Insights into Structure-Activity Relationships of Somatostatin Analogs Containing Mesitylalanine


Abstract
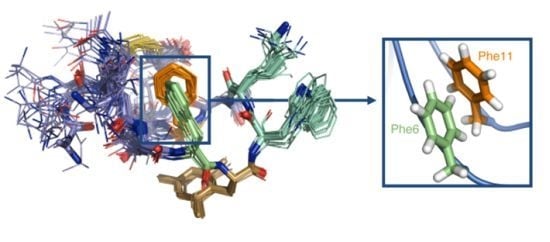
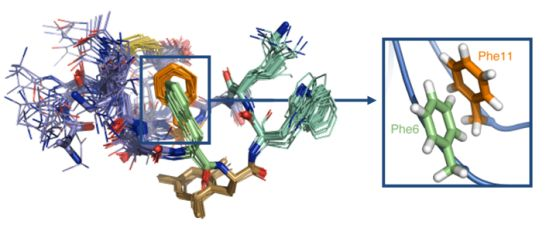
:
1. Introduction
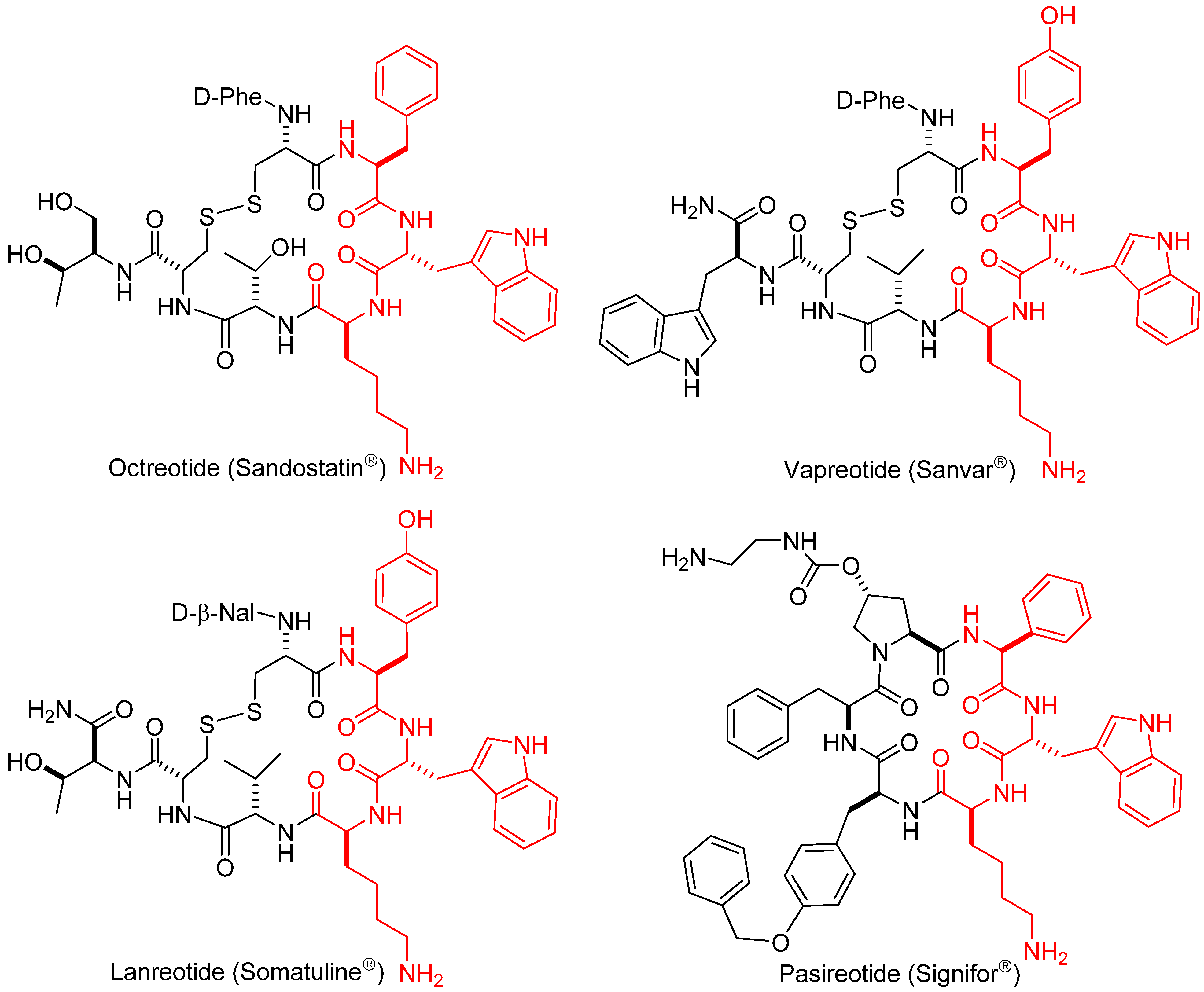
1.1. Somatostatin Analogs

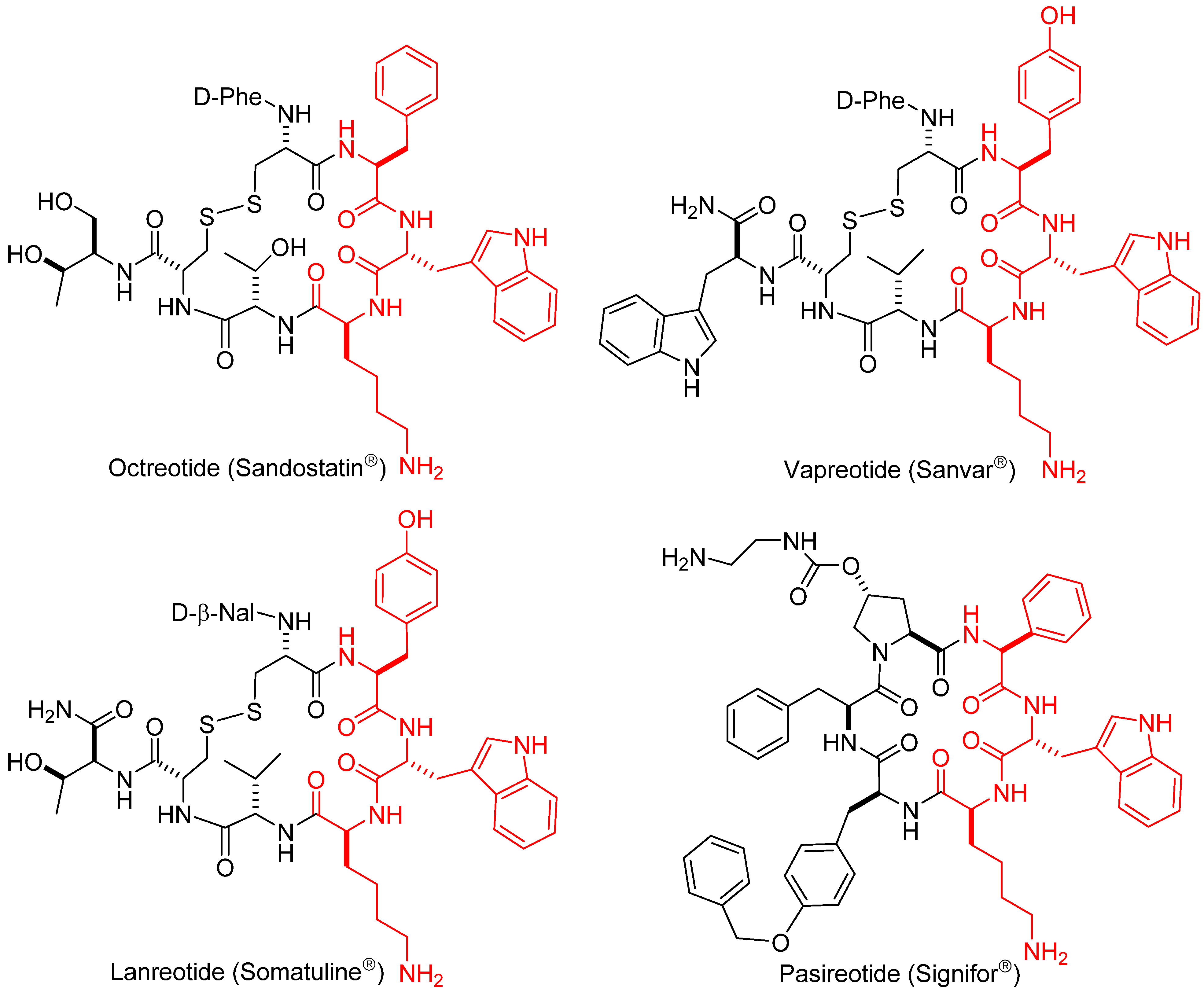
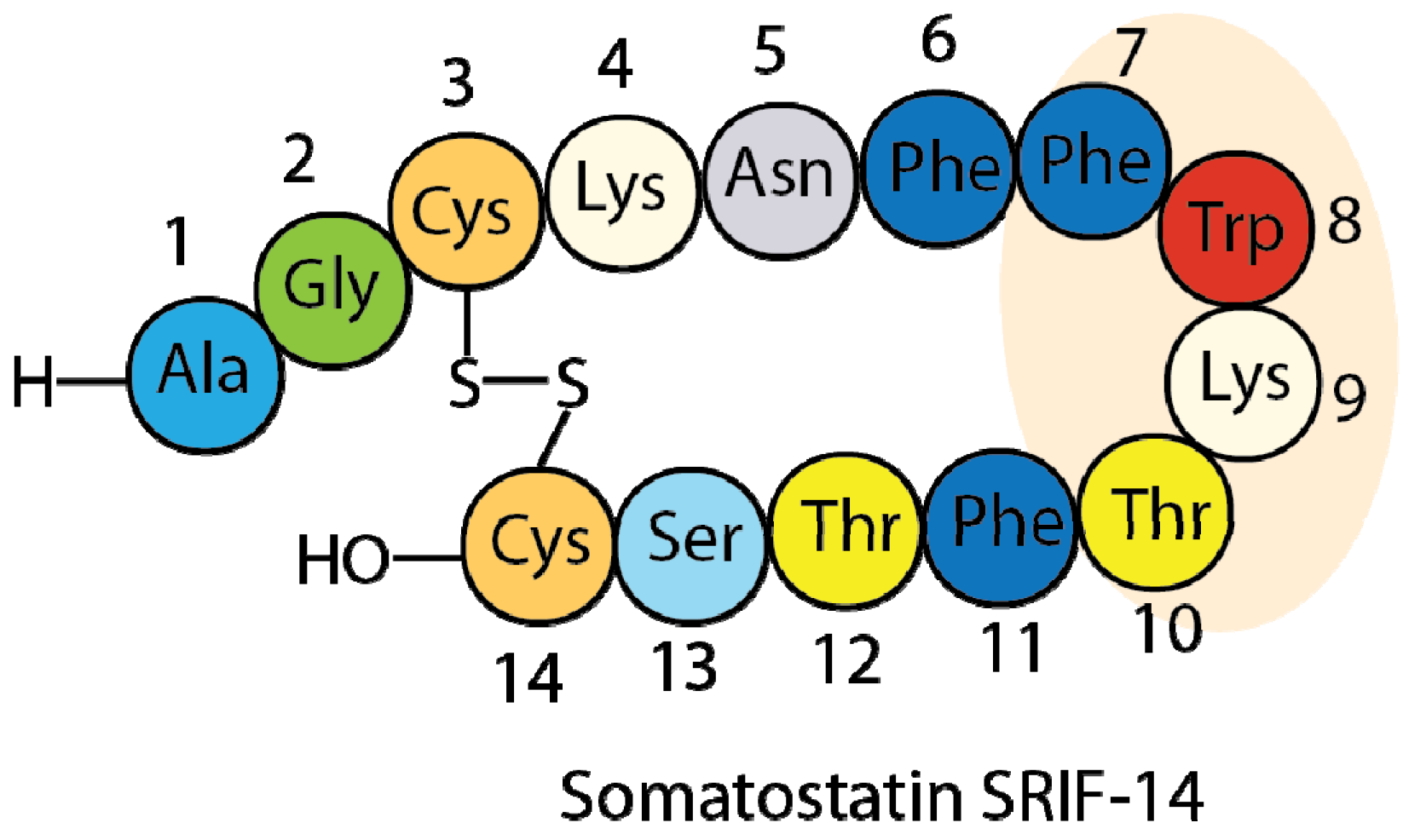
1.2. Somatostatin Structure
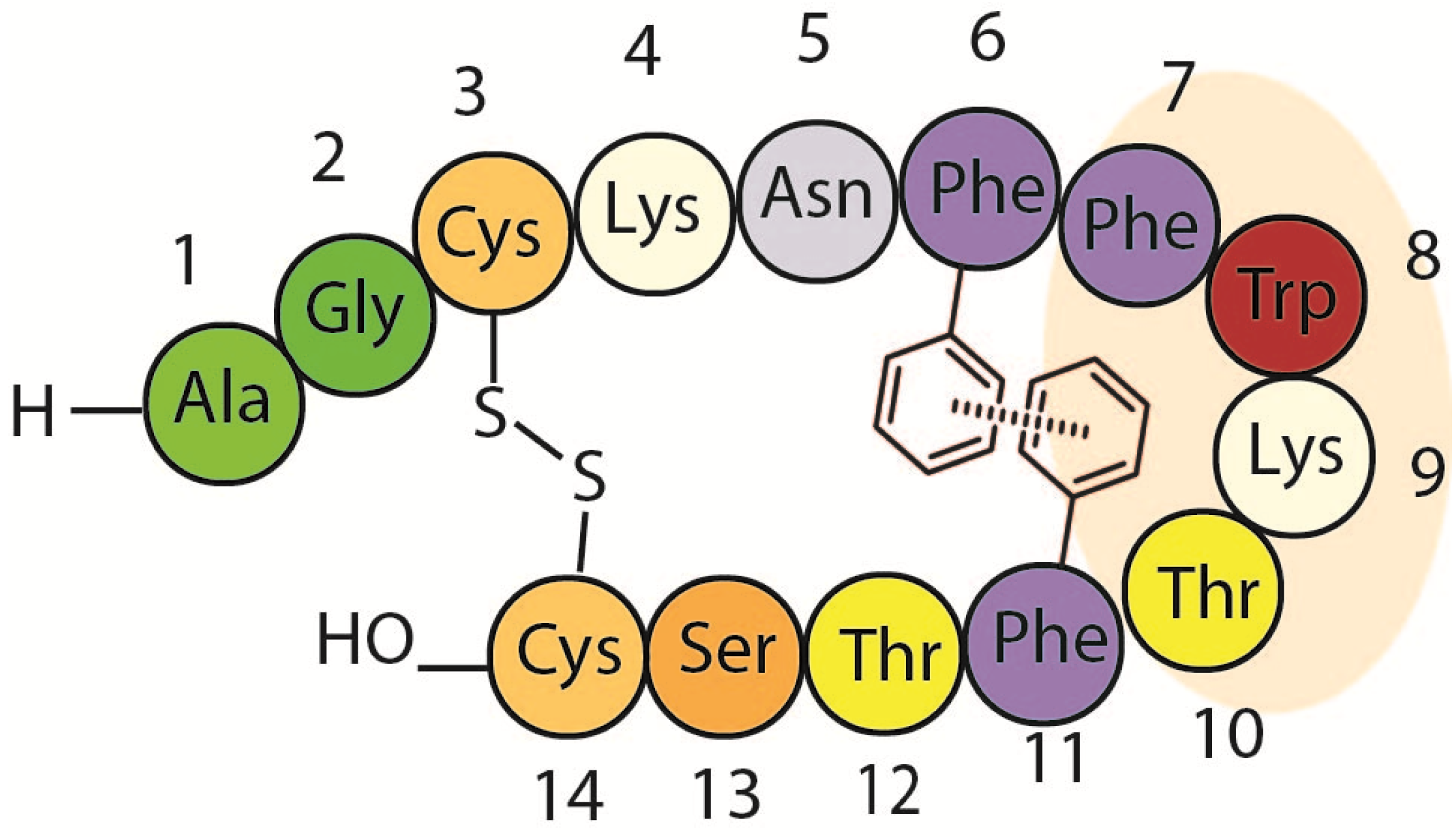
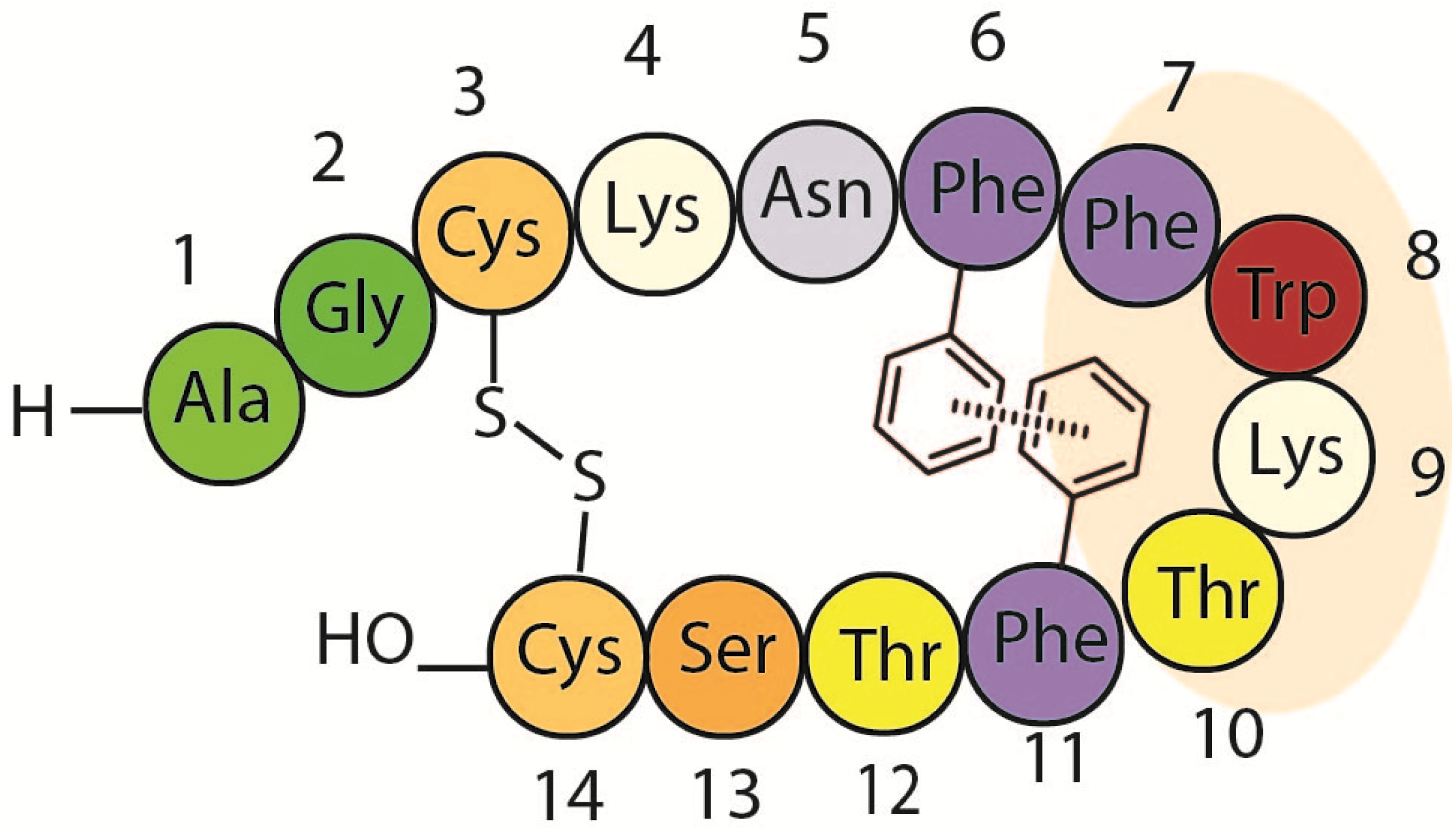
1.3. Somatostatin Analogs with Mesitylalanine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
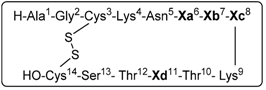
| Entry | Xa(6) | Xb(7) | Xc(8) | Xd(11) | Peptide analog |
|---|---|---|---|---|---|
| 1 | Msa | Phe | L-Trp | Phe | [L-Msa6]-SRIF, 1 |
| 2 | Phe | Msa | L-Trp | Phe | [L-Msa7]-SRIF, 2 a |
| 3 | Phe | Phe | L-Trp | Msa | [L-Msa11]-SRIF, 3 |
| 4 | Msa | Phe | D-Trp | Phe | [L-Msa6,D-Trp8]-SRIF, 4 a |
| 5 | Phe | Msa | D-Trp | Phe | [L-Msa7,D-Trp8]-SRIF, 5 a |
| 6 | Phe | Phe | D-Trp | Msa | [L-Msa11,D-Trp8]-SRIF, 6 a |
| 7 | Msa | Msa | L-Trp | Phe | [L-Msa6,7]-SRIF, 7 |
| 8 | Msa | Phe | L-Trp | Msa | [L-Msa6,11]-SRIF, 8 |
| 9 | Phe | Msa | L-Trp | Msa | [L-Msa7,11]-SRIF, 9 |
| 10 | Msa | Msa | L-Trp | Msa | [L-Msa6,7,11]-SRIF, 10 |
2. Results and Discussion
2.1. Synthesis of Tetradecapeptides with Mesitylalanine
2.2. Serum Stability
2.3. Binding Activity
| Peptide | SSTR1 | SSTR2 | SSTR3 | SSTR4 | SSTR5 | t1/2 (h) a |
|---|---|---|---|---|---|---|
| Somatostatin, SRIF | 0.43 ± 0.08 | 0.0016 ± 0.0005 | 0.53 ± 0.21 | 0.74 ± 0.07 | 0.23 ± 0.04 | 2.75 |
| [D-Trp8]-SRIF | 0.32 ± 0.11 | 0.001 ± 0.0007 | 0.61 ± 0.02 | 5.83 ± 0.44 | 0.46 ± 0.24 | 19.7 |
| Octreotide | 300 ± 85 | 0.053 ± 0.011 | 15.2 ± 5.9 | >103 | 11.53 ± 1.91 | 200 |
| [L-Msa6]-SRIF, 1 | 8.52 ± 1.45 | 1.49 ± 1.45 | 1.36 ± 1.45 | 3.62 ± 1.45 | 0.91 ± 1.45 | 2.1 |
| [L-Msa7]-SRIF, 2 c | 4.17 ± 1.45 | 0.019 ± 0.009 | >103 | 28.72 ± 6.9 | >103 | 5.2 |
| [L-Msa11]-SRIF, 3 | 19.97 ± 5.26 | 0.024 ± 0.004 | 2.8 ± 0.22 | 6.45 ± 2.23 | 2.1 ± 0.70 | 1.7 |
| [L-Msa6_D-Trp8]-SRIF, 4 c | 3.08 ± 0.9 | 4.55 ± 0.66 | 0.78 ± 0.1 | 4.70 ± 0.92 | 0.36 ± 0.003 | 26 |
| [L-Msa7_D-Trp8]-SRIF, 5 c | 0.33 ± 0.09 | 0.0024 ± 0.001 | 7.49 ± 0.63 | >103 | >103 | 25 |
