trans-Resveratrol as A Neuroprotectant


Abstract
:1. Introduction
2. RES as a Neuroprotectant—Experimental Evidence

{kind=link}
| Model | Treatment | Stressor | Effect | Reference |
|---|---|---|---|---|
| Cerebellar granule neurons | 24 h of 1-100 µM RES | MPP+ | Antiapoptotic | [1] |
| PC12 cells | 3 h of 100 µM RES | MPP+ | Antiapoptotic | [2] |
| SH-SY5Y cells | 1 h of 5 µM RES | Dopamine | Antiapoptotic | [3] |
| SH-SY5Y cells | 24–72 h of 1–50 uM RES | H2O2, paraquat or menadione | Reduced incidence of cell death | Robb and Stuart unpublished |
| PC12 cells co-cultured with N9 microglia | 27h of 100 µM RES | LPS treatment | Antiapoptotic | [4] |
| Rat hippocampal slices | Acute 100 µM RES | Oxygen-glucose deprivation | Neuroprotective | [5] |
| Mice | 50 mg/kg/day gavage | Middle cerebral artery occlusion | Neuroprotective | [6] |
| Mice | 50–100 mg/kg/day | MPTP | Prevented loss of DA neurons | [7] |
| Mice | Acute RES at 30 mg/kg i.v. | MPTP | Reduced oxidative damage; protected DA neurons | [8] |
| Mice | 10–40 mg/kg/d for 10 wks | 6-OHDA | Reduced neuronal damage | [9] |
| Inducible p25 Alzheimer’s mouse model | 2.5 µg RES injected into lateral brain ventricles | N.A. | Neuroprotective | [10] |
| Tg 19959 mouse Alzheimer’s model | 300 mg/kg/d RES in diet | N.A. | Reduced plaque pathology | [11] |
| Rats | 10–100 µM RES by i.p. injection | Asphyxial cardiac arrest | Neuroprotective | [12] |
2.1. RES ameliorates oxidative stress
2.2. RES as a chemical antioxidant
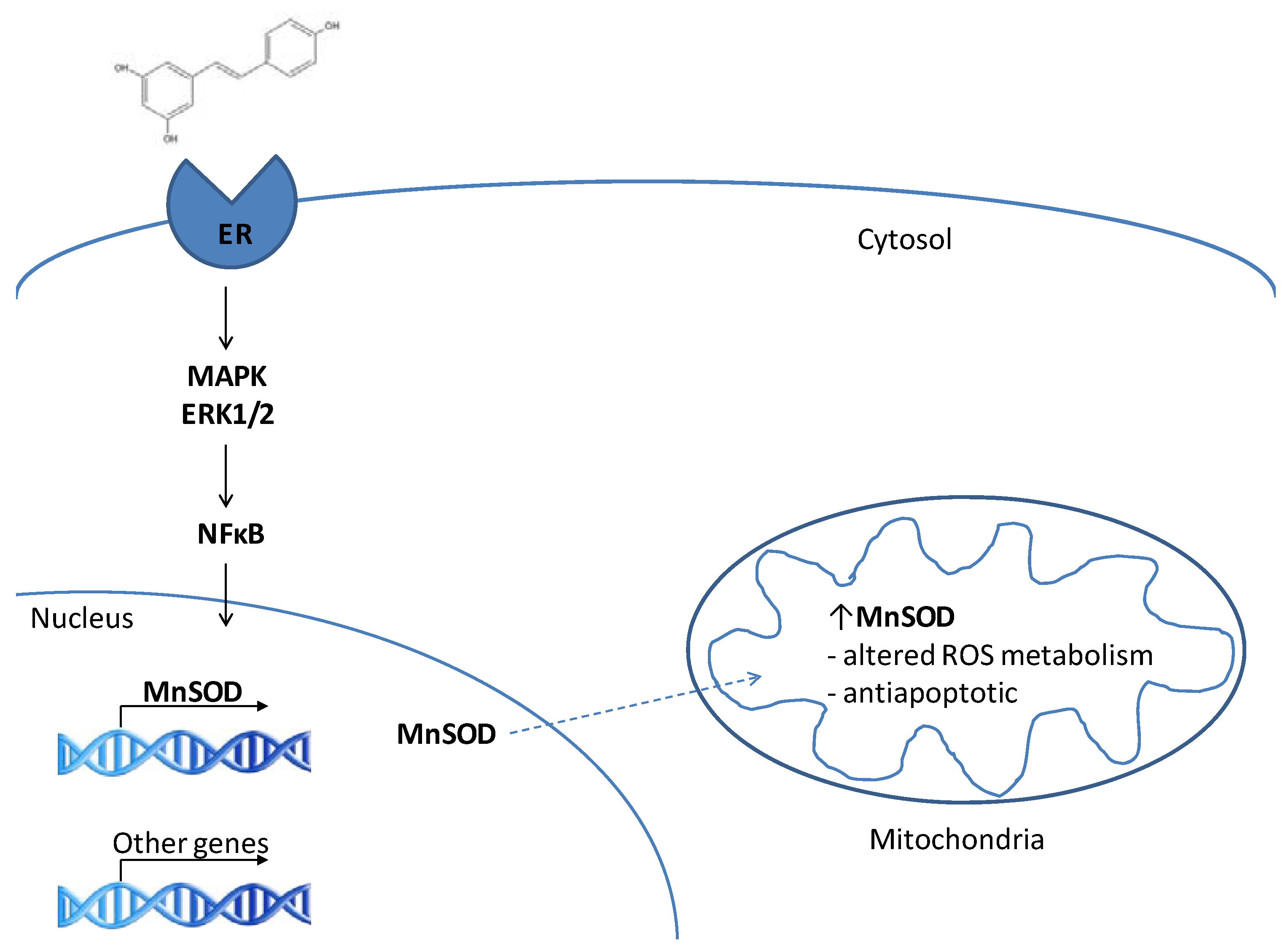
