3. Experimental
3.2. General Procedure A for the Synthesis of Boron-Containing Primary Alcohols 2a–g
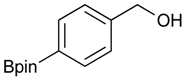
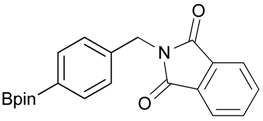
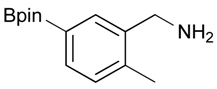
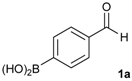
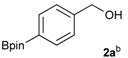
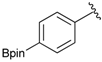
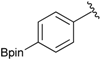
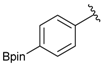
[4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2a). To a mixture of a 4-formylbenzenboronic acid (1a, 375 mg, 2.50 mmol), pinacol (355 mg, 3.00 mmol) and anhydrous magnesium sulfate (625 mg, 5.00 mmol), methanol was added (12.50 mL). The mixture was stirred at room temperature for 6 h. After the reaction was completed, the crude solution was filtered, and then sodium borohydride (47 mg, 1.25 mmol) was added to the filtrate. Afterwards, the reaction mixture was stirred for an additional 5 h. Once the reaction was completed, the reaction mixture was filtered and the filtrate was concentrated in vacuo to give the desired product 2a as a white solid (m.p. 75–77 °C) in 88% yield (513 mg); 1H-NMR (CD3OD-d4) δ ppm 7.71 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 7.8 Hz, 2H), 4.62 (s, 2H), 1.34 (s, 12H); 13C-NMR (CD3OD-d4) δ ppm 146.23, 135.93, 127.26, 85.19, 65.24, 25.34; 11B-NMR (CDCl3) δ ppm 34.82.
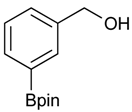
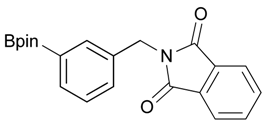
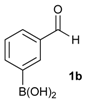
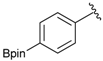
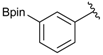
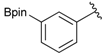
[3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2b). Following the General Procedure A, the desired compound was synthesized utilizing 3-formylbenzenboronic acid (1b, 375 mg, 2.50 mmol), pinacol (355 mg, 3.00 mmol), anhydrous magnesium sulfate (601 mg, 5.00 mmol), sodium borohydride (47 mg, 1.25 mmol), and methanol (12.50 mL) giving compound 2b as a white solid (m.p. 48–50 °C) in 86% yield (502 mg); 1H-NMR (CD3OD-d4) δ ppm 7.74 (s, 1 H), 7.64 (d, J = 7.2 Hz, 1H), 7.46 (d, J = 7.4 Hz, 1H), 7.34 (t, J = 7.4 Hz, 1H), 4.60 (s, 2H), 1.34 (s, 12H); 13C-NMR (CD3OD-d4) δ ppm 142.11, 134.76, 134.50, 131.23, 128.93, 85.23, 65.35, 25.35; 11B-NMR (CDCl3) δ ppm 30.97.
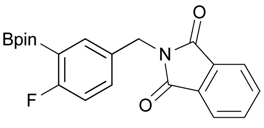
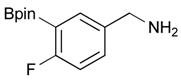
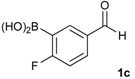
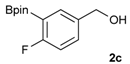
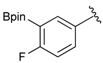
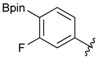
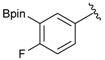
[4-Fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl]phenyl)methanol (2c). Following the General Procedure A, the desired compound was synthesized utilizing 2-fluoro-3-formylbenzenboronic acid (1c, 420 mg, 2.50 mmol), pinacol (355 mg, 3.00 mmol), anhydrous magnesium sulfate (601 mg, 5.00 mmol), sodium borohydride (47 mg, 1.25 mmol), and methanol (12.50 mL) giving compound 2c as a white solid (m.p. 80–82 °C) in 69% yield (435 mg); 1H-NMR (CD3OD-d4) δ ppm 7.68 (t, J = 3.4 Hz, 1H), 7.49–7.44 (m, 1H), 7.01 (t, J = 6.5 Hz, 1H), 4.57 (s, 2H), 1.34 (s, 12H); 13C-NMR (CD3OD-d4) δ ppm 166.42 (d, J = 249.0 Hz), 136.7, 135.1, 135.0, 131.9, 131.9, 114.7, 114.5, 114.2, 83.7, 62.9, 23.7, 23.7, 23.6; 11B-NMR (CDCl3) δ ppm 30.2.
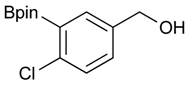
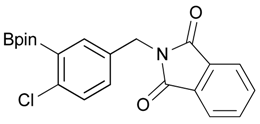
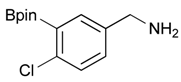
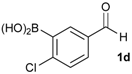
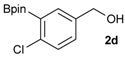
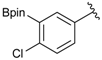
[4-Chloro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2d). Following the General Procedure A, the desired compound was synthesized utilizing 2-chloro-5-(hydroxymethyl)phenylboronic acid (1d, 242 mg, 1.30 mmol), pinacol (184 mg, 1.56 mmol), anhydrous magnesium sulfate (313 mg, 2.60 mmol), sodium borohydride (25 mg, 0.65 mmol), and methanol (6.50 mL) giving compound 2d as a oil in 90% yield (314 mg); 1H-NMR (CD3OD-d4) δ ppm 7.66 (d, J = 1.8 Hz, 1H), 7.39–7.31 (m, 2H), 4.57 (s, 2H), 1.39 (t, J = 18.1 Hz, 12H); 13C-NMR (CDCl3) δ ppm 138.4, 138.1, 134.5, 130.2, 129.2, 84.0, 63.7, 24.5, 24.3; 11B-NMR (CDCl3) δ ppm 30.6.
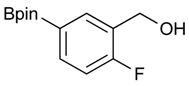
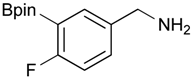
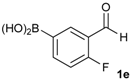
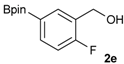
[2-Fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2e). Following the General Procedure A, the desired compound was synthesized utilizing 4-fluoro-3-formylphenylboronic acid (1e, 218 mg, 1.30 mmol), pinacol (184 mg, 1.56 mmol), anhydrous magnesium sulfate (313 mg, 2.60 mmol), sodium borohydride (25 mg, 0.65 mmol), and methanol (6.50 mL) giving compound 2e as a white solid (m.p. 44–47 °C) in 95% yield (312 mg); 1H-NMR (CD3OD-d4) δ ppm 7.87 (d, J = 8.1 Hz, 1H), 7.71–7.09 (m, 1H), 7.05 (t, J = 10.4 Hz, 1H), 4.67 (s, 2H), 1.34 (s, 12H); 13C-NMR (CD3OD-d4) δ ppm 164.4(d, J = 250.5 Hz), 137.6, 137.2, 137.1, 129.2, 129.1, 115.8, 115.7, 85.4, 58.9, 25.3, 25.2; 11B-NMR (CDCl3) δ ppm 30.6.
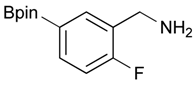
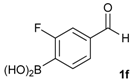
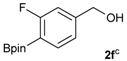
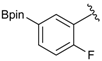
[3-Fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2f). Following the General Procedure A, the desired compound was synthesized utilizing 2-fluoro-4-formylphenylboronic acid (1f, 218 mg, 1.30 mmol), pinacol (184 mg, 1.56 mmol), anhydrous magnesium sulfate (313 mg, 2.60 mmol), sodium borohydride (25 mg, 0.65 mmol), and methanol (6.50 mL) giving compound 2f as a white solid (m.p. 35–38 °C) in 94% yield (308 mg); 1H-NMR (CDCl3) δ ppm 7.60 (t, J = 6.8 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.93 (d, J = 10.4 Hz, 1H), 4.53 (s, 2H), 1.29 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.4 (d, J = 250.5 Hz), 147.2, 136.9, 136.8, 121.5, 113.3, 113.1, 83.9, 64.2, 30.3, 24.8; 11B-NMR (CDCl3) δ ppm 29.9.
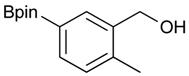
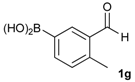
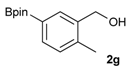
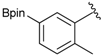
[2-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanol (2g). Following the General Procedure A, the desired compound was synthesized utilizing 3-formylbenzenboronic acid (1g, 213 mg, 1.30 mmol), pinacol (184 mg, 1.56 mmol), anhydrous magnesium sulfate (313 mg, 2.60 mmol), sodium borohydride (25 mg, 0.65 mmol), and methanol (6.50 mL) giving compound 2g as a white solid (m.p. 60–63 °C) in 97% yield (312 mg); 1H-NMR (CD3OD-d4) δ ppm 7.74 (s, 1H), 7.55 (d, J = 7.4 Hz, 1H), 7.16 (d, J = 7.4 Hz, 1H), 4.62 (s, 2H), 2.36 (s, 3H), 1.34 (s, 12H); 13C-NMR (CD3OD-d4) δ ppm 141.1, 139.8, 135.4, 135.1, 130.8, 85.1, 63.5, 25.3, 25.2, 19.1; 11B-NMR (CDCl3) δ ppm 30.9.