| [L-Msa11_D-Trp8]-SRIF, 6 c | 3.35 ± 1.32 | 0.14 ± 0.06 | 1.31 ± 0.2 | >103 | 0.73 ± 0.19 | 41 |
| [L-Msa6,7]-SRIF, 7 | >103 | 14.69 ± 0.82 | >103 | >103 | >103 | 43.9 |
| [L-Msa6,11]-SRIF, 8 | >103 | >103 | 13.34 ± 2.92 | >103 | 9.12 ± 0.61 | nm b |
| [L-Msa7,11]-SRIF, 9 | 105.75 ± 30.6 | 1.37 ± 0.32 | >103 | >103 | >103 | 10 |
| [L-Msa6,7,11]-SRIF, 10 | >103 | >103 | >103 | >103 | >103 | 93.3 |
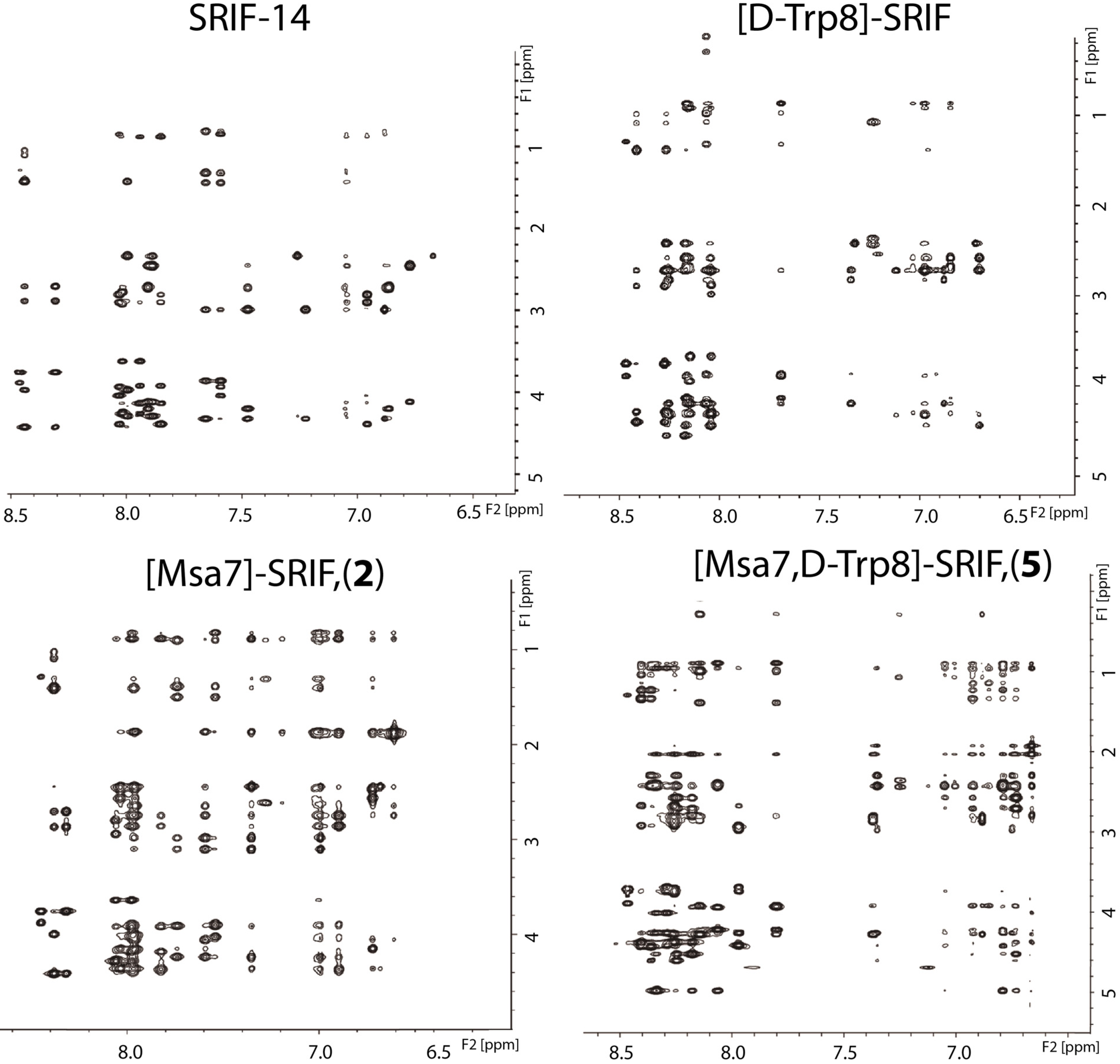
2.4. NMR Structure
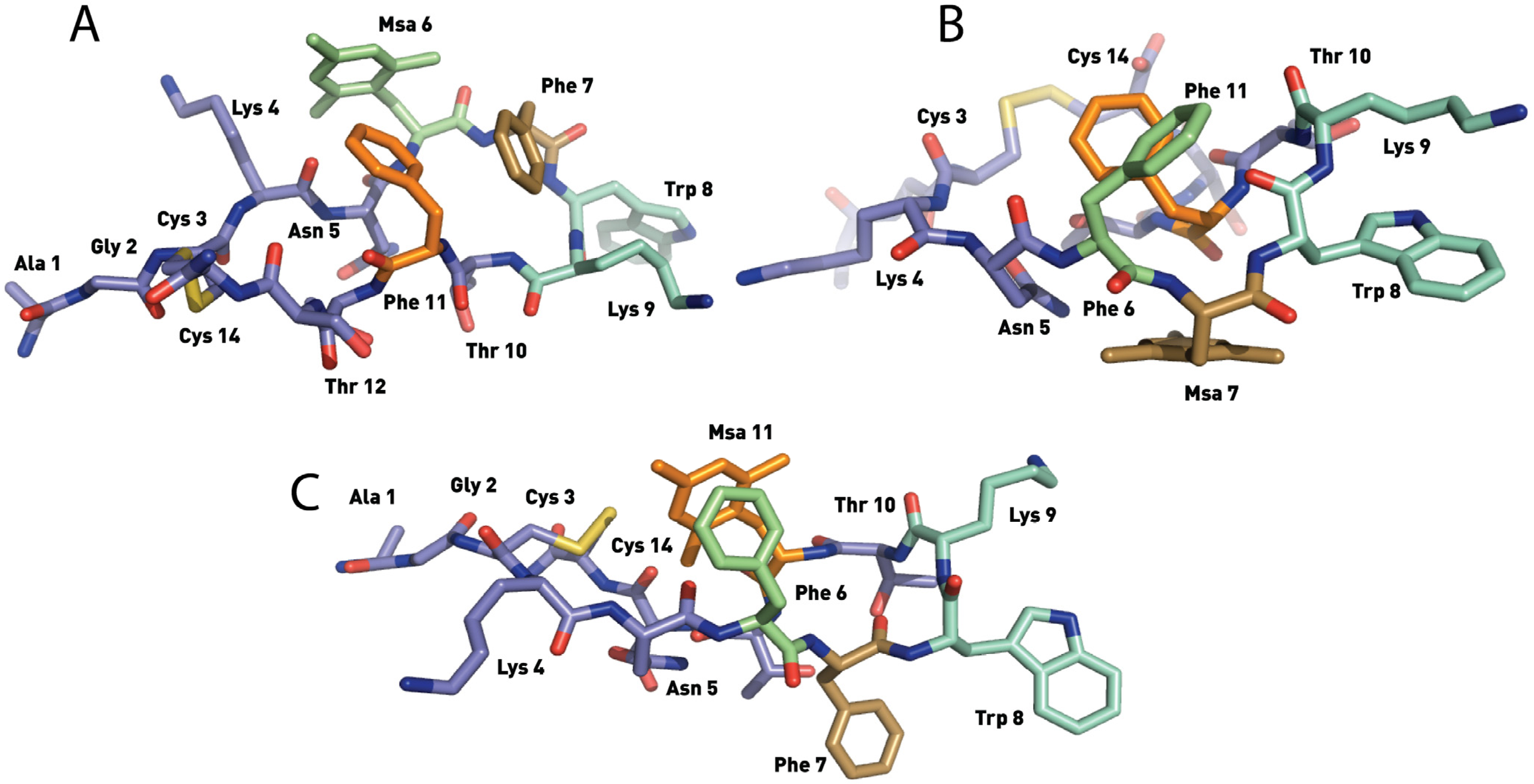
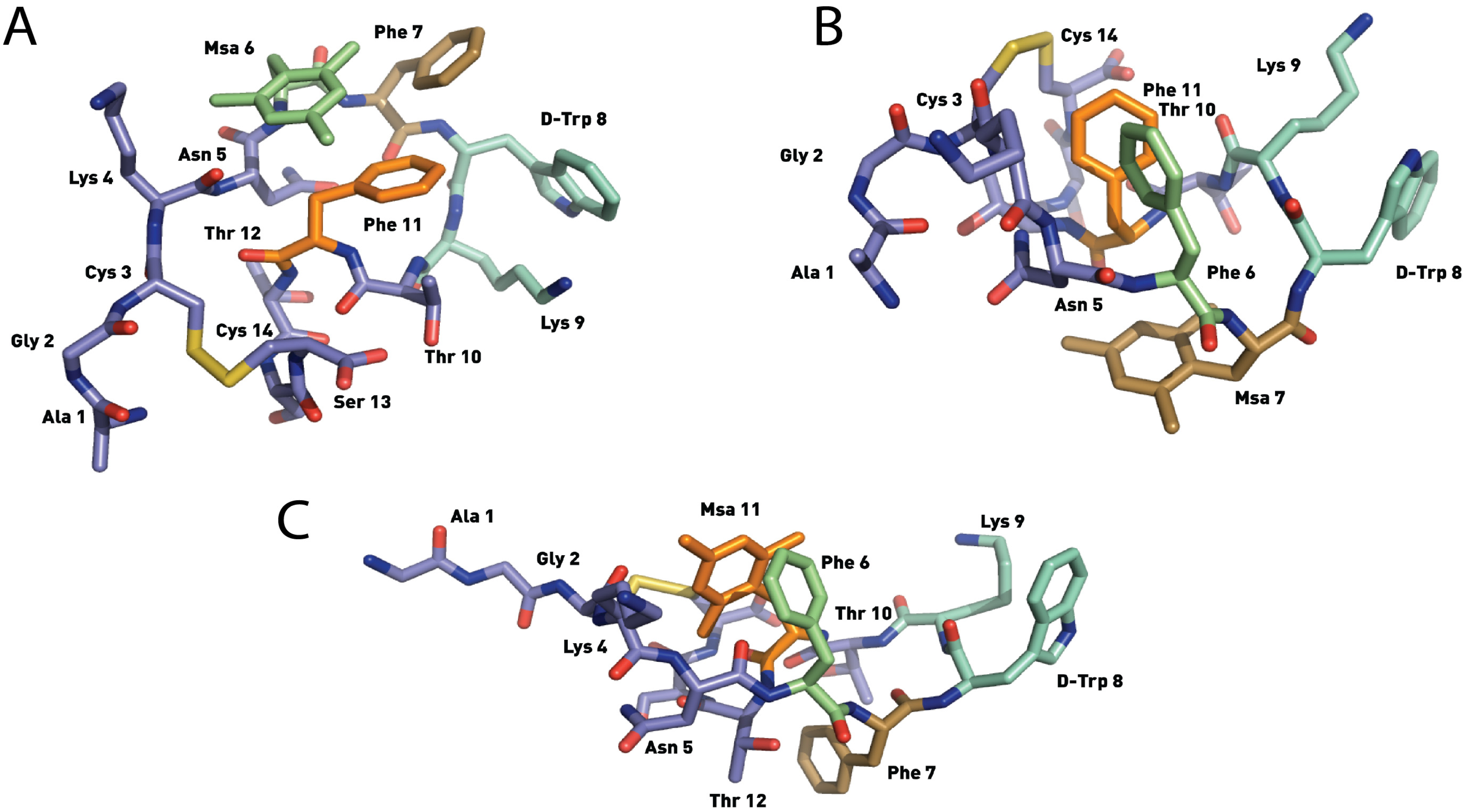
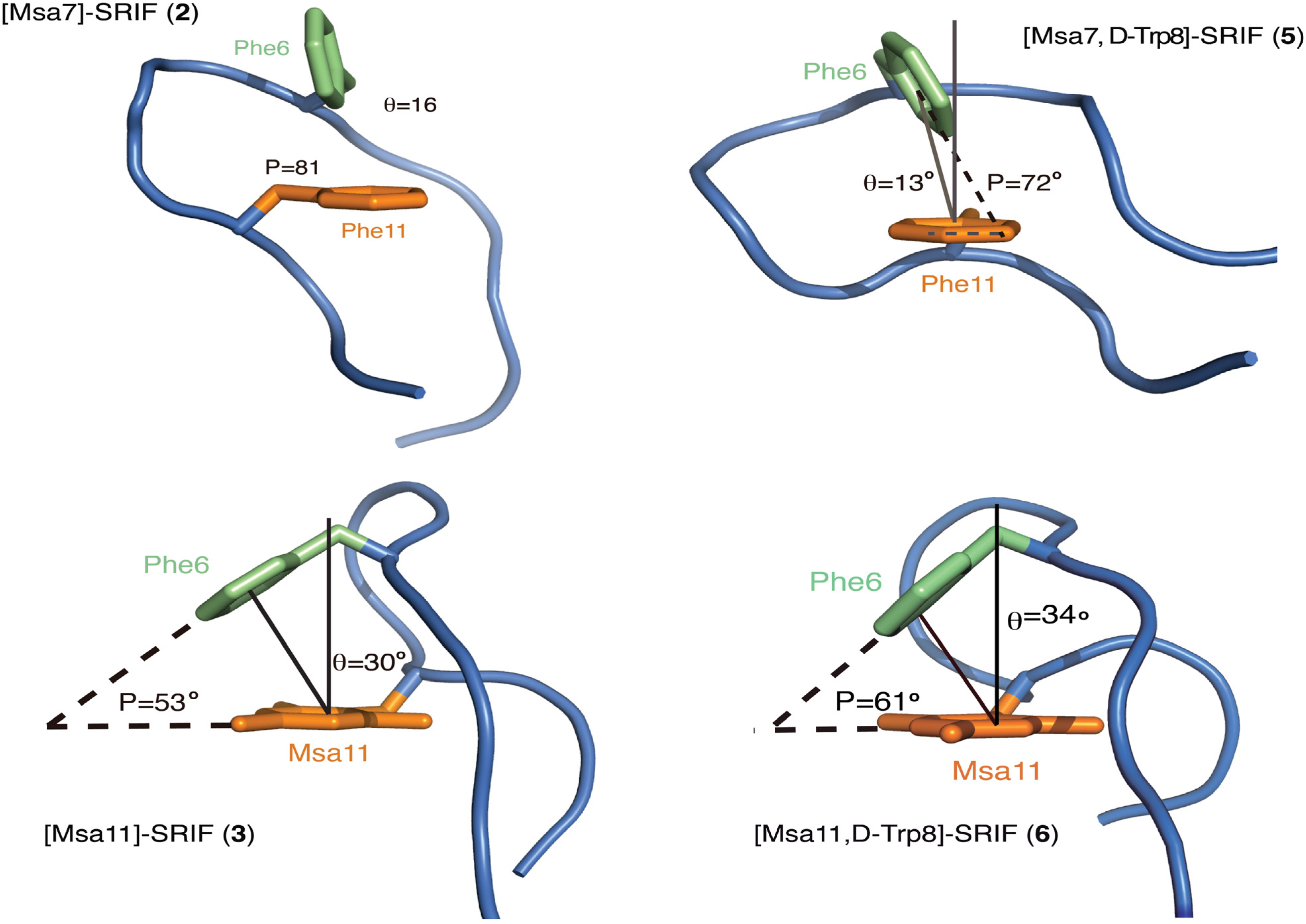
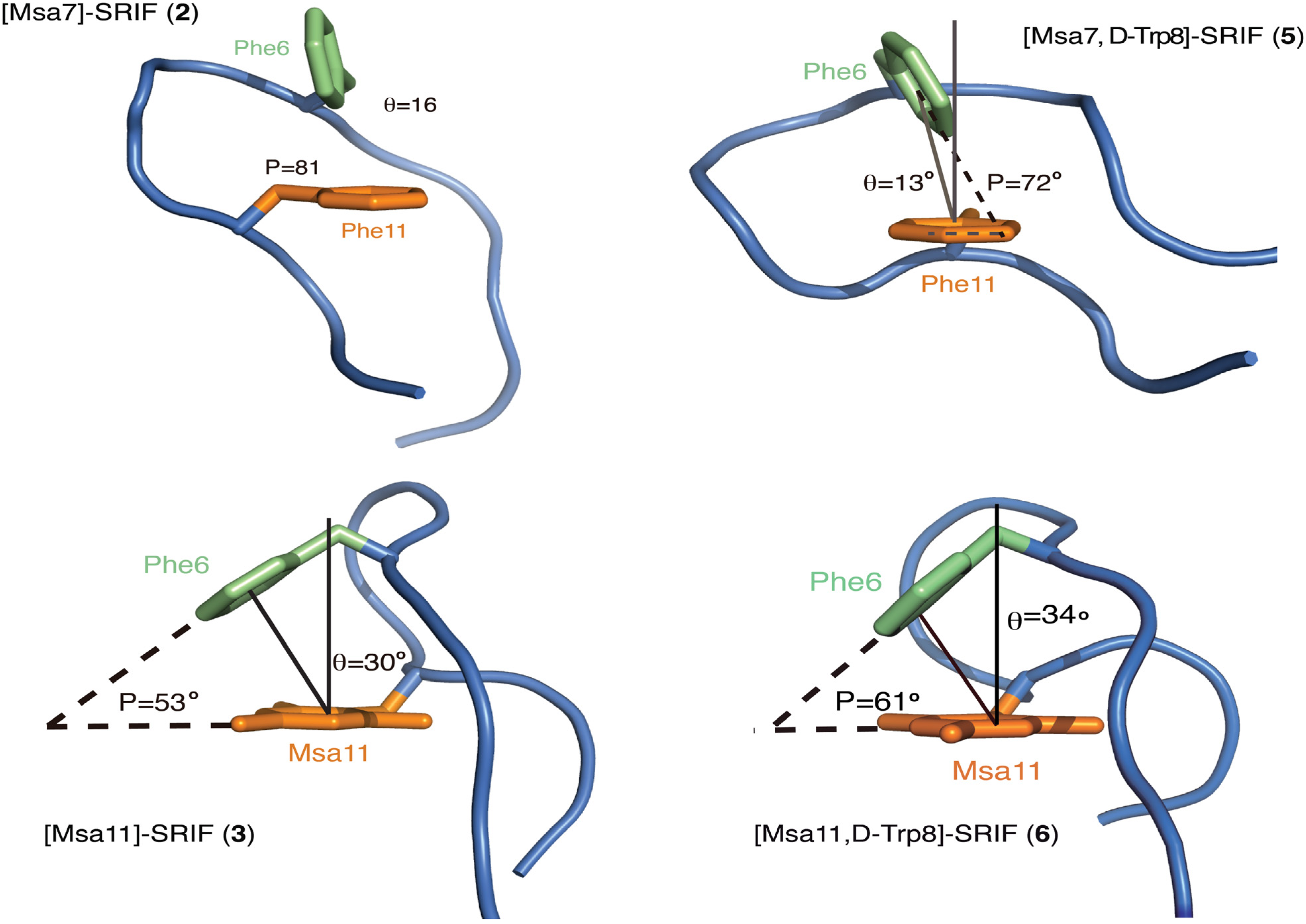
2.4.1. Compounds 1–3 with One Msa Residue
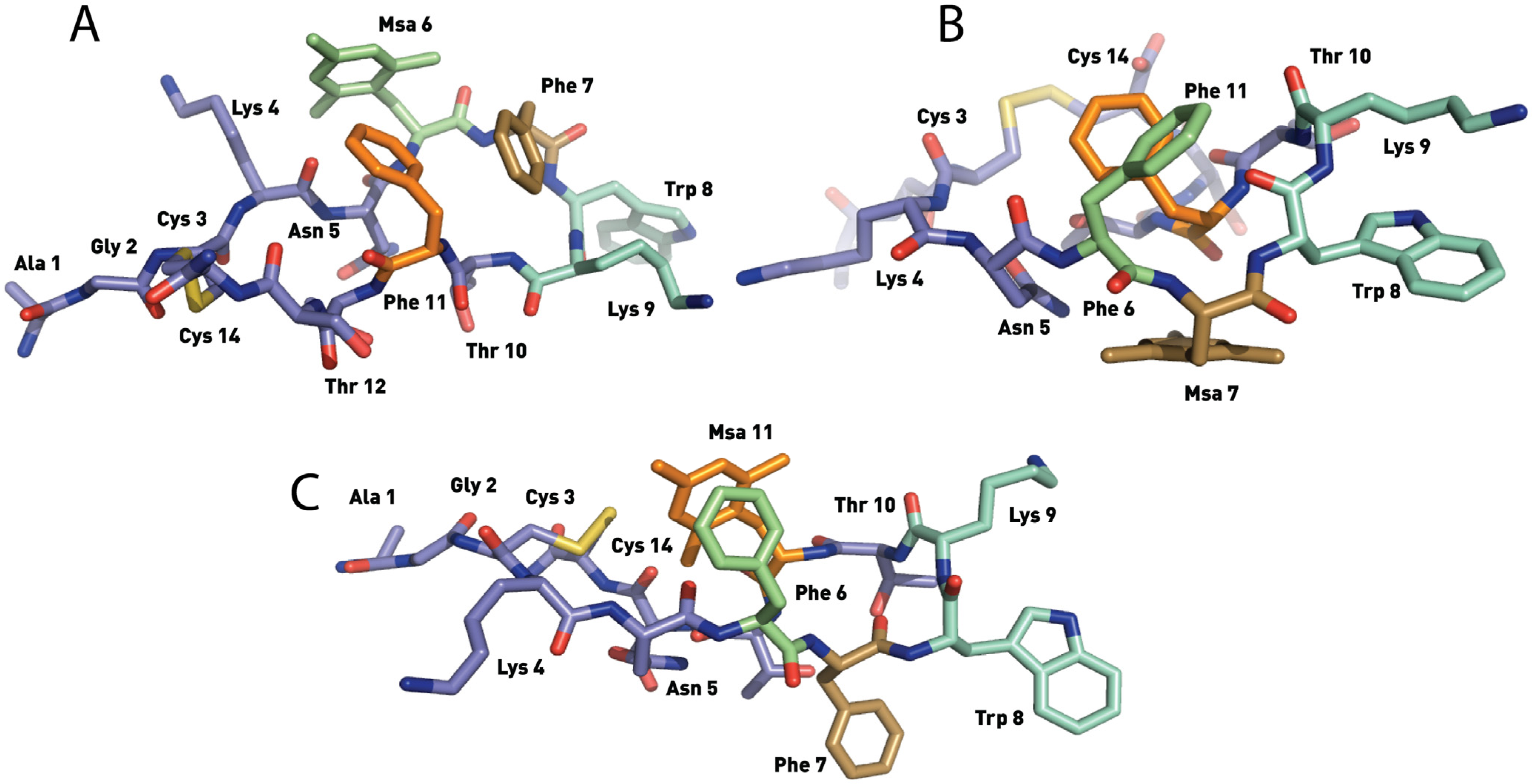
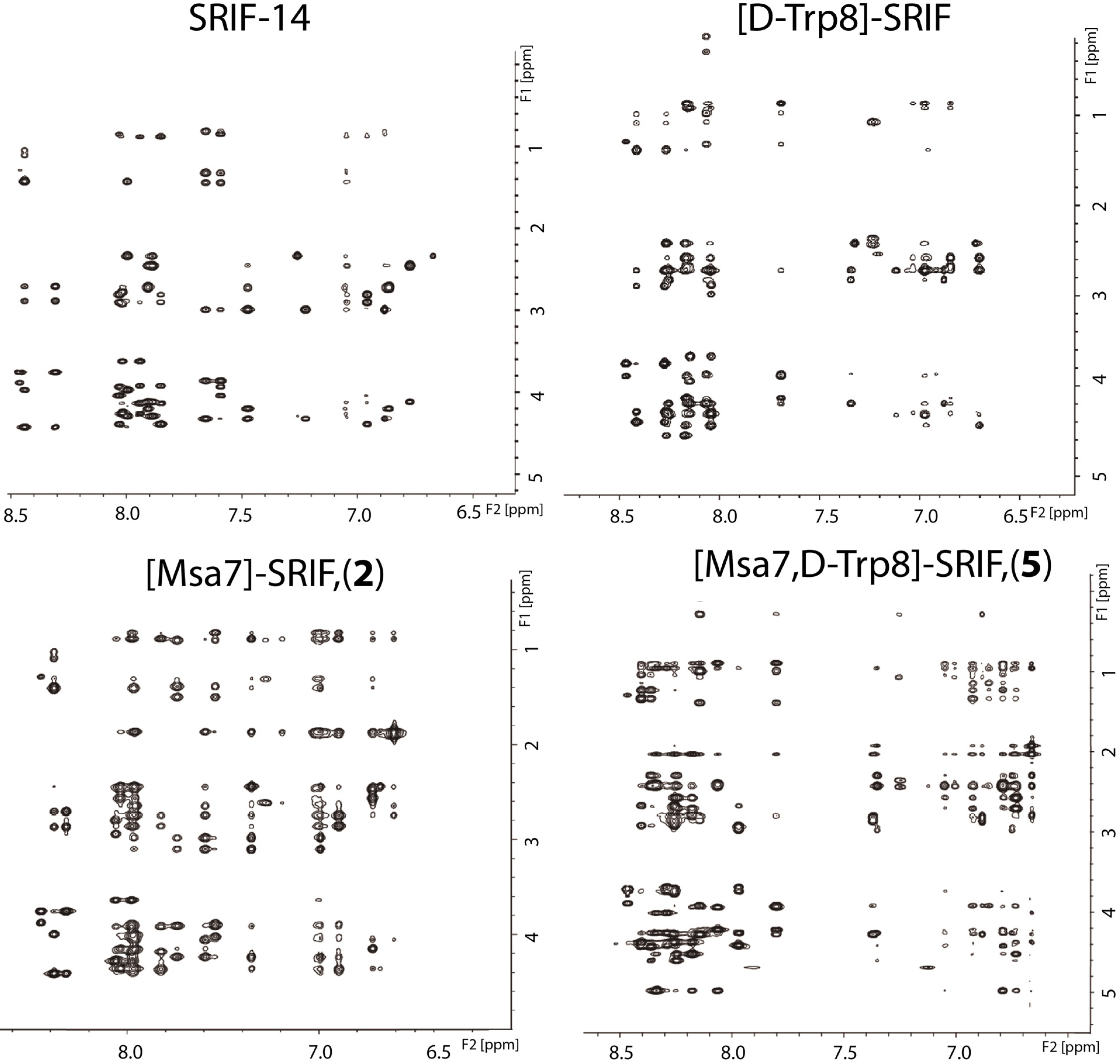
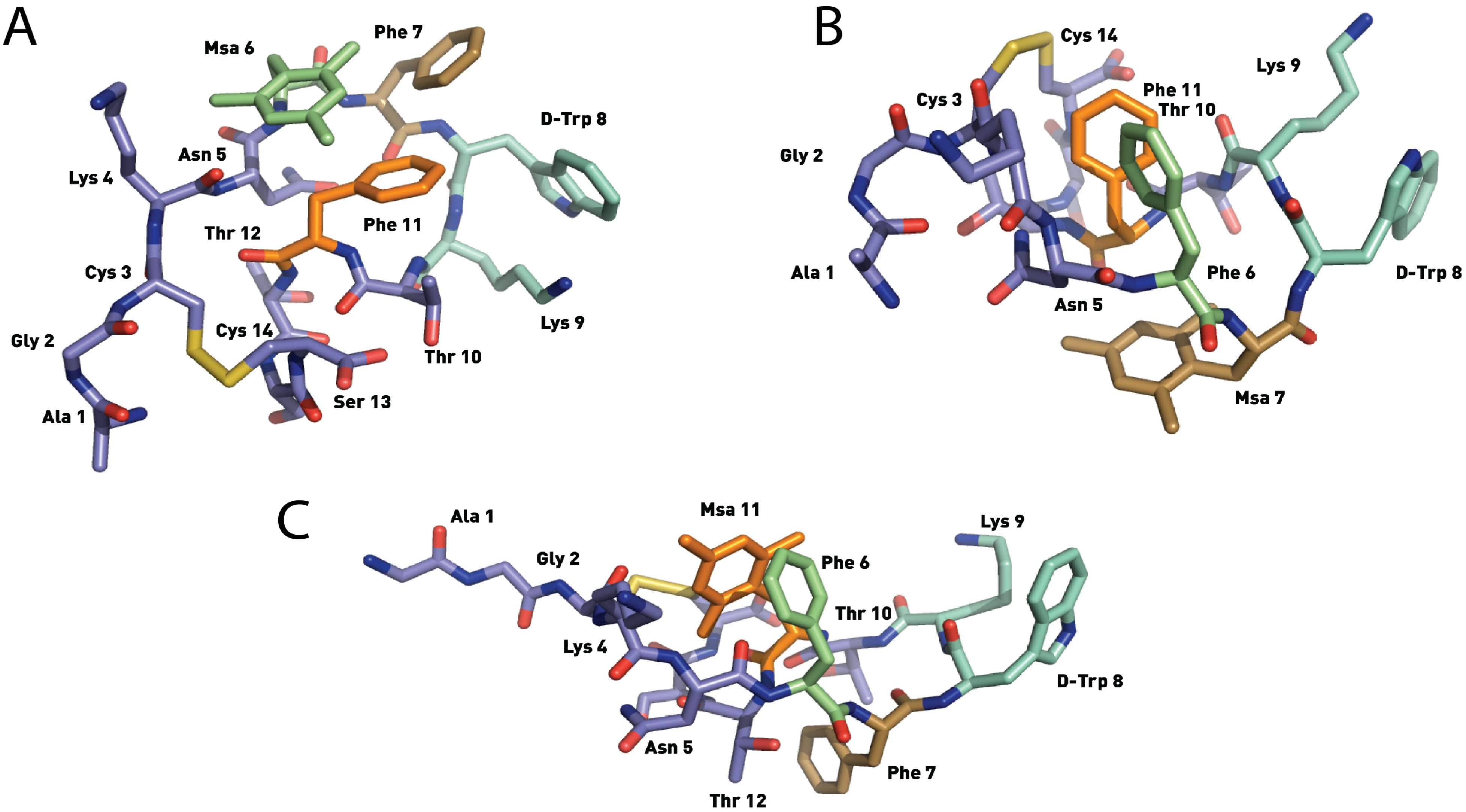
2.4.2. Compounds 4–6 with One Msa Residue and D-Trp8
2.4.3. Compounds 7–10 with Multiple Incorporations of Msa Residues
3. Experimental
3.1. General Syntheses of Peptides
3.2. NMR and Computational Methods
3.3. Preparation of Cells Stably Expressing the SRIF-14 Receptor
3.4. Receptor Ligand-Binding Assay
3.5. Serum Stability Assay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hruby, V.J. Design in topographical space of peptide and peptidomimetic ligands that affect behavior. A chemist’s glimpse at the mind-body problem. Acc. Chem. Res. 2001, 34, 389–397. [Google Scholar] [CrossRef]
- Caporale, A.; Gesiot, L.; Sturlese, M.; Wittelsberger, A.; Mammi, S.; Peggion, E. Design, conformational studies and analysis of structure-function relationships of PTH (1-11) analogues: The essential role of Val in position 2. Amino Acids 2012, 43, 207–218. [Google Scholar] [CrossRef]
- Maity, P.; König, B. Enantio- and diastereoselective syntheses of cyclic Calpha-tetrasubstituted alpha-amino acids and their use to induce stable conformations in short peptides. Biopolymers 2008, 90, 8–27. [Google Scholar] [CrossRef]
- Fiori, N.; Caporale, A.; Schievano, E.; Mammi, S.; Geyer, A.; Tremmel, P.; Wittelsberger, A.; Woznica, I.; Chorev, M.; Peggion, E. Structure-function relationship studies of PTH(1-11) analogues containing sterically hindered dipeptide mimetics. J. Pept. Sci. 2007, 13, 504–512. [Google Scholar] [CrossRef]