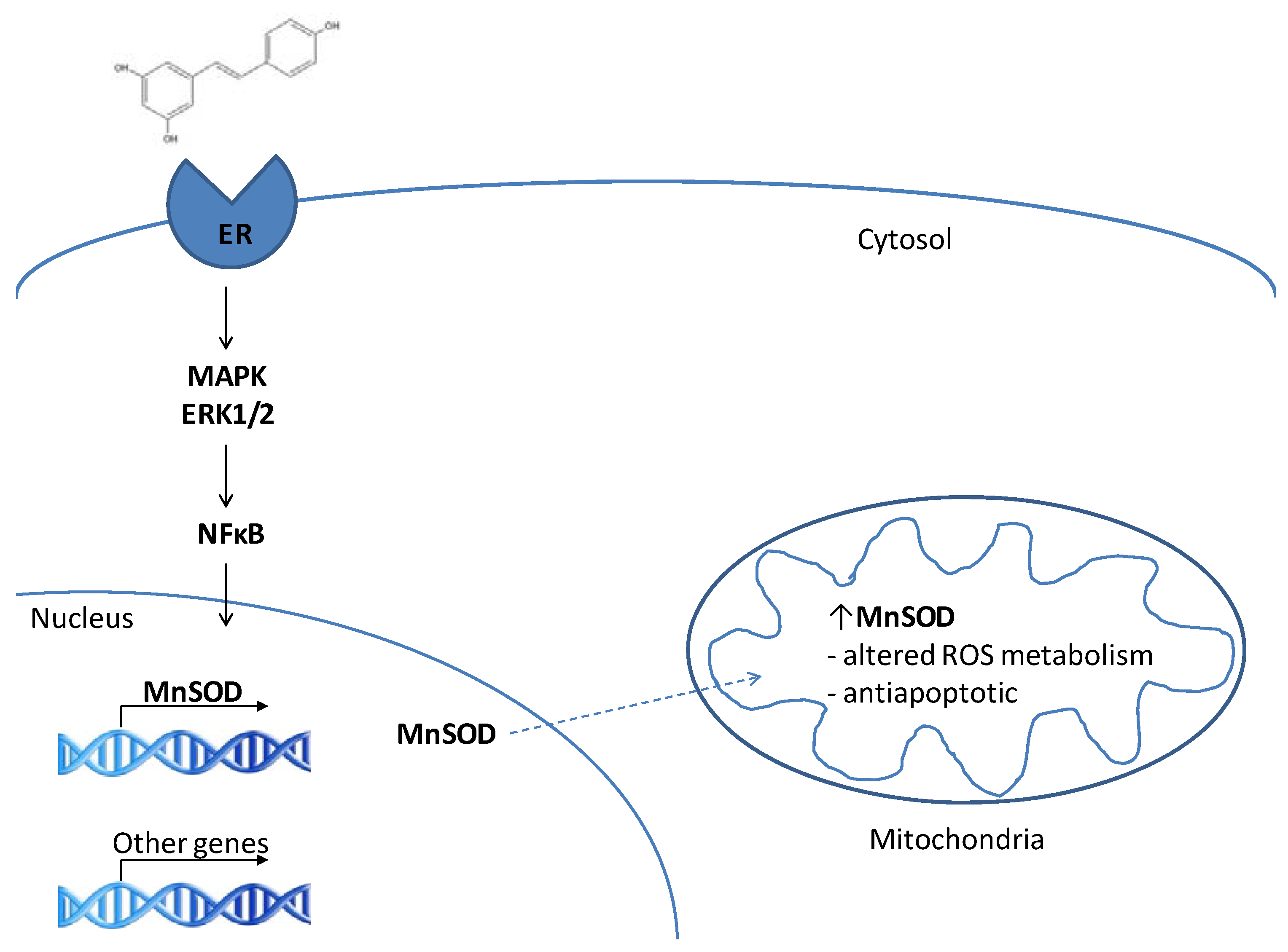
3. RES and MnSOD
3.1. MnSOD is a target of resveratrol
3.2. RES as a phytoestrogen – induction of MnSOD
4. RES and Sirtuins
5. Increasing RES Bioavailability
6. Conclusions
References
- Alvira, D.; Yeste-Velasco, M.; Folch, J.; Verdaguer, E.; Canudas, A.M.; Pallàs, M.; Camins, A. Comparative analysis of the effects of resveratrol in two apoptotic models: inhibition of complex I and potassium deprivation in cerebellar neurons. Neuroscience 2007, 147, 746–756. [Google Scholar] [CrossRef]
- Bournival, J.; Quessy, P.; Martinoli, M.G. Protective Effects of Resveratrol and Quercetin Against MPP(+) -Induced Oxidative Stress Act by Modulating Markers of Apoptotic Death in Dopaminergic Neurons. Cell. Mol. Neurobiol. 2009, 29, 1169–1180. [Google Scholar] [CrossRef]
- Lee, M.K.; Kang, S.J.; Poncz, M.; Song, K.J.; Park, K.S. Resveratrol protects SH-SY5Y neuroblastoma cells from apoptosis induced by dopamine. Exp. Mol. Med. 2007, 39, 376–384. [Google Scholar]
- Bureau, G.; Longpré, F.; Martinoli, M.G. Resveratrol and quercetin, two natural polyphenols, reduce apoptotic neuronal cell death induced by neuroinflammation. J. Neurosci. Res. 2008, 86, 403–410. [Google Scholar] [CrossRef]
- Zhang, H.; Schools, G.P.; Lei, T.; Wang, W.; Kimelberg, H.K.; Zhou, M. Resveratrol attenuates early pyramidal neuron excitability impairment and death in acute rat hippocampal slices caused by oxygen-glucose deprivation. Exp. Neurol. 2008, 212, 44–52. [Google Scholar] [CrossRef]