3.3. General Procedure B for the Synthesis of Boron-Containing Primary Phthalimides 3a–g
2-[4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3a). Compound 2a (585 mg, 2.50 mmol), and dichloromethane (25.00 mL) were added to a dry flask containing a magnetic stir bar under a nitrogen atmosphere. The flask was cooled to 0 °C, then methanesulfonyl chloroide (0.29 mL, 3.75 mmol) and N,N-diisopropylethylamine (DIPEA, 0.87 mL, 5.00 mmol) were slowly added to the flask. The reaction mixture was stirred at 0 °C for 3 h. After the reaction was completed, the reaction mixture was diluted with dichloromethane (25.00 mL) before H2O (25.00 mL) was added. The organic layer was then washed with brine and dried with MgSO4. The resulting organic layer was then filtered and the filtrate was concentrated in vacuo. The resulting crude material was re-dissolved in DMF (4.68 mL) after which both potassium phthalimide salt (695 mg, 3.75 mmol), and K2CO3 (1,036 mg, 7.50 mmol) were added to the solution. The reaction was then allowed to stir at room temperature for 3 days. After the reaction was completed, the distilled H2O (20.00 mL) was slowly added to the reaction mixture to afford the formation of a solid precipitate. The reaction mixture was then filtered and the filtered cake was collected. The filtered cake was re-dissolved in tert-butanol/H2O (4:1, v/v) (10.00 mL) before a freeze-drying process was applied to remove the remaining DMF. The desired product 3a was obtained as a white solid (m.p. 166–169 °C) in 96% yield (872 mg); 1H-NMR (CDCl3) δ ppm 7.84–7.82 (m, 2 H), 7.74 (d, J = 7.85 Hz, 2 H), 7.71–7.68 (m, 2H), 7.42 (d, J = 7.85 Hz, 2H), 4.85 (s, 2H), 1.34 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.90, 139.26, 135.12, 133.93, 132.04, 127.76, 123.29, 83.73, 41.60, 24.77; 11B-NMR (CDCl3) δ ppm 30.94.
2-[3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3b). Following the General Procedure B, the desired compound was synthesized utilizing 2b (767 mg, 3.28 mmol), methanesulfonyl chloride (0.38 mL, 4.92 mmol), DIPEA (1.14 mL, 6.56 mmol), potassium phthalimide salt (911 mg, 4.92 mmol), and potassium carbonate (1,360 mg, 9.84 mmol) giving compound 3b as a white solid (m.p. 170–173 °C) in 80% yield (953 mg); 1H-NMR (CDCl3) δ ppm 7.76–7.83 (m, 3H), 7.72–7.68 (m, 3H), 7.51 (d, J = 7.70 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 4.85 (s, 2H), 1.33 (s, 12H); 13C-NMR (CDCl3) δ ppm 135.60, 134.78, 134.25, 133.91, 132.13, 131.36, 128.05, 123.31, 83.82, 41.59, 24.84; 11B-NMR (CDCl3) δ ppm 30.88.
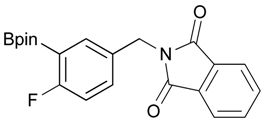
2-[4-Fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3c). Following the General Procedure B, the desired compound was synthesized utilizing 2c (378 mg, 1.50 mmol), methanesulfonyl chloroide (0.17 mL, 2.25 mmol), DIPEA (0.52 mL, 3.00 mmol), potassium phthalimide salt (417 mg, 2.25 mmol), and potassium carbonate (622 mg, 4.5 mmol) giving compound 3c as a white solid (m.p. 55–58 °C) in 75% yield (429 mg); 1H-NMR (CDCl3) δ ppm 7.84–7.78 (m, 3H), 7.78–7.67 (m, 2H), 7.52–7.7.47 (m, 1H), 6.95 (t, J = 8.8 Hz, 1H), 4.80 (s, 2H), 1.34 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.93, 137.12, 137.07, 133.96, 133.73, 133,67, 132.03, 131.63, 123.47, 123.32, 115.61, 115.45, 83.95, 40.80, 24.75; 11B-NMR (CDCl3) δ ppm 30.19.
2-[4-Chloro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3d). Following the General Procedure B, the desired compound was synthesized utilizing 2d (348 mg, 1.30 mmol), methanesulfonyl chloroide (0.15 mL, 1.95 mmol), DIPEA (0.45 mL, 2.60 mmol), potassium phthalimide salt (360 mg, 1.95 mmol), and potassium carbonate (539 mg, 3.90 mmol). Compound 3d was obtained as a white solid (m.p. 102–104 °C) in 85% yield (439 mg); 1H-NMR (CDCl3) δ ppm 7.78–7.74 (m, 2H), 7.71 (d, J = 2.0 Hz, 1H), 7.66–7.62 (m, 2H), 7.35 (q, J1 = 6.1 Hz, J2 = 2.1 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 4.75 (s, 2H), 1.32 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.8, 139.0, 136.5, 134.1, 134.0, 132.1, 131.9, 129.6, 123.3, 84.2, 40.9, 24.8; 11B-NMR (CDCl3) δ ppm 30.6.
2-[2-Fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3e). Following the General Procedure B, the desired compound was synthesized utilizing 2e (272 mg, 1.08 mmol), methanesulfonyl chloroide (0.13 mL, 1.62 mmol), DIPEA (0.38 mL, 2.16 mmol), potassium phthalimide salt(300 mg, 1.62 mmol), and potassium carbonate (447 mg, 3.24 mmol) giving 3e as a white solid (m.p. 139–142 °C) in 53% yield (218 mg); 1H-NMR (CDCl3) δ ppm 7.85–7.79 (m, 3H), 7.73–768 (m, 3H), 7.02 (q, J1 = 1.8 Hz, J2 = 8.2 Hz, 1H), 4.91 (s, 2H), 1.32 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.8, 163.0 (d, J = 252.0 Hz), 137.2, 136.7, 136.6, 133.9, 132.1, 123.4, 122.5, 122.4, 115.1, 114.9, 83.9, 35.6, 35.5, 24.8; 11B-NMR (CDCl3) δ ppm 30.6.
2-[3-Fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3f). Following the General Procedure B, the desired compound was synthesized utilizing 2f (293 mg, 1.16 mmol), methanesulfonyl chloroide (0.14 mL, 1.74 mmol), DIPEA (0.39 mL, 2.32 mmol), potassium phthalimide salt (322 mg, 1.74 mmol), and potassium carbonate (480 mg, 3.48 mmol) giving compound 3f as a white solid (m.p. 101–103 °C) in 97% yield (429 mg); 1H-NMR (CDCl3) δ ppm 7.85–7.81 (m, 2H), 7.73–7.66 (m, 3H), 7.17 (d, J = 7.7 Hz, 1H), 7.06 (d, J = 9.9 Hz, 1H), 4.82 (s, 2H), 1.32 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.7, 167.2 (d, J = 252.0 Hz), 142.0, 141.9, 137.2, 137.2, 134.1, 131.9, 123.5, 123.4, 115.2, 114.9, 83.8, 40.9, 24.7; 11B-NMR (CDCl3) δ ppm 30.06.
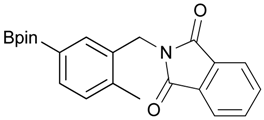
2-[2-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]isoindoline-1,3-dione (3g). Following the General Procedure B, the desired compound was synthesized utilizing 2g (598 mg, 2.41 mmol), methanesulfonyl chloroide (0.28 mL, 3.62 mmol), DIPEA (0.83 mL, 4.83 mmol), potassium phthalimide salt (670 mg, 3.62 mmol), and potassium carbonate (1,000 mg, 7.24 mmol) giving compound 3g as a white solid (m.p. 168–171 °C) in 48% yield (438 mg); 1H-NMR (CDCl3) δ ppm 7.81–7.76 (m, 3H), 7.68–7.61 (m, 3H), 7.16 (d, J = 7.4 Hz, 1H), 4.86 (s, 2H), 2.49 (s, 3H), 1.29 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.8, 139.5, 134.9, 134.2, 133.7, 133.4, 131.8, 129.7, 126.5, 122.9, 83.4, 39.1, 24.6, 19.6; 11B-NMR (CDCl3) δ ppm 31.1.
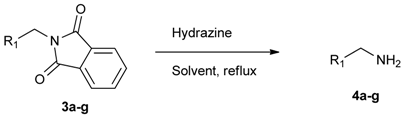
3.4. General Procedure C for the Synthesis of Boron-Containing Primary Amines 4a–g
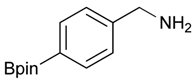
[4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4a). Compound 3a (196 mg, 0.54 mmol) was added to a dry flask containing magnetic stir and dissolved in THF (5.50 mL). Afterwards, hydrazine hydrate (0.08 mL, 1.62 mmol) was added to the reaction mixture. The reaction was stirred under a reflux for 12 h. The resulting dried crude material was then re-suspended with chloroform (50.00 mL), filtered, and the filtrate was concentrated in vacuo. The crude material was purified by High Performance Liquid Chromatography (HPLC) to give the desired product 4a as a white solid (m.p. 85 °C) in 87% yield (111 mg); 1H-NMR (CDCl3) δ ppm 7.78 (d, J = 7.9 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 3.88 (s, 2H), 1.34 (s, 12H); 13C-NMR (CDCl3) δ ppm 136.72, 135.56, 127.18, 84.11, 43.94, 25.02; 11B-NMR (CD3OD-d4) δ ppm 30.90; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C13H20BNO2, 234.1669; found, 234.1651.
