Pyrene: A Probe to Study Protein Conformation and Conformational Changes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
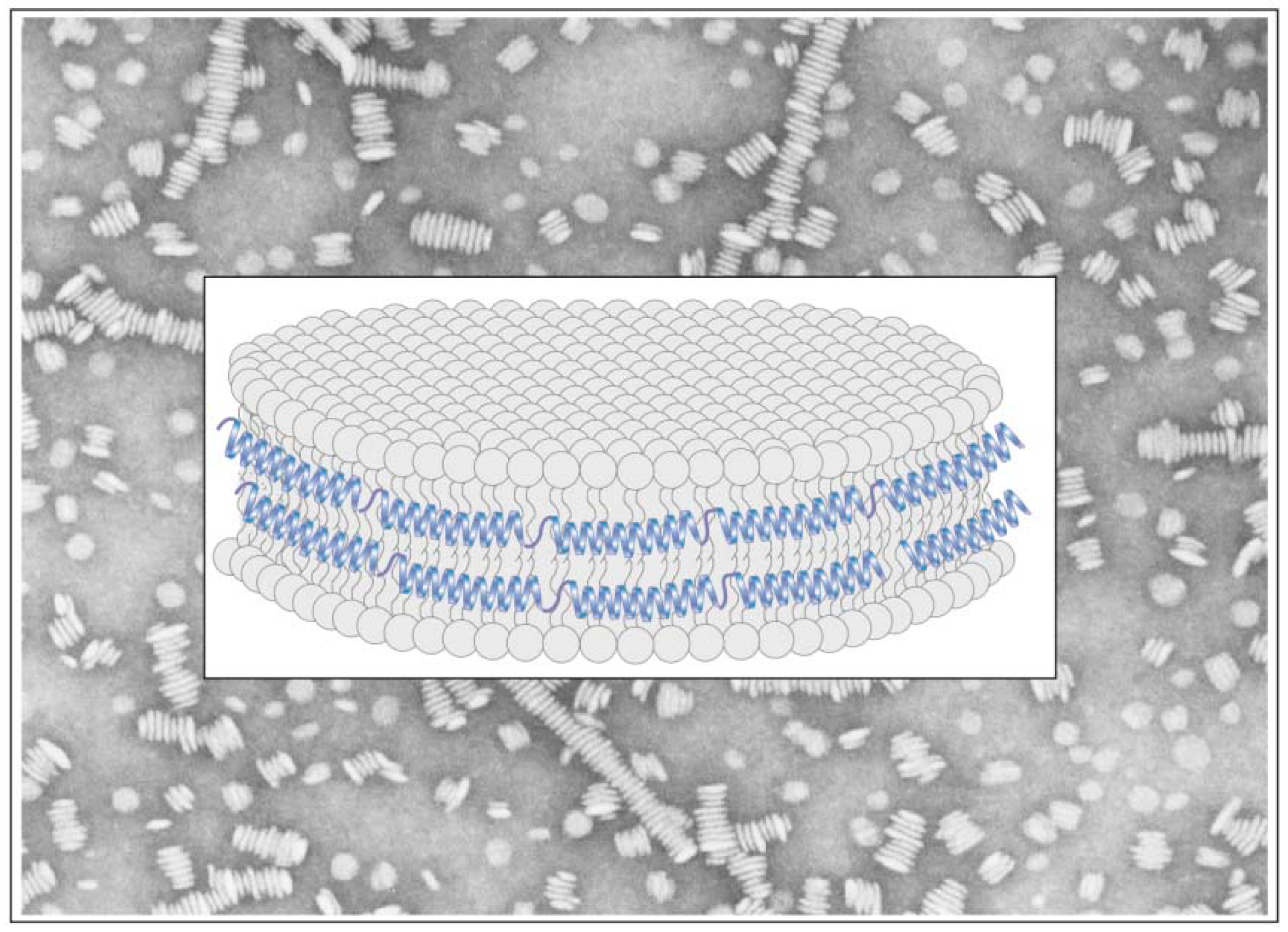{kind=link}
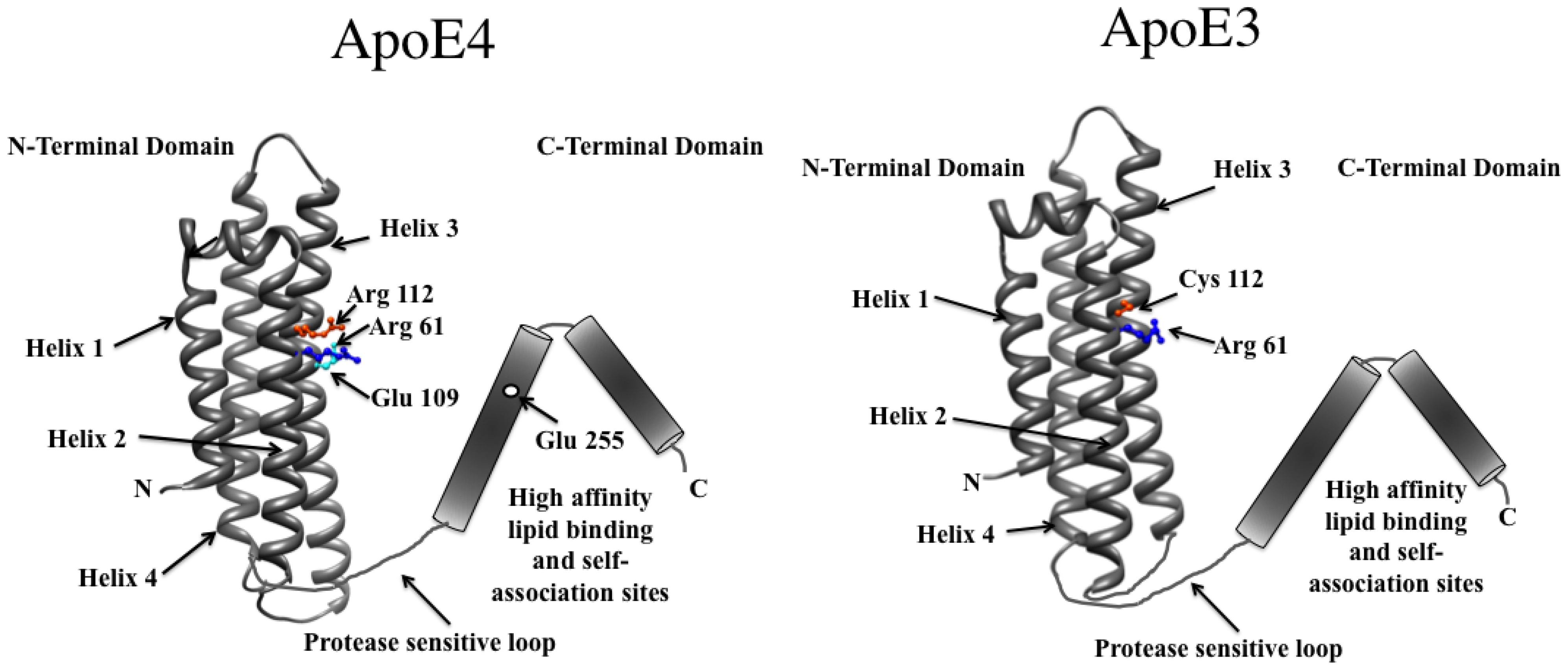{kind=link}
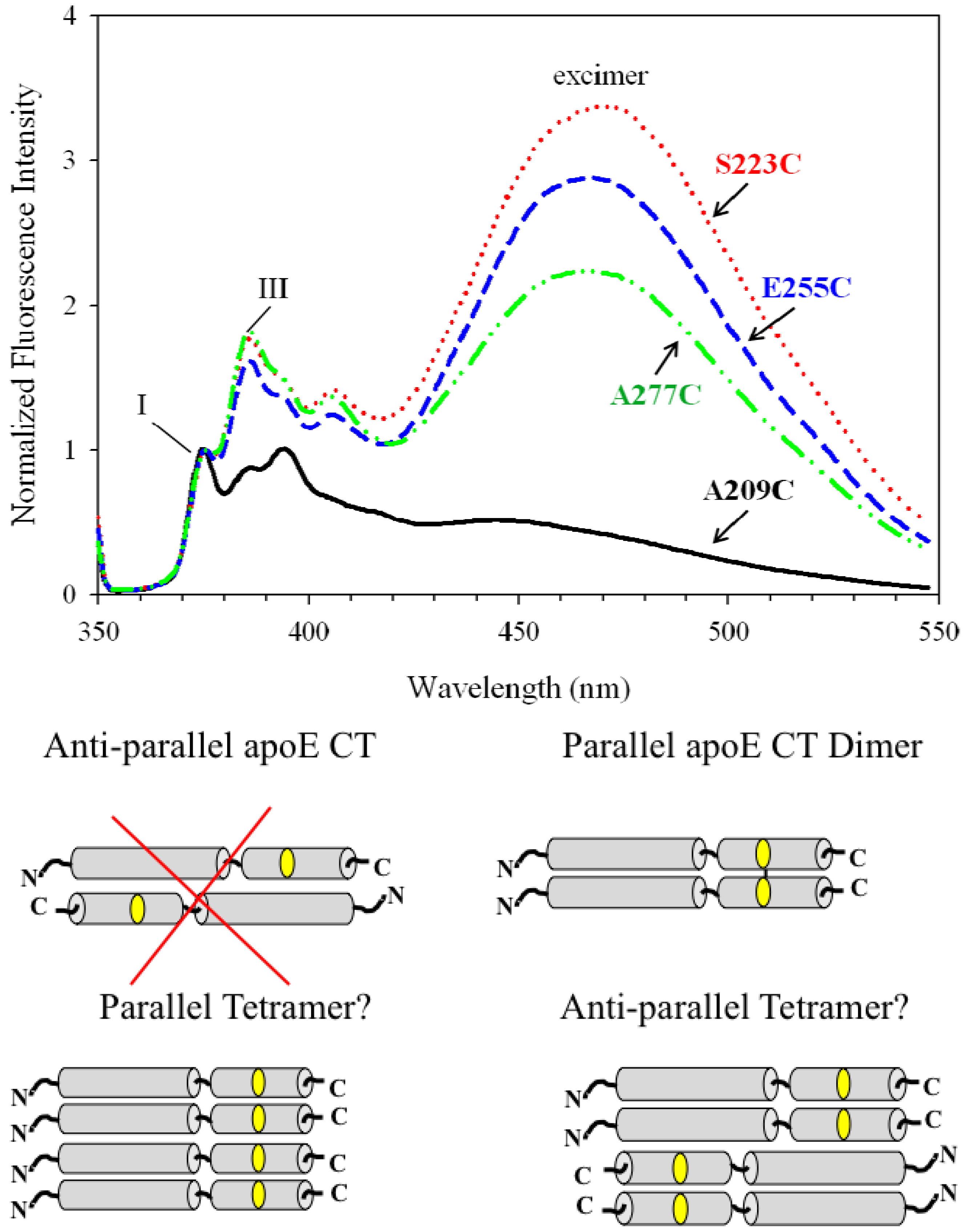{kind=link}
Abstract
:1. Introduction
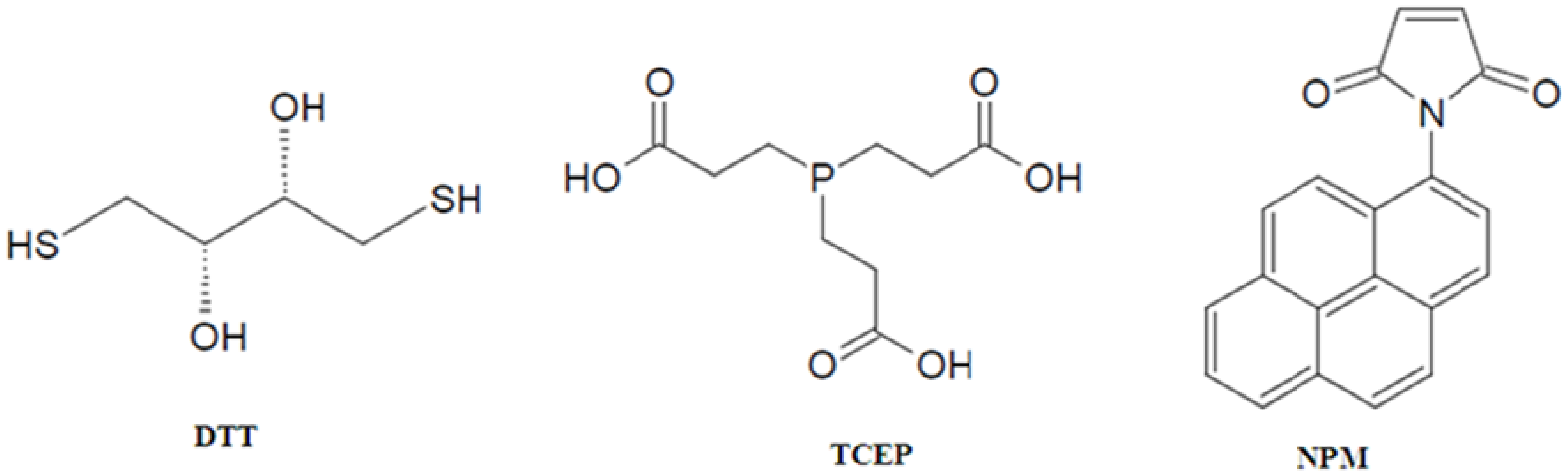
2. Site-Specific Labeling of Proteins with Pyrene
3. Photophysics of Pyrene Fluorescence Emission
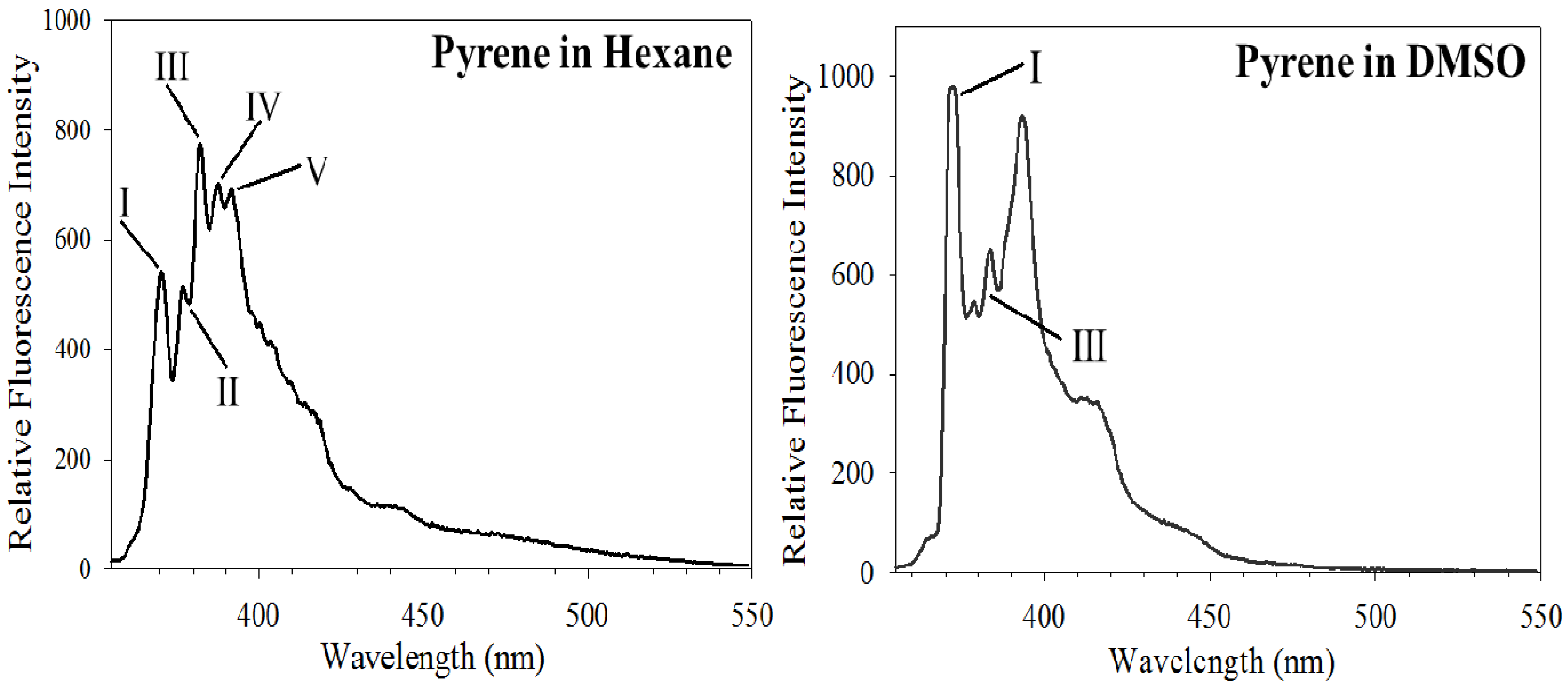
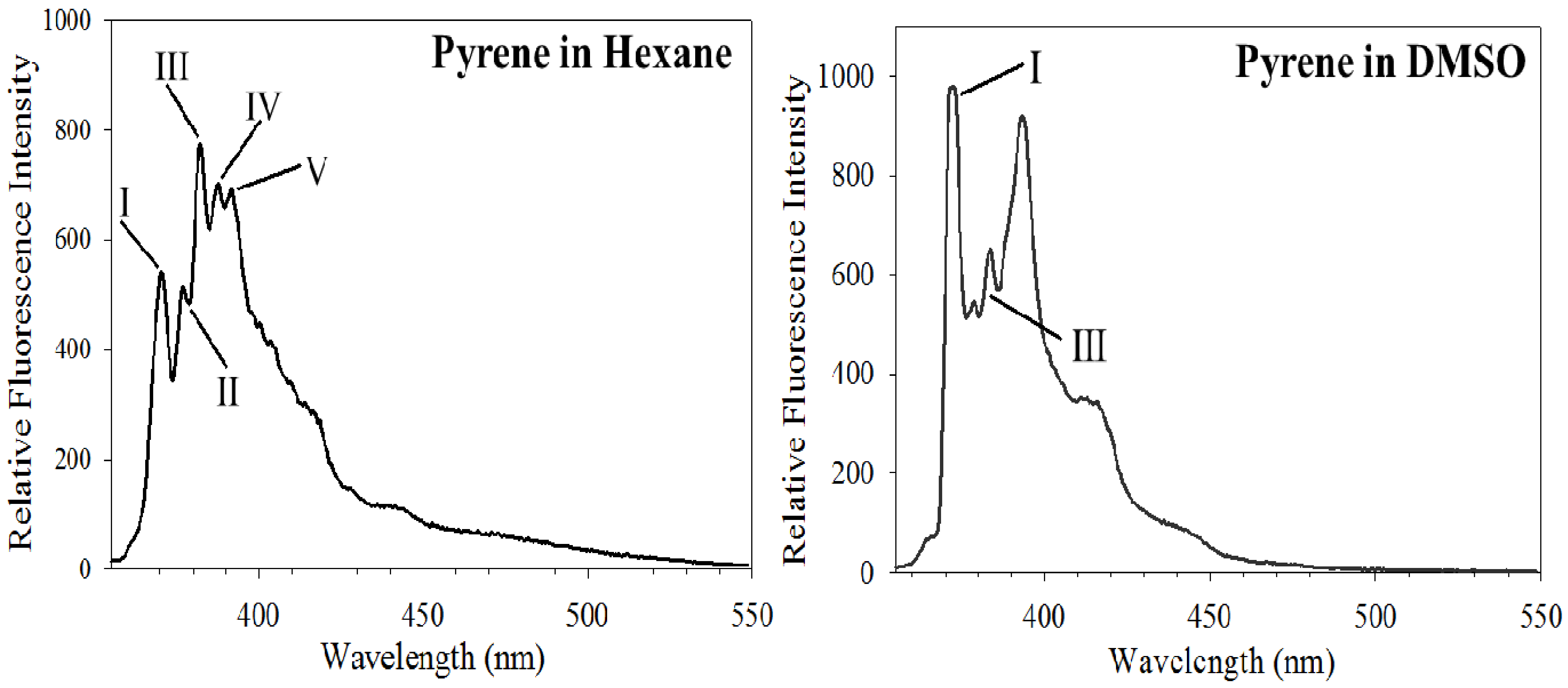
3.1. Py Value
3.2. Excimer Emission
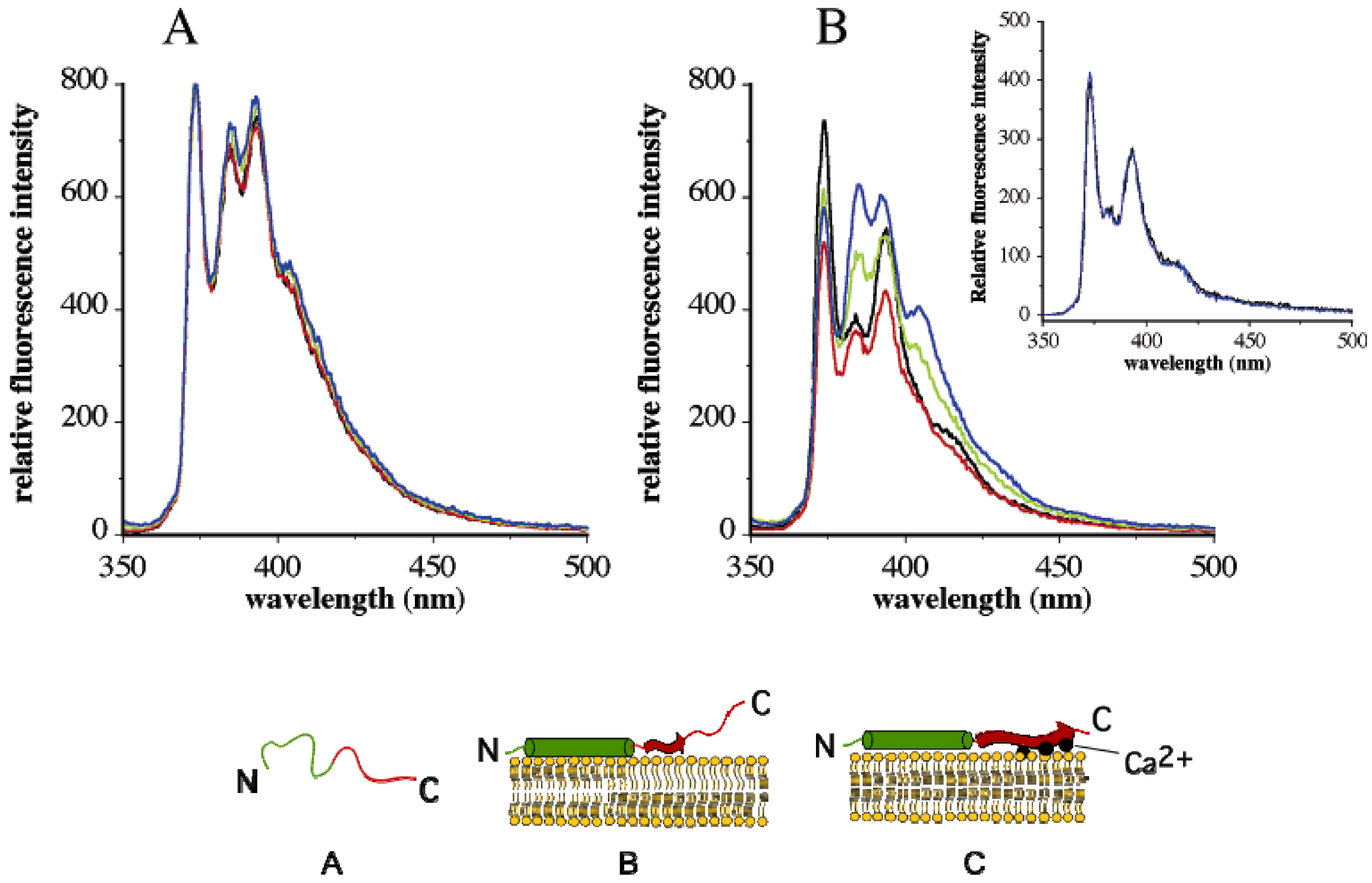
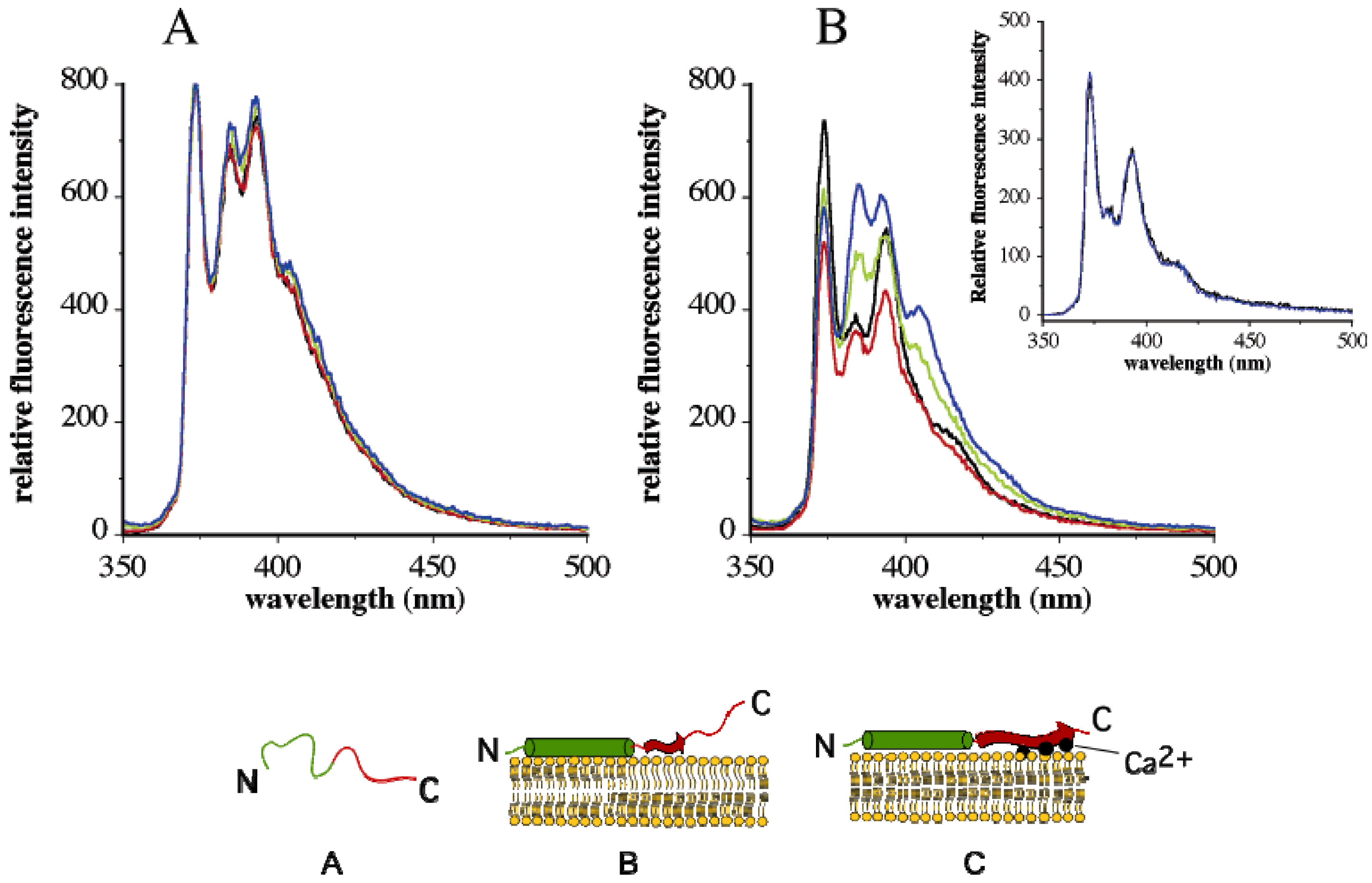
4. Change in Py Value Reflects Protein-Membrane Interaction
5. Excimer Emission Is an Indicator of Spatial Proximity in Proteins
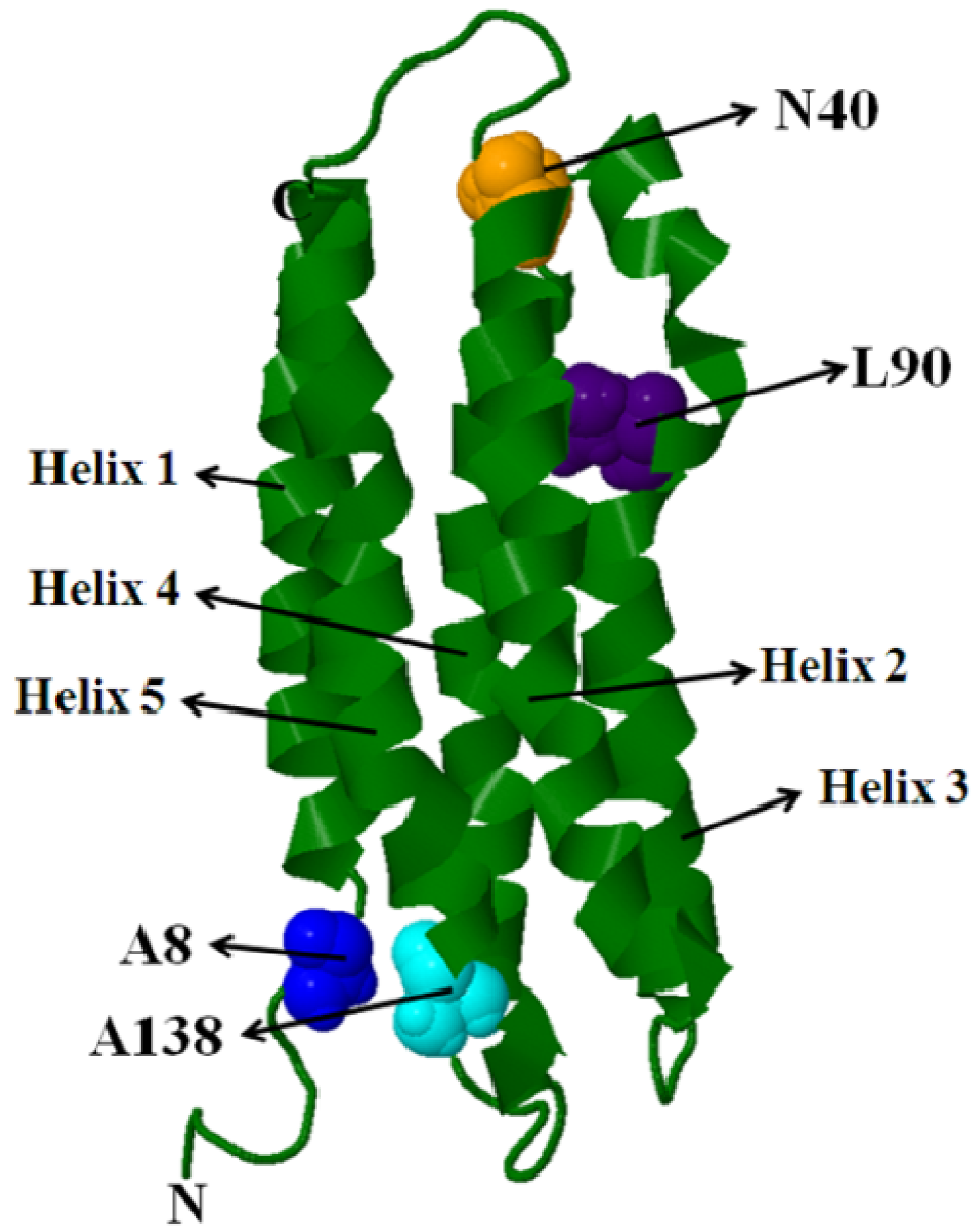
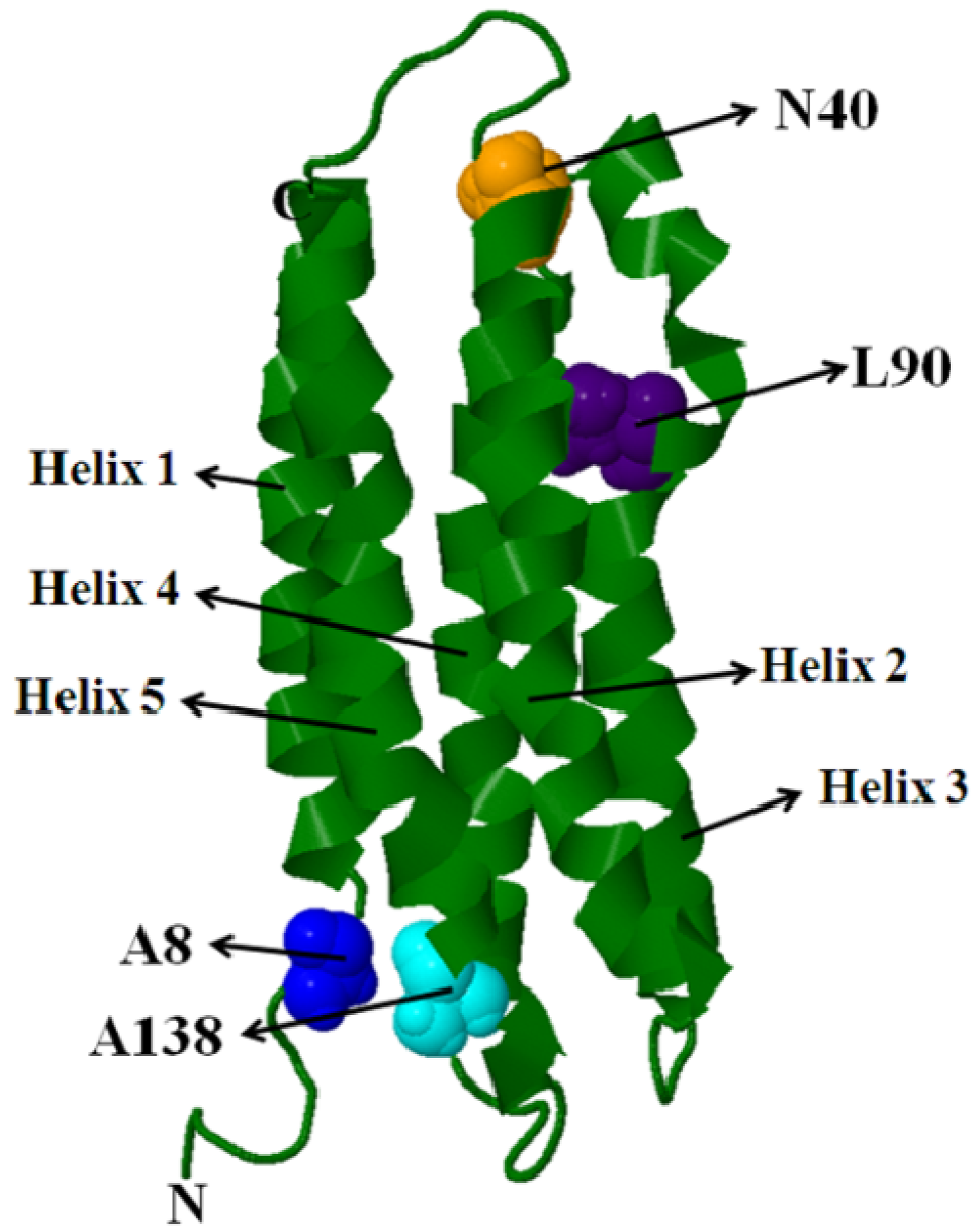
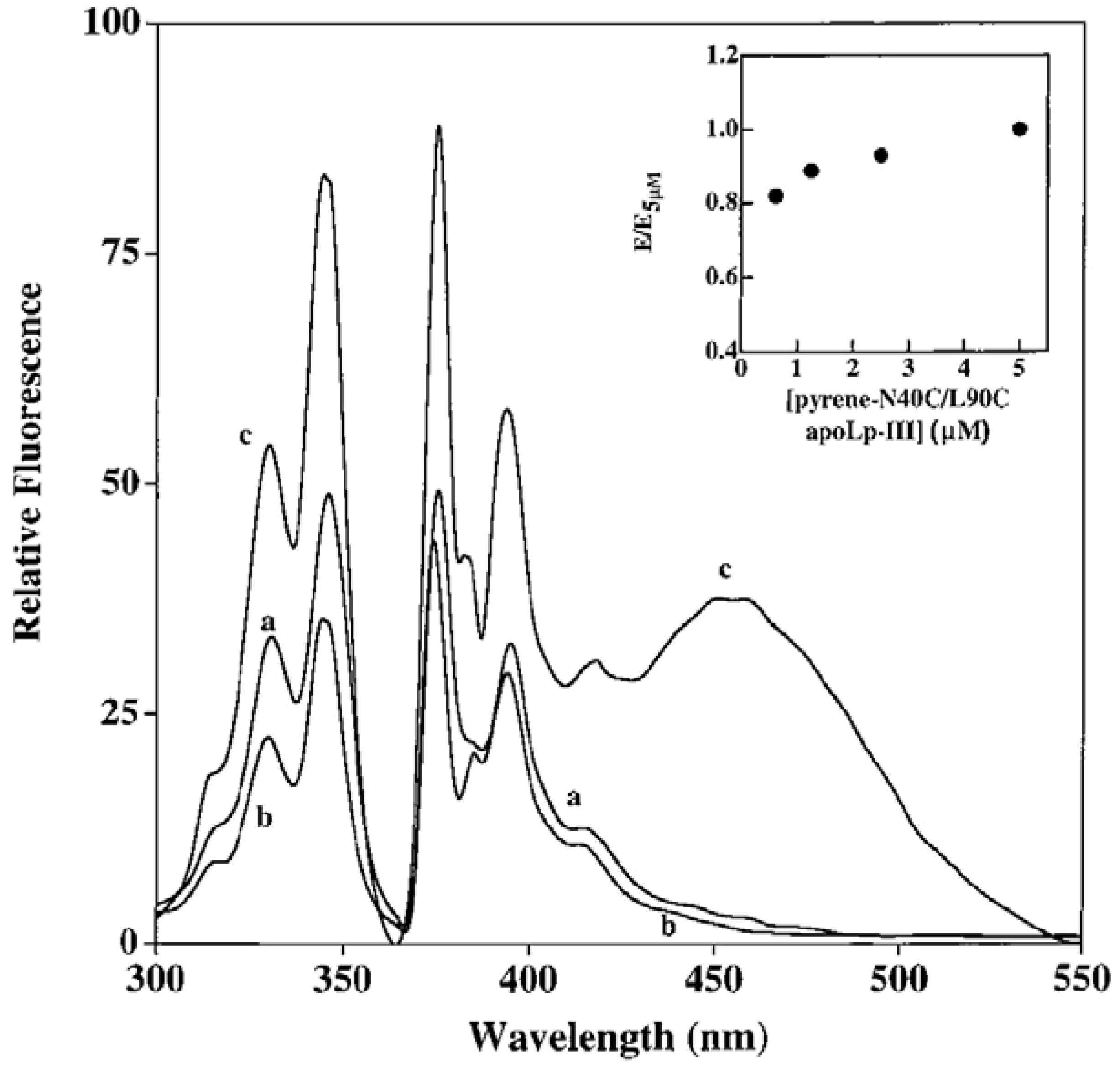
5.1. Intra-Molecular Distance Relationships
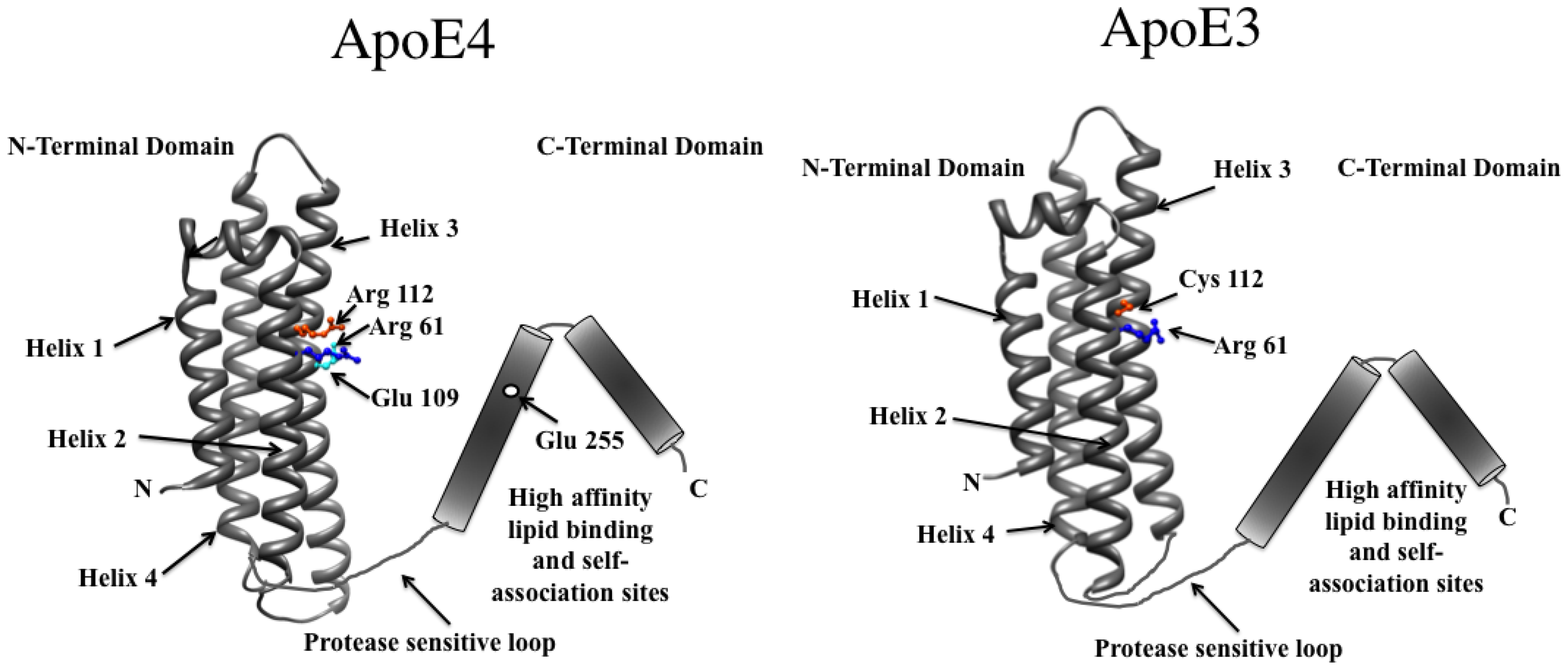
5.2. Domain-Domain Distance Relationships
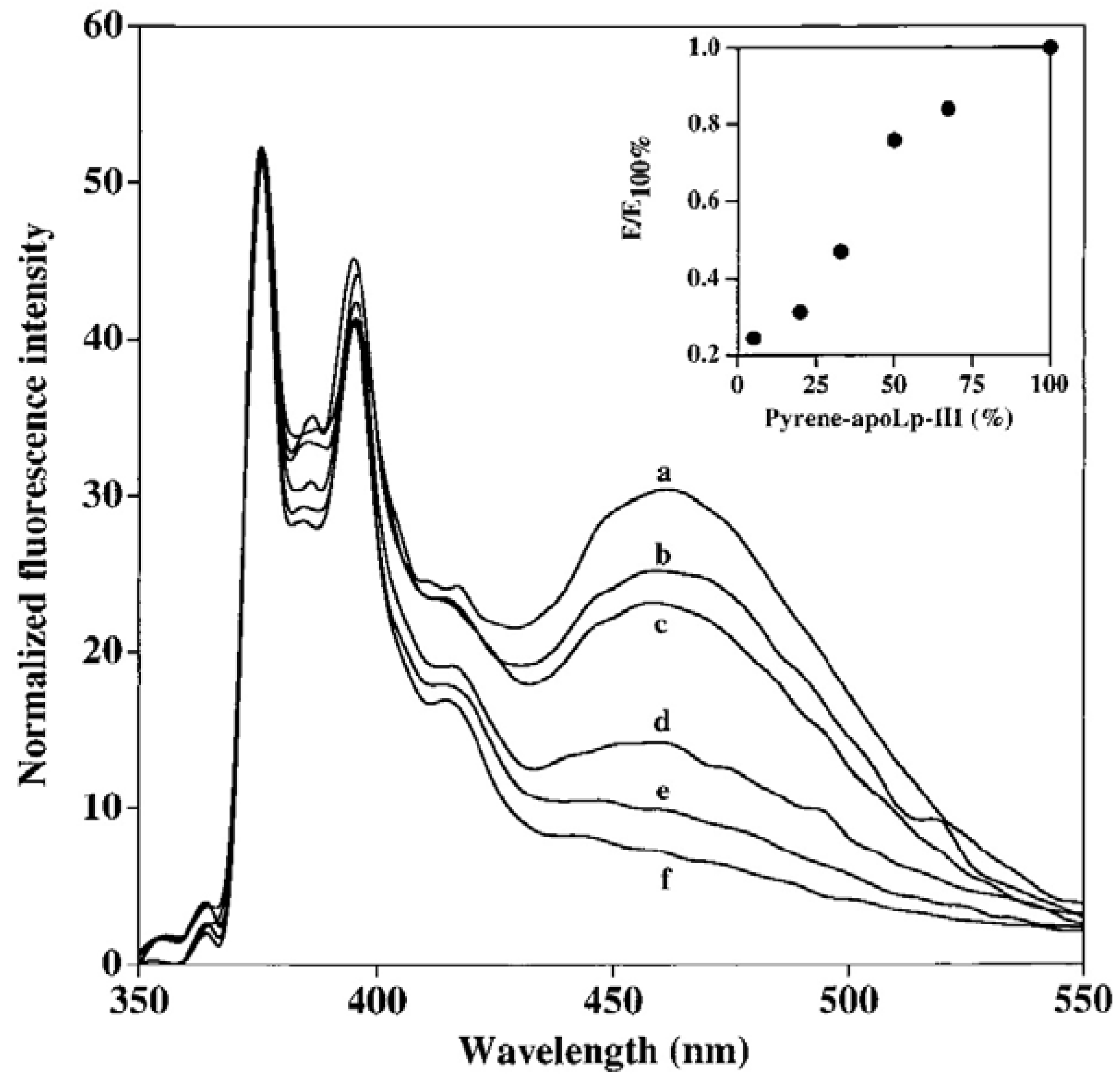
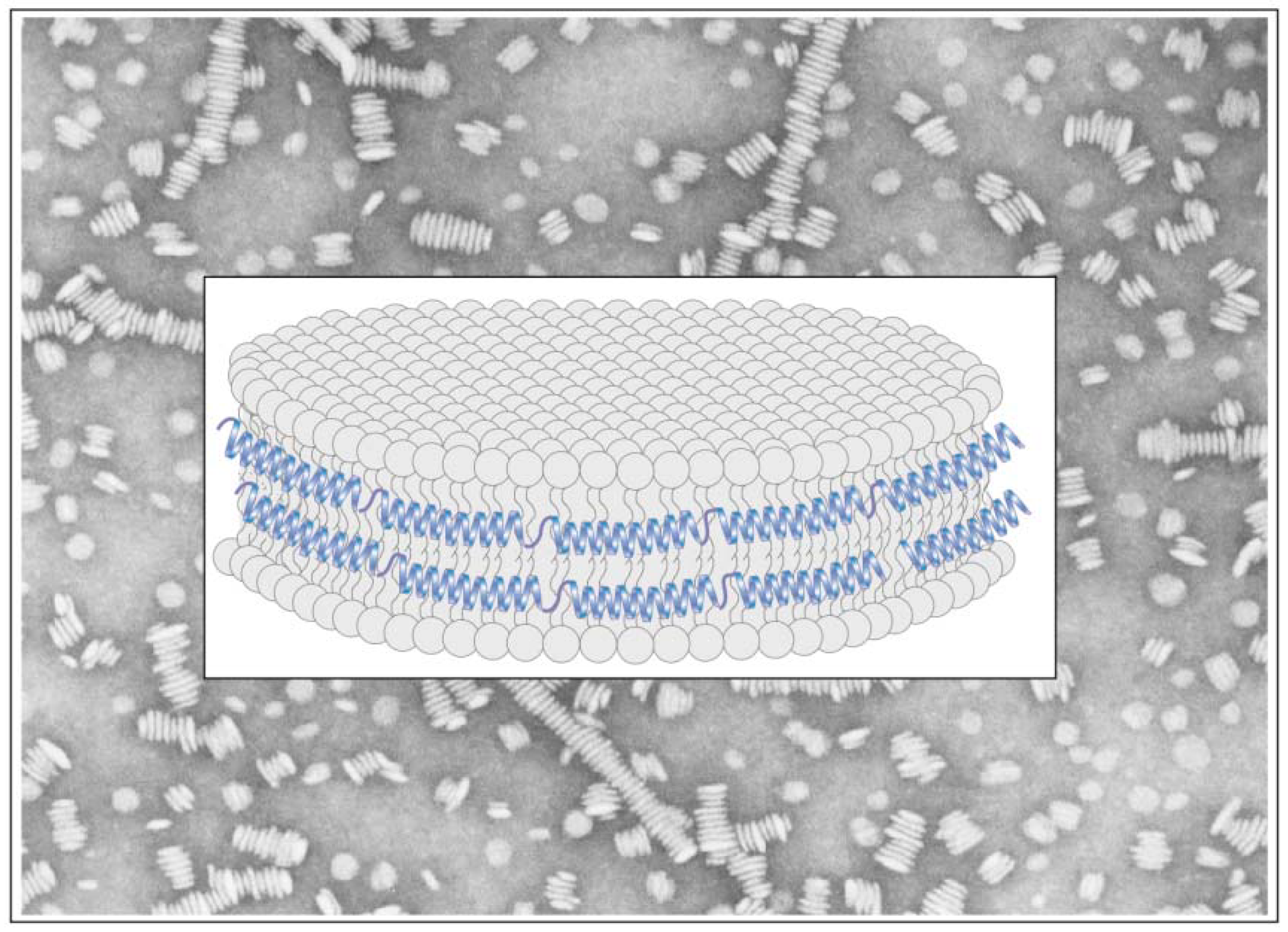
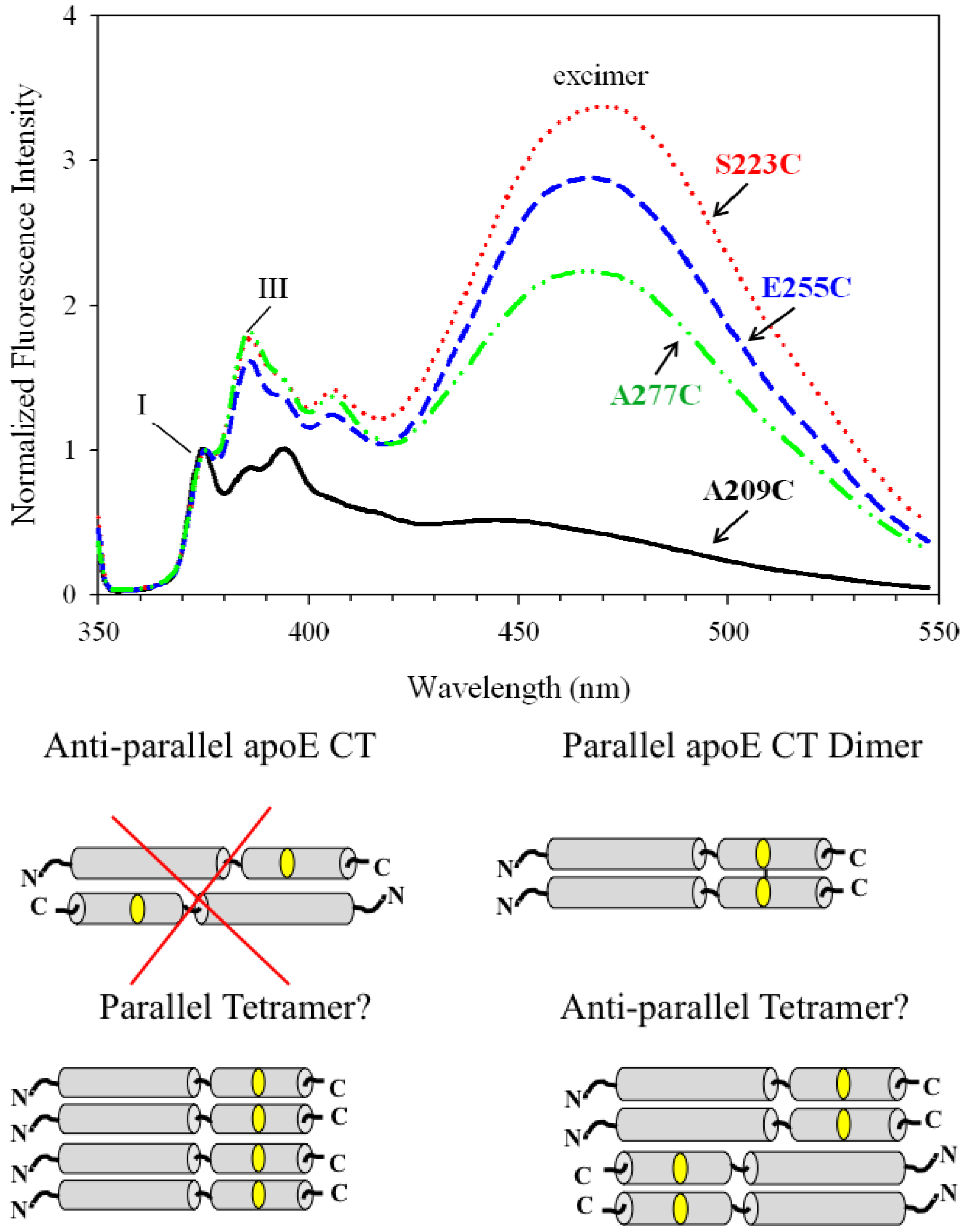
5.3. Excimer Emission Reveals Inter-Molecular Interactions: Protein Oligomerization, Aggregation and Polymerization
5.4. Pyrene Fluorescence to Decipher Transmembrane Organization and Protein Dynamics
5.5. Protein Unfolding
6. Advantages and Limitations of Using Pyrene
7. Summary and Perspectives
Acknowledgements
References
- Van der Meer, B.W. Biomembrane structure and dynamics viewed by fluorescence. Subcell. Biochem. 1988, 13, 1–53. [Google Scholar] [PubMed]
- Somerharju, P. Pyrene-labeled lipids as tools in membrane biophysics and cell biology. Chem. Phys. Lipids 2002, 116, 57–74. [Google Scholar] [CrossRef]
- Wu, C.; Wang, C.; Yan, L.; Yang, C.J. Pyrene excimer nucleic acid probes for biomolecule signaling. J. Biomed. Nanotechnol. 2009, 5, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Conlon, P.; Yang, C.J.; Wu, Y.; Chen, Y.; Martinez, K.; Kim, Y.; Stevens, N.; Marti, A.A.; Jockusch, S.; Turro, N.J.; Tan, W. Pyrene excimer signaling molecular beacons for probing nucleic acids. J. Am. Chem. Soc. 2008, 130, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.N.; Kool, E.T. Fluorescent DNA base replacements: Reporters and sensors for biological systems. Org. Biomol. Chem. 2006, 4, 4265–4274. [Google Scholar] [CrossRef] [PubMed]
- Pownall, H.J.; Smith, L.C. Pyrene-labeled lipids: Versatile probes of membrane dynamics in vitro and in living cells. Chem. Phys. Lipids 1989, 50, 191–211. [Google Scholar] [CrossRef]
- Linnertz, H.; Miksik, I.; Kvasnicka, P.; Bertoli, E.; Mazzanti, L.; Schoner, W.; Amler, E. Binding of pyrene isothiocyanate to the E1ATP site makes the H4-H5 cytoplasmic loop of Na+/K+-ATPase rigid. Eur. J. Biochem. 1998, 251, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Zolese, G.; Rabini, R.A.; Fumelli, P.; Staffolani, R.; Curatola, A.; Kvasnicka, P.; Kotyk, A.; Cester, N.; Mazzanti, L. Modifications induced by insulin-dependent diabetes mellitus on human placental Na+/K+-adenosine triphosphatase. J. Lab. Clin. Med. 1997, 130, 374–380. [Google Scholar] [CrossRef]
- Benecky, M.J.; Wine, R.W.; Kolvenbach, C.G.; Mosesson, M.W. Ionic-strength- and pH-dependent conformational states of human plasma fibronectin. Biochemistry 1991, 30, 4298–4306. [Google Scholar] [CrossRef] [PubMed]