- Dong, W.; Li, N.; Gao, D.; Zhen, H.; Zhang, X.; Li, F. Resveratrol attenuates ischemic brain damage in the delayed phase after stroke and induces messenger RNA and protein express for angiogenic factors. J. Vasc. Surg. 2008, 48, 709–714. [Google Scholar] [CrossRef]
- Blanchet, J.; Longpré, F.; Bureau, G.; Morissette, M.; DiPaolo, T.; Bronchti, G.; Martinoli, M.G. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1243–1250. [Google Scholar] [CrossRef]
- Lu, K.T.; Ko, M.C.; Chen, B.Y.; Huang, J.C.; Hsieh, C.W.; Lee, M.C.; Chiou, R.Y.; Wung, B.S.; Peng, C.H.; Yang, Y.L. Neuroprotective effects of resveratrol on MPTP-induced neuron loss mediated by free radical scavenging. J. Agric. Food Chem. 2008, 56, 6910–6913. [Google Scholar]
- Jin, F.; Wu, Q.; Lu, Y.F.; Gong, Q.H.; Shi, J.S. Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson's disease in rats. Eur. J. Pharmacol. 2008, 600, 78–82. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; Puigserver, P.; Sinclair, D.A.; Tsai, L.H. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef]
- Della-Morte, D.; Dave, K.R.; DeFazio, R.A.; Bao, Y.C.; Raval, A.P.; Perez-Pinzon, M.A. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 2009, 159, 993–1002. [Google Scholar] [CrossRef]
- Dal-Pan, A.; Guignard, A.; George, E.; Aujard, F.; Picq, J.L. Resveratrol supplementation modified memory and physical performances in a primate. In Proceedings of the 4th International Conference on Polyphenols and Health, Yorkshire, UK, 7-10 December 2009; p. 387.