- Brazeau, P.; Vale, W.; Burgus, R.; Ling, N.; Butcher, M.; Rivier, J.; Guillemin, R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973, 179, 77–79. [Google Scholar]
- Burgus, R.; Ling, N.; Butcher, M.; Guillemin, R. Primary structure of somatostatin, a hypothalamic peptide that inhibits the secretion of pituitary growth hormone. Proc. Natl. Acad. Sci. USA 1973, 70, 684–688. [Google Scholar] [CrossRef]
- Hoyer, D.; Bell, G.I.; Berelowitz, M.; Epelbaum, J.; Feniuk, W.; Humphrey, P.P.A.; O’Carroll, A.-M.; Patel, Y.C.; Schonbrunn, A.; Taylor, J.E.; et al. Classification and nomenclature of somatostatin receptors. Trends Pharmacol. Sci. 1995, 16, 86–88. [Google Scholar]
- Patel, Y.C.; Srikant, C.B. Subtype selectivity of peptide analogs for all five cloned human somatostatin receptors (hsstr 1-5). Endocrinology 1994, 135, 2814–2817. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Sanyal, A.J.; Grace, N.D.; Carey, W. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007, 46, 922–938. [Google Scholar] [CrossRef]
- Ayuk, J.; Sheppard, M.C. Growth hormone and its disorders. Postgrad. Med. J. 2006, 82, 24–30. [Google Scholar] [CrossRef]
- Pawlikowski, M.; Melen-Mucha, G. Somatostatin analogs - from new molecules to new applications. Curr. Opin. Pharmacol. 2004, 4, 608–613. [Google Scholar] [CrossRef]
- De Herder, W.W.; Hofland, L.J.; van der Lely, A.J.; Lamberts, S.W.J. Somatostatin receptors in gastroentero-pancreatic neuroendocrine tumours. Endocr. Relat. Cancer 2003, 10, 451–458. [Google Scholar] [CrossRef]
- Grace, C.R.R.; Erchegyi, J.; Koerber, S.C.; Reubi, J.C.; Rivier, J.; Riek, R. Novel sst2-selective somatostatin agonists. Three-dimensional consensus structure by NMR. J. Med. Chem. 2006, 49, 4487–4496. [Google Scholar] [CrossRef]
- Di Cianni, A.; Carotenuto, A.; Brancaccio, D.; Novellino, E.; Reubi, J.C.; Beetschen, K.; Papini, A.M.; Ginanneschi, M. Novel octreotide dicarba-analogs with high affinity and different selectivity for somatostatin receptors. J. Med. Chem. 2010, 53, 6188–6197. [Google Scholar] [CrossRef]
- Chatterjee, J.; Laufer, B.; Beck, J.G.; Helyes, Z.; Pintér, E.; Szolcsányi, J.; Horvath, A.; Mandl, J.; Reubi, J.C.; Kéri, G.; et al. N -methylated sst 2 selective somatostatin cyclic peptide analogue as a potent candidate for treating neurogenic inflammation. ACS Med. Chem. Lett. 2011, 2, 509–514. [Google Scholar] [CrossRef]
