Molecular Recognition Studies on Naphthyridine Derivatives



Abstract
:
1. Introduction
2. Results and Discussion
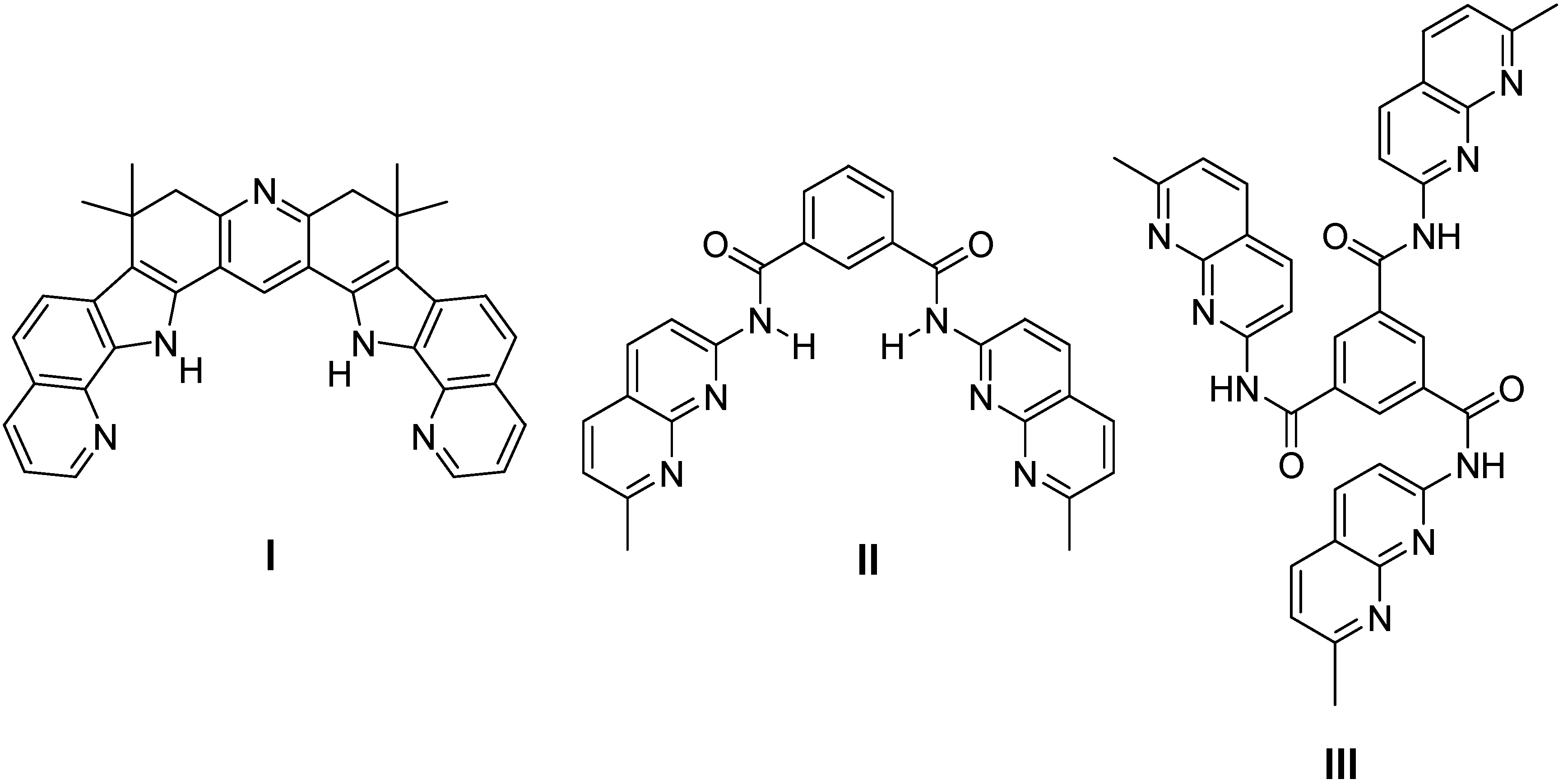
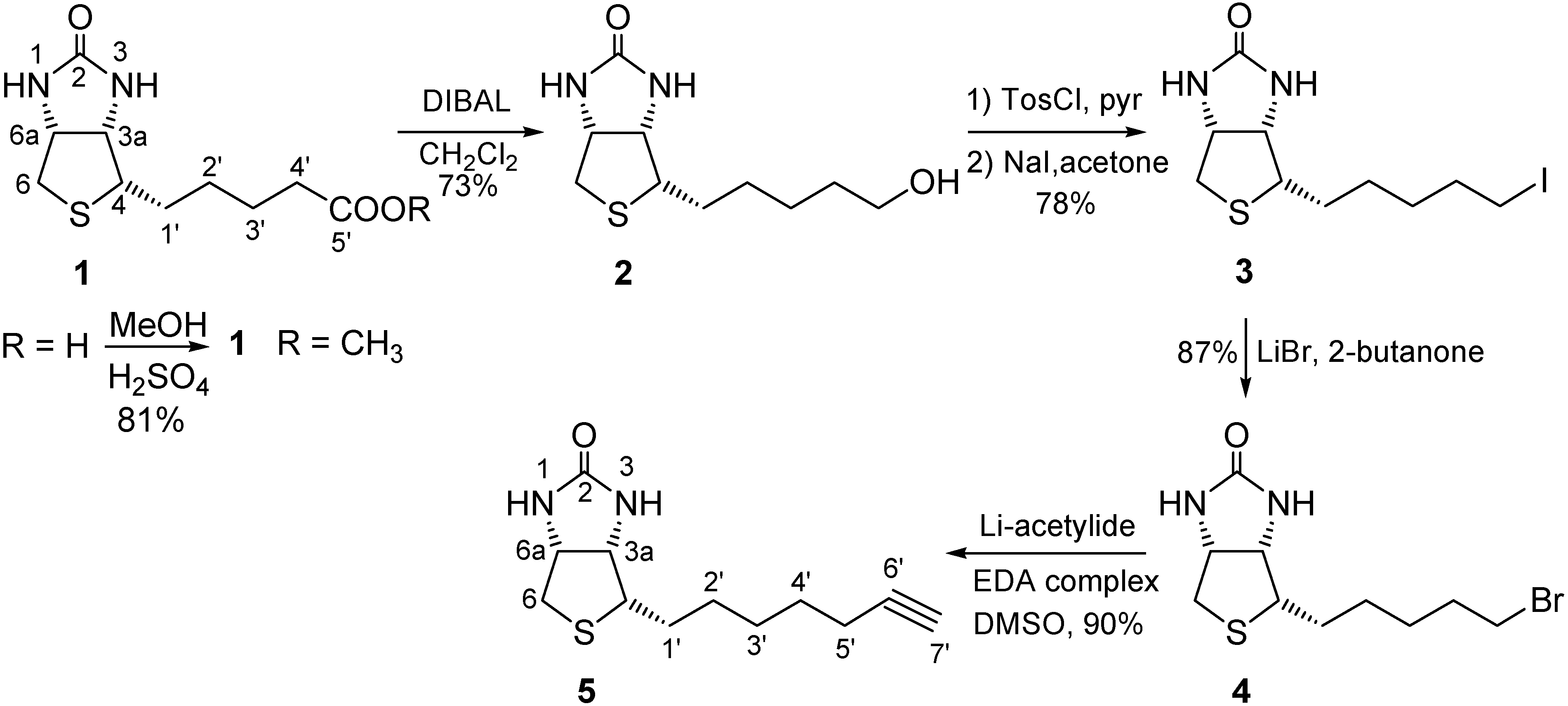
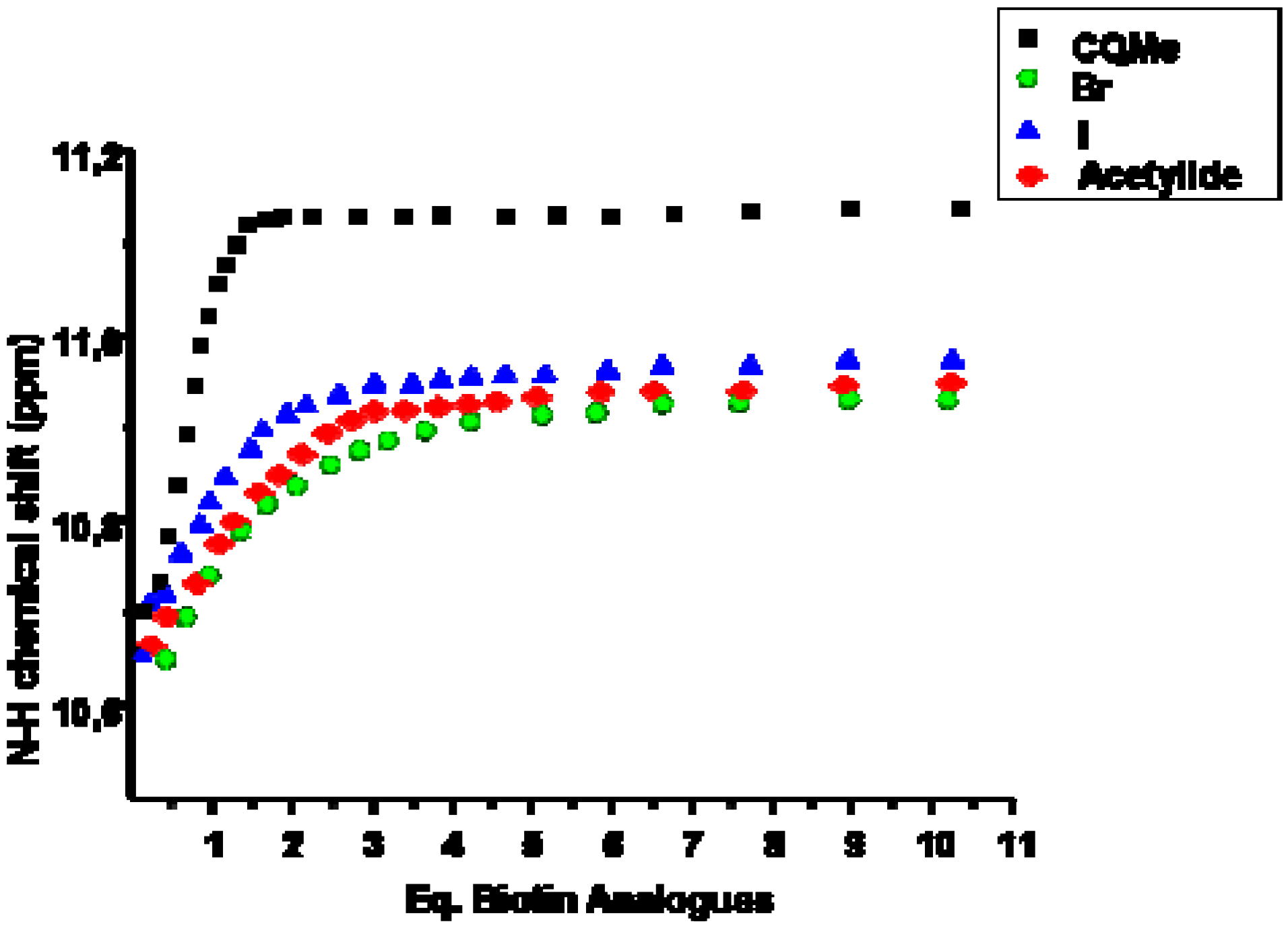
2.1. Binding Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | ||||
|---|---|---|---|---|---|---|
| Kb[a] | Kb[b] | Kb[a] | Kb[b] | Kb[a] | Kb[b] | |
| 1 | 2,700 | 4,200 | 67,000 | 27,200 | 250,000 | 148,000 |
| 100[c] | ||||||
| 2 | [d] | [d] | [d] | [d] | [d] | [d] |
| 110[c] | ||||||
| 3 | 1,500 | 600 | 5,600 | 1,500 | 800 | 150 |
| 4 | 2,000 | 630 | 6,020 | 1,900 | 1,700 | 540 |
| 5 | 1,900 | 244 | 4,700 | 1,000 | 1,000 | 180 |
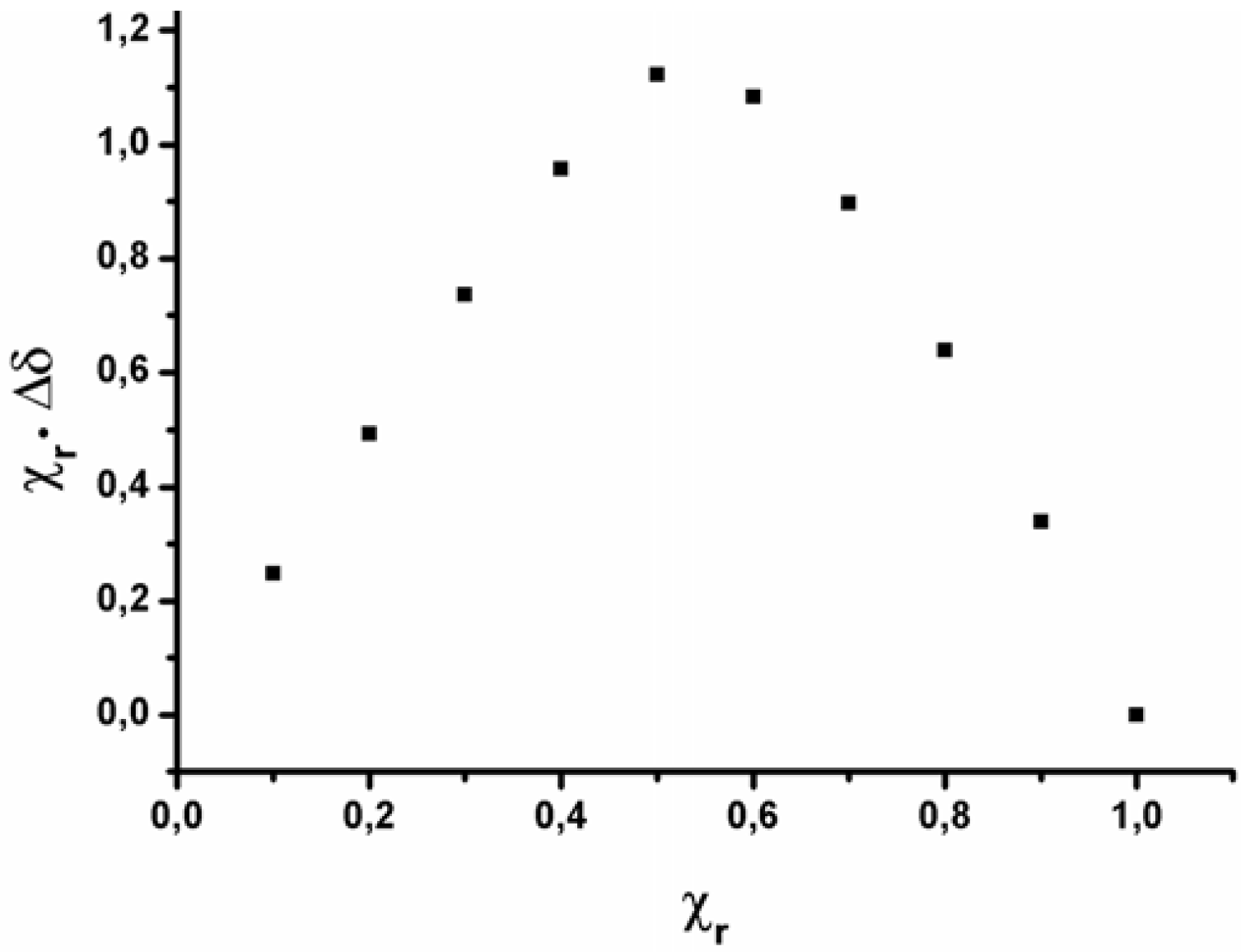
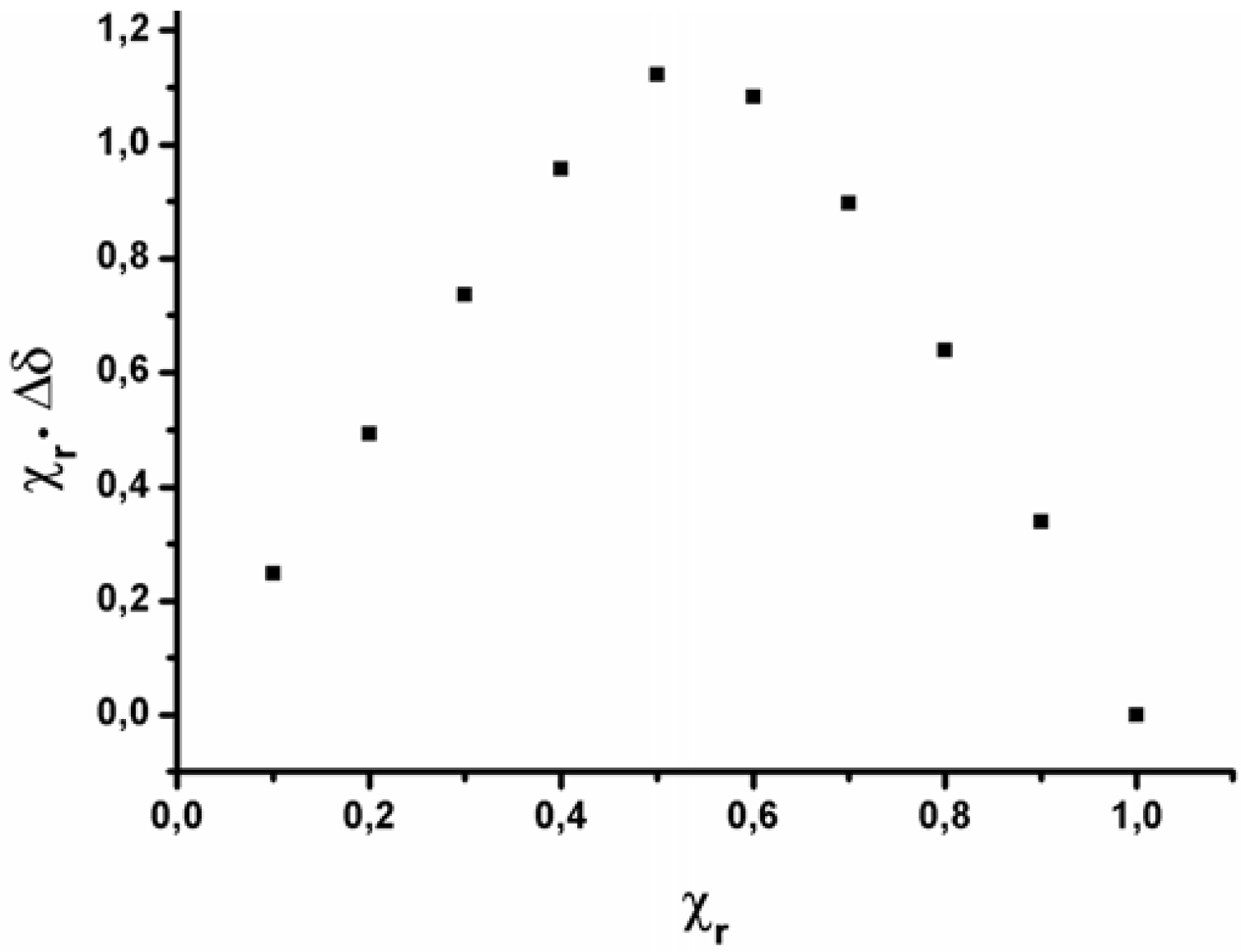
2.2. Data analysis
| Compound | Kb | HI | HII | HIII | G1 | G2 | G3 | G4 | ln Kb |
|---|---|---|---|---|---|---|---|---|---|
| I·1 | 10,447 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 9.25 |
| II·1 | 49,277 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 10.80 |
| III·1 | 253,219 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 12.44 |
| I·2 | 11,156 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 9.32 |
| II·2 | 53,869 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 10.89 |