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef]
- Tsang, A.H.; Chung, K.K. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 643–650. [Google Scholar] [CrossRef]
- Javitch, J.A.; D’Amato, R.H.; Strittmatter, S.M.; Snyder, S.H. Parkinsonism-inducing neurotoxin N-methyl-4-phenyl-1,2,3,6-terahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc. Natl. Acad. Sci. USA 1985, 82, 2173–2177. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035. [Google Scholar]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N.Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Szeto, H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006, 8, E227–E283. [Google Scholar]
- Zhao, K.; Luo, G.; Gianneli, S.; Szeto, H.H. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar]
- Quincozes-Santos, A.; Andreazza, A.C.; Nardin, P.; Funchal, C.; Gonçalves, C.A.; Gottfried, C. Resveratrol attenuates oxidative-induced DNA damage in C6 glioma cells. Neurotoxicity 2007, 28, 886–891. [Google Scholar] [CrossRef]
- Wenzel, E.; Soldo, T.; Erbersdobler, H.; Somoza, V. Bioactivity and metabolism of trans-resveratrol orally administered to Wistar rats. Mol. Nutr. Food Res. 2005, 49, 482–494. [Google Scholar] [CrossRef]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef]
- Guo, L.; Wang, L.H.; Sun, B.; Yang, J.Y.; Zhao, Y.Q.; Dong, Y.X.; Spranger, M.I.; Wu, C.F. Direct in vivo evidence of protective effects of grape seed procyanidin fractions and other antioxidants against ethanol-induced oxidative DNA damage in mouse brain cells. J. Agric. Food Chem. 2007, 5881–5891. [Google Scholar]
- Yousuf, S.; Atif, F.; Ahmad, M.; Hoda, N.; Ishrat, T.; Khan, B.; Islam, F. Resveratrol exerts its neuroprotective effect by modulating mitochondrial dysfunctions and associated cell death during cerebral ischemia. Brain Res. 2009, 1250, 242–253. [Google Scholar] [CrossRef]
- Velioğlu-Oğunc, A.; Sehirli, O.; Toklu, H.Z.; Ozyurt, H.; Mayadağli, A.; Eksioğlu-Demiralp, E.; Erzik, C.; Cetinel, S.; Yeğen, B.C.; Sener, G. Resveratrol protects against irradiation-induced hepatic and ileal damage via its anti-oxidative activity. Free Radic. Res. 2009, 43, 1060–1071. [Google Scholar] [CrossRef]
- Wong, Y.T.; Gruber, J.; Jenner, A.M.; Ng, M.P.; Ruan, R.; Tay, F.E. Elevation of oxidative-damage biomarkers during aging in F2 hybrid mice: protection by chronic oral intake of resveratrol. Free Radic Biol Med. 2009, 46, 799–809. [Google Scholar]
- Yu, W.; Fu, Y.C.; Zhou, X.H.; Chen, C.J.; Wang, X.; Lin, R.B.; Wang, W. Effects of resveratrol on H2O2-induced apoptosis and expression of SIRTs in H9c2 cells. J. Cell Biochem. 2009, 107, 741–747. [Google Scholar] [CrossRef]
- De Almeida, L.M.; Leite, M.C.; Thomazi, A.P.; Battu, C.; Nardin, P.; Tortorelli, L.S.; Zanotto, C.; Posser, T.; Wofchuk, S.T.; Leal, R.B.; Gonçalves, C.A.; Gottfried, C. Resveratrol protects against oxidative injury induced by H2O2 in acute hippocampal slice preparations from Wistar rats. Arch. Biochem. Biophys. 2008, 480, 27–32. [Google Scholar] [CrossRef]
- Vieira de Almeida, L.M.; Pineiro, C.C.; Leite, M.C.; Brolese, G.; Leal, R.B.; Gottfried, C.; Conçalves, C.A. Protective effects of resveratrol on hydrogen peroxide induced toxicity in primary cortical astrocyte cultures. Neurochem. Res. 2008, 33, 8–15. [Google Scholar] [CrossRef]
- Halliwell, B. Are polyphenols antioxidants or pro-oxidants? What do we learn from cell culture and in vivo studies? Arch. Biochem. Biophys. 2008, 476, 1070112. [Google Scholar]