- Morpurgo, M.; Monfardini, C.; Hofland, L.J.; Sergi, M.; Orsolini, P.; Dumont, J.M.; Veronese, F.M. Selective alkylation and acylation of α and ε amino groups with PEG in a somatostatin analogue: Tailored chemistry for optimized bioconjugates. Bioconjug. Chem. 2002, 13, 1238–1243. [Google Scholar] [CrossRef]
- Bauer, W.; Briner, U.; Doepfner, W.; Haller, R.; Huguenin, R.; Marbach, P.; Petcher, T.J.; Pless, J. SMS 201-995: A very potent and selective octapeptide analog of somatostatin with prolonged action. Life Sci. 1982, 31, 1133–1140. [Google Scholar] [CrossRef]
- Van der Lely, A.J.; de Herder, W.W.; Krenning, E.P.; Kwekkeboom, D.J. Octreoscan radioreceptor imaging. Endocrine 2003, 20, 307–311. [Google Scholar] [CrossRef]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef]
- Ramon, R.; Martin-Gago, P.; Verdaguer, X.; Macias, M.J.; Martin-Malpartida, P.; Fernandez-Carneado, J.; Gomez-Caminals, M.; Ponsati, B.; Lopez-Ruiz, P.; Cortes, M.A.; et al. SSTR1- and SSTR3-selective Somatostatin analogs. ChemBioChem 2011, 12, 625–632. [Google Scholar] [CrossRef]
- Martin-Gago, P.; Gomez-Caminals, M.; Ramon, R.; Verdaguer, X.; Martin-Malpartida, P.; Aragon, E.; Fernandez-Carneado, J.; Ponsati, B.; Lopez-Ruiz, P.; Cortes, M.A.; et al. Fine-tuning the pi-pi aromatic interactions in peptides: Somatostatin analogs containing mesitylalanine. Angew. Chem. Int. Ed. 2012, 51, 1820–1825. [Google Scholar] [CrossRef]
- Knappenberg, M.; Michel, A.; Scarso, A.; Brison, J.; Zanen, J.; Hallenga, K.; Deschrijver, P.; van Binst, G. The conformational properties of somatostatin. IV. The conformers contributing to the conformational equilibrium of somatostatin in aqueous solution as found by semi-empirical energy calculations and high-resolution NMR experiments. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1982, 700, 229–246. [Google Scholar] [CrossRef]
- Hallenga, K.; van Binst, G.; Scarso, A.; Michel, A.; Knappenberg, M.; Dremier, C.; Brison, J.; Dirkx, J. The conformational properties of the peptide hormone somatostatin. III. Assignment and analysis of the proton and carbon-13 high resolution NMR spectra of somatostatin in aqueous solution. FEBS Lett. 1980, 119, 47–52. [Google Scholar] [CrossRef]