| III·2 | 278,206 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 12.54 |
| I·3 | 4,370 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 8.38 |
| II·3 | 4,707 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 8.46 |
| III·3 | 3,610 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 8.19 |
| I·4 | 4,420 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 8.39 |
| II·4 | 6,564 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 8.79 |
| III·4 | 4,268 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 8.36 |
| I·5 | 3,769 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 8.23 |
| II·5 | 5,045 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 8.53 |
| III·5 | 3,661 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 8.20 |
3. Experimental
3.1. General
3.2. Synthesis
3.3. NMR titrations
| Volume of added guest (I) (μL) | Total-Volume (μL) | [host I] (10-3 M) | [guest 1] (10-3 M) | Equivalents of added guest | δ (NH) (ppm) |
|---|---|---|---|---|---|
| 0 | 500 | 8.40 | 0 | 0 | 10.7474 |
| 5 | 505 | 8.32 | 0.77 | 0.09268 | 10.8176 |
| 10 | 510 | 8.24 | 1.53 | 0.18537 | 10.8729 |
| 20 | 520 | 8.08 | 3.00 | 0.37074 | 10.9920 |
| 30 | 530 | 7.93 | 4.41 | 0.55611 | 11.0745 |
| 40 | 540 | 7.78 | 5.77 | 0.74147 | 11.1484 |
| 65 | 565 | 7.44 | 8.96 | 1.20489 | 11.2488 |
| 90 | 590 | 7.12 | 11.88 | 1.66832 | 11.2949 |
| 115 | 615 | 6.83 | 14.56 | 2.13174 | 11.3352 |
| 215 | 715 | 5.88 | 23.42 | 3.98542 | 11.4600 |
| 315 | 815 | 5.15 | 30.10 | 5.83910 | 11.5452 |
| 565 | 1,065 | 3.94 | 41.31 | 10.47332 | 11.6432 |
| 815 | 1,315 | 3.19 | 48.27 | 15.10753 | 11.6570 |
| 915 | 1,415 | 2.97 | 50.36 | 16.96121 | 11.6582 |
| Volume of added guest (I) (μL) | Total-Volume (μL) | [host I] (10-4 M) | [guest 1] (10-4 M) | Equivalents of added guest | δ (NH) (ppm) |
|---|---|---|---|---|---|
| 20 | 520 | 4.14 | 0.355 | 0.08588 | 10.8521 |
| 30 | 530 | 4.07 | 0.523 | 0.12882 | 11.0125 |
| 40 | 540 | 3.99 | 0.686 | 0.17176 | 11.1025 |
| 65 | 565 | 3.81 | 1.060 | 0.27911 | 11.1730 |
| 90 | 590 | 3.65 | 1.410 | 0.38646 | 11.2587 |
| 115 | 615 | 3.50 | 1.730 | 0.49380 | 11.2925 |
| 215 | 715 | 3.01 | 2.780 | 0.92320 | 11.3014 |
| 315 | 815 | 2.64 | 3.580 | 1.35259 | 11.3514 |
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds, hosts I, II, III and guests 1–5 are available from authors.
References and Notes
- Kumar, S.; Singh, H.; Sharma, R. Role of amide and urea moieties in molecular recognition. J. Indian Chem. Soc. 2003, 80, 1111–1128. [Google Scholar]
- Bell, T.W.; Hext, N.M. Supramolecular optical chemosensors for organic analytes. Chem. Soc. Rev. 2004, 33, 589–598. [Google Scholar]
- Gale, P.A.; Quesada, R. Anion coordination and anion-templated assembly: highlights from 2002 to 2004. Coord. Chem. Rev. 2006, 250, 3219–3244. [Google Scholar] [CrossRef]
- Kang, S.O.; Begum, R.A.; Bowman-James, K. Amide-based ligands for anion coordination. Angew. Chem. Int. Ed. 2006, 45, 7882–7894. [Google Scholar]
- Steed, J.W.; Turner, D.R.; Wallace, K.J. The host component is defined as an organic molecule or ion whose binding sites converge in the complex. The guest component is any molecule or ion whose binding sites diverge in the complex. In Core Concepts in Supramolecular Chemistry and Nanochemistry; John Wiley & Sons: Chichester, UK, 2007; p. 2. [Google Scholar]
- Claramunt, R.M.; Herranz, F.; Santa María, M.D.; Jaime, C.; de Federico, M.; Elguero, J. Towards the design of host–guest complexes: biotin and urea derivatives versus artificial receptors. Biosens. Bioelectron. 2004, 20, 1242–1249. [Google Scholar] [CrossRef]
- Claramunt, R.M.; Herranz, F.; Santa María, M.D.; Pinilla, E.; Torres, M.R.; Elguero, J. Molecular recognition of biotin, barbital and tolbutamide with new synthetic receptors. Tetrahedron 2005, 61, 5089–5100. [Google Scholar] [CrossRef]
- Herranz, F.; Santa María, M.D.; Claramunt, R.M. Molecular recognition: improved binding of biotin derivatives with synthetic receptors. J. Org. Chem. 2006, 71, 2944–2951. [Google Scholar]
- Herranz, F.; Santa María, M.D.; Claramunt, R.M. Host-guest chemistry of tolbutamide. Molecules 2006, 11, 478–485. [Google Scholar] [CrossRef]
- Herranz, F.; Santa María, M.D.; Claramunt, R.M. A new tool for the rational design of methylbiotin hosts. Tetrahedron Lett. 2006, 47, 9017–9020. [Google Scholar] [CrossRef]
- Santa María, D.; Claramunt, R.M.; Herranz, F.; Alkorta, I.; Elguero, J. A theoretical and experimental NMR study of (+)-biotin methyl ester. J. Mol. Struct. 2009, 920, 323–326. [Google Scholar] [CrossRef]
- Hedge, V.; Hung, C.-Y.; Madhukar, P.; Cunningham, R.; Höpfner, T.; Thummel, R.P. Design of receptors for urea derivatives based on the pyrido[3,2-g]indole subunit. J. Am. Chem. Soc. 1993, 115, 872–878. [Google Scholar] [CrossRef]
- Goswami, S.; Mukherjee, R. Molecular recognition: a simple dinaphthyridine receptor for urea. Tetrahedron Lett. 1997, 38, 1619–1622. [Google Scholar] [CrossRef]