- Ungvari, Z.; Labinskyy, N.; Mukhopadhyay, P.; Pinto, J.T.; Bagi, Z.; Ballabh, P.; Zhang, C.; Pacher, P.; Csiszar, A. Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1876–H1881. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant an antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Robb, E.L.; Stuart, J.A. Resveratrol targets the mitochondrial antioxidant enzyme manganese superoxide dismutase and increases cellular stress resistance. In Proceedings of the 4th International Conference on Polyphenols and Health, Yorkshire, UK, 7-10 December 2009; p. 393.
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: the in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar]
- Hu, Y.; Rahlfs, S.; Mersch-Sundermann, V.; Becker, K. Resveratrol modulates mRNA transcripts of gene related to redox metabolism and cell proliferation in on-small-cell lung carcinoma cells. Biol. Chem. 2007, 388, 207–219. [Google Scholar]
- Leonard, S.S.; Xia, C.; Jiang, B.H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef]
- Wenzel, E.; Somoza, V. metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 2005, 49, 472–481. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Williamson, G.; Barron, D.; Shimoi, K.; Terao, J. In vitro biological properties of flavonoid conjugates found in vivo. Free Radic. Res. 2005, 39, 457–469. [Google Scholar] [CrossRef]
- Keller, J.N.; Kindy, M.S.; Holtsberg, F.W.; St Clair, D.K.; Yen, H.C.; Germeyer, A.; Steiner, S.M.; Bruce-Keller, A.J.; Hutchins, J.B.; Mattson, M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998, 18, 687–697. [Google Scholar]
- Lee, Y.J.; Cho, H.N.; Jeoung, D.I.; Soh, J.W.; Cho, C.K.; Bae, S.; Chung, H.Y.; Lee, S.J.; Lee, Y.S. HSP25 overexpression attenuates oxidative stress-induced apoptosis: roles of ERK1/2 signaling and manganese superoxide dismutase. Free Radic. Biol. Med. 2004, 36, 429–444. [Google Scholar] [CrossRef]
- Cruthirds, D.L.; Saba, H.; MacMillan-Crow, L.A. Overexpression of manganese superoxide dismutase protects against ATP depletion-mediated cell death of proximal tubule cells. Arch. Biochem. Biophys. 2005, 437, 96–105. [Google Scholar] [CrossRef]
- Silva, J.P.; Shabalina, I.G.; Dufour, E.; Petrovic, N.; Backlund, E.C.; Hultenby, K.; Wibom, R.; Nedergaard, J.; Cannon, B.; Larsson, N.G. SOD2 overexpression: enhanced mitochondrial tolerance by absence of effect on UCP activity. EMBO J. 2005, 24, 4061–4070. [Google Scholar] [CrossRef]
- Mattson, M.P.; Goodman, Y.; Luo, H.; Fu, W.; Furukawa, K. Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J. Neurosci. Res. 1997, 49, 681–697. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; Wallace, D.C.; Epstein, C.J. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J. Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; Nelson, J.; Strong, R.; Richardson, A. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate again. Physiol. Genomics 2003, 16, 29–37. [Google Scholar] [CrossRef]
- Loch, T.; Vakhrusheva, O.; Piotrowska, I.; Ziolkowski, W.; Ebelt, H.; Braun, T.; Bober, E. Different extent of cardiac malfunction and resistance to oxidative stress in heterozygous and homozygous manganese-dependent superoxide dismutase-mutant mice. Cardiovasc. Res. 2009, 82, 448–457. [Google Scholar] [CrossRef]
- Klivenyi, P.; St Clair, D.; Wermer, M.; Yen, H.C.; Oberley, T.; Yang, L.; Flint Beal, M. Manganese superoxide dismutase overexpression attenuates MPTP toxicity. Neurobiol. Dis. 1998, 5, 253–258. [Google Scholar] [CrossRef]