- Buffington, L.A.; Garsky, V.; Rivier, J.; Gibbons, W.A. Assignments of the 270 MHz PMR spectrum of somatostatin using pH titration, synthetic analogs and double resonance difference spectroscopy. Int. J. Pept. Protein Res. 1983, 21, 231–241. [Google Scholar]
- Buffington, L.A.; Garsky, V.; Rivier, J.; Gibbons, W.A. Conformation of somatostatin using scalar coupling constants from 270 and 600 MHz simulated proton magnetic resonance spectra. Biophys. J. 1983, 41, 299–304. [Google Scholar] [CrossRef]
- Kaerner, A.; Weaver, K.H.; Rabenstein, D.L. 1H-NMR studies of the conformational properties of somatostatin, reduced somatostatin and somatostatin-glutathione mixed disulfides. Magn. Reson. Chem. 1996, 34, 587–594. [Google Scholar] [CrossRef]
- Erchegyi, J.; Cescato, R.; Grace, C.R.R.; Waser, B.; Piccand, V.; Hoyer, D.; Riek, R.; Rivier, J.E.; Reubi, J.C. Novel, potent, and radio-iodinatable somatostatin receptor 1 (sst1) selective analogs. J. Med. Chem. 2009, 52, 2733–2746. [Google Scholar] [CrossRef]
- Grace, C.R.R.; Koerber, S.C.; Erchegyi, J.; Reubi, J.C.; Rivier, J.; Riek, R. Novel sst4-selective somatostatin (SRIF) agonists in relation to three-dimensional consensus structure by NMR. J. Med. Chem. 2003, 46, 5606–5618. [Google Scholar] [CrossRef]
- Melacini, G.; Zhu, Q.; Osapay, G.; Goodman, M. A refined model for the somatostatin pharmacophore: Conformational analysis of lanthionine-sandostatin analogs. J. Med. Chem. 1997, 40, 2252–2258. [Google Scholar] [CrossRef]
- Lewis, I.; Bauer, W.; Albert, R.; Chandramouli, N.; Pless, J.; Weckbecker, G.; Bruns, C. A novel somatostatin mimic with broad somatotropin release inhibitory factor receptor binding and superior therapeutic potential. J. Med. Chem. 2003, 46, 2334–2344. [Google Scholar] [CrossRef]
- Caumes, C.; Hjelmgaard, T.; Roy, O.; Reynaud, M.; Servent, D.; Taillefumier, C.; Faure, S. Synthesis and binding affinities for sst receptors of cyclic peptoid SRIF-mimetics. Med. Chem. Commun. 2012, 3, 1531–1535. [Google Scholar] [CrossRef]
- Rivier, J.; Brown, M.; Rivier, C.; Ling, N.; Vale, W. Hypothalamic hypophysiotropic hormones. review on the design of synthetic analogs. Peptides 1976, 1, 427–449. [Google Scholar]