- Benecky, M.J.; Kolvenbach, C.G.; Wine, R.W.; DiOrio, J.P.; Mosesson, M.W. Human plasma fibronectin structure probed by steady-state fluorescence polarization: Evidence for a rigid oblate structure. Biochemistry 1990, 29, 3082–3091. [Google Scholar] [CrossRef] [PubMed]
- Haugland, R.P. Thiol-reactive probes. In The Handbook: A Guide to Fluorescent Probes and Labeling Technologies, 10th ed.; Spence, M.T.Z., Ed.; Invitrogen Corp.: Carlsbad, CA, USA, 2005; pp. 93–116. [Google Scholar]
- Mims, M.P.; Sturgis, C.B.; Sparrow, J.T.; Morrisett, J.D. Acrylodan can label amino as well as sulfhydryl groups: Results with low-density lipoprotein, lipoprotein[a], and lipid-free proteins. Biochemistry 1993, 32, 9215–9220. [Google Scholar] [CrossRef] [PubMed]
- West, J.M.; Tsuruta, H.; Kantrowitz, E.R. A fluorescent probe-labeled Escherichia coli aspartate transcarbamoylase that monitors the allosteric conformational state. J. Biol. Chem. 2004, 279, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Narayanaswami, V.; Budamagunta, M.S.; Yamamato, T.; Voss, J.C.; Ryan, R.O. Lipid-induced extension of apolipoprotein E helix 4 correlates with low density lipoprotein receptor binding ability. J. Biol. Chem. 2006, 281, 39294–39299. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, D.; Narayanaswami, V.; Kay, C.M.; Ryan, R.O. Pyrene excimer fluorescence: A spatially sensitive probe to monitor lipid-induced helical rearrangement of apolipophorin III. Biochemistry 2000, 39, 6594–6601. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.; Narayanaswami, V. Examination of lipid-bound conformation of apolipoprotein E4 by pyrene excimer fluorescence. J. Biol. Chem. 2005, 280, 14605–14610. [Google Scholar] [CrossRef] [PubMed]
- Tamamizu-Kato, S.; Kosaraju, M.G.; Kato, H.; Raussens, V.; Ruysschaert, J.M.; Narayanaswami, V. Calcium-triggered membrane interaction of the alpha-synuclein acidic tail. Biochemistry 2006, 45, 10947–10956. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Khumsupan, P.; Narayanaswami, V. Pyrene fluorescence analysis offers new insights into the conformation of the lipoprotein-binding domain of human apolipoprotein E. Biochemistry 2010, 49, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Excimers. Angew. Chem. Int. Ed. Engl. 1969, 8, 333–343. [Google Scholar] [CrossRef]
- Winnik, F.M. Photophysics of preassociated pyrenes in aqueous polymer solutions and in other organized media. Chem. Rev. 1993, 93, 587–614. [Google Scholar] [CrossRef]
- Lehrer, S.S. Pyrene excimer fluorescence as a probe of protein conformational change. Subcell. Biochem. 1995, 24, 115–132. [Google Scholar] [PubMed]
- Lehrer, S.S. Intramolecular pyrene excimer fluorescence: A probe of proximity and protein conformational change. Meth. Enzymol. 1997, 278, 286–295. [Google Scholar] [PubMed]
- Hara, K.; Ware, W.R. Influence of solvent perturbation on the radiative transition probability from the B1u state of pyrene. Chem. Phys. 1980, 51, 61–68. [Google Scholar] [CrossRef]
- Karpovich, D.S.; Blanchard, G.J. Relating the polarity-dependent fluorescence response of pyrene to vibronic Coupling. Achieving a fundamental understanding of the py polarity scale. J. Phys. Chem. 1995, 99, 3951–3958. [Google Scholar] [CrossRef]
- Peter Geigle, K.; Wolf, J.; Hohlneicher, G. Franck-Condon/Herzberg-Teller interferences in the 1Lb transitions of pyrene and chrysene. J. Photochem. Photobiol. A: Chem. 1997, 105, 183–187. [Google Scholar] [CrossRef]
- Nakajima, A. Solvent effect on the vibrational structures of the fluorescence and absorption spectra of pyrene. Bull. Chem. Soc. Jpn. 1971, 44, 3272–3277. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Thomas, J.K. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Solvent effects on emission spectra. In Principles of Fluorescence Spectroscopy, 2nd ed.; Lakowicz, J.R., Ed.; Kluwer Academic/ Plenum Publisher: New York, NY, USA, 1999; pp. 185–189. [Google Scholar]
- Nakajima, A. Effects of isomeric solvents on vibronic band intensities in fluorescence spectrum of pyrene. J. Mol. Spectrosc. 1976, 61, 467–469. [Google Scholar] [CrossRef]
- Tedeschi, C.; Mohwald, H.; Kirstein, S. Polarity of layer-by-layer deposited polyelectrolyte films as determined by pyrene fluorescence. J. Am. Chem. Soc. 2001, 123, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.C.; Winnik, M.A. The Py scale of solvent polarities. Can. J. Chem. 1984, 62, 2560–2565. [Google Scholar] [CrossRef]
- Birks, J.B. Excimers. In Photophysics of Aromatic Molecules; JohnWiley & Sons, Inc: New York, NY, USA, 1970; pp. 301–371. [Google Scholar]
- Lakowicz, J.R. Introduction to fluorescence. In Principles of Fluorescence Spectroscopy, 2nd ed.; Lakowicz, J.R., Ed.; Kluwer Academic/ Plenum Publisher: New York, NY, USA, 1999; pp. 1–23. [Google Scholar]
- Birks, J.B. Triplet-triplet interaction of aromatic molecules in solution. Chem. Phys. Lett. 1968, 2, 417–419. [Google Scholar] [CrossRef]
- Newcomb, L.F.; Gellman, S.H. Aromatic stacking interactions in aqueous solution: Evidence that neither classical hydrophobic effects nor dispersion forces are important. J. Am. Chem. Soc. 1994, 116, 4993–4994. [Google Scholar] [CrossRef]
- Betcher-Lange, S.L.; Lehrer, S.S. Pyrene excimer fluorescence in rabbit skeletal alpha-tropomyosin labeled with N-(1-pyrene)maleimide. A probe of sulfhydryl proximity and local chain separation. J. Biol. Chem. 1978, 253, 3757–3760. [Google Scholar] [PubMed]
- Zama, M.; Bryan, P.N.; Harrington, R.E.; Olins, A.L.; Olins, D.E. Conformational states of chromatin. Cold Spring Harb. Symp. Quant. Biol. 1978, 42 Pt 1, 31–41. [Google Scholar] [CrossRef]
- Warshel, A.; Huler, E. Theoretical evaluation of potential surfaces, equilibrium geometries and vibronic transition intensities of excimers: The pyrene crystal excimer. Chem. Phys. 1974, 6, 463–468. [Google Scholar] [CrossRef]
- Ishii, Y.; Lehrer, S.S. Intramolecular excimer fluorescence of pyrene maleimide-labeled dithiothreitol. In Fluorescent Biomolecules; Jameson, D.M., Reinhart, G.D., Eds.; Plenum Press: New York, NY, USA, 1989; pp. 286–295. [Google Scholar]
- Bussell, R., Jr.; Ramlall, T.F.; Eliezer, D. Helix periodicity, topology, and dynamics of membrane-associated alpha-synuclein. Prot. Sci. 2005, 14, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Islam, K.; Kenney, J.; Nielsen, M.S.; Power, J.; Gai, W.P. Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds alpha-synuclein filaments. J. Biol. Chem. 2000, 275, 21500–21507. [Google Scholar] [CrossRef] [PubMed]
- Bussell, R., Jr.; Eliezer, D. Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated alpha-synuclein. Biochemistry 2004, 43, 4810–4818. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.Y.; Karlin, S. Clusters of charged residues in protein three-dimensional structures. Proc. Natl. Acad. Sci. USA 1996, 93, 8350–8355. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswami, V.; Kiss, R.S.; Weers, P.M. The helix bundle: A reversible lipid binding motif. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 2010, 155, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Breiter, D.R.; Kanost, M.R.; Benning, M.M.; Wesenberg, G.; Law, J.H.; Wells, M.A.; Rayment, I.; Holden, H.M. Molecular structure of an apolipoprotein determined at 2.5 Å resolution. Biochemistry 1991, 30, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswami, V.; Wang, J.; Kay, C.M.; Scraba, D.G.; Ryan, R.O. Disulfide bond engineering to monitor conformational opening of apolipophorin III during lipid binding. J. Biol. Chem. 1996, 271, 26855–26862. [Google Scholar] [CrossRef] [PubMed]
- Wientzek, M.; Kay, C.M.; Oikawa, K.; Ryan, R.O. Binding of insect apolipophorin III to dimyristoylphosphatidylcholine vesicles. Evidence for a conformational change. J. Biol. Chem. 1994, 269, 4605–4612. [Google Scholar] [PubMed]
- Sahoo, D.; Weers, P.M.; Ryan, R.O.; Narayanaswami, V. Lipid-triggered conformational switch of apolipophorin III helix bundle to an extended helix organization. J. Mol. Biol. 2002, 321, 201–214. [Google Scholar] [CrossRef]
- Wang, J.; Gagne, S.M.; Sykes, B.D.; Ryan, R.O. Insight into lipid surface recognition and reversible conformational adaptations of an exchangeable apolipoprotein by multidimensional heteronuclear NMR techniques. J. Biol. Chem. 1997, 272, 17912–17920. [Google Scholar] [CrossRef] [PubMed]
- Raussens, V.; Narayanaswami, V.; Goormaghtigh, E.; Ryan, R.O.; Ruysschaert, J.M. Alignment of the apolipophorin-III alpha-helices in complex with dimyristoylphosphatidylcholine. A unique spatial orientation. J. Biol. Chem. 1995, 270, 12542–12547. [Google Scholar] [CrossRef] [PubMed]
- Raussens, V.; Narayanaswami, V.; Goormaghtigh, E.; Ryan, R.O.; Ruysschaert, J.M. Hydrogen/deuterium exchange kinetics of apolipophorin-III in lipid-free and phospholipid-bound states. An analysis by Fourier transform infrared spectroscopy. J. Biol. Chem. 1996, 271, 23089–23095. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Wardell, M.R.; Weisgraber, K.H.; Mahley, R.W.; Agard, D.A. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 1991, 252, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Wilson, C.; Wardell, M.R.; Simmons, T.; Mahley, R.W.; Weisgraber, K.H.; Agard, D.A. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 1994, 269, 22358–22365. [Google Scholar] [PubMed]
- Posse de Chaves, E.; Narayanaswami, V. Apolipoprotein E and cholesterol transport in aging and disease. Future Lipidol. 2008, 3, 505–530. [Google Scholar] [CrossRef]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid. Res. 2010, 50, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Weisgraber, K.H. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J. Biol. Chem. 1996, 271, 19053–19057. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E structure: Insights into function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M.; Budamagunta, M.S.; Voss, J.C.; Weisgraber, K.H. Modulation of apolipoprotein E structure by domain interaction: Differences in lipid-bound and lipid-free forms. J. Biol. Chem. 2005, 280, 34288–34295. [Google Scholar] [CrossRef] [PubMed]
- Choy, N.; Raussens, V.; Narayanaswami, V. Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J. Mol. Biol. 2003, 334, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Graceffa, P.; Lehrer, S.S. The excimer fluorescence of pyrene-labeled tropomyosin. A probe of conformational dynamics. J. Biol. Chem. 1980, 255, 11296–11300. [Google Scholar] [PubMed]
- Ishii, Y.; Lehrer, S.S. Two-site attachment of troponin to pyrene-labeled tropomyosin. J. Biol. Chem. 1991, 266, 6894–6903. [Google Scholar] [PubMed]
- Ishii, Y.; Lehrer, S.S. Fluorescence studies of the conformation of pyrene-labeled tropomyosin: Effects of F-actin and myosin subfragment 1. Biochemistry 1985, 24, 6631–6638. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Lehrer, S.S. Excimer fluorescence of pyrenyliodoacetamide-labeled tropomyosin: A probe of the state of tropomyosin in reconstituted muscle thin filaments. Biochemistry 1990, 29, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Wang, X.; Sykes, B.D. Structural based insights into the role of troponin in cardiac muscle pathophysiology. J. Muscle Res. Cell Motil. 2004, 25, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, I. Troponin: Structure, function and dysfunction. Adv. Exp. Med. Biol. 2007, 592, 21–36. [Google Scholar] [PubMed]
- Hoffman, R.M.; Sykes, B.D. Disposition and dynamics: Interdomain orientations in troponin. Adv. Exp. Med. Biol. 2007, 592, 59–70. [Google Scholar] [PubMed]
- Filatov, V.L.; Katrukha, A.G.; Bulargina, T.V.; Gusev, N.B. Troponin: Structure, properties, and mechanism of functioning. Biochemistry (Mosc) 1999, 64, 969–985. [Google Scholar] [PubMed]
- Lin, T.I. Excimer fluorescence of pyrene-tropomyosin adducts. Biophys. Chem. 1982, 15, 277–288. [Google Scholar] [CrossRef]
- Lehrer, S.S.; Stafford, W.F., 3rd. Preferential assembly of the tropomyosin heterodimer: Equilibrium studies. Biochemistry 1991, 30, 5682–5688. [Google Scholar] [CrossRef] [PubMed]
- Burtnick, L.D.; Sanders, C.; Smillie, L.B. Fluorescence from pyrene-labeled native and reconstituted chicken gizzard tropomyosins. Arch. Biochem. Biophys. 1988, 266, 622–627. [Google Scholar] [CrossRef]
- Burtnick, L.D.; Stewart, D.I.; Clark, I.D.; Smillie, L.B. Excimer fluorescence of equine platelet tropomyosin labeled with N-(1-pyrenyl)iodoacetamide. Biochemistry 1986, 25, 3875–3880. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, O.; James, M.N. Refined crystal structure of troponin C from turkey skeletal muscle at 2.0 Å resolution. J. Mol. Biol. 1988, 203, 761–779. [Google Scholar] [CrossRef]
- Slupsky, C.M.; Sykes, B.D. NMR solution structure of calcium-saturated skeletal muscle troponin C. Biochemistry 1995, 34, 15953–15964. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.M.; Fuchs, F. Pyrene-labeled cardiac troponin C. Effect of Ca2+ on monomer and excimer fluorescence in solution and in myofibrils. Biophys. J. 1992, 61, 892–901. [Google Scholar] [CrossRef]
- Verin, A.D.; Gusev, N.B. Ca2+-induced conformational changes in cardiac troponin C as measured by N-(1-pyrene)maleimide fluorescence. Biochim. Biophys. Acta 1988, 956, 197–208. [Google Scholar] [CrossRef]
- Wang, Z.; Gergely, J.; Tao, T. Characterization of the Ca(2+)-triggered conformational transition in troponin C. Proc. Natl. Acad. Sci. USA 1992, 89, 11814–11817. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.M.; Fuchs, F. Pyrene-labeled cardiac troponin C. Effect of Ca2+ on monomer and excimer fluorescence in solution and in myofibrils. Biophys. J. 1992, 61, 892–901. [Google Scholar] [CrossRef]
- Strasburg, G.M.; Leavis, P.C.; Gergely, J. Troponin-C-mediated calcium-sensitive changes in the conformation of troponin I detected by pyrene excimer fluorescence. J. Biol. Chem. 1985, 260, 366–370. [Google Scholar] [PubMed]
- Vassylyev, D.G.; Takeda, S.; Wakatsuki, S.; Maeda, K.; Maeda, Y. Crystal structure of troponin C in complex with troponin I fragment at 2.3 Å resolution. Proc. Natl. Acad. Sci. USA 1998, 95, 4847–4852. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.S.; Wall, M.E.; Gallagher, S.C.; Trewhella, J. A model of troponin-I in complex with troponin-C using hybrid experimental data: The inhibitory region is a beta-hairpin. Prot. Sci. 2000, 9, 1312–1326. [Google Scholar]
- Liou, Y.M.; Chen, M.W. Calcium-dependent protein-protein interactions induce changes in proximity relationships of Cys48 and Cys64 in chicken skeletal troponin I. Eur. J. Biochem. 2003, 270, 3092–3100. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.C.; Hodges, R.S. Proximity of sulfhydryl groups to the sites of interaction between components of the troponin complex from rabbit skeletal muscle. J. Biol. Chem. 1982, 257, 2549–2555. [Google Scholar] [PubMed]
- Takeda, S.; Yamashita, A.; Maeda, K.; Maeda, Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature 2003, 424, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Ishii, Y. Fluorescence properties of acrylodan-labeled tropomyosin and tropomyosin-actin: Evidence for myosin subfragment 1 induced changes in geometry between tropomyosin and actin. Biochemistry 1988, 27, 5899–5906. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Diaz, E.; Chalovich, J.M. Kinetics of regulated actin transitions measured by probes on tropomyosin. Biophys. J. 2010, 98, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Coulton, A.; Lehrer, S.S.; Geeves, M.A. Functional homodimers and heterodimers of recombinant smooth muscle tropomyosin. Biochemistry 2006, 45, 12853–12858. [Google Scholar] [CrossRef] [PubMed]
- Golitsina, N.L.; Kordowska, J.; Wang, C.L.; Lehrer, S.S. Ca2+-dependent binding of calcyclin to muscle tropomyosin. Biochem. Biophys. Res. Commun. 1996, 220, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Alahyan, M.; Marston, S.B.; El-Mezgueldi, M. Role of caldesmon in the Ca2+ regulation of smooth muscle thin filaments: Evidence for a cooperative switching mechanism. J. Biol. Chem. 2008, 283, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Segrest, J.P.; De Loof, H.; Dohlman, J.G.; Brouillette, C.G.; Anantharamaiah, G.M. Amphipathic helix motif: Classes and properties. Proteins 1990, 8, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Brouillette, C.G.; Venkatachalapathi, Y.V.; Anantharamaiah, G.M. The amphipathic helix in the exchangeable apolipoproteins: A review of secondary structure and function. J. Lipid Res. 1992, 33, 141–166. [Google Scholar] [PubMed]
- Segrest, J.P.; Garber, D.W.; Brouillette, C.G.; Harvey, S.C.; Anantharamaiah, G.M. The amphipathic alpha helix: A multifunctional structural motif in plasma apolipoproteins. Adv. Prot. Chem. 1994, 45, 303–369. [Google Scholar]
- Westerlund, J.A.; Weisgraber, K.H. Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J. Biol. Chem. 1993, 268, 15745–15750. [Google Scholar] [PubMed]
- Sakamoto, T.; Tanaka, M.; Vedhachalam, C.; Nickel, M.; Nguyen, D.; Dhanasekaran, P.; Phillips, M.C.; Lund-Katz, S.; Saito, H. Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry 2008, 47, 2968–2977. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Li, Q.; Korando, L.; Jerome, W.G.; Wang, J. A monomeric human apolipoprotein E carboxyl-terminal domain. Biochemistry 2004, 43, 5055–5064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Vasudevan, S.; Sojitrawala, R.; Zhao, W.; Cui, C.; Xu, C.; Fan, D.; Newhouse, Y.; Balestra, R.; Jerome, W.G.; Weisgraber, K.; Li, Q.; Wang, J. A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry 2007, 46, 10722–10732. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, F.; Caccuri, A.M.; Morgenstern, R.; Rosato, N.; Federici, G. Aggregation of pyrene-labeled microsomal glutathione S-transferase. Effect of concentration. Eur. J. Biochem. 1993, 217, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, R.; Guthenberg, C.; Depierre, J.W. Microsomal glutathione S-transferase. Purification, initial characterization and demonstration that it is not identical to the cytosolic glutathione S-transferases A, B and C. Eur. J. Biochem. 1982, 128, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, R.; DePierre, J.W.; Jornvall, H. Microsomal glutathione transferase. Primary structure. J. Biol. Chem. 1985, 260, 13976–13983. [Google Scholar] [PubMed]
- Singh, A.; Hitchcock-Degregori, S.E. A peek into tropomyosin binding and unfolding on the actin filament. PLoS One 2009, 4, e6336. [Google Scholar] [CrossRef] [PubMed]
- Coulton, A.T.; Koka, K.; Lehrer, S.S.; Geeves, M.A. Role of the head-to-tail overlap region in smooth and skeletal muscle beta-tropomyosin. Biochemistry 2008, 47, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Coates, J.H.; Criddle, A.H.; Geeves, M.A. Pressure-relaxation studies of pyrene-labelled actin and myosin subfragment 1 from rabbit skeletal muscle. Evidence for two states of acto-subfragment 1. Biochem. J. 1985, 232, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Criddle, A.H.; Geeves, M.A.; Jeffries, T. The use of actin labelled with N-(1-pyrenyl)iodoacetamide to study the interaction of actin with myosin subfragments and troponin/tropomyosin. Biochem. J. 1985, 232, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kouyama, T.; Mihashi, K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labelled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur. J. Biochem. 1981, 114, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Kouyama, T.; Mihashi, K. Pulse-fluorometry study on actin and heavy meromyosin using F-actin labelled with N-(1-pyrene)maleimide. Eur. J. Biochem. 1980, 105, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Maytum, R.; Lehrer, S.S.; Geeves, M.A. Cooperativity and switching within the three-state model of muscle regulation. Biochemistry 1999, 38, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Grabarek, Z.; Wang, C.L. Differential effects of caldesmon on the intermediate conformational states of polymerizing actin. J. Biol. Chem. 2010, 285, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Kim, E.; Lee, W.L.; Miller, C.J.; Kuang, B.; Reisler, E.; Rubenstein, P.A. Fluorescence probing of yeast actin subdomain 3/4 hydrophobic loop 262-274. Actin-actin and actin-myosin interactions in actin filaments. J. Biol. Chem. 1997, 272, 16829–16837. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.K.; Rubenstein, P.A. Differential regulation of actin polymerization and structure by yeast formin isoforms. J. Biol. Chem. 2009, 284, 16776–16783. [Google Scholar] [CrossRef] [PubMed]
- Eads, J.C.; Mahoney, N.M.; Vorobiev, S.; Bresnick, A.R.; Wen, K.K.; Rubenstein, P.A.; Haarer, B.K.; Almo, S.C. Structure determination and characterization of Saccharomyces cerevisiae profilin. Biochemistry 1998, 37, 11171–11181. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.K.; Giardina, P.C.; Blake, M.S.; Edwards, J.; Apicella, M.A.; Rubenstein, P.A. Interaction of the gonococcal porin PIB with G- and F-actin. Biochemistry 2000, 39, 8638–8647. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.K.; Rubenstein, P.A.; DeMali, K.A. Vinculin nucleates actin polymerization and modifies actin filament structure. J. Biol. Chem. 2009, 284, 30463–30473. [Google Scholar] [CrossRef] [PubMed]
- Bakolitsa, C.; de Pereda, J.M.; Bagshaw, C.R.; Critchley, D.R.; Liddington, R.C. Crystal structure of the vinculin tail suggests a pathway for activation. Cell 1999, 99, 603–613. [Google Scholar] [CrossRef]
- Jung, K.; Jung, H.; Wu, J.; Prive, G.G.; Kaback, H.R. Use of site-directed fluorescence labeling to study proximity relationships in the lactose permease of Escherichia coli. Biochemistry 1993, 32, 12273–12278. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Jung, H.; Kaback, H.R. Dynamics of lactose permease of Escherichia coli determined by site-directed fluorescence labeling. Biochemistry 1994, 33, 3980–3985. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Smirnova, I.; Kasho, V.; Verner, G.; Kaback, H.R.; Iwata, S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003, 301, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Valeva, A.; Pongs, J.; Bhakdi, S.; Palmer, M. Staphylococcal alpha-toxin: The role of the N-terminus in formation of the heptameric pore—A fluorescence study. Biochim. Biophys. Acta 1997, 1325, 281–286. [Google Scholar] [CrossRef]
- Hammarstrom, P.; Kalman, B.; Jonsson, B.H.; Carlsson, U. Pyrene excimer fluorescence as a proximity probe for investigation of residual structure in the unfolded state of human carbonic anhydrase II. FEBS Lett. 1997, 420, 63–68. [Google Scholar] [CrossRef]
- Hakansson, K.; Carlsson, M.; Svensson, L.A.; Liljas, A. Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol. 1992, 227, 1192–1204. [Google Scholar] [CrossRef]
- Eriksson, A.E.; Jones, T.A.; Liljas, A. Refined structure of human carbonic anhydrase II at 2.0 Å resolution. Proteins 1988, 4, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L. Seeding protein folding. Trends Biochem. Sci. 1986, 11, 6–9. [Google Scholar] [CrossRef]
- Dill, K.A.; Shortle, D. Denatured states of proteins. Annu. Rev. Biochem. 1991, 60, 795–825. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.L.; Thompson, T.E. Oxygen quenching of pyrene-lipid fluorescence in phosphatidylcholine vesicles. A probe for membrane organization. Biophys. J. 1985, 47, 613–621. [Google Scholar] [CrossRef]
- Yang, C.J.; Jockusch, S.; Vicens, M.; Turro, N.J.; Tan, W. Light-switching excimer probes for rapid protein monitoring in complex biological fluids. Proc. Natl. Acad. Sci. USA 2005, 102, 17278–17283. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Shimizu, H.; Inouye, M. Unambiguous detection of target DNAs by excimer-monomer switching molecular beacons. J. Org. Chem. 2004, 69, 3271–3275. [Google Scholar] [CrossRef] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bains, G.; Patel, A.B.; Narayanaswami, V. Pyrene: A Probe to Study Protein Conformation and Conformational Changes. Molecules 2011, 16, 7909-7935. https://doi.org/10.3390/molecules16097909
Bains G, Patel AB, Narayanaswami V. Pyrene: A Probe to Study Protein Conformation and Conformational Changes. Molecules. 2011; 16(9):7909-7935. https://doi.org/10.3390/molecules16097909
Chicago/Turabian StyleBains, Gursharan, Arti B. Patel, and Vasanthy Narayanaswami. 2011. "Pyrene: A Probe to Study Protein Conformation and Conformational Changes" Molecules 16, no. 9: 7909-7935. https://doi.org/10.3390/molecules16097909
APA StyleBains, G., Patel, A. B., & Narayanaswami, V. (2011). Pyrene: A Probe to Study Protein Conformation and Conformational Changes. Molecules, 16(9), 7909-7935. https://doi.org/10.3390/molecules16097909