- Callio, J.; Oury, T.D.; Chu, C.T. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J. Biol. Chem. 2005, 280, 18536–18542. [Google Scholar]
- Shan, X.; Chi, L.; Ke, Y.; Luo, C.; Qian, S.; Gozal, D.; Liu, R. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiol. Dis. 2007, 28, 206–215. [Google Scholar] [CrossRef]
- Dumont, M.; Wille, E.; Stack, C.; Calingasan, N.Y.; Beal, M.F., Lin. Reduction of oxidative stress, amyloid deposition and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2009, 23, 2459–2466. [Google Scholar] [CrossRef]
- Robb, E.L.; Page, M.M.; Wiens, B.E.; Stuart, J.A. Molecular mechanisms of oxidative stress resistance induced by resveratrol: Specific and progressive induction of MnSOD. Biochem. Biophys. Res. Commun. 2008, 367, 406–412. [Google Scholar] [CrossRef]
- Li, Y.; Cao, Z.; Zhu, H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol. Res. 2006, 53, 6–15. [Google Scholar] [CrossRef]
- Robb, E.L.; Winkelmolen, L.; Visanji, N.; Brotchie, J.; Stuart, J.A. Dietary resveratrol administration increases MnSOD expression and activiyt in mouse brain. Biochem. Biophys. Res. Commun. 2008, 372, 254–259. [Google Scholar] [CrossRef]
- Le Bail, J.C.; Champavier, Y.; Chulia, A.J.; Habrioux, G. Effects of phytoestrogens on aromatase, 3beta and 17beta-hydroxy-steroid dehydrogenase activities and human breast cancer cells. Life Sci. 2000, 66, 1281–1291. [Google Scholar] [CrossRef]
- Bowers, J.L.; Tyulmenkov, V.V.; Jernigan, S.C.; Klinge, C.M. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology 2000, 141, 3657–3667. [Google Scholar]
- Mense, S.M.; Hei, T.K.; Ganju, R.K.; Bhat, H.K. Phytoestrogens and breast cancer prevention: possible mechanisms of action. Environ. Health Perspect. 2008, 116, 426–433. [Google Scholar] [CrossRef]
- Borrás, C.; Gambini, J.; Gómez-Cabrera, M.C.; Sastre, J.; Pallardó, F.V.; Mann, G.E.; Vina, J. 17beta-oestradiol up-regulates longevity-related, antioxidant enzyme expression via the ERK1 and ERK2 MAPK/NFkappaB cascade. Aging Cell 2005, 4, 113–118. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef]
- Borrás, C.; Sastre, J.; Garcia-Sala, D.; Lloret, A.; Pallardó, F.V.; Vina, J. Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic. Biol. Med. 2003, 34, 546–552. [Google Scholar] [CrossRef]
- Chen, J.Q.; Cammarata, P.R.; Baines, C.P.; Yager, J.D. Regulation of mitochondrial respiratory chain biogenesis by estrogens/estrogen receptors and physiological, pathological and pharmacological implications. Biochim. Biophys. Acta 2009, 1793, 1540–1570. [Google Scholar] [CrossRef]
- Sawada, H.; Ibi, M.; Kihara, T.; Urushitani, M.; Akaike, A.; Shimohama, S. Estradiol protects mesencephalic dopaminergic neurons from oxidative stress-induced neuronal death. J. Neurosci. Res. 1998, 54, 717–719. [Google Scholar]
- Razmara, A.; Duckles, S.P.; Krause, D.N.; Procaccio, V. Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007, 1176, 71–81. [Google Scholar] [CrossRef]
- Dali-Youcef, N.; Lagouge, M.; Froelich, S.; Koehl, C.; Schoonjans, K.; Auwerx, J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann. Med. 2007, 39, 335–345. [Google Scholar] [CrossRef]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 1003–1009. [Google Scholar]
- Howitz, K.T.; Bitterman, K.K.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; Scherer, B.; Sinclair, D.A. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; Geny, B.; