Studies on Log Po/w of Quinoxaline di-N-Oxides: A Comparison of RP-HPLC Experimental and Predictive Approaches


Abstract
:1. Introduction
2. Results and Discussion
2.1. Experimental log Po/w: Shake-Flask vs. RP-HPLC Method
2.1.1. Correlation between log Po/w and log k’0

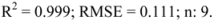

{kind=link}
| Name | Code | log Po/wa | log k’0 | RP-HPLC log k’ | LOO-predicted RP-HPLC log Po/w |
|---|---|---|---|---|---|
| 2-Butanone | 1 | 0.3 | −0.06 | 0.21 | 0.15 |
| Aniline | 2 | 0.9 | 0.67 | 0.94 | 0.96 |
| Acetanilide | 3 | 1.0 | 0.88 | 1.15 | 1.19 |
| Acetophenone | 4 | 1.7 | 1.27 | 1.54 | 1.52 |
| Benzene | 5 | 2.1 | 1.87 | 2.14 | 2.14 |
| Chlorobenzene | 6 | 2.8 | 2.62 | 2.89 | 2.90 |
| Bromobenzene | 7 | 3.0 | 2.84 | 3.10 | 3.12 |
| Naphthalene | 8 | 3.6 | 3.15 | 3.42 | 3.36 |
| Benzyl benzoate | 9 | 4.0 | 3.74 | 4.01 | 4.01 |
2.1.2. log Po/w of Quinoxalines di-N-Oxide
 | Capacity factors | RP-HPLC log Po/w | ||||||
|---|---|---|---|---|---|---|---|---|
| R | R7/R6 | Comp. | log k’70 | log k’60 | log k’50 | log k’40 | log k’0 | |
| C6H5-CH2 | H/H | 10 | −0.64 | −0.34 | −0.05 | 0.28 | 1.49 | 1.76 |
| CH3/H | 11 | −0.56 | −0.24 | 0.07 | 0.35 | 1.58 | 1.85 | |
| Cl/H | 12 | −0.36 | −0.05 | 0.30 | 0.65 | 1.98 | 2.25 | |
| Cl/Cl | 13 | −0.11 | 0.27 | 0.66 | 0.98 | 2.46 | 2.72 | |
| 2-phenylethyl | H/H | 14 | −0.58 | −0.26 | 0.05 | 0.36 | 1.61 | 1.88 |
| CH3/H | 15 | −0.45 | −0.13 | 0.21 | 0.47 | 1.73 | 2.00 | |
| Cl/H | 16 | −0.37 | 0.00 | 0.36 | 0.69 | 2.11 | 2.38 | |
| Cl/Cl | 17 | −0.06 | 0.34 | 0.74 | 1.07 | 2.60 | 2.87 | |
| p-BrC6H4-CH2 | H/H | 18 | −0.24 | 0.11 | 0.48 | 0.85 | 2.30 | 2.57 |
| CH3/H | 19 | −0.16 | 0.21 | 0.60 | 0.94 | 2.43 | 2.69 | |
| Cl/H | 20 | 0.02 | 0.42 | 0.84 | 1.22 | 2.83 | 3.09 | |
| Cl/Cl | 21 | 0.19 | 0.65 | 1.15 | 1.60 | 3.51 | 3.78 | |
| p-CH3C6H4-CH2 | H/H | 22 | −0.44 | −0.10 | 0.25 | 0.62 | 2.02 | 2.29 |
| CH3/H | 23 | −0.36 | −0.01 | 0.34 | 0.66 | 2.02 | 2.29 | |
| Cl/H | 24 | −0.25 | 0.15 | 0.54 | 0.93 | 2.51 | 2.77 | |
| Cl/Cl | 25 | 0.14 | 0.54 | 0.93 | 1.33 | 2.91 | 3.18 | |
| 2,2-diphenylethyl | H/H | 26 | −0.33 | 0.14 | 0.62 | 1.06 | 2.92 | 3.18 |
| CH3/H | 27 | −0.12 | 0.33 | 0.80 | 1.21 | 3.02 | 3.28 | |
| Cl/H | 28 | −0.13 | 0.38 | 0.91 | 1.38 | 3.42 | 3.69 | |
| Cl/Cl | 29 | 0.25 | 0.76 | 1.31 | 1.78 | 3.86 | 4.12 | |
2.2. Calculated log Po/w
| Comp. | RP-HPLC log Po/w | Calculated log Po/w | ||||||
|---|---|---|---|---|---|---|---|---|
| ALOGPs | miLOGP | ALOGP | MLOGP | LogKOW | XLOGP2 | XLOGP3 | ||
| 10 | 1.76 | 0.43 | −0.48 | 1.11 | −1.71 | 0.55 | 5.03 | 1.28 |
| 11 | 1.85 | 0.50 | −0.05 | 1.60 | −1.47 | 1.10 | 5.46 | 1.64 |
| 12 | 2.25 | 1.03 | 0.18 | 1.77 | −1.20 | 1.20 | 5.65 | 1.90 |
| 13 | 2.72 | 1.72 | 0.78 | 2.44 | −0.70 | 1.84 | 6.27 | 2.53 |
| 14 | 1.88 | 0.63 | −0.07 | 1.43 | −1.47 | 1.04 | 5.18 | 1.74 |
| 15 | 2.00 | 0.80 | 0.35 | 1.92 | −1.24 | 1.59 | 5.62 | 2.10 |
| 16 | 2.38 | 1.25 | 0.58 | 2.09 | −0.97 | 1.69 | 5.80 | 2.37 |
| 17 | 2.87 | 2.03 | 1.19 | 2.76 | −0.47 | 2.33 | 6.43 | 2.99 |
| 18 | 2.57 | 1.14 | 0.33 | 1.86 | −1.08 | 1.44 | 5.82 | 1.97 |
| 19 | 2.69 | 1.29 | 0.76 | 2.34 | −0.85 | 1.99 | 6.26 | 2.33 |
| 20 | 3.09 | 1.82 | 0.99 | 2.52 | −0.59 | 2.09 | 6.44 | 2.60 |
| 21 | 3.78 | 2.74 | 1.59 | 3.19 | −0.09 | 2.73 | 7.07 | 3.22 |
| 22 | 2.29 | 0.56 | −0.03 | 1.60 | −1.47 | 1.10 | 5.46 | 1.64 |
| 23 | 2.29 | 0.75 | 0.40 | 2.08 | −1.24 | 1.65 | 5.90 | 2.01 |
| 24 | 2.77 | 1.22 | 0.63 | 2.26 | −0.97 | 1.74 | 6.08 | 2.27 |
| 25 | 3.18 | 2.31 | 1.07 | 2.71 | −0.36 | 2.16 | 6.71 | 2.90 |
| 26 | 3.18 | 1.78 | 1.31 | 2.78 | −0.35 | 2.16 | 6.67 | 3.19 |
| 27 | 3.28 | 1.98 | 1.74 | 3.27 | −0.14 | 2.71 | 7.11 | 3.56 |
| 28 | 3.69 | 2.36 | 1.97 | 3.45 | 0.12 | 2.81 | 7.29 | 3.82 |
| 29 | 4.12 | 3.19 | 2.57 | 4.11 | 0.60 | 3.45 | 7.92 | 4.45 |
| RMSE | 1.28 | 1.96 | 0.43 | 3.52 | 0.89 | 3.47 | 0.36 | |
| Comp. | RP-HPLC log Po/w | ALOGPs | ALOGPs LIBRARY |
|---|---|---|---|
| 14 | 1.88 | 0.63 | 1.93 |
| 15 | 2.38 | 1.25 | 2.43 |
| 16 | 2.00 | 0.80 | 2.08 |
| 17 | 2.87 | 2.03 | 3.04 |
| 18 | 2.57 | 1.14 | 2.40 |
| 19 | 3.09 | 1.82 | 2.92 |
| 20 | 2.69 | 1.29 | 2.54 |
| 21 | 3.78 | 2.74 | 3.54 |
| 22 | 2.29 | 0.56 | 1.90 |
| 23 | 2.77 | 1.22 | 2.41 |
| 24 | 2.29 | 0.75 | 2.06 |
| 25 | 3.18 | 2.31 | 3.19 |
| 26 | 3.18 | 1.78 | 3.18 |
| 27 | 3.69 | 2.36 | 3.67 |
| 28 | 3.28 | 1.98 | 3.33 |
| 29 | 4.12 | 3.19 | 4.32 |
| RMSE | 1.29 | 0.19 |
In Silico Screening of the QSAR Relationship
3. Experimental Section
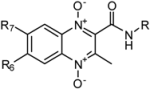
3.1. Chemical Synthesis
3.2. RP-HPLC Method
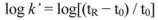

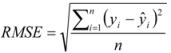
3.3. Cross-Validation of the RP-HPLC Method

 is the mean of the reference shake-flask log Po/w value and n is the number of reference compounds. A high value of R2LOO indicates a good predictive ability of the model. The root mean squared error in prediction (RMSELOO) is calculated with Equation (6):
is the mean of the reference shake-flask log Po/w value and n is the number of reference compounds. A high value of R2LOO indicates a good predictive ability of the model. The root mean squared error in prediction (RMSELOO) is calculated with Equation (6):
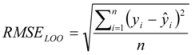
3.4. log Po/w Predictive Approaches
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflict of Interest
References
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef]
- Carta, A.; Corona, P.; Loriga, M. Quinoxaline 1,4-dioxide: A versatile scaffold endowed with manifold activities. Curr. Med. Chem. 2005, 12, 2259–2272. [Google Scholar] [CrossRef]
- Amin, K.M.; Ismail, M.F.; Noaman, E.; Soliman, D.H.; Ammar, Y.A. New quinoxaline 1,4-di-N-oxides. Part 1: Hypoxia-selective cytotoxins and anticancer agents derived from quinoxaline 1,4-di-N-oxides. Bioorg. Med. Chem. 2006, 14, 6917–6923. [Google Scholar]
- Solano, B.; Junnotula, V.; Marín, A.; Villar, R.; Burguete, A.; Vicente, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A.; Dutta, S.; et al. Synthesis and biological evaluation of new 2-arylcarbonyl-3-trifluoromethylquinoxaline 1,4-di-N-oxide derivatives and their reduced analogs. J. Med. Chem. 2007, 50, 5485–5492. [Google Scholar] [CrossRef]
- Carta, A.; Loriga, M.; Paglietti, G.; Mattana, A.; Fiori, P.L.; Mollicotti, P.; Sechi, L.; Zanetti, S. Synthesis, anti-mycobacterial, anti-trichomonas and anti-candida in vitro activities of 2-substituted-6,7-difluoro-3-methylquinoxaline 1,4-dioxides. Eur. J. Med. Chem. 2004, 39, 195–203. [Google Scholar] [CrossRef]
- Carta, A.; Paglietti, G.; Nikookar, M.E.R.; Sanna, P.; Sechi, L.; Zanetti, S. Novel substituted quinoxaline 1,4-dioxides with in vitro antimycobacterial and anticandida activity. Eur. J. Med. Chem. 2002, 37, 355–366. [Google Scholar] [CrossRef]
- Kurt, L.; Ulrich, E. U.S. Patent 3,660,398, 1972.
- Kurt, L.; Ulrich, E. U.S. Patent 3,686,401, 1972.
- Abu El-Haj, M.J. DE Patent 19732316765, 1973.
- Pfizer. FR Patent 19750000544, 1975.
- Cronin, T.; Richardson, K. U.S. Patent 19,710,135,792, 19710420 1974.
- Aguirre, G.; Cerecetto, H.; Di Maio, R.; González, M.; Montoya, M.E.; Jaso, A.; Zarranz, B.; Ortega, M.A.; Aldana, I.; Monge, A. Quinoxaline N,N′-dioxide derivatives and related compounds as growth inhibitors of Trypanosoma cruzi. Structure-activity relationships. Bioorg. Med. Chem. Lett. 2004, 14, 3835–3839. [Google Scholar]
- Urquiola, C.; Vieites, M.; Aguirre, G.; Marin, A.; Solano, B.; Arrambide, G.; Noblía, P.; Lavaggi, M.L.; Torre, M.H.; González, M.; et al. Improving anti-trypanosomal activity of 3-aminoquinoxaline-2-carbonitrile N-1, N-4-dioxide derivatives by complexation with vanadium. Bioorg. Med. Chem. 2006, 14, 5503–5509. [Google Scholar] [CrossRef]
- Deel, P. EG Patent 19,730,000,352, 1977.
- Lepant, M.; Laruelle, C. FR Patent 19,850,000,777, 1986.
- Berthod, A.; Carda-Broch, S. Determination of liquid-liquid partition coefficients by separation methods. J. Chromatogr. A 2004, 1037, 3–14. [Google Scholar]
- Fernandez, L.; Santo, M.; Reta, M.; Giacomelli, L.; Cattana, R.; Silber, J.; Risso, M.; Cerecetto, H.; González, M.; Olea-Azar, C. Relationship between physicochemical properties and herbicidal activity of 1,2,5-oxadiazole N-oxide derivatives. Molecules 2005, 10, 1197–1208. [Google Scholar] [CrossRef]
- Poole, S.K.; Poole, C.F. Separation methods for estimating octanol-water partition coefficients. J. Chromatogr. B 2003, 797, 3–19. [Google Scholar] [CrossRef]
- Vrakas, D.; Tsantili-Kakoulidou, A.; Hadjipavlou-Litina, D. Exploring the consistency of log P estimation for substituted coumarins. QSAR Comb. Sci. 2003, 22, 622–629. [Google Scholar] [CrossRef]
- Stella, C.; Galland, A.; Liu, X.; Testa, B.; Rudaz, S.; Veuthey, J.L.; Carrupt, P.A. Novel RPLC stationary phases for lipophilicity measurement: Solvatochromic analysis of retention mechanisms for neutral and basic compounds. J. Sep. Sci. 2005, 28, 2350–2362. [Google Scholar] [CrossRef]
- Tetko, I.V.; Poda, G.I. Application of ALOGPS 2.1 to predict log D distribution coeffficient for Pfizer propietary compounds. J. Med. Chem. 2004, 47, 5601–5604. [Google Scholar] [CrossRef]
- Dias, N.C.; Nawas, M.I.; Poole, C.F. Evaluation of a reversed-phase column (Supelcosil LC-ABZ) under isocratic and gradient elution conditions for estimating octanol-water partition coefficients. Analyst 2003, 128, 427–433. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- OECD. OECD Guideline for the Testing of Chemicals. Partition Coefficient (n-octanol/water): Shake Flask Method; OECD: Paris, France, 1995.
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Valkó, K. Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J. Chromatogr. A 2004, 1037, 299–310. [Google Scholar] [CrossRef]
- Platts, J.A.; Ermondi, G.; Caron, G.; Ravera, M.; Gabano, E.; Pelosi, G.; Osella, D. Molecular and statistical modeling of reduction peak potential and lipophilicity of platinum (IV) complexes. J. Biol. Inor. Chem. 2011, 16, 361–372. [Google Scholar] [CrossRef]
- Platts, J.A.; Hibbs, D.E.; Hambley, T.W.; Hall, M.D. Calculation of the hydrophobicity of platinum drugs. J. Med. Chem. 2001, 44, 472–474. [Google Scholar] [CrossRef]
- Hansch, C.; Björkroth, J.P.; Leo, A.J. Hydrophobicity and central nervous system agents: On the principle of minimal hydrophobicity in drug design. J. Pharm. Sci. 1987, 76, 663–687. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. ρ-σ-π Analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626. [Google Scholar] [CrossRef]
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients. Nature 1962, 194, 178–180. [Google Scholar]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Wang, R.; Gao, Y.; Lai, L. Calculating partition coefficient by atom-additive method. Perspect. Drug Discov. Des. 2000, 19, 47–66. [Google Scholar] [CrossRef]
- Tetko, I.V.; Tanchuk, V.Y.; Kasheva, T.N.; Villa, A.E.P. Internet software for the calcualtion of the lipophilicity and aqueous solubility of chemical compounds. J. Chem. Inf. Comput. Sci. 2001, 41, 246–252. [Google Scholar] [CrossRef]
- Haddadin, M.; Issidorides, C. The Beirut reaction. Heterocycles 1993, 35, 1503–1523. [Google Scholar] [CrossRef]
- Albini, A.; Alpegiani, M. The photochemistry of the N-oxide function. Chem. Rev. 1984, 84, 43–71. [Google Scholar]
- Kawata, H.; Kikuchi, K.; Kokubun, H. Studies of the photoreactions of heterocyclic N,N-oxides: identifiaction of the oxaziridine of quinoxaline 1,4-dioxide. J. Photochem. 1983, 21, 343–349. [Google Scholar]
- OECD. OECD Guideline for Testig of Chemicals. Partition Coefficient (n-Octanol/Water), High Performance Liquid Chromatography (HPLC) Method; OECD: Paris, France, 1989.
- Ancizu, S.; Moreno, E.; Solano, B.; Villar, R.; Burguete, A.; Torres, E.; Pérez-Silanes, S.; Aldana, I.; Monge, A. New 3-methylquinoxaline-2-carboxamide 1,4-di-N-oxide derivatives as anti-Mycobacterium tuberculosis agents. Bioorg. Med. Chem. 2010, 18, 2713–2719. [Google Scholar]
- Moreno, E.; Ancizu, S.; Pérez-Silanes, S.; Torres, E.; Aldana, I.; Monge, A. Synthesis and antimycobacterial activity of new quinoxaline-2-carboxamide 1,4-di-N-oxide derivatives. Eur. J. Med. Chem. 2010, 45, 4418–4426. [Google Scholar] [CrossRef]
- Virtual Computational Chemistry Laboratory. Available online: http://www.vcclab.org/ (accessed on 17 November 2010).
- Ducati, R.G.; Ruffino-Netto, A.; Basso, L.A.; Santos, D.S. The resumption of consumption-A review on tuberculosis. Mem. Inst. Oswaldo Cruz. 2006, 101, 697–714. [Google Scholar]
- Ekins, S.; Freundlich, J.S.; Choi, I.; Sarker, M.; Talcott, C. Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends. Microbiol. 2011, 19, 65–74. [Google Scholar] [CrossRef]
- Moreno, E.; Pérez-Silanes, S.; Gouravaram, S.; Macharam, A.; Ancizu, S.; Torres, E.; Aldana, I.; Monge, A.; Crawford, P.W. 1,4-di-N-oxide quinoxaline-2-carboxamide: Cyclic voltammetry and relationship between electrochemical behavior, structure and anti-tuberculosis activity. Electrochim. Acta 2011, 56, 3270–3275. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE). Available online: http://www.chemcomp.com/software.htm/ (accessed on 23 November 2010).
- Carey, F.A.; Sundeberg, R.J. Aromatic Substitution Reactions. In Advanced Organic Chemistry. Part B: Reactions and Synthesis, 5th ed; Springer: New York, NY, USA, 2007; Volume 2. [Google Scholar]
- Haddadin, M.; Issidorides, C. Enamines with isobenzofuroxan: a novel synthesis of quinoxaline-di-N-oxides. Tetrahedron Lett. 1965, 36, 3253–3256. [Google Scholar] [CrossRef]
- Haddadin, M.; Taha, M.; Jarrar, A.; Issidorides, C. Reaction of benzofurazan oxide with unsymmetrical 1,3-diketones; Steric and polar effects. Tetrahedron 1976, 32, 719–724. [Google Scholar] [CrossRef]
- Gasco, A.; Boulton, J. Furoxans and benzofuroxans. Adv. Heterocycl. Chem. 1981, 29, 251–340. [Google Scholar] [CrossRef]
- Stumm, G.; Niclas, H.J. An improved and efficient synthesis of quinoxalinecarboxamide 1,4-dioxides from benzofuroxan and acetoacetamides in the presence of calcium salts. J. fur Prakt. Chemie. 1989, 331, 736–744. [Google Scholar] [CrossRef]
- Minick, D.J.; Frenz, J.H.; Patrick, M.A.; Brent, D.A. A comprehensive method for determining hydrophobicity constants by reversed-phase high-performance liquid chromatography. J. Med. Chem. 1988, 31, 1923–1933. [Google Scholar] [CrossRef]
- Lombardo, F.; Shalaeva, M.Y.; Tupper, K.A.; Gao, F.; Abraham, M.H. ElogPoct: A tool for lipophilicity determination in drug discovery. Med. Chem. 2000, 43, 2922–2928. [Google Scholar] [CrossRef]
- Pomper, M.G.; van Brocklin, H.; Thieme, A.M.; Thomas, R.D.; Kiesewetter, D.O.; Carlson, K.E.; Mathias, C.J.; Welch, M.J.; Katzenellenbogen, J.A. 11β-Methoxy-, 11 β -ethyl, and 17α-ethynyl-substituted 16.alpha.-fluoroestradiols: Receptor-based imaging agents with enhanced uptake efficiency and selectivity. Med. Chem. 1990, 33, 3143–3155. [Google Scholar] [CrossRef]
- Gasco, A.; Boulton, J. Furoxans and benzofuroxans. Adv. Heterocycl. Chem. 1981, 29, 251–340. [Google Scholar] [CrossRef]
- Platts, J.A.; Oldfield, S.P.; Reif, M.M.; Palmucci, A.; Gabano, E.; Osella, D. The RP-HPLC measurement and QSPR analysis of log Po/w values of several Pt(II) complexes. J. Inorg. Biochem. 2006, 100, 1199–1207. [Google Scholar] [CrossRef]
- SRC. Interactive LogKow (KowWin). Available online: http://www.syrres.com/what-we-do/product.aspx?id=854/ (accessed on 18 November 2010).
- Molinspiration cheminformatics. Cheminformatics on the web. Available online: http://www.molinspiration.com/ (accessed on 18 November 2011).
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, 1st ed; WILEY-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Sample Availability: Samples of all compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moreno, E.; Gabano, E.; Torres, E.; Platts, J.A.; Ravera, M.; Aldana, I.; Monge, A.; Pérez-Silanes, S. Studies on Log Po/w of Quinoxaline di-N-Oxides: A Comparison of RP-HPLC Experimental and Predictive Approaches. Molecules 2011, 16, 7893-7908. https://doi.org/10.3390/molecules16097893
Moreno E, Gabano E, Torres E, Platts JA, Ravera M, Aldana I, Monge A, Pérez-Silanes S. Studies on Log Po/w of Quinoxaline di-N-Oxides: A Comparison of RP-HPLC Experimental and Predictive Approaches. Molecules. 2011; 16(9):7893-7908. https://doi.org/10.3390/molecules16097893
Chicago/Turabian StyleMoreno, Elsa, Elisabetta Gabano, Enrique Torres, James A. Platts, Mauro Ravera, Ignacio Aldana, Antonio Monge, and Silvia Pérez-Silanes. 2011. "Studies on Log Po/w of Quinoxaline di-N-Oxides: A Comparison of RP-HPLC Experimental and Predictive Approaches" Molecules 16, no. 9: 7893-7908. https://doi.org/10.3390/molecules16097893
APA StyleMoreno, E., Gabano, E., Torres, E., Platts, J. A., Ravera, M., Aldana, I., Monge, A., & Pérez-Silanes, S. (2011). Studies on Log Po/w of Quinoxaline di-N-Oxides: A Comparison of RP-HPLC Experimental and Predictive Approaches. Molecules, 16(9), 7893-7908. https://doi.org/10.3390/molecules16097893