- Veber, D.F.; Holly, F.W.; Paleveda, W.J.; Nutt, R.F.; Bergstrand, S.J.; Torchiana, M.; Glitzer, M.S.; Saperstein, R.; Hirschmann, R. Conformationally restricted bicyclic analogs of somatostatin. Proc. Natl. Acad. Sci. USA 1978, 75, 2636–2640. [Google Scholar] [CrossRef]
- Arison, B.H.; Hirschmann, R.; Paleveda, W.J.; Brady, S.F.; Veber, D.F. On the low energy solution conformation of somatostatin. Biochem. Biophys. Res. Commun. 1981, 100, 1148–1153. [Google Scholar] [CrossRef]
- Cutnell, J.D.; la Mar, G.N.; Dallas, J.L.; Hug, P.; Ring, H.; Rist, G. High-field proton NMR studies of synthetic analogs of somatostatin. Structural features involving aromatic residues in an active eight-membered ring analog. Biochim. Protein Struct. Mol. Enzymol. 1982, 700, 59–66. [Google Scholar] [CrossRef]
- Jans, A.W.H.; Hallenga, K.; van Binst, G.; Michel, A.; Scarso, A.; Zanen, J. The conformational properties of somatostatin V. Side-chain interaction in aqueous solution as studied by one-dimensional and two-dimensional NMR methods. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1985, 827, 447–452. [Google Scholar] [CrossRef]
- Van den Berg, E.M.; Jans, A.W.; Van Binst, G. Conformational properties of somatostatin. VI. In a methanol solution. Biopolymers 1986, 25, 1895–1908. [Google Scholar] [CrossRef]
- Medina, E.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective syntheses of conformationally rigid, highly lipophilic mesityl-substituted amino acids. Helv. Chim. Acta 2000, 83, 972–988. [Google Scholar] [CrossRef]
- Rivier, J.; Brown, M.; Vale, W. D-Trp8-somatostatin: An analog of somatostatin more potent than the native molecule. Biochem. Biophys. Res. Commun. 1975, 65, 746–751. [Google Scholar] [CrossRef]
- Arison, B.H.; Hirschmann, R.; Veber, D.F. Inferences about the conformation of somatostatin at a biologic receptor based on NMR studies. Bioorg. Chem. 1978, 7, 447–451. [Google Scholar] [CrossRef]
- Ovadia, O.; Greenberg, S.; Laufer, B.; Gilon, C.; Hoffman, A.; Kessler, H. Improvement of drug-like properties of peptides: The somatostatin paradigm. Expert Opin. Drug Discov. 2010, 5, 655–671. [Google Scholar] [CrossRef]
- Ramon, R.; Alonso, M.M.; Riera, A.; Ramón, R. A unified approach to mesityl amino acids based on Sharpless dihydroxylation. Tetrahedron Asymmetry 2007, 18, 2797–2802. [Google Scholar]
- Li, T.; Tsuda, Y.; Minoura, K.; In, Y.; Ishida, T.; Lazarus, L.H.; Okada, Y. Enantioselective synthesis of a phenylalanine library containing alkyl groups on the aromatic moiety: Confirmation of stereostructure by X-ray analysis. Chem. Pharm. Bull. 2006, 54, 873–877. [Google Scholar] [CrossRef]
- Cristóbal-Lecina, E.; Etayo, P.; Doran, S.; Revés, M.; Martín-Gago, P.; Grabulosa, A.; Costantino, A.R.; Vidal-Ferran, A.; Riera, A.; Verdaguer, X. MaxPHOS Ligand: PH/NH tautomerism and rhodium-catalyzed asymmetric hydrogenations. unpublished.
- Wuthrich, K.; Wider, G.; Wagner, G.; Braun, W. Sequential resonance assignments as a basis for determination of spatial protein structures by high resolution proton nuclear magnetic resonance. J. Mol. Biol. 1982, 155, 311–319. [Google Scholar] [CrossRef]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar] [CrossRef]
- Neelamkavil, S.; Arison, B.; Birzin, E.; Feng, J.-J.; Chen, K.-H.; Lin, A.; Cheng, F.-C.; Taylor, L.; Thornton, E.R.; Smith, A.B.; et al. Replacement of Phe6, Phe7, and Phe11 of D-Trp8-somatostatin-14 with L-pyrazinylalanine. Predicted and observed effects on binding affinities at hSST2 and hSST4. An unexpected effect of the chirality of Trp8 on NMR spectra in methanol. J. Med. Chem. 2005, 48, 4025–4030. [Google Scholar] [CrossRef]
- Samanta, U.; Pal, D.; Chakrabarti, P. Packing of aromatic rings against tryptophan residues in proteins. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, D55, 1421–1427. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Samanta, U.; Chakrabarti, P. Aromatic-aromatic interactions in and around α-helices. Protein Eng. 2002, 15, 91–100. [Google Scholar] [CrossRef]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Keller, R. The Computer Aided Resonance Assignment Tutorial, 1st ed.; CANTINA Verlag: Goldau, Switzerland, 2004. [Google Scholar]
- Rens-Domiano, S.; Law, S.F.; Yamada, Y.; Seino, S.; Bell, G.I.; Reisine, T. Pharmacological properties of two cloned somatostatin receptors. Mol. Pharmacol. 1992, 42, 28–34. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Mezo, G.; Manea, M. Receptor-mediated tumor targeting based on peptide hormones. Expert Opin. Drug Deliv. 2010, 7, 79–96. [Google Scholar] [CrossRef]
- Sun, L.-C.; Coy, D.H. Somatostatin receptor-targeted anti-cancer therapy. Curr. Drug Deliv. 2011, 8, 2–10. [Google Scholar] [CrossRef]
- Sun, L.-C.; Mackey, L.V.; Luo, J.; Fuselier, J.A.; Coy, D.H. Targeted chemotherapy using a cytotoxic somatostatin conjugate to inhibit tumor growth and metastasis in nude mice. Clin. Med. Oncol. 2008, 2, 491–499. [Google Scholar]
- Barbieri, F.; Bajetto, A.; Pattarozzi, A.; Gatti, M.; Würth, R.; Thellung, S.; Corsaro, A.; Villa, V.; Nizzari, M.; Florio, T. Peptide receptor targeting in cancer: The somatostatin paradigm. Int. J. Pept. 2013. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–10 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martín-Gago, P.; Aragón, E.; Gomez-Caminals, M.; Fernández-Carneado, J.; Ramón, R.; Martin-Malpartida, P.; Verdaguer, X.; López-Ruiz, P.; Colás, B.; Cortes, M.A.; et al. Insights into Structure-Activity Relationships of Somatostatin Analogs Containing Mesitylalanine. Molecules 2013, 18, 14564-14584. https://doi.org/10.3390/molecules181214564
Martín-Gago P, Aragón E, Gomez-Caminals M, Fernández-Carneado J, Ramón R, Martin-Malpartida P, Verdaguer X, López-Ruiz P, Colás B, Cortes MA, et al. Insights into Structure-Activity Relationships of Somatostatin Analogs Containing Mesitylalanine. Molecules. 2013; 18(12):14564-14584. https://doi.org/10.3390/molecules181214564
Chicago/Turabian StyleMartín-Gago, Pablo, Eric Aragón, Marc Gomez-Caminals, Jimena Fernández-Carneado, Rosario Ramón, Pau Martin-Malpartida, Xavier Verdaguer, Pilar López-Ruiz, Begoña Colás, María Alicia Cortes, and et al. 2013. "Insights into Structure-Activity Relationships of Somatostatin Analogs Containing Mesitylalanine" Molecules 18, no. 12: 14564-14584. https://doi.org/10.3390/molecules181214564
APA StyleMartín-Gago, P., Aragón, E., Gomez-Caminals, M., Fernández-Carneado, J., Ramón, R., Martin-Malpartida, P., Verdaguer, X., López-Ruiz, P., Colás, B., Cortes, M. A., Ponsati, B., Macias, M. J., & Riera, A. (2013). Insights into Structure-Activity Relationships of Somatostatin Analogs Containing Mesitylalanine. Molecules, 18(12), 14564-14584. https://doi.org/10.3390/molecules181214564