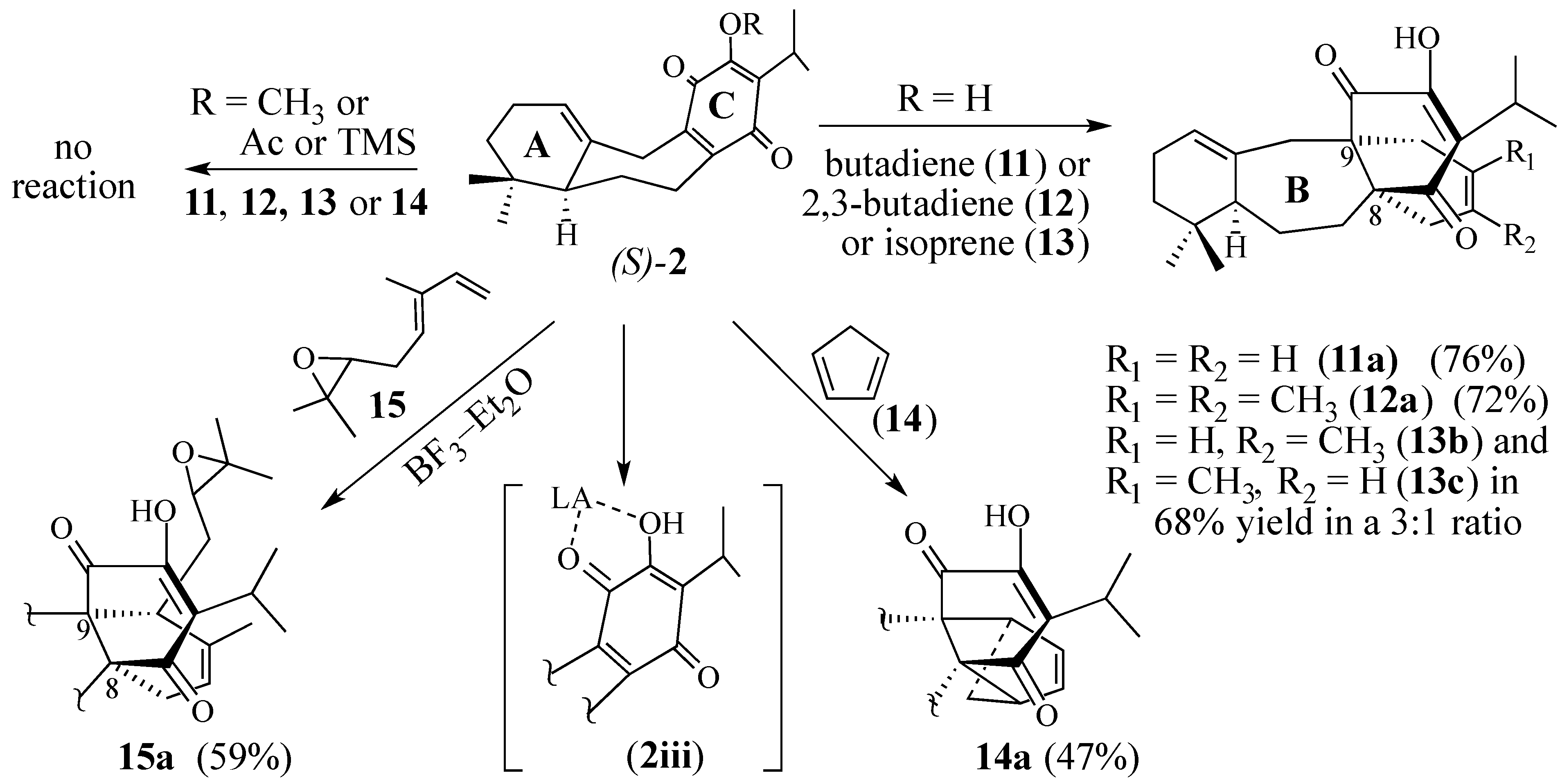
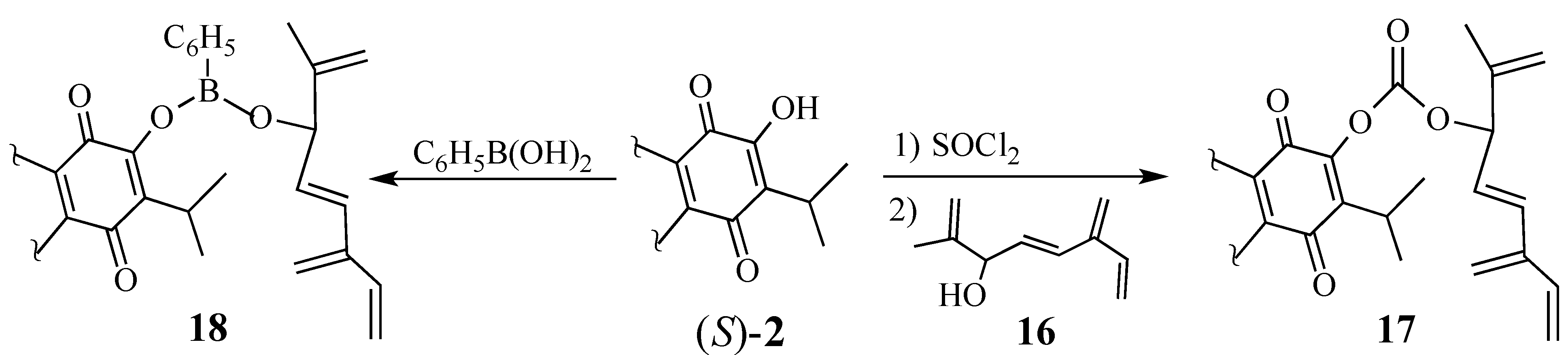
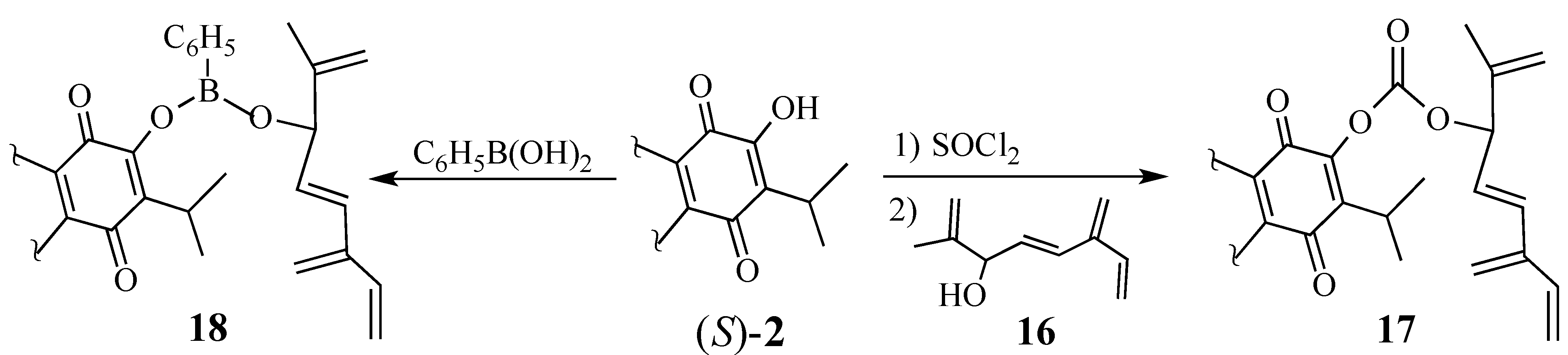
2.1.3. Modifications of the Dienophile
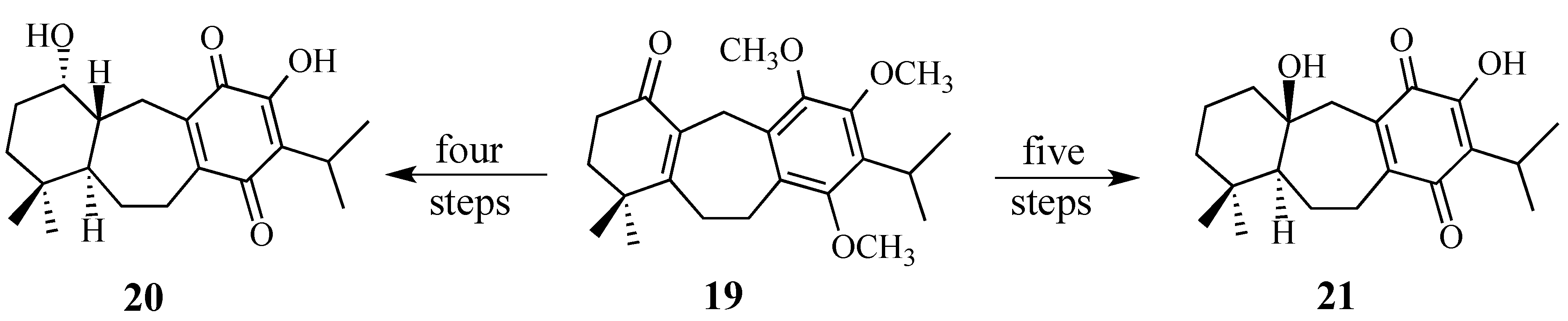
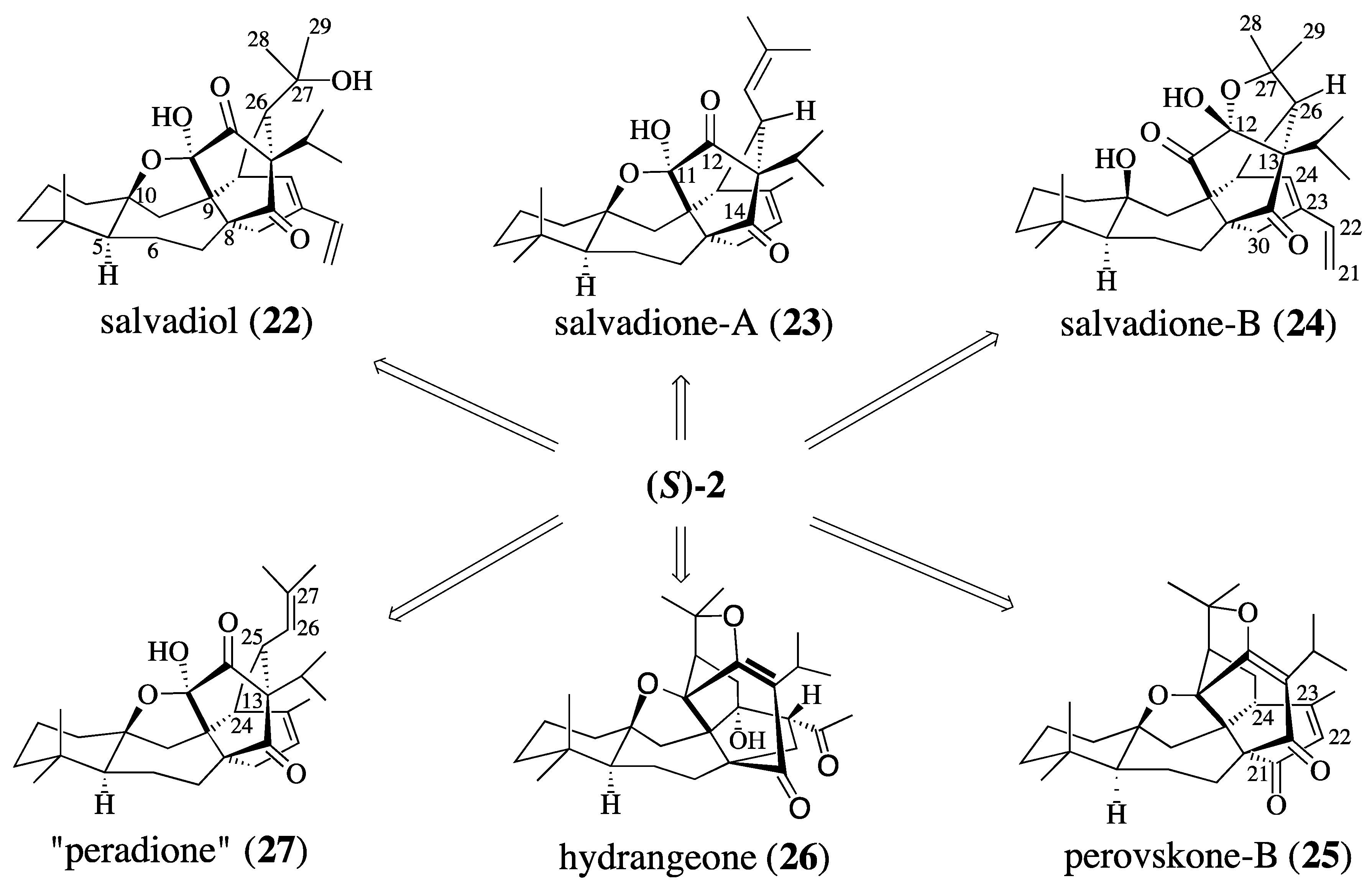
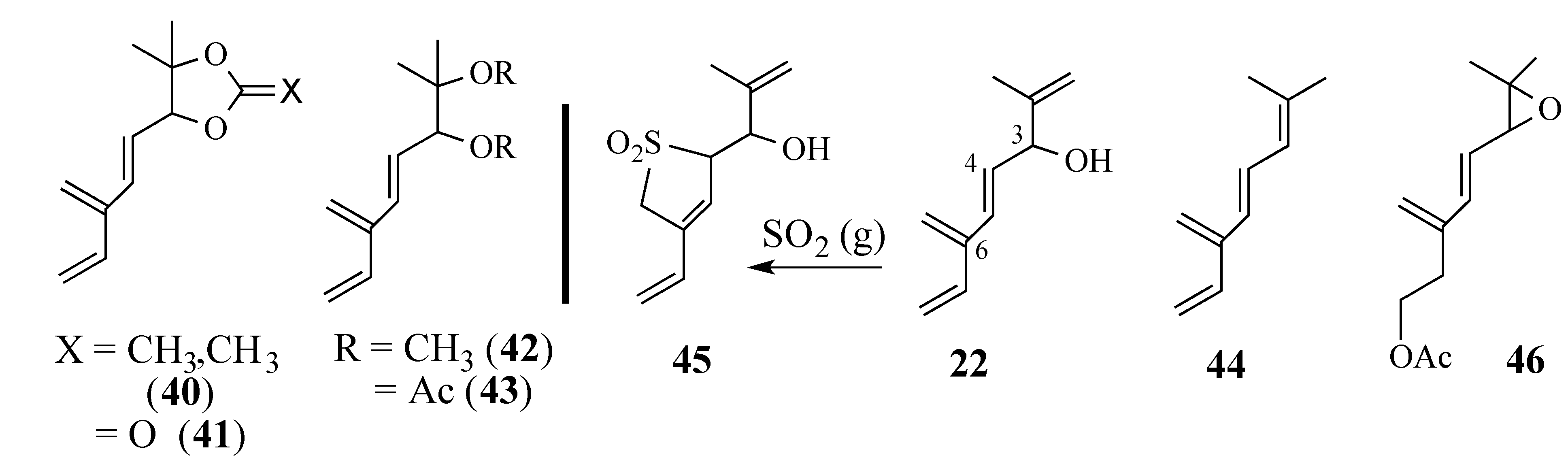
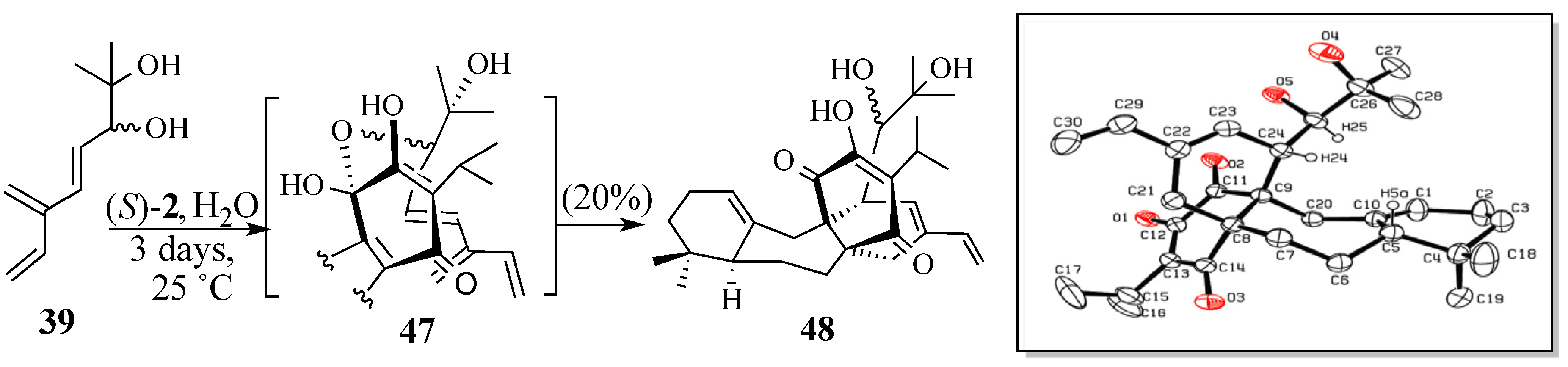
2.2. Studies Directed Toward the Synthesis of Salvadione-B and Salvadiol
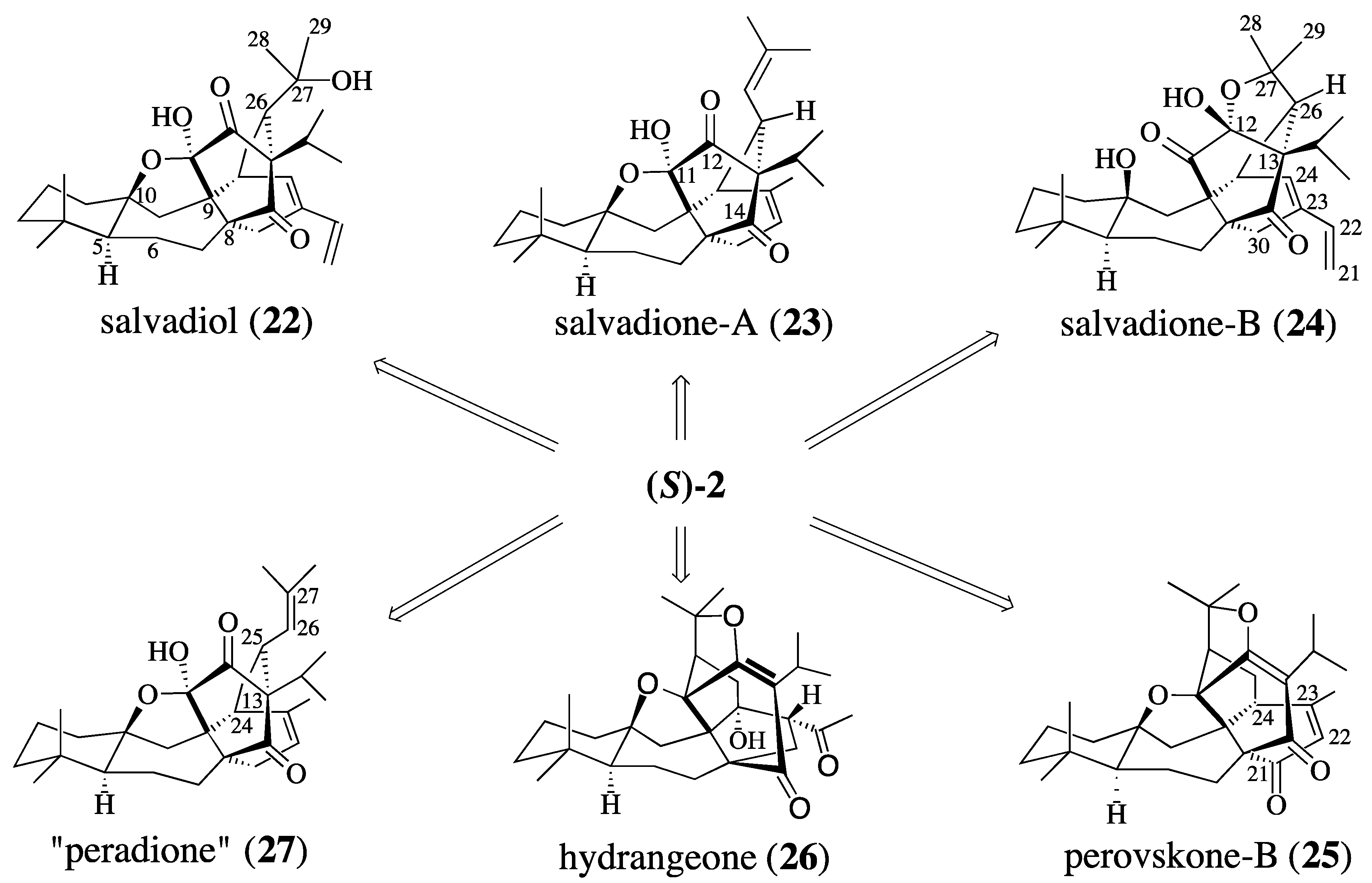
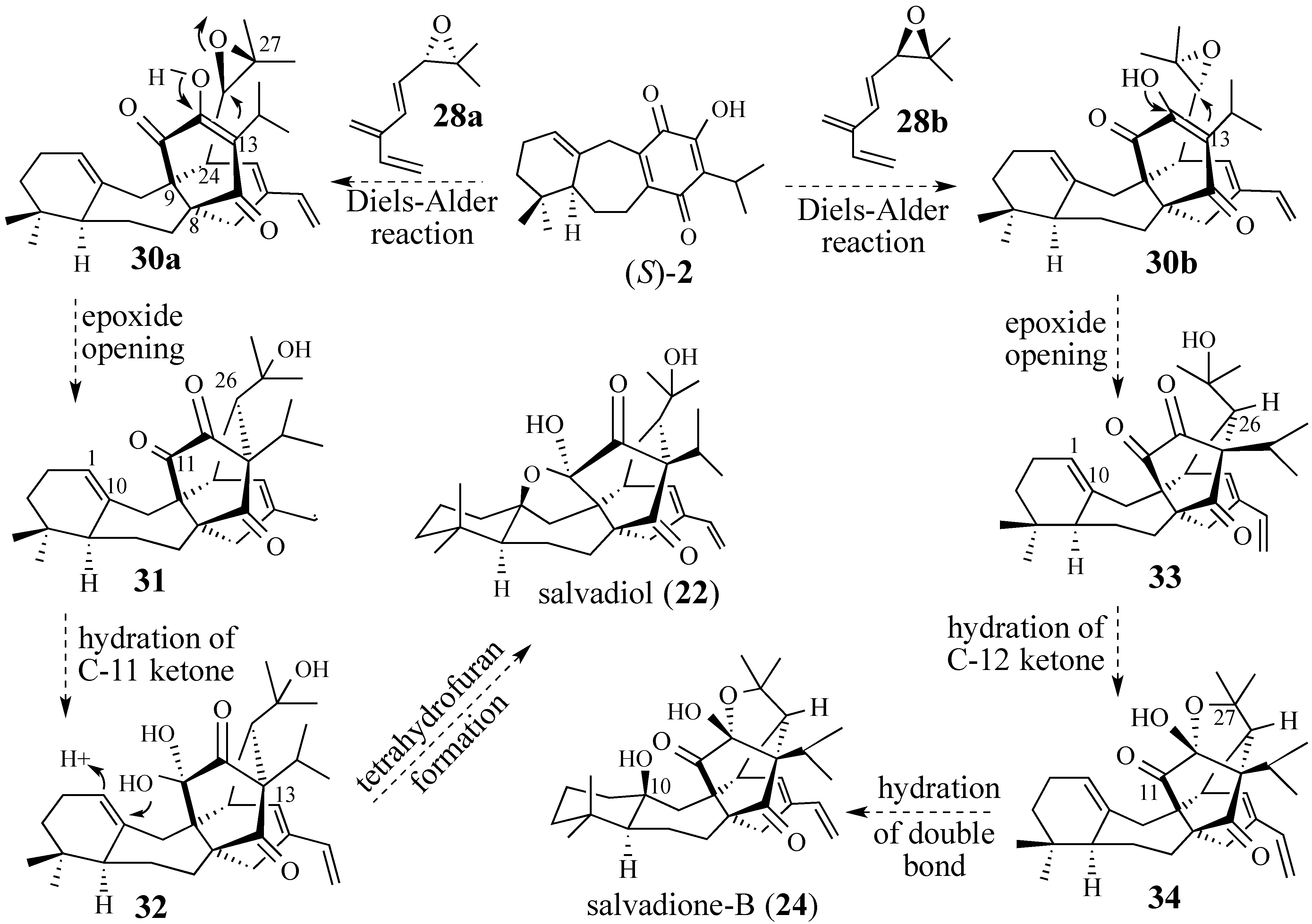
2.2.1. Epoxide-Based Synthesis of Salvadione-B (24) and Salvadiol (22)
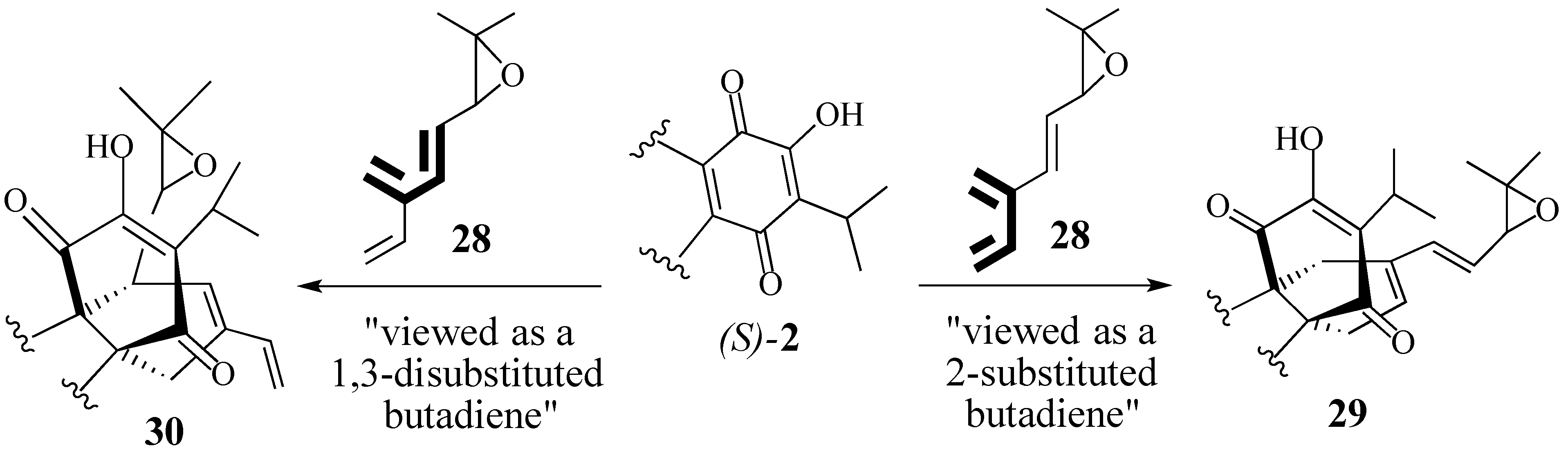
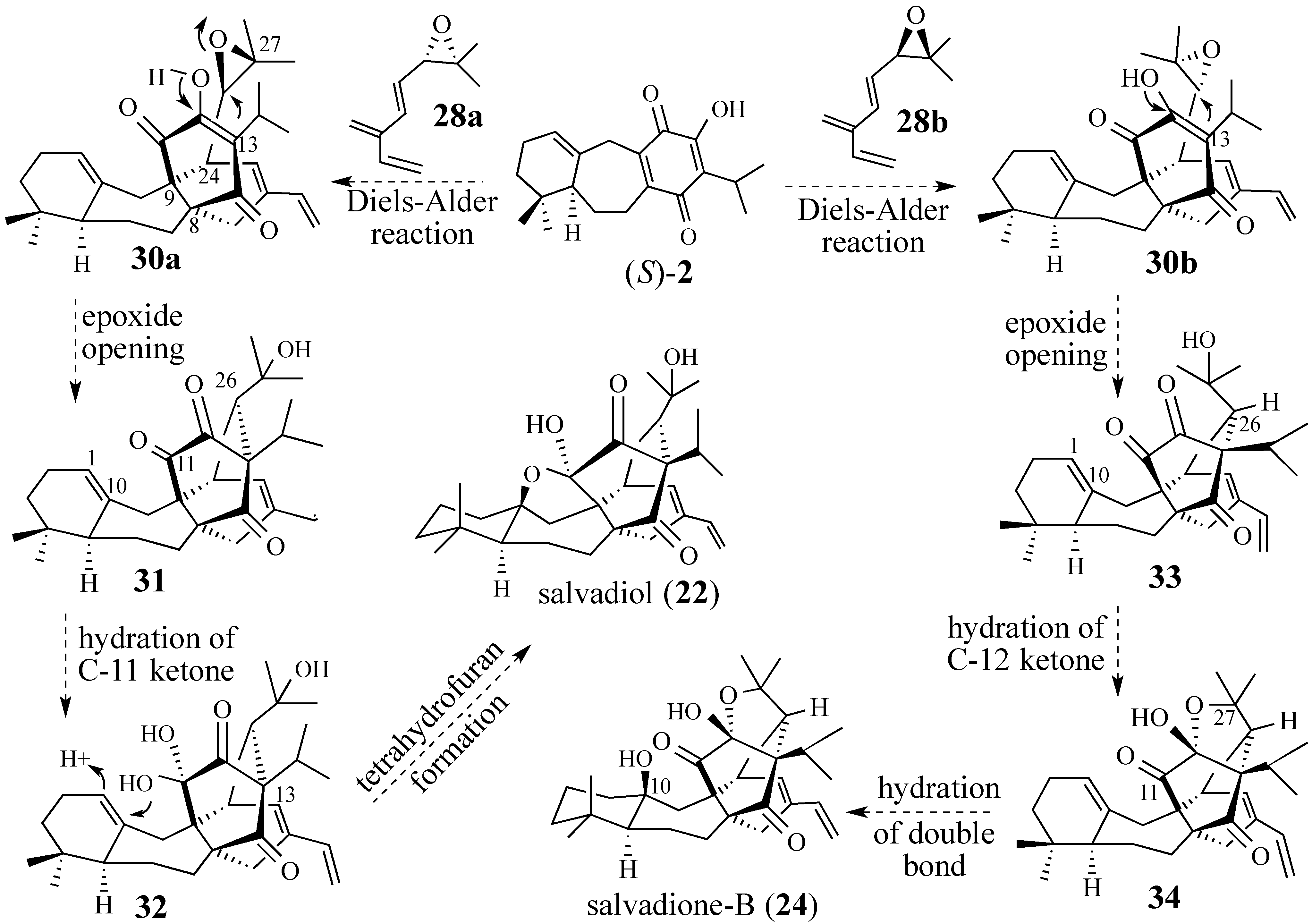
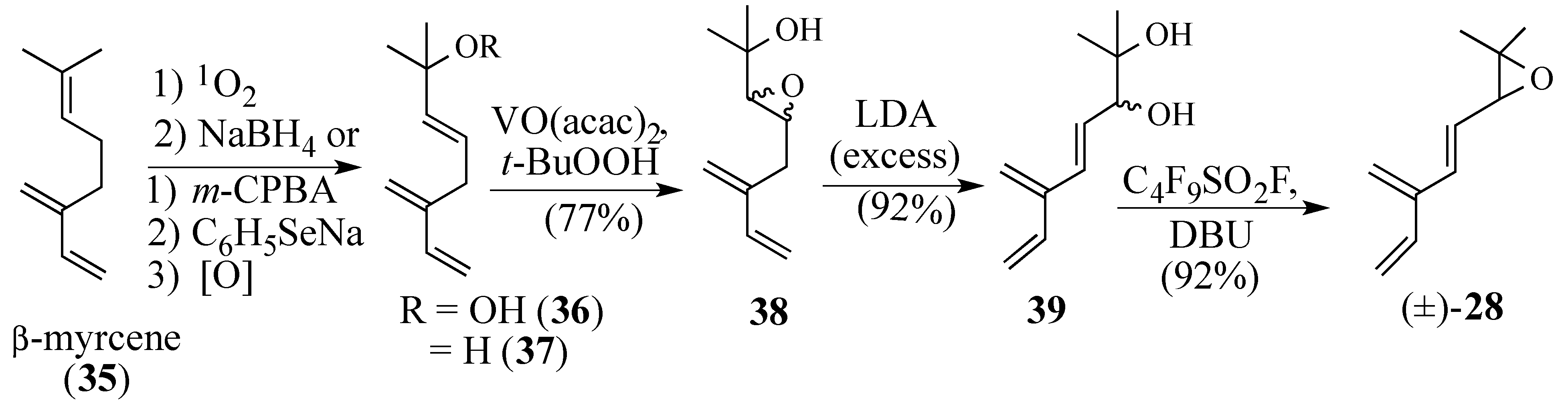
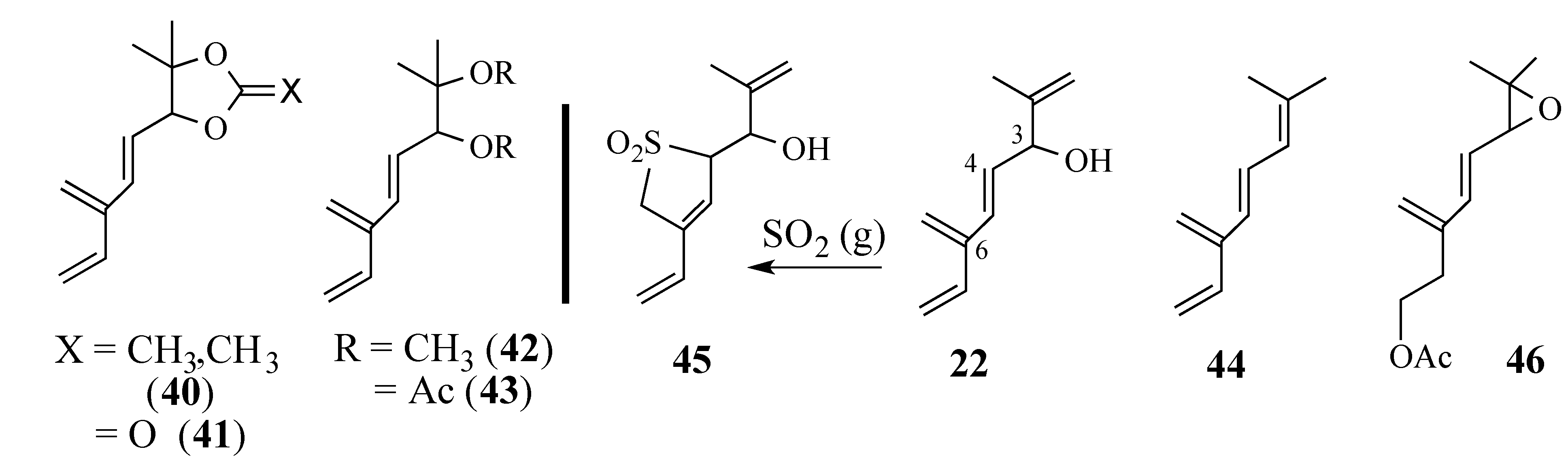
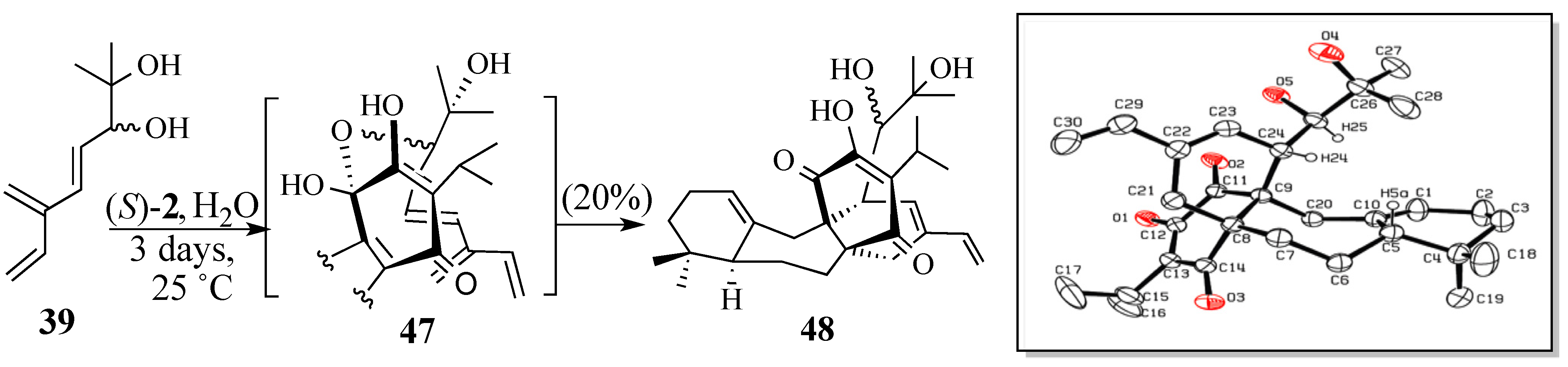
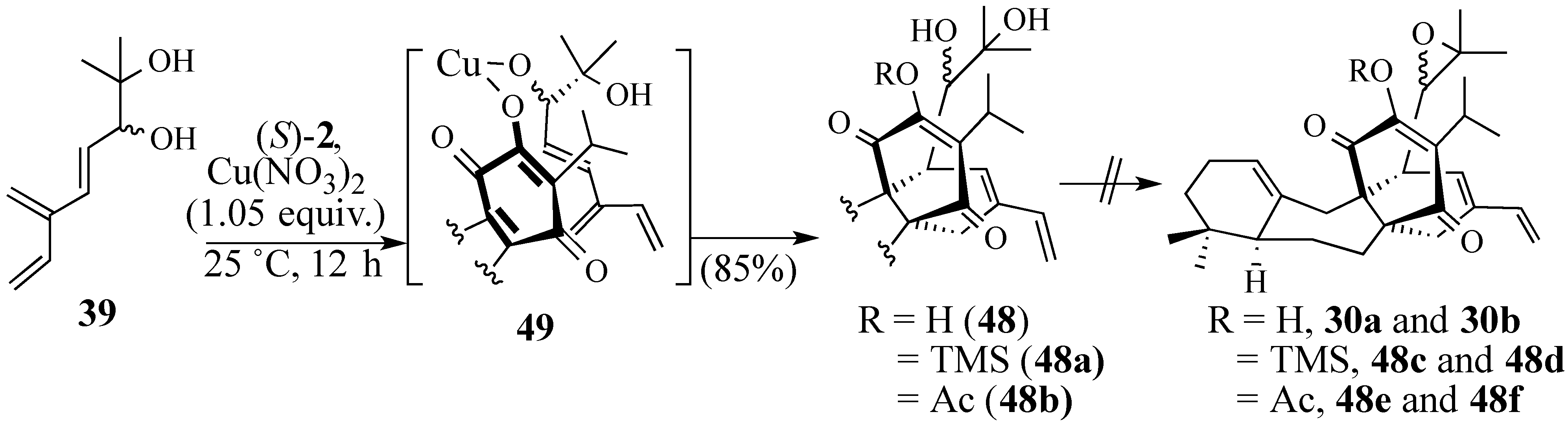
2.2.2. An Alternative Diels-Alder Strategy to Synthesize Salvadione-B (24)
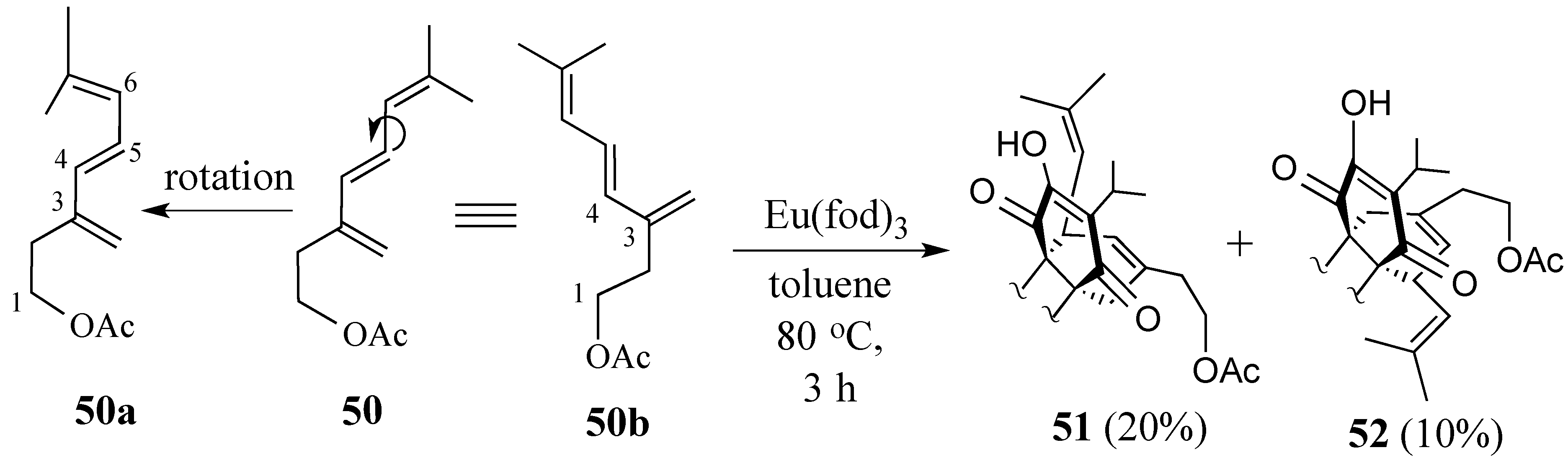
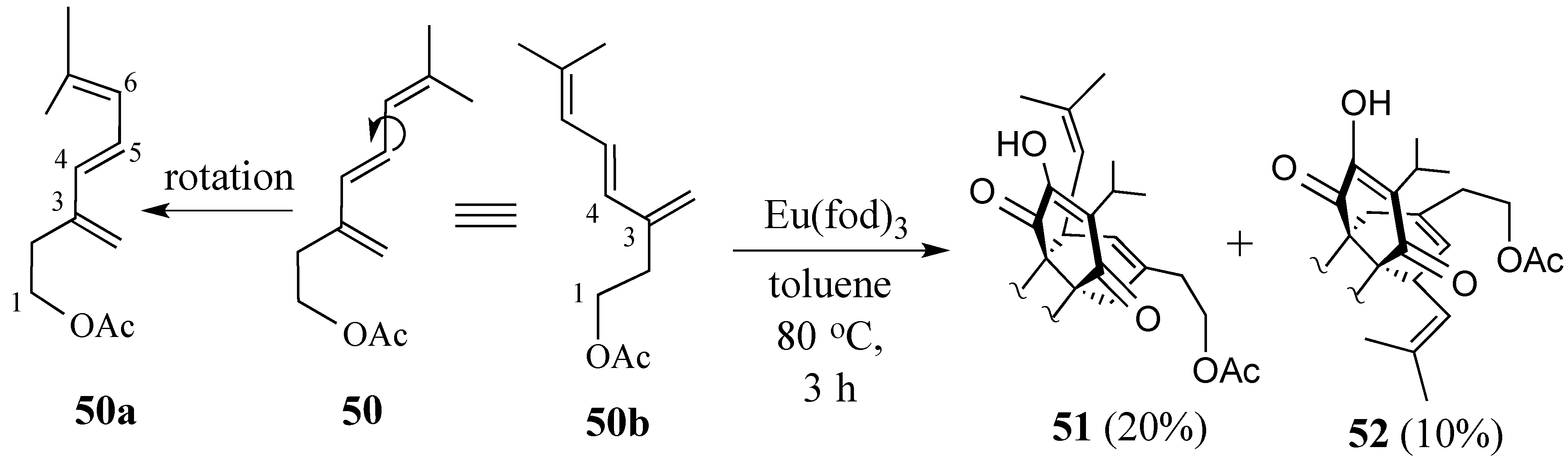
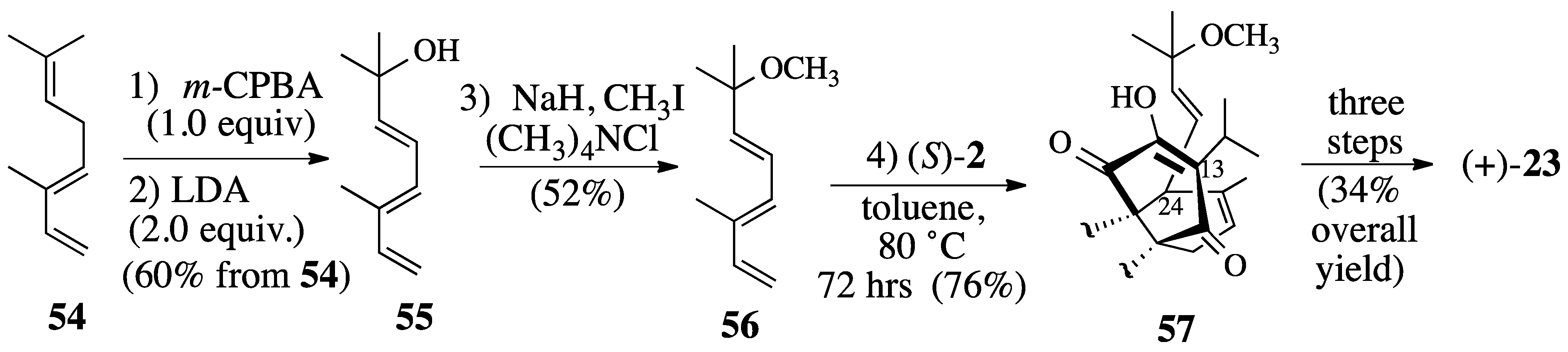
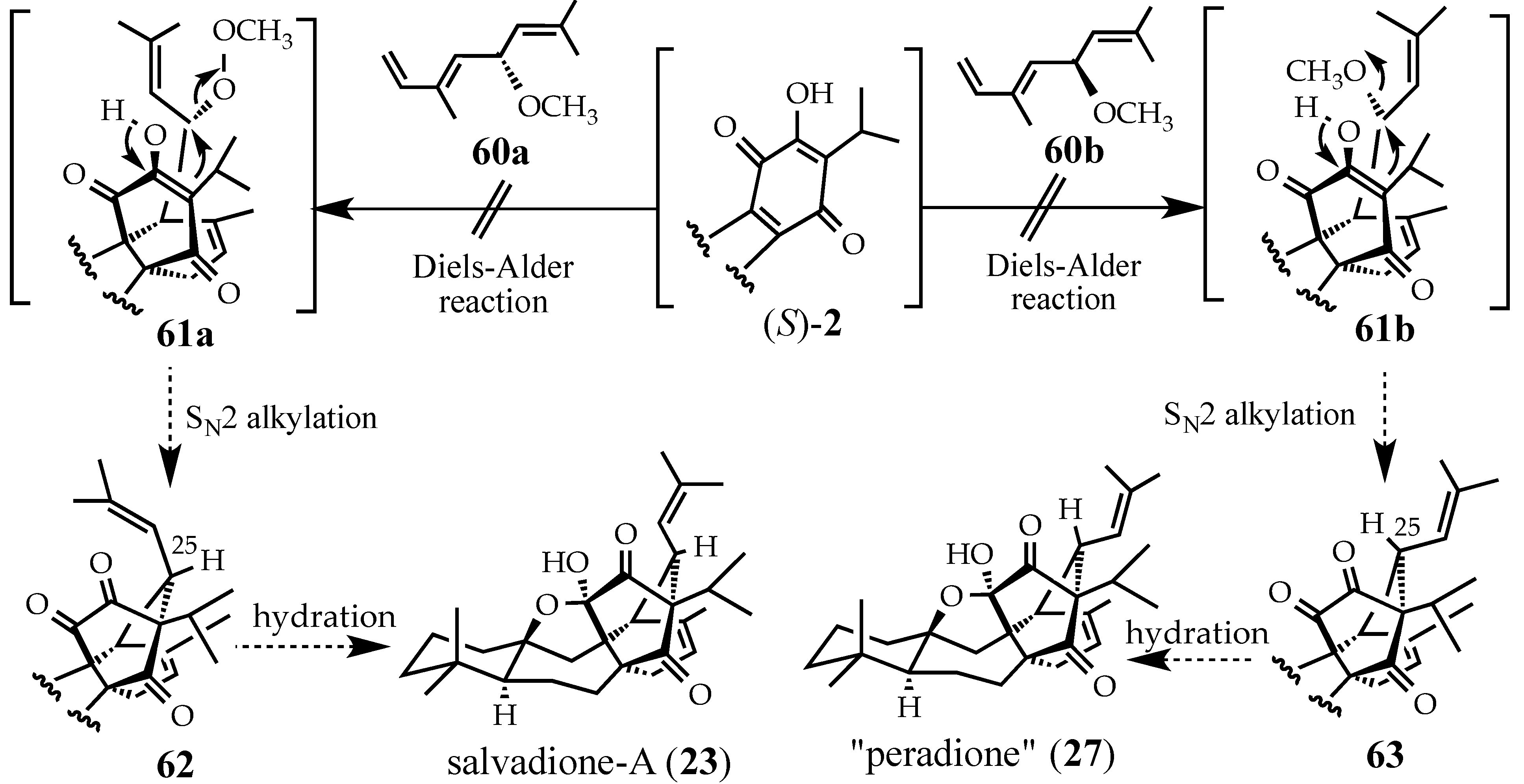
2.3. Synthesis of (+)-Salvadione-A (23)
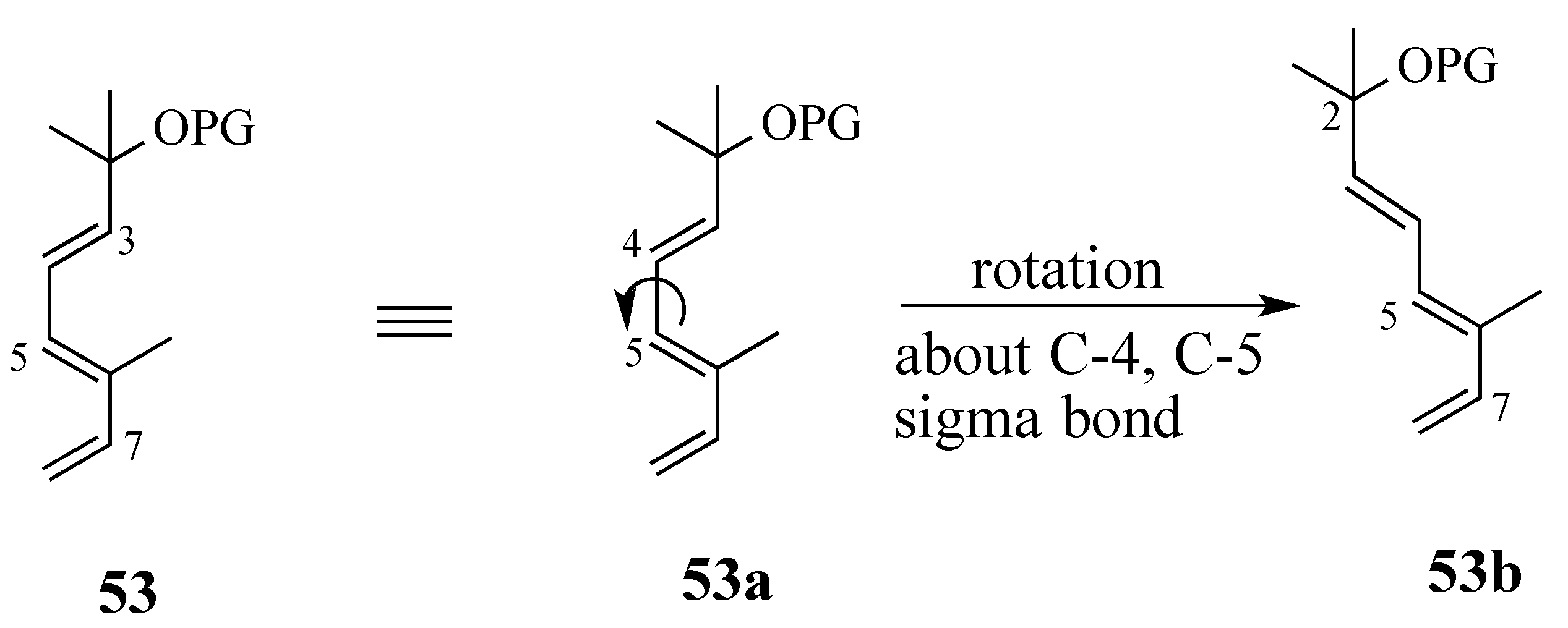
2.3.1. Diels-Alder reaction of triene ether 53 and quinone (S)-2
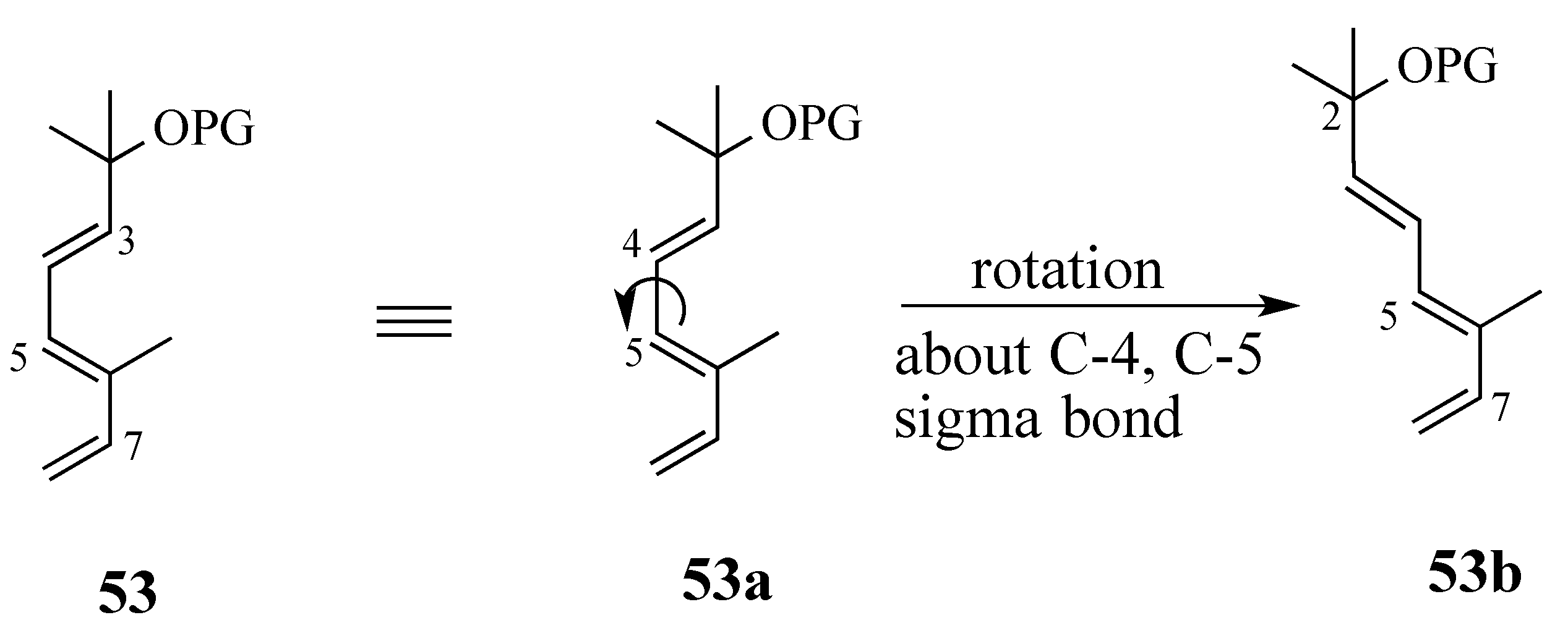
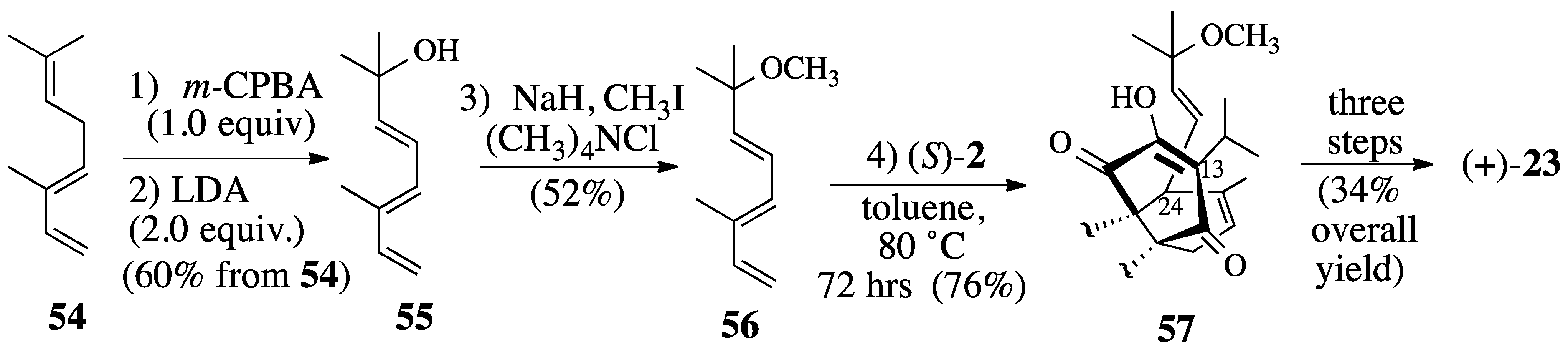
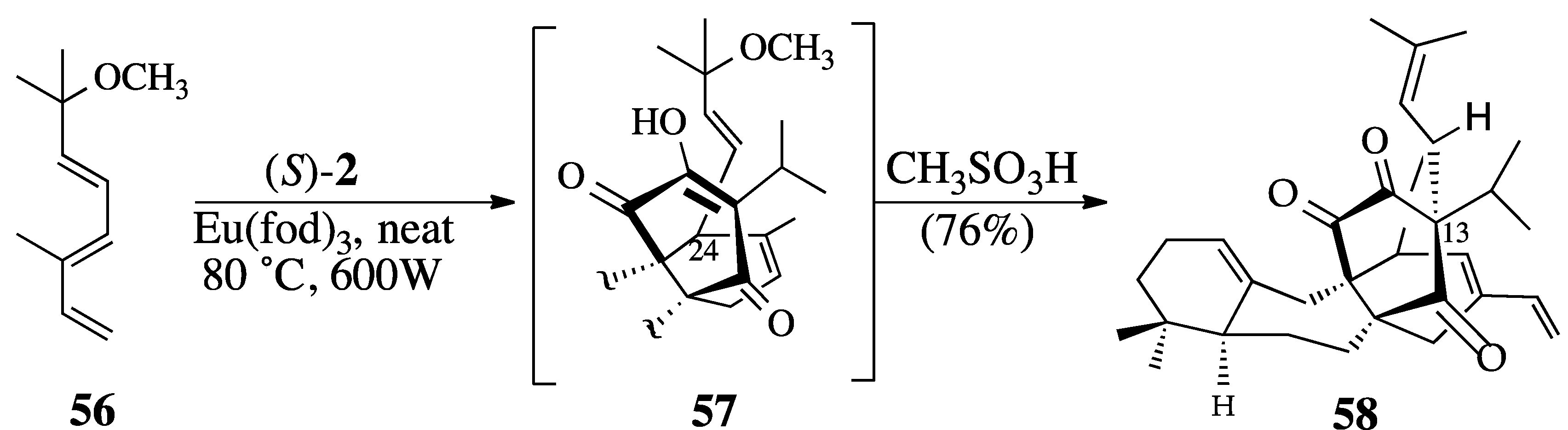
2.3.2. The Microwave-Promoted Diels-Alder Reaction of triene ether 56 and (S)-2

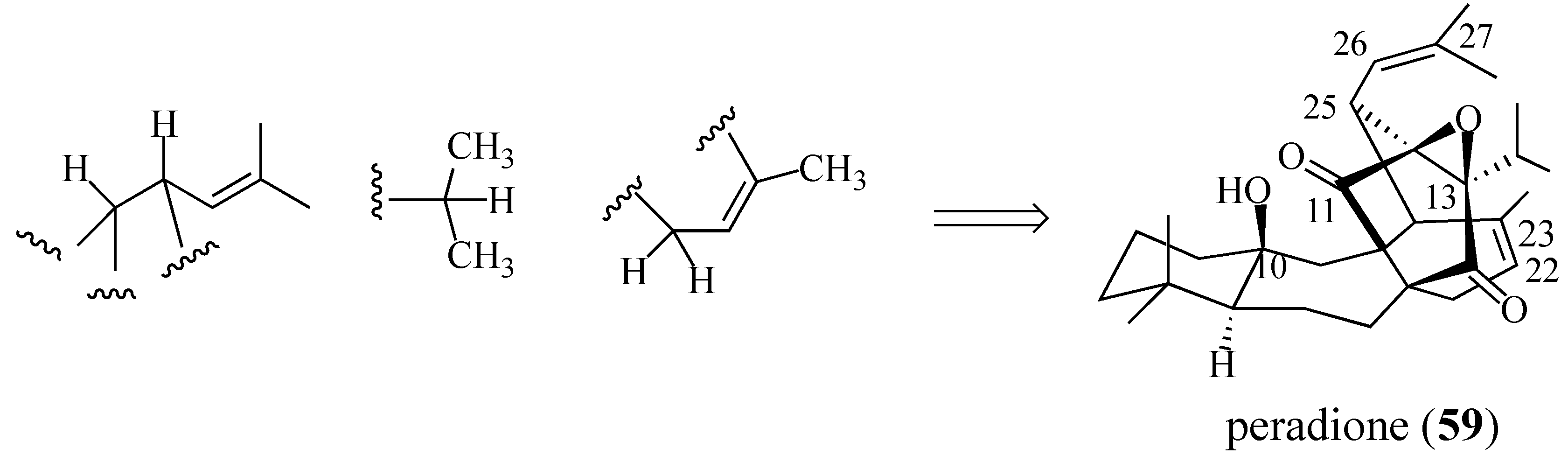
2.4. Studies Directed toward a Synthesis of “peradione” (27)
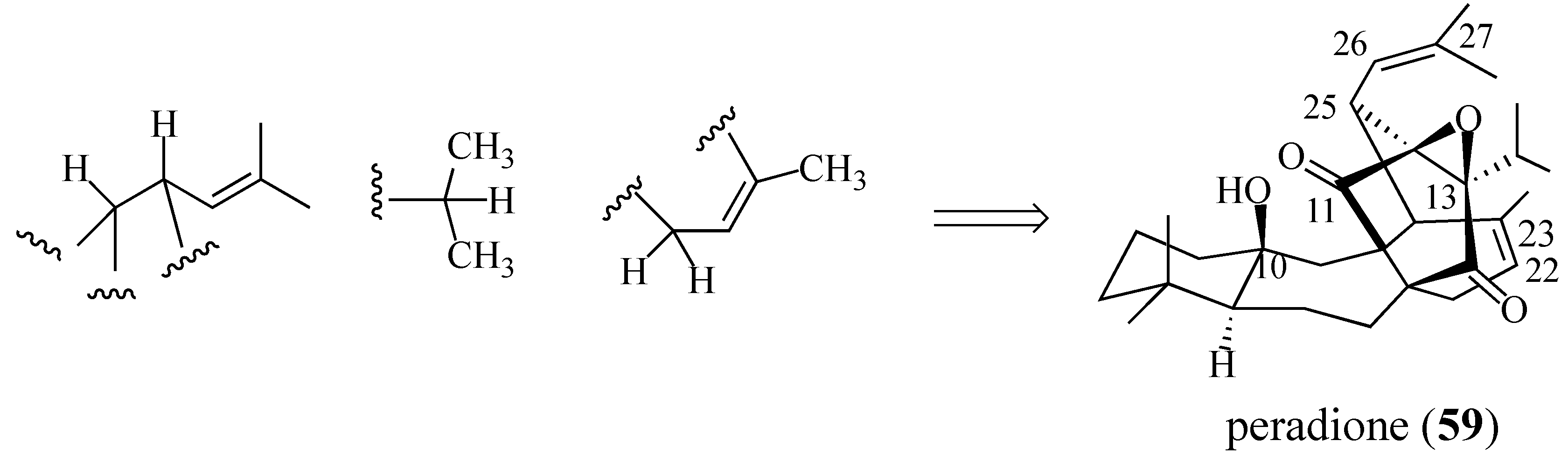
2.4.1. A Wrong Structural Assignment
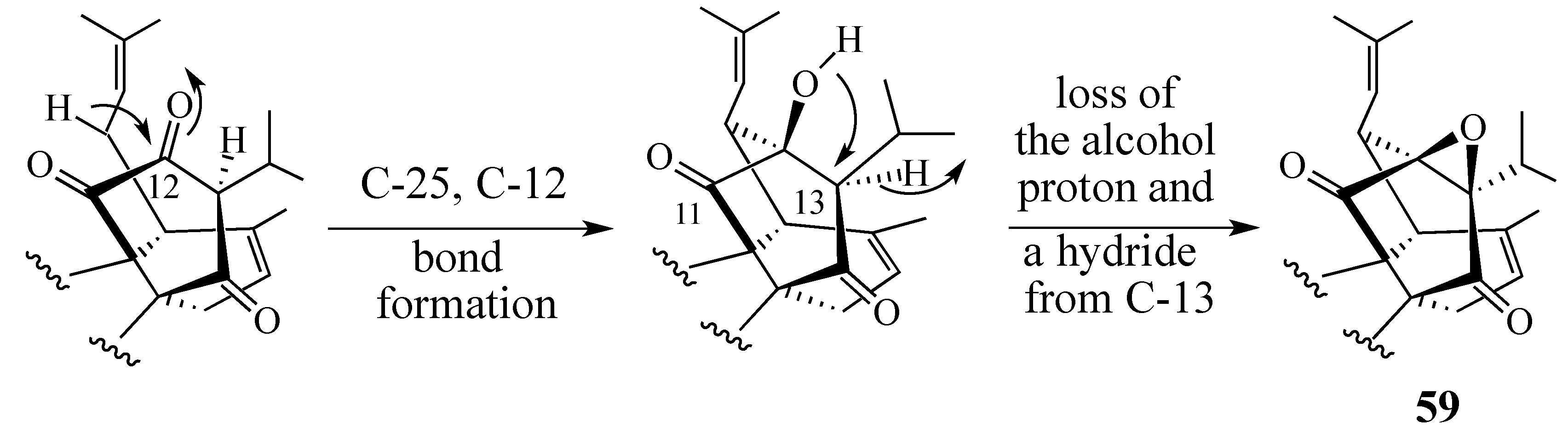
2.4.2. A Displacement-Based Strategy to Prepare Salvadione-A (23) and Our Proposed Structure for Peradione (27)
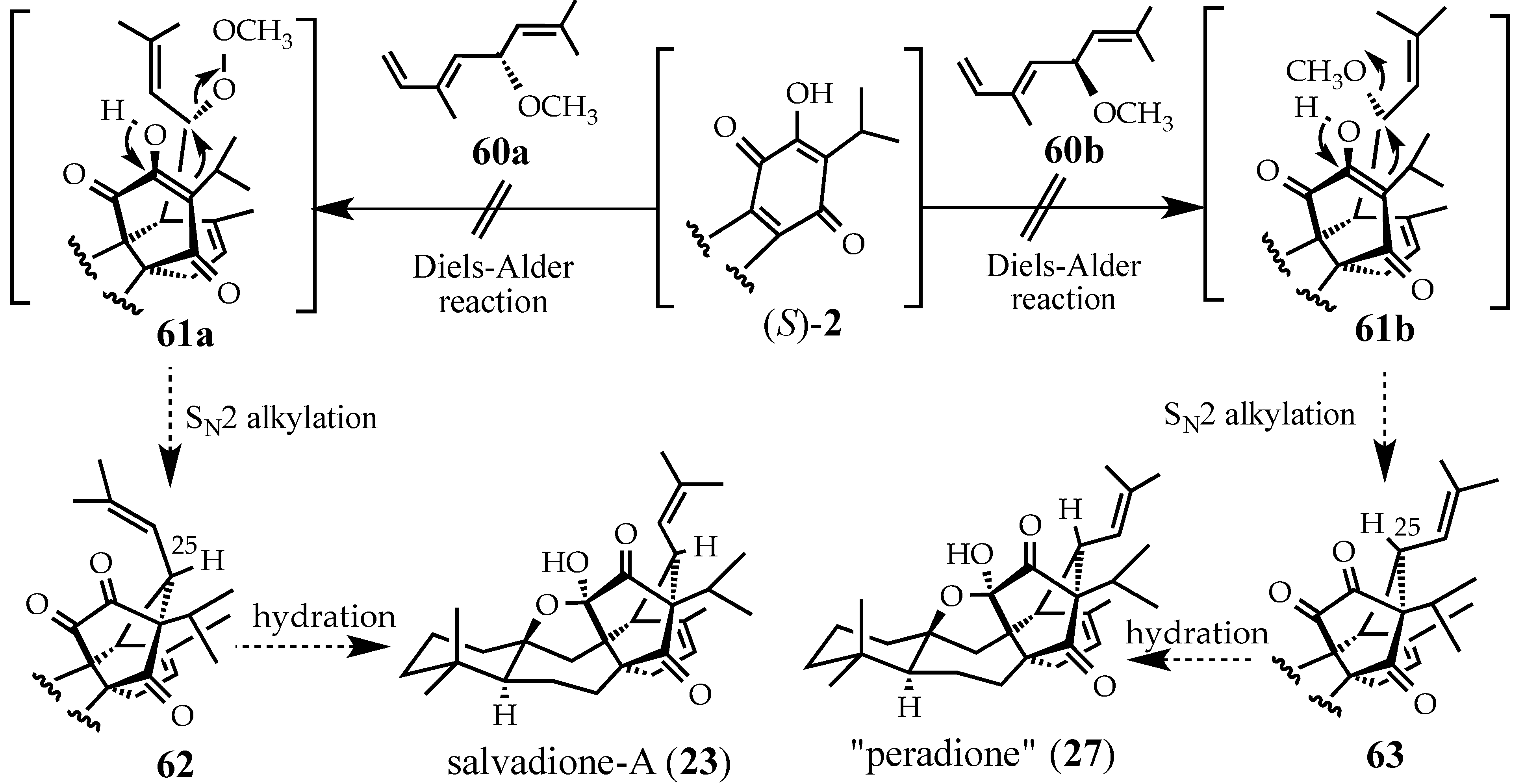
3. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Danishefsky, S.; Schuda, P.F.; Kitahara, T.; Etheredge, S.J. The Total Synthesis of dl-Vernolepin and dl-Vernomenin. J. Am. Chem. Soc. 1977, 99, 6066–6075. [Google Scholar]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Justus Libigs Ann. Chem. 1928, 460, 98–122. [Google Scholar]
- Smith, M.B. Organic Synthesis, 3rd ed.; Wavefunction, Inc.: Irvine, CA, USA, 2010; pp. 1013–1075. [Google Scholar]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry. Reactions, Mechanisms and Structure, 6th ed.; Wiley-Interscience: New York, NY, USA, 2007; pp. 1194–1218. [Google Scholar]
- Briegar, G.; Bennett, J.N. The Intramolecular Diels-Alder Reaction. Chem. Rev. 1980, 80, 63–97. [Google Scholar]
- Taber, D.F. Intramolecular Diels-Alder and Alder Ene Reactions; Springer-Verlag: Berlin, Germany, 1984. [Google Scholar]
- Ciganek, E. The intramolecular Diels-Alder reaction. Org. React. 1984, 32, 1–374. [Google Scholar]
- Bolger, D.L.; Weinred, S.M. Hetero Diels-Alder Methodology in Organic Synthesis; Academic Press: San Diego, CA, USA, 1987. [Google Scholar]
- Shen, J.; Tan, C.-H. Brönsted-acid and Brönsted-base catalyzed Diels-Alder reactions. Org. Biomol. Chem. 2007, 6, 3229–3236. [Google Scholar]
- Danishefsky, S.; Kerwin, J.F., Jr.; Kobayashi, S. Lewis acid catalyzed cyclocondensations of functionalized dienes with aldehydes. J. Am. Chem. Soc. 1982, 104, 358–360. [Google Scholar]
- Heintzelman, G.R.; Weinreb, S.M. Imino Diels-Alder-Based construction of a piperidine a-ring unit for total synthesis of the marine hepatoxin cylindrospermopsin. J. Org. Chem. 1996, 61, 4594–4499. [Google Scholar]
- Memeo, M.G.; Quadrelli, P. Iminium ions as dienophiles in Aza-Diels-Alder reactions: A closer look. Chem.-A Eur. J. 2012, 18, 12554–12582. [Google Scholar]
- Majumdar, K.C.; Taher, A.; Nandi, R.K. Synthesis of heterocycles by domino-Knoevenagel-hetero-Diels-Alder reactions. Tetrahedron 2012, 68, 5693–5718. [Google Scholar]
- Gosselin, P.; Bonfand, E.; Hayes, P.; Retoux, R.; Maignan, C. Asymmetric intermolecular Diels-Alder reactions of enantiopure sulfinyl-homo- and -hetero-dienes: Preliminary results. Tetrahedron: Asymmetry 1994, 5, 781–784. [Google Scholar]
- Ullman, E.F. Diels-Alder reaction of an unconjugated diene. Chem. Ind. (London) 1958, 1173–1174. [Google Scholar]
- Taschner, M.J. Asymmetric Diels-Alder Reactions. In Organic Synthesis: Theory and Applications; Hudlicky, T., Ed.; Jai Press: Greenwich, CT, USA, 1989; Vol. 1 , pp. 1–102. [Google Scholar]
- Kagan, H.B.; Riant, O. Catalytic Asymmetric Diels-Alder Reactions. Chem. Rev. 1992, 92, 1007–1019. [Google Scholar]
- Pellissier, H. Asymmetric hetero-Diels-Alder reactions of carbonyl compounds. Tetrahedron 2009, 65, 2839–2877. [Google Scholar]
- Fringuelli, F.; Taticchi, A. The Diels-Alder Reaction. Selected Practical Methods; Wiley-Interscience: New York, NY, USA, 2002. [Google Scholar]
- Cycloaddition Reactions in Organic Synthesis; Kobayashi, S.; Jorgensen, K.A. (Eds.) Wiley-VCH Verlag: New York, NY, USA, 2002.
- Fringuelli, F.; Taticchi, A. Dienes in the Diels-Alder Reaction; Wiley & Sons: New York, NY, USA, 1990. [Google Scholar]
- Ohba, M. Synthesis of natural products via intramolecular oxazole-olefin Diels-Alder reaction. Trends Org. Chem. 2010, 14, 1–12. [Google Scholar]
- Sapeta, K.; Leobold, T.P.; Kerr, M.A. Synthesis of highly substituted indoles via Diels-Alder/Plieninger indolization sequence: Applications in total synthesis. Synlett 2011, 11, 1495–1514. [Google Scholar]
- Brocksom, T.J.; Donatoni, M.C.; Marciana, P.U.; Viera, Y.W. The Diels-Alder reaction at the beginning of the twenty-first century. Quimica Nova 2010, 33, 2211–2218. [Google Scholar]
- Shea, K.M. Diels-Alder Reaction. In Name Reactions for Carbocyclic Ring Formation; Li, J.J., Ed.; Wiley: Hoboken, NJ, USA, 2010; pp. 275–308. [Google Scholar]
- For recent reviews of tandem reactions in organic synthesis, see ref. 26–32: Asymmetric tandem reactions: New synthetic strategies. Pure Appl. Chem. 2010, 82, 669–677.
- Grondal, C.; Jeanty, M.; Ender, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar]
- Li, X.-W.; Wang, Z.-Y.; Zheng, L.-Y. Recent progress on the application of tandem reactions in organic synthesis. Youji Huaxue 2006, 26, 1144–1149. [Google Scholar]
- McCarrol, A.J.; Walton, J.C. Programming organic molecules: Design and management of organic syntheses through free-radical cascade processes. Angew. Chem. Int. Ed. 2001, 40, 2224–2248. [Google Scholar]
- Tietze, L.F. Domino reactions in organic synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar]
- Bunce, R.A. Recent advances in the use of tandem reactions for organic synthesis. Tetrahedron 1995, 51, 13103–13159. [Google Scholar]
- Ho, T.-L. Tandem Organic Reactions; Wiley-Interscience: New York, NY, USA, 1992. [Google Scholar]
- A cascade-Diels-Alder reaction is a one-pot reaction involving at least two Diels-Alder reactions occurring in sequence, independent of any nascent functionality, see: Timed Diels-Alder Reactions. J. Am. Chem. Soc. 1980, 102, 1974–1977.
- A domino-Diels-Alder reaction is a reaction in which two or more Diels-Alder reactions take place in a one-pot reaction in which the subsequent Diels-Alder reaction occurs as consequence of the functionality generated by the initial Diels-Alder, see: Domino Diels-Alder reactions. 6. Domino Diels-Alder cycloadditions to 9,10-dihydrofulvalene and 11, 12-dihydrosesquifulvalene. A synthetic tool for the elaboration of polycondensed alicyclic systems. J. Am. Chem. Soc. 1978, 100, 5845–5855.
- A tandem-Diels-Alder reaction refers to two operating units that are distinct but take place at the same time, see: C-Aryl Glycosides via Tandem Intramolecular Benzyne–Furan Cycloadditions. Total Synthesis of Vineomycinone B2 Methyl Ester. J. Am. Chem. Soc. 2006, 128, 13696–13697.
- A consecutive-Diels-Alder reaction is a one-pot process in which the initial Diels-Alder reaction does not promote the second Diels-Alder reaction, thus requiring a change in the reaction conditions or the addition or reagents, see: Tandem pericyclic reactions. Novel and efficient methodology for the rapid assembly of complex polycyclic systems. Tetrahedron Lett. 1991, 32, 2549–2552.
- Parvez, A.; Choudhary, M.I.; Akhter, F.; Noorwala, M.; Mohammad, F.V.; Hasan, N.M.; Zamir, T.; Ahmad, V.U. Perovskone: a triterpene with a novel carbon skeleton from Perovskia abrotanoides. J. Org. Chem. 1992, 57, 4339–4340. [Google Scholar]
- Majetich, G.; Zhang, Y. Concise Synthesis of (±)-Perovskone. J. Am. Chem. Soc. 1994, 116, 4979–4980. [Google Scholar]
- Gandhi, R.P.; Ishar, M.P.S.; Wali, A. Diels-Alder addition to cyclopentadiene to allenic esters: catalysis by lanthanide complexes. J. Chem. Soc., Chem. Commun. 1988, 1074–1075. [Google Scholar]
- Lamy-Schelkens, H.; Giomi, D.; Ghosez, L. Stereochemical variations in the Diels-Alder reactions of siloxydienes with N,N-dimethyl acrylamide. A remarkable influence of the Lewis acid catalyst. Tetrahedron Lett. 1989, 5887–5890. [Google Scholar]
- Castellino, S.; Sims, J.J. The total synthesis of (±)-kawain via a hetero-Diels-Alder cycloaddition. Tetrahedron Lett. 1984, 25, 4059–4062. [Google Scholar]
- Majetich, G.; Zhang, Y.; Tian, X.; Britton, J.E.; Li, Y.; Phillips, R. Synthesis of (±)- and (+)-perovskone. Tetrahedron 2011, 67, 10129–10146. [Google Scholar]
- Li, Y.I. Stereoselective synthesis of (+)-Perovskone and (+)-Salvadione-A. II. Total synthesis of (±)-komaroviquinone and studies toward the synthesis of (+)-Komaroviquinone. III. The regiochemistry of the o-claisen rearrangement of bis-(Allyloxy)-polycyclic aromatics. IV. The use of β-bromodimethylalkoxysulonium Ions for olefin epoxidation. Ph.D. thesis, University of Georgia, Athens, GA, USA, 2006. [Google Scholar]
- Britton, J.E. Studies directed toward the synthesis of (+)-perovskone. M.Sc thesis, University of Georgia, Athens, GA, USA, 2002. [Google Scholar]
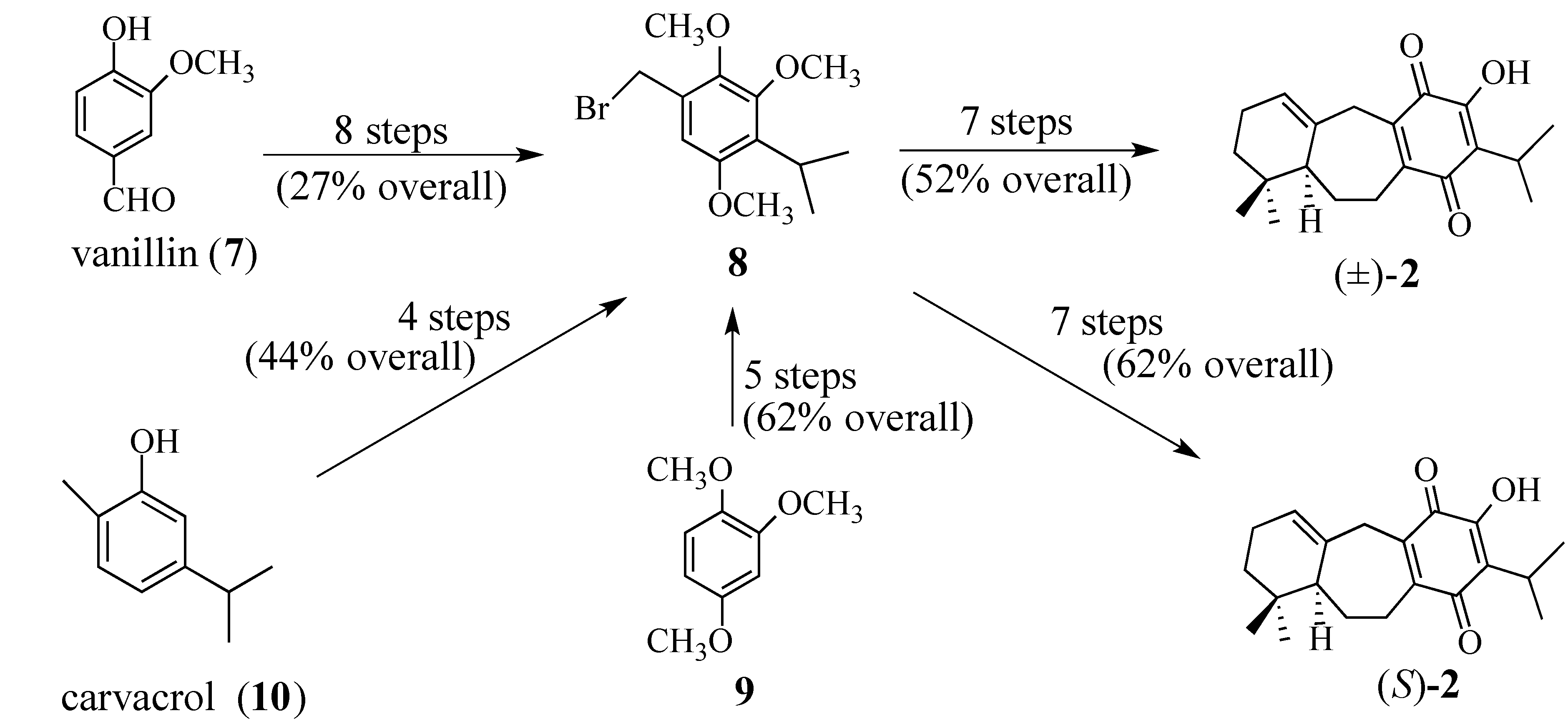
- For the first use of (S)-2, see: A Synthesis of (+)-Salvadione-A. Org. Lett. 2003, 5, 3847–3850.. For a detailed discussion of the preparation of (S)-2 see reference 42.
- Majetich, G.; Grove, J.L. Total synthesis of (+)-19-deoxyicetexone, (–)-icetexone and (+)-5-epi-icetexone. Org. Lett. 2009, 11, 2904–2907. [Google Scholar]
- Lindberg, T. Strategies and Tactics in Organic Synthesis; Academic Press Inc.: Orlando, FL, USA, 1984; p. xiii. [Google Scholar]
- Benchikn le-Hocine, M.; Do Khac, D.; Fetizon, M.; Guir, F.; Guo, Y.; Prange, T. A new method for preparing taxane derivatives. Tetrahedron Lett. 1992, 33, 1443–1446. [Google Scholar]
- Engler, T.A.; Sampath, N.; Naganathan, S.; Vander Velde, D.; Takusagawa, F.; Yohannes, D. A new general synthetic approach to diterpenes: Application to syntheses of (±)-taxodione and (±)-royleanone. J. Org. Chem. 1989, 54, 5712–5727. [Google Scholar]
- Allinger, N.L.; Yuh, Y.; Lii, J.-H. The MM3 force field for hydrocarbons. J. Am. Chem. Soc. 1989, 111, 8551–8582. [Google Scholar]
- Hollinshead, D.M.; Howell, S.C.; Ley, S.V.; Mahon, M.; Ratcliffe, N.M.; Worthington, P.A. The Diels-Alder route to drimane related sesquiterpenes. Synthesis of cinnamolide, Polygodial, Isodrimeninol, Drimenin, And warburganal. J. Chem. Soc., Perkin Trans. 1 1983, 1579–1586. [Google Scholar]
- Yates, D.; Eaton, P.E. Acceleration of the Diels-Alder reaction by aluminum chloride. J. Am. Chem. Soc. 1960, 82, 4436–4437. [Google Scholar]
- Jurczak, J.; Tkacz, M. Stereochemistry of Diels-Alder reactions at high pressure. 4. Asymmetric induction in high-pressure cycloadditions of (R)-(–)-menthyl glyoxylate and symmetric 1,3-dienes. J. Org. Chem. 1979, 3347–3352. [Google Scholar]
- Dauben, W.G.; Bunce, R.A. Organic reactions at high pressure. Asymmetric induction in the Diels-Alder reaction of p-benzoquinone with chiral 2,4-pentadienoic acid derivatives. Tetrahedron Lett. 1982, 4875–4878. [Google Scholar]
- Takeshita, A.H.; Liu, J.F.; Kato, N. High-pressure Diels-Alder reaction of [60] fullerene with several tropones. Characterization of the 1:1-cycloadducts. J. Chem. Soc. Perkin 1 1994, 1433–1437. [Google Scholar]
- Bols, M.; Skrydstrup, T. Silicon-Tethered Reactions. Chem. Rev. 1995, 95, 1253–1277. [Google Scholar]
- Fensterbank, L.; Malacria, M.; Sieburth, S.M. Intramolecular reactions of temporarily silicon-tethered molecules. Synthesis 1997, 813–854. [Google Scholar]
- Gauthier, D.R.; Zandi, K.S.; Shea, K.J. Disposable tethers in synthetic organic chemistry. Tetrahedron 1998, 54, 2289–2292. [Google Scholar]
- Shea, K.J.; Fruscella, W.M.; Carr, R.C.; Burke, L.D.; Cooper, D.K. Synthesis of Bridgehead Enol Lactones via Type 2 Intramolecular Diels-Alder cycloaddition. New intermediates for steeocontrolled organic synthesis. J. Am. Chem. Soc. 1987, 109, 447–452. [Google Scholar]
- Batey, R.A.; Tadani, A.V.; Lough, A.J. Alkenylboronate tethered intramolecular Diels-Alder reactions. J. Am. Chem. Soc. 1999, 121, 450–451. [Google Scholar]
- Stoltz and Greene have attempted to synthesize salvadione-A via a clever tandem Claisen rearrangement / Cope rearrangement / intramolecular Diels-Alder reaction strategy, see: Progress toward the total synthesis of salvadione-A and related diterpenoids. M.Sc thesis, California Institute of Technology, Pasadena, CA, USA, 2004.
- Trost, B.M.; Ippen, J.; Vladuchick, W.C. The regioselectivity of the catalyzed and uncatalyzed Diels-Alder reaction. J. Am. Chem. Soc. 1977, 99, 8116–8118. [Google Scholar]
- For the formation of a chelate of o-methoxyquinones with titanium-based Lewis acids, see: Preparation of α-thiolated carbonyl compounds. Chem. Ber. 1976, 109, 1601–1616..
- Domingo, L.R.; Saez, J.A. Understanding the mechanism of polar Diels-Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar]
- Soto-Delgado, J.; Domingo, L.R.; Contreras, R. Understanding the influence of Lewis acids in the regioselectivity of the Diels-Alder reactions of 2-methoxy-5-methyl-1,4-benzoquinone: A DFT study. J. Mol. Struct. (Theochem.) 2009, 902, 103–108. [Google Scholar]
- Soto-Delgado, J.; Aizman, A.; Contreras, R.; Domingo, L.R. On the catalytic effect of water in the intramolecular Diels-Alder reaction of quinone systems: A theoretical Study. Molecules 2012, 17, 13687–13703. [Google Scholar]
- Fujita, E.; Node, M. Diterpenoids of Rabdosia Species. Prog. Chem. Org. Nat. Prod. 1986, 46, 77–157. [Google Scholar]
- Nasir, E.; Ali, S.I. Flora of Pakistan; Fakhri Printing Press: Karachi, Pakistan, 1986; Volume 56, p. 156. [Google Scholar]
- Chopra, R.N.; Nayar, S.L.; Chopra, I.C. Glossary of Indian Medicinal Plants; CSIR: New Delhi, India, 1956; p. 189. [Google Scholar]
- Kirtikar, K.R.; Basu, B.D. dian Medicinal Plants; Indian Press: Allahabad, India, 1918; p. 1031. [Google Scholar]
- Ahmad, V.U.; Zahid, M.; Ali, M.S.; Choudhary, M.I.; Akhtar, F.; Ali, Z.; Iqbal, M.Z. Salvadiol: A novel triterpenoid from Salvia bucharica. Tetrahedron Lett. 1999, 40, 7561–7564. [Google Scholar]
- Ahmad, V.U.; Zahid, M.; Ali, M.S.; Jassbi, A.R.; Abbas, M.; Clardy, J.; Lobkovsky, E.; Tareen, R.B.; Iqbal, M.Z. Salvadiones-A and -B: Two terpenoids having novel carbon skeleta from Salvia bucharica. J. Org. Chem. 1999, 64, 8465–8467. [Google Scholar]
- For a stepwise-based synthesis of salvadione-B, see: Synthesis of (+)-Salvadione-B. Org. Lett.Submitted.
- For a two-pot cascade-based synthesis of salvadione-B, see: A Cascade-based Synthesis of (+)-Salvadione-B. Org. Lett.Submitted.
- Farimani, M.M.; Bahadori, M.B.; Taheri, S.; Ebrahimi, S.N.; Zimmermann, S.; Brun, R.; Amin, G.; Hamburger, M. Triterpenoids with Rare Carbon Skeletons from Salvia hydrangea: Antiprotozoal Activity and Absolute Configurations. J. Nat. Prod. 2011, 74, 2200–2205. [Google Scholar]
- Perovskone-B has been synthesized by G. Majetich and G. Zou, University of Georgia, (unpublished results).
- Farimani, M.M.; Taheri, S.; Ebrahimi, S.N.; Bahadori, M.B.; Khavasi, H.R.; Zimmermann, S.; Brun, R.; Hamburger, M. Hydrangenone, a new isoprenoid with an unprecedented skeleton from Salvia hydrangea. Org. Lett. 2012, 14, 166–169. [Google Scholar]
- On a biogenetic basis, triterpenes 1, 22, 23, 24, 25, 26 and 27 should be regarded as “pseudo-triterpenes” since they are not derived from squalene but are the result of the coupling of a mono- and diterpene.
- Ahmad, V.U.; Parvez, A.; Hassan, N.M. Isolation and structure determination of peradione A. Novel triterpene with a rearranged perovskane skeleton from Perovskia abrotanoides. Tetrahedron Lett. 1993, 34, 5337–5340. [Google Scholar]
- Maier, M.E.; Bayer, A. A formal total synthesis of Salvadione. Eur. J. Org. Chem. 2006, 4034–4043. [Google Scholar]
- House, H.O. Modern Synthetic Reactions, 2nd ed.; Benjamin, Inc.: Menlo Park, CA, USA, 1972; p. 534. [Google Scholar]
- Dr. Yangyang Wang, who synthesized (+)-salvadione-A (23), died in child-birth in 2005.
- Zou, G. The Total Synthesis of Salvia Terpenes: (+)-Grandione, (-)-Brussonol, (-)-Salviasperanol and (+)-Salvadione-B. Ph.D. thesis, University of Georgia, Athens, GA, USA, 2009. [Google Scholar]
- Kenney, R.L.; Fisher, G.S. Photosensitized oxidation of Myrcene. J. Am. Chem. Soc. 1959, 81, 4288–4291. [Google Scholar]
- Matsumoto, M.; Kondo, K. Sensitized photooxygenation of linear monoterpenes bearing conjugated double bonds. J. Org. Chem. 1975, 40, 2259–2260. [Google Scholar]
- Reich, H.J.; Reich, I.L.; Renga, J.M. Organoselenium chemistry. α-Phenylseleno carbonyl compounds as precursors for α,β-unsaturated ketones and esters. J. Am. Chem. Soc. 1973, 95, 5813–5815. [Google Scholar]
- Sharpless, K.B.; Michaelson, R.C. High Stereo- and regioselectivities in the transition metal catalyzed epoxidation of olefinic alcohols by tert-Butyl hydroperoxide. J. Am. Chem. Soc. 1973, 95, 6136–6137. [Google Scholar]
- Gao, Y.; Hanson, R.M.; Klunder, J.M.; Ko, S.Y.; Masamune, H.; Sharpless, K.B. Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in Situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar]
- Scettri, A.; Bonadies, F.; Lattanzi, A. Asymmetric oxidation with Furylhydroperoxides. Tetrahedron Asymm. 1996, 7, 629–632. [Google Scholar]
- Klar, U.; Neef, G.; Vorbrüggen, H. Perfluorobutanesulfonyl Fluoride—1,8-Diazabicyclo-[5.4.0]undec-7-ene. Tetrahedron Lett. 1996, 37, 7497–7498. [Google Scholar]
- Rideout, D.C.; Breslow, R. Hydrophobic acceleration of Diels-Alder Reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar]
- Grieco, P.A.; Garner, P.; He, Z.-M. Micellar catalysis in the aqueous intermolecular Diels-Alder Reaction: Rate acceleration and enhanced selectivity. Tetrahedron Lett. 1983, 24, 1897–1900. [Google Scholar]
- Crystal data for C30H42O5; MW = 482.64, monoclinic, C2/c, a = 30.544(10) Å, b = 13.568(4) Å, c = 13.581(4) Å, B = 106.248(5) º, V = 5403(3) Å3, Z = 8, T = 293(2) K, μ(Mo Kα) = 0.71073 Å, Dcalcd = 1.187 g/cm3, R(1) = 0.0608 for 3502 observed independent reflections (I > 2σ(I)). All non-hydrogen atoms were refined anisotrpically. Hydrogen atoms were treated as idealized contributions.
- Pai, C.K.; Smith, M.B. Rate enhancement in dilute salt solutions of aqueous ethanol: The Diels-Alder Reaction. J. Org. Chem. 1995, 60, 3731–3735. [Google Scholar]
- Smith, M.B.; Fay, J.N.; Son, Y.C. Rate enhancement in ethanolic aqueous buffer. The Diels-Alder reaction. Chem. Lett. 1992, 12, 2451–2454. [Google Scholar]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar]
- Gedye, R.N.; Smith, F.E.; Westaway, K. The rapid synthesis of organic compounds in microwave ovens. Can. J. Chem. 1987, 66, 17–26. [Google Scholar]
- Giguere, R.J.; Bray, T.; Duncan, S.M.; Majetich, G. Application of Commercial Microwave Ovens to Organic Synthesis. Tetrahedron Lett. 1986, 27, 4995–4998. [Google Scholar]
- Giguere, R.J.; Namen, A.; Lopez, B.O.; Arepally, A.; Ramos, D.E.; Majetich, G.; Defauw, J. Studies on Tandem Ene / Intramolecular Diels-Alder Reactions. Tetrahedron Lett. 1987, 28, 6553–6556. [Google Scholar]
- Majetich, G.; Wheless, K. Microwave Heating in Organic Chemistry: An Update. In Microwave Enhanced Chemistry, Fundamentals, Sample Preparation and Applications; Kingston, H.M., Haswell, S.J., Eds.; ACS: Washington, DC, USA, 1997; pp. 455–501. [Google Scholar]
- Majetich, G.; Hicks, R. The Use of Microwave Heating in Undergraduate Organic Laboratories. In Microwave Enhanced Chemistry, Fundamentals, Sample Preparation and Applications; Kingston, H.M., Haswell, S.J., Eds.; ACS: Washington, DC, USA, 1997; pp. 507–522. [Google Scholar]
- van Dort, H.M.; Jägers, P.P.; Heide, R.T.; van der Weerdt, A.J.A. Narcissus trevithian and Narcississ geranium: Analysis and Synthesis of Compounds. J. Agric. Food Chem. 1993, 41, 2063–2075. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Majetich, G.; Zhang, Y.; Tian, X.; Zou, G.; Li, Y.; Wang, Y.; Hu, S.; Huddleston, E. Diels-Alder Reactions of 12-Hydroxy-9(10®20)-5aH-abeo-abieta-1(10),8(9),12(13)-triene-11,14-dione. Molecules 2013, 18, 6969-6989. https://doi.org/10.3390/molecules18066969
Majetich G, Zhang Y, Tian X, Zou G, Li Y, Wang Y, Hu S, Huddleston E. Diels-Alder Reactions of 12-Hydroxy-9(10®20)-5aH-abeo-abieta-1(10),8(9),12(13)-triene-11,14-dione. Molecules. 2013; 18(6):6969-6989. https://doi.org/10.3390/molecules18066969
Chicago/Turabian StyleMajetich, George, Yong Zhang, Xinrong Tian, Ge Zou, Yang Li, Yangyang Wang, Shougang Hu, and Eric Huddleston. 2013. "Diels-Alder Reactions of 12-Hydroxy-9(10®20)-5aH-abeo-abieta-1(10),8(9),12(13)-triene-11,14-dione" Molecules 18, no. 6: 6969-6989. https://doi.org/10.3390/molecules18066969
APA StyleMajetich, G., Zhang, Y., Tian, X., Zou, G., Li, Y., Wang, Y., Hu, S., & Huddleston, E. (2013). Diels-Alder Reactions of 12-Hydroxy-9(10®20)-5aH-abeo-abieta-1(10),8(9),12(13)-triene-11,14-dione. Molecules, 18(6), 6969-6989. https://doi.org/10.3390/molecules18066969