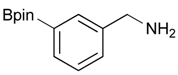
[3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4b). Following the General Procedure C, the desired compound was synthesized utilizing 3b (250 mg, 0.69 mmol), THF (7.00 mL), and hydrazine hydrate (0.10 mL, 2.07 mmol) giving compound 4b as a white solid (m.p. 91 °C) in 82% yield (131 mg); 1H-NMR (CDCl3) δ ppm 7.74–7.68 (m, 2H), 7.42–7.32 (m, 2H), 3.86 (s, 2H), 1.34 (s, 12H); 13C-NMR (CDCl3) δ ppm 142.78, 133.58, 133.48, 130.30, 128.23, 84.03, 46.69, 25.07; 11B-NMR (CD3OD-d4) δ ppm 30.94; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C13H20BNO2, 234.1669; found, 234.1644.
[4-Fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4c). Following the General Procedure C, the desired compound was synthesized utilizing 3c (953 mg, 2.50 mmol), THF (25.00 mL), and hydrazine hydrate(0.36 mL, 7.50 mmol) giving compound 4c as a white solid (m.p. 62 °C) with 98% yield (616 mg); 1H-NMR (CDCl3) δ ppm 7.66–7.63 (m, 1H), 7.40–7.34 (m, 1H), 6.99 (t, J = 5.9 Hz, 1H), 3.83 (s, 2H), 1.34 (s, 12H); 13C-NMR (CDCl3) δ ppm 166.44 (d, J = 249.9 Hz), 138.64, 135.58, 135.47, 132.37, 132.25, 116.48, 116.31, 84.13, 45.92, 25.03; 11B-NMR (CD3OD-d4) δ ppm 30.12; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C13H19BFNO2, 252.1574; found, 252.1548.
[4-Chloro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4d). Following the General Procedure C, the desired compound was synthesized utilizing 3d (524 mg, 1.32 mmol), THF (13.00 mL), and hydrazine hydrate (0.20 mL, 3.96 mmol) giving compound 4d as a white solid (m.p. 56 °C) in 36% yield (128 mg); 1H-NMR (CDCl3) δ ppm 7.59 (s, 1H), 7.31–7.26 (m, 2H), 4.19 (s, 2H), 1.27 (s, 12H); 13C-NMR (CDCl3) δ ppm 135.26, 134.97, 131.96, 130.91, 130.59, 84.37, 45.76, 24.95; 11B-NMR (CD3OD-d4) δ ppm 30.45; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C13H19BClNO2, 268.1279; found, 268.1153.
[2-Fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4e). Following the General Procedure C, the desired compound was synthesized utilizing 3e (679 mg, 1.78 mmol), THF (18.00 mL), and hydrazine hydrate (0.26 mL, 5.34 mmol) giving compound 4e as a white solid (m.p. 78 °C) in 91% yield (405 mg); 1H-NMR (CDCl3) δ ppm 7.76–7.66 (m, 2H), 7.02 (t, 1H), 3.91 (s, 2H), 1.33 (s, 12H); 13C-NMR (CDCl3) δ ppm 163.27 (d, J = 126.8 Hz), 138.35, 136.29, 136.24, 124.02, 123.93, 83.96, 36.59, 24.88; 11B-NMR (CD3OD-d4) δ ppm 30.05; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C13H19BFNO2, 252.1574; found, 252.1549.