- Dixon, R.W.; Radmer, R.J.; Kuhn, B.; Kollman, P.A. Theoretical and experimental studies of biotin analogues that bind almost as tightly to streptavidin as biotin. J. Org. Chem. 2002, 67, 1827–1837. [Google Scholar] [CrossRef]
- Corona, C.; Bryant, B.K.; Arterburn, J.B. Synthesis of a biotin-derived alkyne for Pd-catalyzed coupling reactions. Org. Lett. 2006, 8, 1883–1886. [Google Scholar] [CrossRef]
- Job, P. Studies on the formation of complex minerals in solution and on their stability. Ann. Chim. 1928, 9, 113–203. [Google Scholar]
- Connors, K.A. Binding Constants: The Measurement of Molecular Complex Stability; Wiley-Interscience: New York, NY, USA, 1987. [Google Scholar]
- Schneider, H.-J.; Yatsimirsky, A.K. Principles and methods in supramolecular chemistry; John Wiley & Sons: The Atrium, Southern Gate, Chichester, West Sussex, UK, 2000. [Google Scholar]
- Hynes, M.J. EQNMR: a computer program for the calculation of stability constants from nuclear magnetic resonance chemical shift data. J. Chem. Soc., Dalton Trans. 1993, 311–312. [Google Scholar] [CrossRef]
- Kubinyi, H. Free Wilson analysis. Theory, applications and its relationship to Hansch analysis. QSAR 1988, 7, 121–133. [Google Scholar]
- Alkorta, I.; Blanco, F.; Elguero, J. Application of Free-Wilson matrices to the analysis of the tautomerism and aromaticity DFT study. Tetrahedron 2008, 19, 3826–3836. [Google Scholar]
- DeLaLuz, P.J.; Golinski, M.; Watt, D.S.; Vanaman, T.C. Synthesis and use of a biotinylated 3-azidophenothiazine to photolabel both amino- and carboxyl-terminal sites in calmodulin. Bioconjugate Chem. 1995, 6, 558. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iglesias-Sánchez, J.C.; María, D.S.; Claramunt, R.M.; Elguero, J. Molecular Recognition Studies on Naphthyridine Derivatives. Molecules 2010, 15, 1213-1222. https://doi.org/10.3390/molecules15031213
Iglesias-Sánchez JC, María DS, Claramunt RM, Elguero J. Molecular Recognition Studies on Naphthyridine Derivatives. Molecules. 2010; 15(3):1213-1222. https://doi.org/10.3390/molecules15031213
Chicago/Turabian StyleIglesias-Sánchez, José Carlos, Dolores Santa María, Rosa M. Claramunt, and José Elguero. 2010. "Molecular Recognition Studies on Naphthyridine Derivatives" Molecules 15, no. 3: 1213-1222. https://doi.org/10.3390/molecules15031213
APA StyleIglesias-Sánchez, J. C., María, D. S., Claramunt, R. M., & Elguero, J. (2010). Molecular Recognition Studies on Naphthyridine Derivatives. Molecules, 15(3), 1213-1222. https://doi.org/10.3390/molecules15031213