[3-Fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4f). Following the General Procedure C, the desired compound was synthesized utilizing 3f (907 mg, 2.38 mmol), THF (24.00 mL), and hydrazine hydrate (0.34 mL, 7.15 mmol) giving compound 4f as a white solid (m.p. 52 °C) in 66% yield (396 mg); 1H-NMR (CDCl3) δ ppm 7.70 (t, J = 6.5 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.04 (d, J = 9.7 Hz, 1H), 3.93 (s, 2H), 1.30 (s, 12H); 13C-NMR (CDCl3) δ ppm 167.24 (d, J = 252.2 Hz), 137.98, 137.87, 124.21, 116.05, 115.72, 84.37, 43.20, 25.02, 24.93; 11B-NMR (CDCl3-d3) δ ppm 30.16; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C13H19BFNO2, 252.1574; found, 252.1549.
[2-Methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanamine (4g). Following the General Procedure C, the desired compound was synthesized utilizing 3g (690 mg, 1.83 mmol), THF (18.00 mL), and hydrazine hydrate (0.26 mL, 5.49 mmol) giving compound 4g as a white solid (m.p. 59 °C) in 58% yield (262 mg); 1H-NMR (CDCl3) δ ppm 7.62–7.60 (m, 2H), 7.17 (d, J = 6.7 Hz, 1H), 3.75 (s, 2H), 2.38 (s, 3 H), 1.34 (s, 12H); 13C-NMR (CDCl3) δ ppm 140.44, 136.90, 134.49, 134.32, 130.10, 83.71, 42.04, 24.91, 19.68; 11B-NMR (CDCl3) δ ppm 31.01; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C14H22BNO2, 248.1489; found, 248.9602.
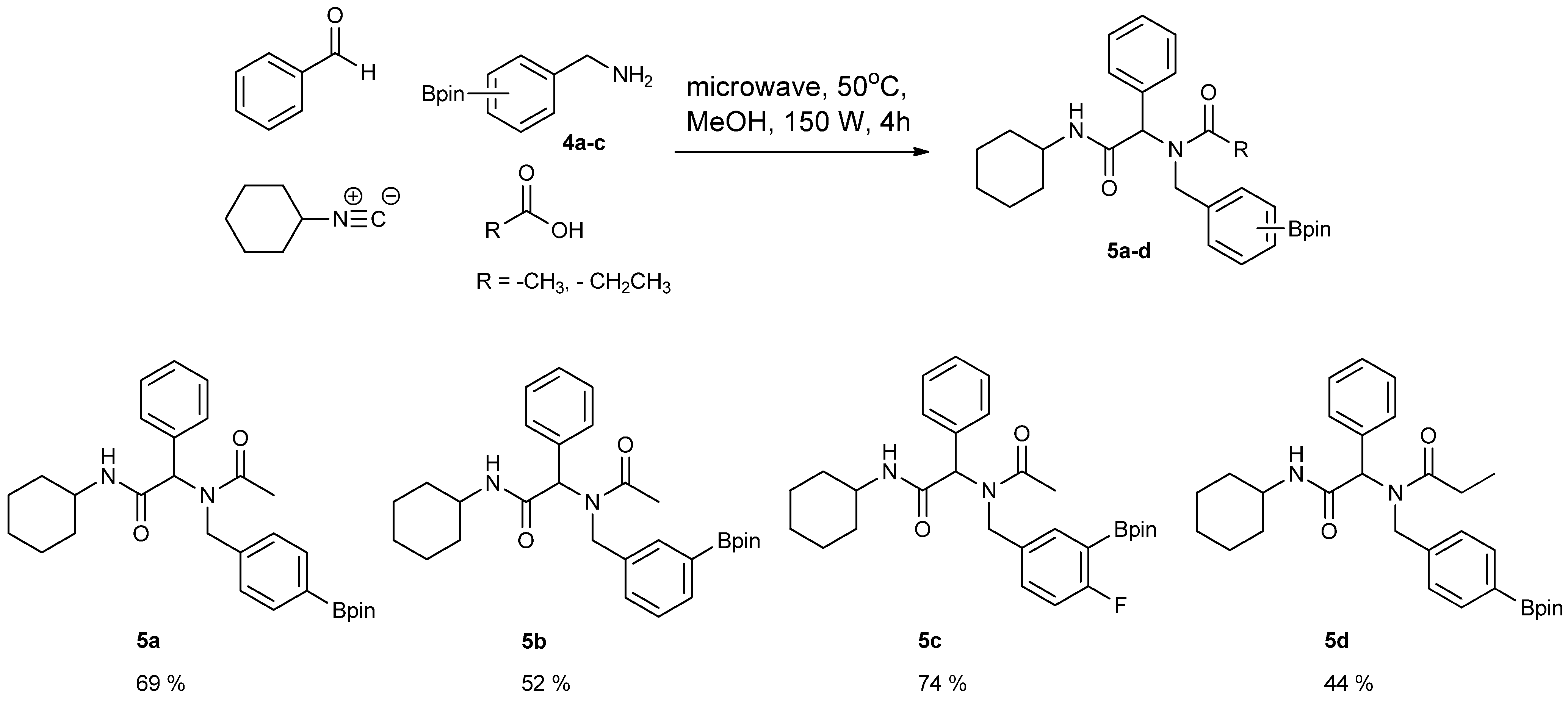
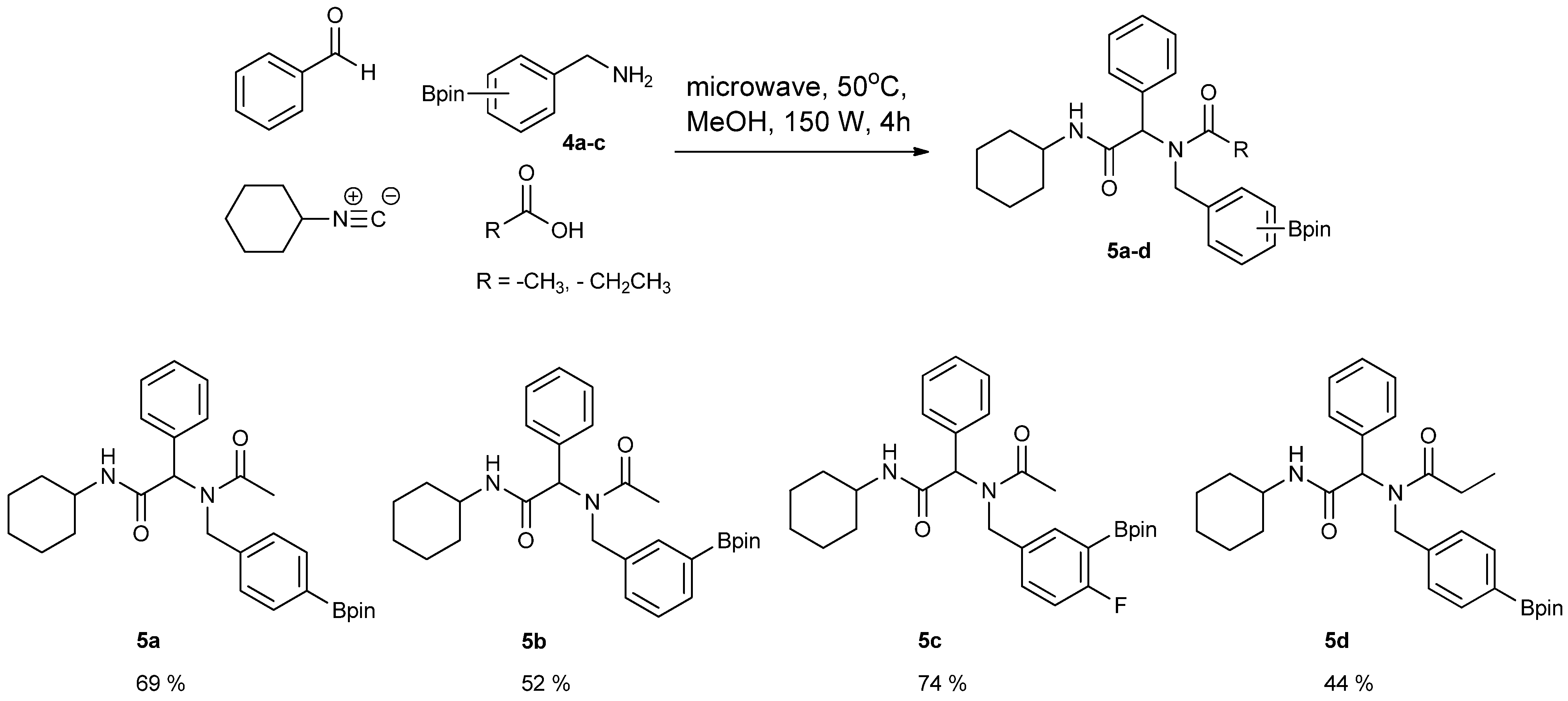
3.5. General Procedure D for the Synthesis of Ugi-4CR Boronate esters 5a–c
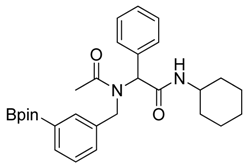
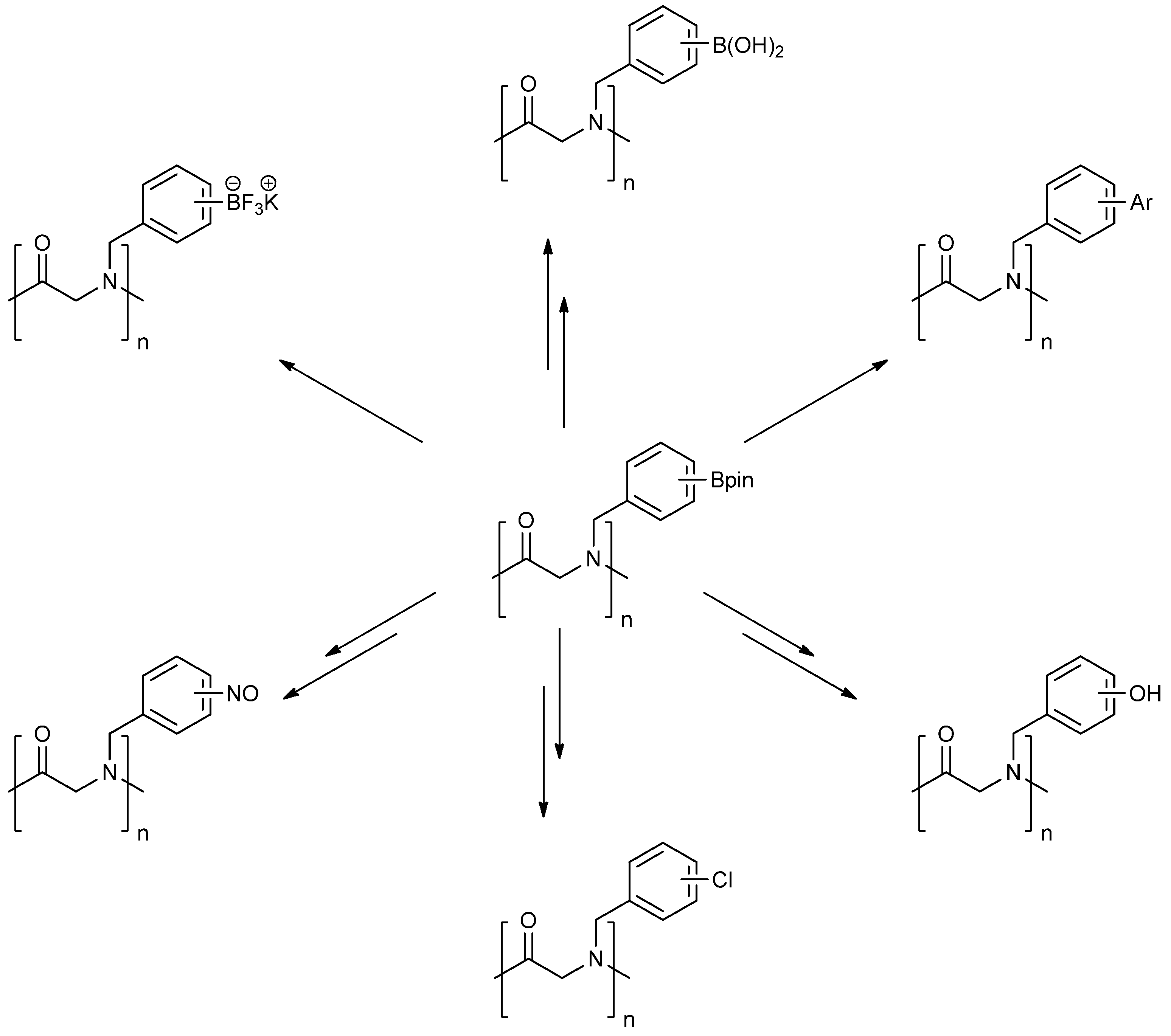
N-Cyclohexyl-2-phenyl-2-{N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl] acetamido}acetamide (5a). A 10 mL glass tube containing 4a (396 mg, 1.70 mmol), benzylaldehyde (0.17 mL, 1.70 mmol) and MeOH (3.40 mL) was first microwave irradiated for 120 min (50 °C, 150 W) under medium speed magnetic stirring. Acetic acid (0.12 mL, 2.04 mmol) and cyclohexyl isocyanide (0.21 mL, 1.70 mmol) were then added to the reaction mixture. Additional microwave irradiation was applied for 120 min (50 °C, 150 W) under moderate magnetic stirring. After the reaction was completed, MeOH was removed in vacuo. The crude material was re-dissolved in ethyl acetate and the resulting organic solution was washed with 1 M aqueous HCl solution. This was followed by adding a saturated aqueous solution of K2CO3 combined with brine. The resulting organic layer was collected, dried by MgSO4, and then concentrated in vacuo. Afterwards, the crude material was purified by flash column chromatography on silica gel using n-hexane/ethyl acetate = 1:1 as the eluent to afford the desired product 5a as a white solid (m.p. 100 °C) in 69% yield (575 mg); 1H-NMR (CDCl3) δ ppm 7.59 (d, J = 7.7 Hz, 2H), 7.44–7.18 (m, 5H), 7.10–6.90 (m, 2H), 6.08 (s, 0.55H), 5.79 (b, 0.45H), 4.66 (q, J1 = 41.9, J2 = 18.1, 2H), 3.77 (br, 1H), 2.00–1.03 (m, 22H); 13C-NMR (CDCl3) δ ppm 172.9, 168.8, 141.1, 135.4, 135.0, 129.8, 128.9, 128.7, 128.6, 127.5, 125.5, 83.9, 62.3, 50.8, 48.7, 32.9, 26.1, 25.6, 25.0, 24.9, 24.9, 22.6; 11B-NMR (CD3OD-d4) δ ppm 31.2; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C29H39BN2O4, 491.3079; found, 491.3067.
N-Cyclohexyl-2-phenyl-2-{N-[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]acetamido}acetamide (5b). Following the General Procedure D, the desired compound was synthesized utilizing 4b (394 mg, 1.69 mmol), benzylaldehyde (0.17 mL, 1.69 mmol), cyclohexyl isocyanide (0.21 mL, 1.69 mmol), acetic acid (0.12 mL, 2.03 mmol), and MeOH (3.40 mL) giving compound 5b as a white solid (m.p. 91 °C) in 52% yield (430 mg); 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CDCl3) δ ppm 7.57 (d, J = 7.2 Hz, 1H), 7.33–7.09 (m, 8 H), 5.92 (s, 1H), 5.67 (br, 0.45H), 4.65 (q, J1 = 46.5, J2 = 17.4, 2H), 3.75 (br, 1H), 2.10 (s, 3H), 1.64–1.05 (m, 22H); 13C-NMR (Bruker AC-600 FT-NMR spectrometer at 150.9 MHz, CDCl3) δ ppm 22.3, 24.8, 25.2, 32.6, 48.7, 50.9, 63.0, 83.8, 127.8, 128.7, 128.9, 129.0, 129.5, 132.4, 133.4, 134.8, 136.3, 168.7, 173.3; 11B-NMR (CD3OD-d4) δ ppm 31.08; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C29H39BN2O4, 491.3079; found, 491.3057.
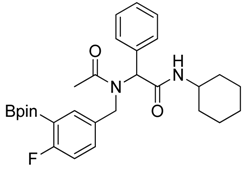
N-Cyclohexyl-2-{N-[4-fluoro-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]acetamido}-2-phenylacetamide (5c). Following the General Procedure D, the desired compound was synthesized utilizing 4c (733 mg, 2.92 mmol), benzylaldehyde (0.30 mL, 2.92 mmol), cyclohexyl isocyanide (0.36 mL, 2.92 mmol), acetic acid (0.20 mL, 3.50 mmol), and MeOH (4.00 mL) giving 5c a white solid (m.p. 110 °C) in 74% yield (1,100 mg); 1H-NMR (CDCl3) δ ppm 7.32–7.06 (m, 7H), 6.81(br, 1H), 6.00 (s, 0.50H), 5.66 (br, 0.50H), 4.62 (q, J1 = 44.0, J2 = 17.3, 2H), 3.76 (m, 1H), 2.06 (s, 3H), 1.85–1.06 (m, 22H); 13C-NMR (CDCl3) δ ppm 172.62, 168.62, 166.97, 165.31, 135.11, 134.32, 134.28, 132.68, 130.92, 130.87, 129.73, 128.76, 128.55, 115.15, 83.91, 62.39, 49.99, 48.61, 31.91, 29.34, 24.82, 24.58, 22.47; 11B-NMR (CDCl3) δ ppm 29.94; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C30H40BFN2O4, 509.2985; found, 509.3139.
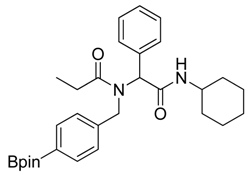
N-Cyclohexyl-2-phenyl-2-{N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]acetamido}propionamide (5d). Following the General Procedure D, the desired compound was synthesized utilizing 4a (487 mg, 2.09 mmol), benzylaldehyde (0.21 mL, 2.09 mmol), cyclohexyl isocyanide (0.26 mL, 2.09 mmol), propionic acid (0.19 mL, 2.50 mmol), and MeOH (4.00 mL) giving 5d as a white solid (m.p. 102 °C) in 44% yield (464 mg); 1H-NMR (CDCl3) δ ppm 7.59 (d, J = 7.3 Hz, 2H), 7.34–7.23 (m, 5H), 6.93 (d, J = 7.2, 2H), 6.09 (s, 0.48H), 5.66 (b, 0.52H), 4.66 (q, J1 = 46.4, J2 = 18.1, 2H), 3.78 (br, 1H), 2.37–1.04 (m, 27H); 13C-NMR (CDCl3) δ ppm 175.9, 168.8, 141.1, 135.2, 134.7, 129.7, 128.7, 128.5, 125.2, 83.7, 62.4, 49.9, 48.5, 32.7, 26.2, 25.4, 24.8, 24.7, 9.2; 11B-NMR (CDCl3) δ ppm 30.6; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C30H41BN2O4, 505.3157; found, 505.3219.
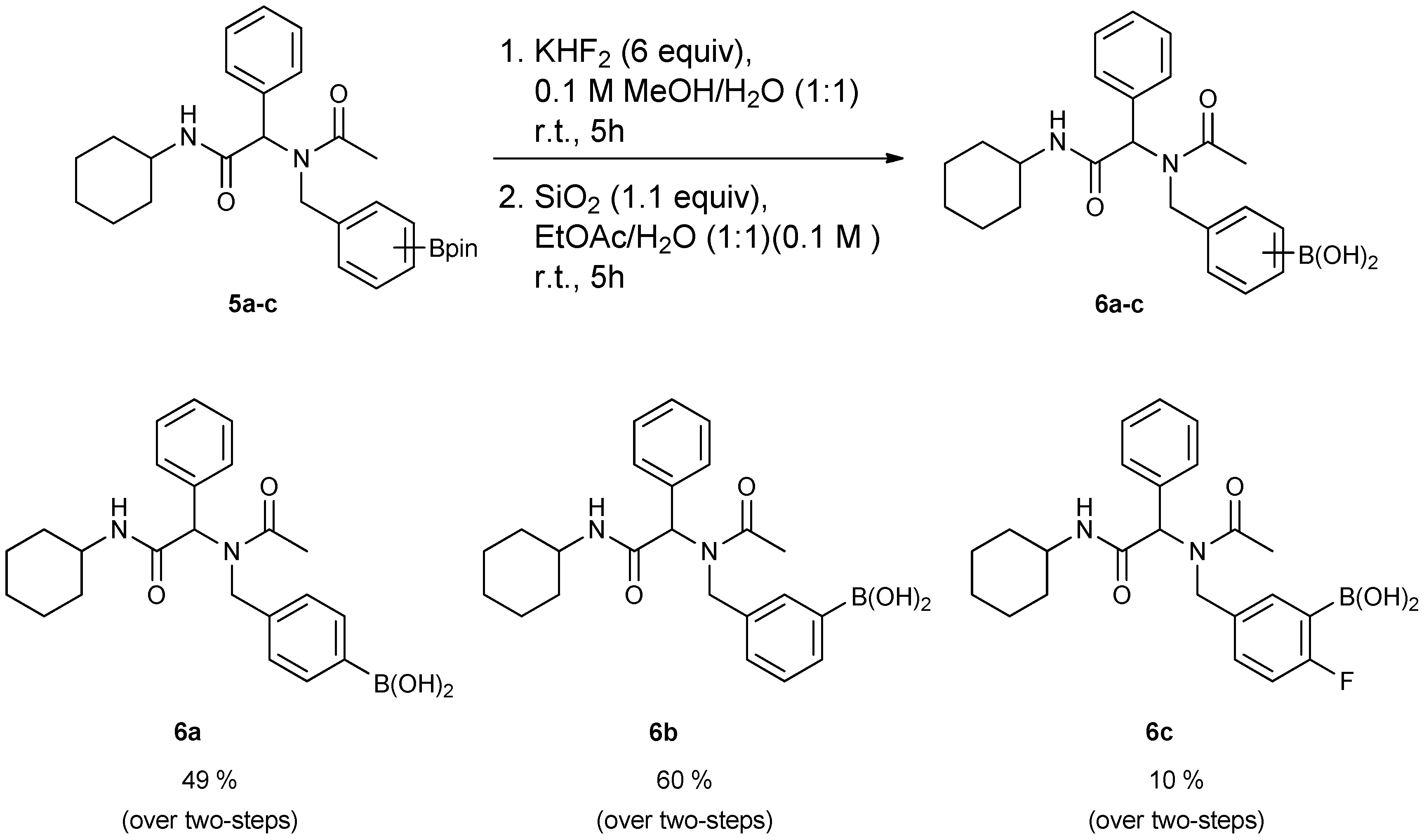
3.6. General Procedure E for the Synthesis of Ugi-4CR Boronic Acid Derivatives 6a–c
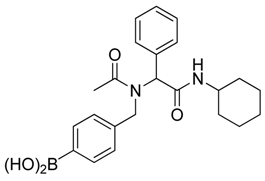
[4-({N-[2-(Cyclohexylamino)-2-oxo-1-phenylethyl]acetamido}methyl)phenyl]boronic acid (6a). Compound 5a (917 mg, 1.87 mmol) and KHF2 (876 mg, 11.22 mmol) were added to a flask containing magnetic stir, which was then suspended in MeOH/H2O (1:1, v/v, 9.35 mL). The reaction mixture was stirred vigorously at room temperature for 6 hours, and the solvents were removed in vacuo. The resulting mixture of solids were dried under a dry-freezing vacuum overnight and then subjected to extraction with hot acetone (100.00 mL). The resulting acetone extracts were concentrated in vacuo. SiO2 (123 mg, 2.05 mmol) was added to this crude material which was re-suspended in H2O/ethyl acetate (1:1, v/v) (5.5 mL). The reaction was stirred at room temperature until 11B-NMR indicated completion of the reaction (5 h). The reaction mixture was filtered to remove SiO2, and the filter cake was thoroughly rinsed with ethyl acetate. The aqueous layer was separated from the organic layer, and the aqueous layer was extracted with ethyl acetate (2 × 15 mL). The organic layer was collected and dried with MgSO4. This organic solution was then filtered, and concentrated in vacuo to afford the desired pure product 6a as a white solid (m.p. 177 °C) in 49% yield (374 mg); 1H-NMR (CD3OD-d4) δ ppm 7.56–7.20 (m, 6H), 6.94–6.89 (m, 3H), 6.09 (s, 0.81H), 5.67 (br, 0.23H), 4.64 (q, J1 = 88.5, J2 = 19.4, 2H), 3.69–3.54 (br, 1H), 2.08 (s., 3H), 1.96–1.06 (m, 10H); 13C-NMR (CD3OD-d4) δ ppm 175.4, 171.6, 136.4, 135.1, 134.7, 131.2, 130.2, 129.8, 129.7, 127.6, 126.3, 63.9, 51.3, 50.2, 33.6, 33.6, 26.7, 26.2, 26.2, 22.7; 11B-NMR (CD3OD-d4) δ ppm 28.09; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C23H29BN2O4, 409.2297; found, 409.2310.
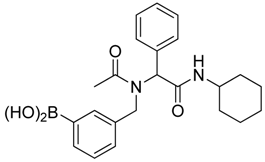
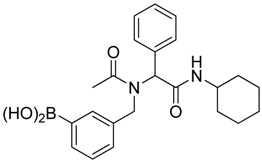
[3-({N-[2-(Cyclohexylamino)-2-oxo-1-phenylethyl]acetamido}methyl)phenyl]boronic acid (6b). Following the General Procedure E, the desired compound was synthesized utilizing 5b (431 mg, 0.88 mmol), KHF2 (412 mg, 5.28 mmol), SiO2 (58 mg, 0.96 mmol), and MeOH/H2O (1:1, v/v, 4.5 mL) giving 6b as a white solid (m.p. 150 °C) in 60% yield (215 mg); 1H-NMR (CD3OD-d4) δ ppm 7.51–6.98 (m, 9H), 6.10 (s, 0.81H), 5.66 (br, 0.19H), 4.69 (q, J1 = 70.7, J2 = 18.1, 2H), 3.66 (br, 1H), 2.09 (s, 3H), 1.82–1.03 (m, 10H); 13C-NMR (CD3OD-d4) δ ppm 175.25, 171.43, 170.26, 138.30, 138.05, 136.65, 136.35, 133.93, 133.37, 133.08, 132.87, 132.46, 131.90, 131.01, 130.36, 130.08, 129.95, 129.65, 129.48, 128.85, 128.61, 128.46, 128.31, 66.41, 63.76, 51.21, 50.11, 33.46, 26.60, 26.09, 26.04, 22.56; 11B-NMR (CD3OD-d4) δ ppm 28.58; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C23H29BN2O4, 409.2318; found, 409.2297.
[5-({N-[2-(Cyclohexylamino)-2-oxo-1-phenylethyl]acetamido}methyl)-2-fluorophenyl]boronic Acid (6c). Following the General Procedure E, the desired compound was synthesized utilizing 5c (46 mg, 0.09 mmol), KHF2 (42 mg; 0.54 mmol), SiO2 (6 mg; 0.10 mmol), and MeOH/H2O (1:1, v/v, 2.0 mL) giving compound 6c as a white solid (m.p. 61 °C) with 10% yield (4 mg); 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CD3OD-d4) δ ppm 7.27–7.20 (m, 5H), 6.97–6.82 (m, 3H), 6.10 (s, 0.72H), 5.66 (br, 0.28H), 4.65 (q, J1 = 127.8, J2 = 17.4, 2H), 3.65 (br, 1H), 2.19 (s, 0.89H), 2.08 (s, 2.11H), 1.85–1.59 (m, 5H), 1.34–1.14 (m, 5H); 13C-NMR (CD3OD-d4) δ ppm 175.21, 174.62, 171.36, 170.35, 136.51, 136.34, 135.62, 134.81, 134.08, 132.57, 131.10, 129.68, 115.57, 115.41, 114.83, 66.27, 63.48, 50.51, 50.02, 33.51, 33.46, 26.60, 26.10, 26.05, 22.49; 11B-NMR (CD3OD-d4) δ ppm 28.46; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C23H28BFN2O4, 427.2202; found, 427.2204
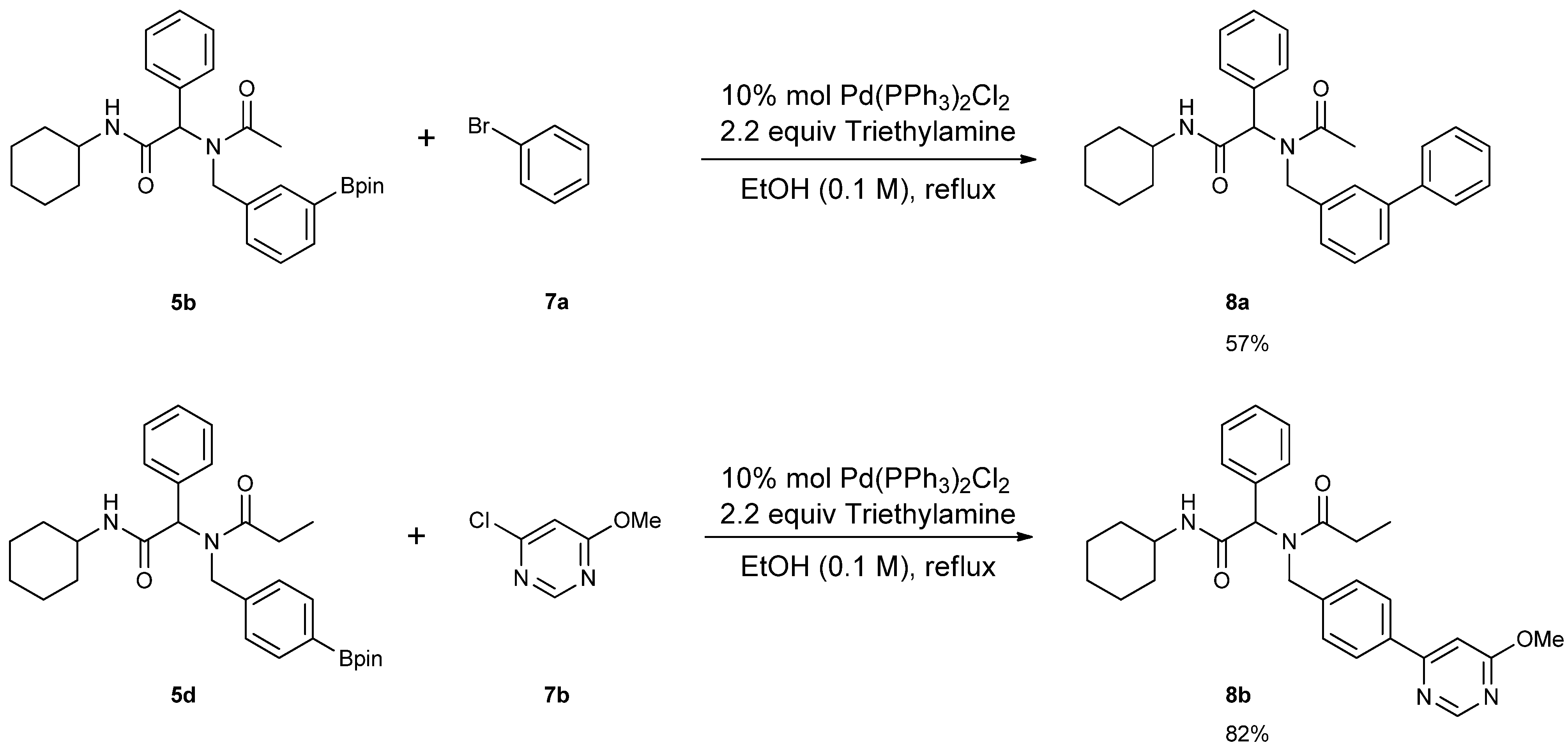
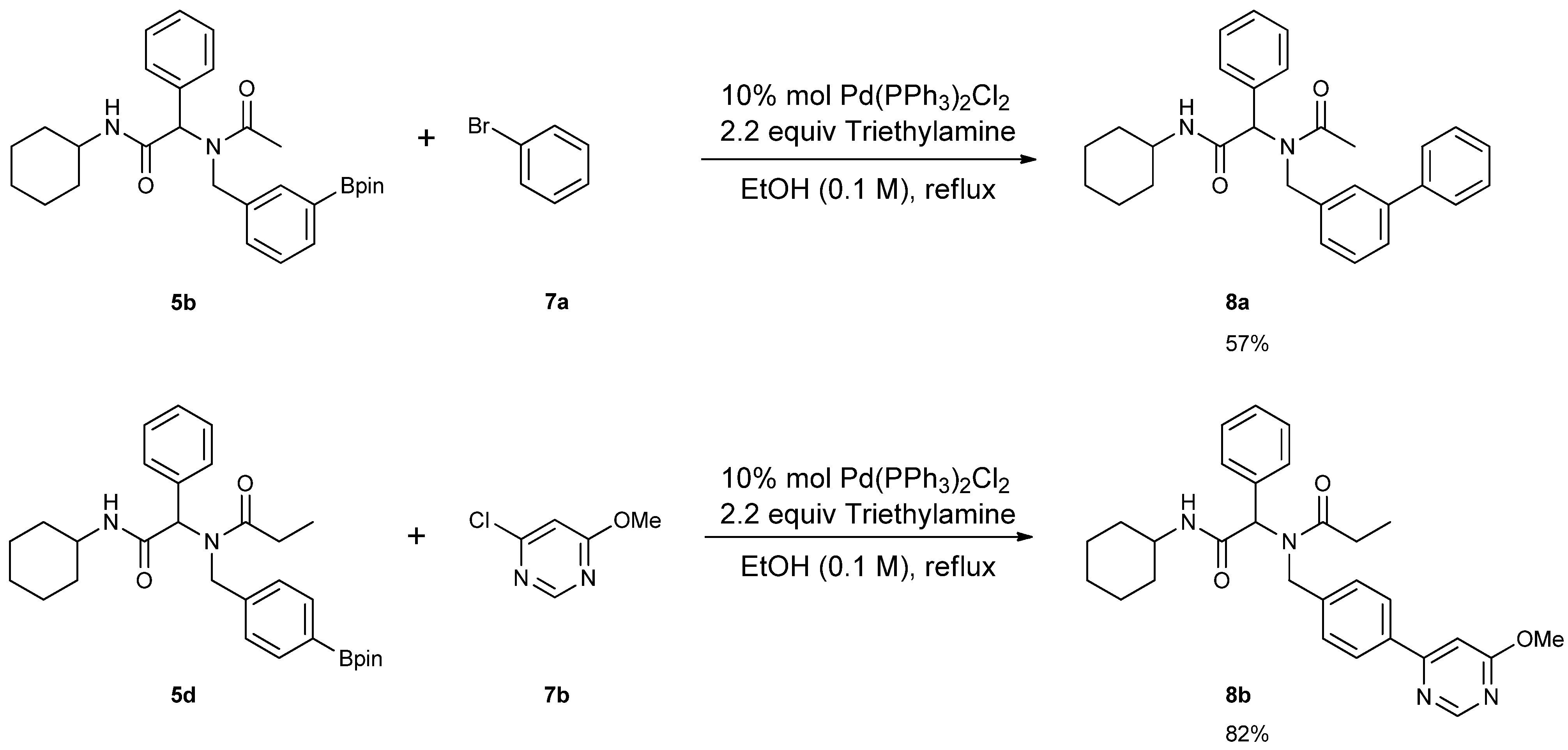
3.7. General Procedure F for the Synthesis 8a–b
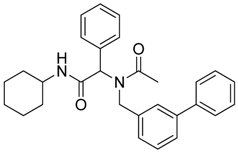
2-[N-([1,1'-Biphenyl]-3-ylmethyl)acetamido]-N-cyclohexyl-2-phenylacetamid (8a). Compound 5b (284 mg, 0.58 mmol), bromobenzene (0.08 mL, 0.75 mmol), and Pd(PPh3)2Cl2 (40 mg, 0.06 mmol) were added to a flask containing a magnetic stir with EtOH (5.80 mL). Triethylamine (0.18 mL, 1.28 mmol) was then added to the reaction mixture and the reaction mixture was stirred under reflux condition for 10 h under a N2 atmosphere. After cooling the mixture to room temperature, solvent was removed in vacuo. The crude material was diluted with ethyl acetate (5.00 mL) and water (5.00 mL). The aqueous layer was separated from the organic layer, and the aqueous layer was extracted with ethyl acetate (2 × 15 mL). Organic layers were then combined, washed with brine solution, and dried with MgSO4, and solvent was removed in vacuo. The crude material was then purified by flash column chromatography on silica gel using n-hexane/ethyl acetate = 1:1 as the eluent to give the desired product 8a as a white solid (m.p. 180 °C) in 57% yield (146 mg); white solid; 1H-NMR (Bruker AC-600 FT-NMR spectrometer at 600 MHz, CD3OD-d4) δ ppm 7.47–7.19 (m, 12H), 7.07 (s, 1H), 6.96 (d, J = 7.8 Hz, 1H), 6.15 (s, 0.77H), 5.68 (br, 0.23H), 4.75 (q, J1 = 115.8, J2 = 18.0, 2H), 3.65 (m, 1H), 2.11 (s, 3H), 1.82–1.08 (m, 10H); 13C-NMR (Bruker AC-600 FT-NMR spectrometer at 150.9 MHz, CD3OD-d4) δ ppm 175.41, 171.51, 142.75, 142.33, 140.03, 136.65, 131.20, 130.26, 130.00, 129.93, 129.72, 128.56, 128.15, 126.67, 126.21, 125.77, 63.73, 51.35, 50.19, 33.63. 26.75, 26.25, 26.20, 22.72; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C29H32N2O2, 441.2543; found, 441.2542.
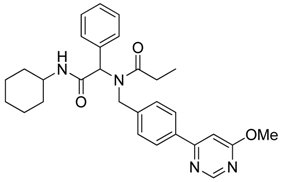
N-[2-(Cyclohexylamino)-2-oxo-1-phenylethyl]-N-[4-(6-methoxypyrimidin-4-yl)benzyl]propionamide (8b). Following the General Procedure F, the desired compound was synthesized, utilizing N-[2-(cyclohexylamino)-2-oxo-1-phenylethyl]-N-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl]-propionamide (5d, 236 mg, 0.52 mmol), 4-chloro-6-methoxypyrimidine (105.24 mg, 0.728 mmol), Pd(PPh3)2Cl2 (15 mg, 0.06 mmol), and triethylamine (0.17 mL, 1.23 mmol) giving compound 8b as a white solid (m.p. 174 °C) in 82% yield (223 mg); 1H-NMR (CDCl3) δ ppm 8.76 (s, 1 H), 7.78 (d, J = 8.2 Hz, 1H), 7.32–6.66 (m, 9H), 6.11 (s, 0.42H), 5.93 (b, 0.58H), 4.68 (q, J1 = 51.4, J2 = 18.2, 2H), 3.97 (s, 3H), 3.77 (m, 1H), 2.40–2.13 (m, 2H), 1.84–1.02 (m, 13H); 13C-NMR (CDCl3) δ ppm 176.29, 175.82, 169.97, 164.83, 158.44, 141.87, 135.43, 129.84, 128.87, 128.79, 128.71, 127.10, 126.57, 117.01, 103.40, 62.73, 54.01, 49.91, 48.76, 32.85, 27.41, 25.56, 24.91, 24.83, 9.46; HRMS (ESI, positive ion) (m/z): [M+H]+ calcd for C29H34N4O3, 487.2710; found, 487.2702.

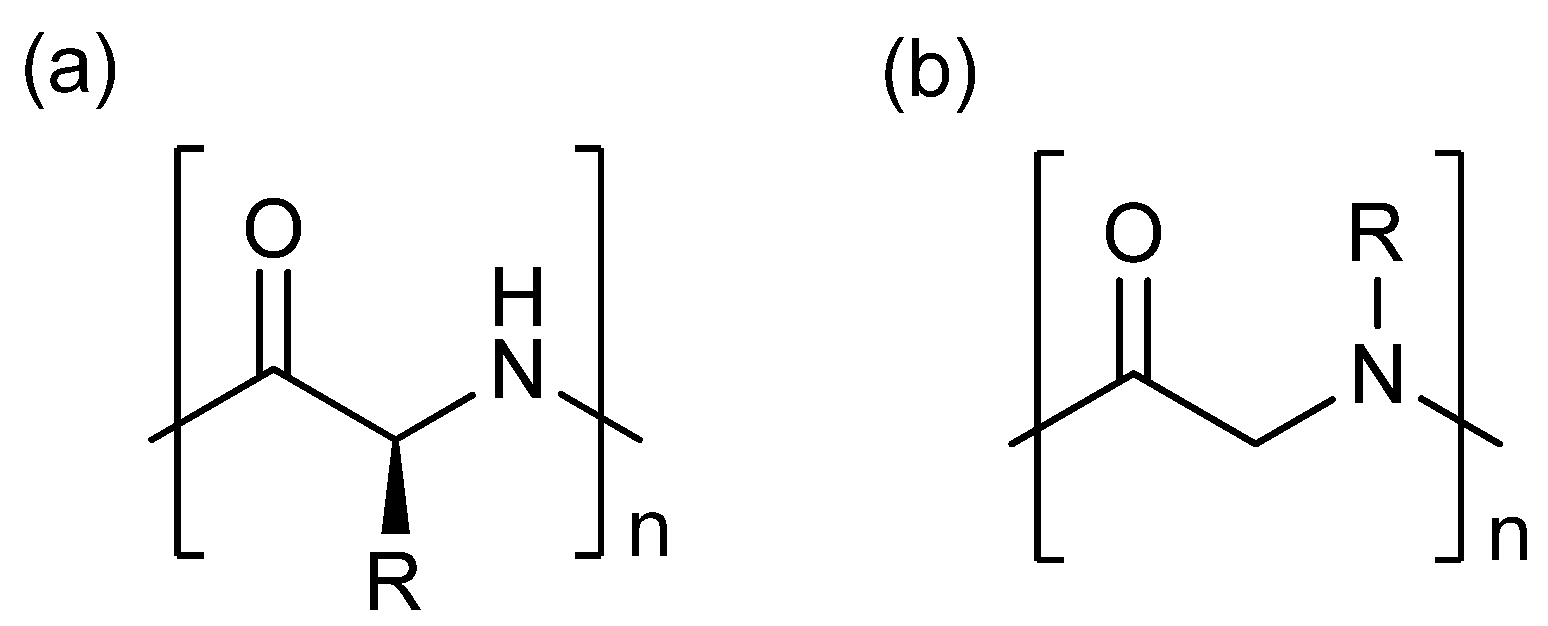
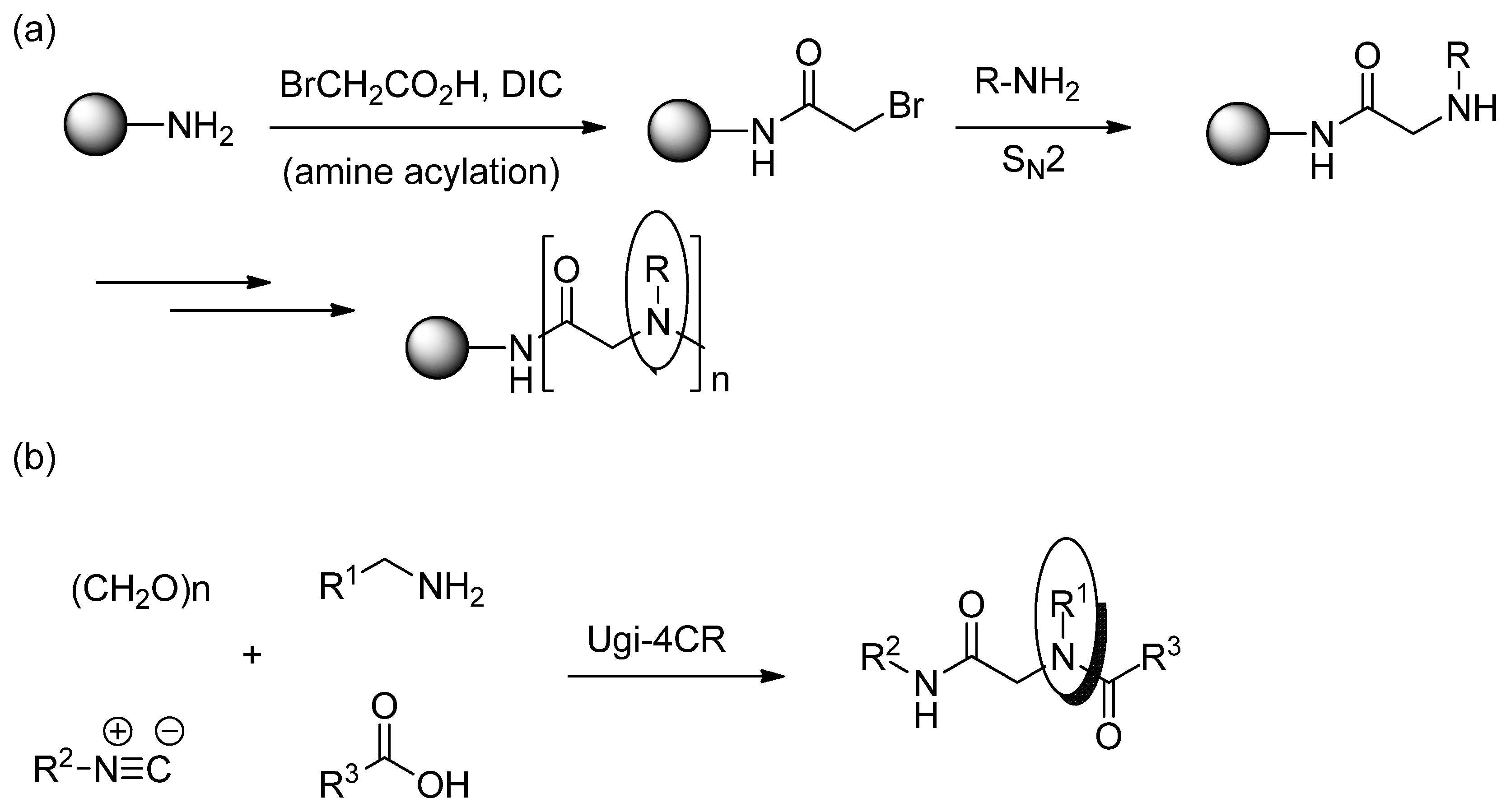
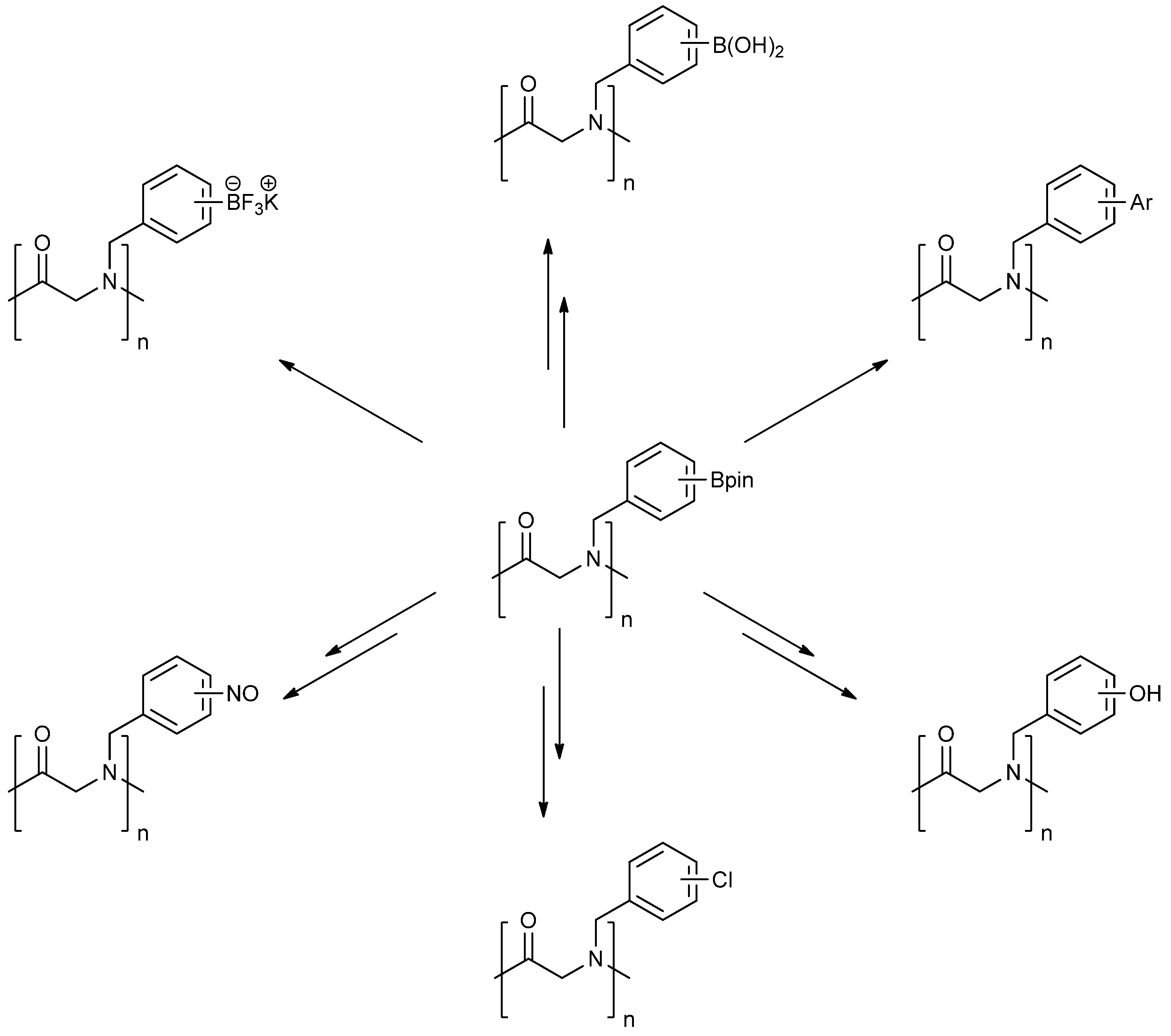








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}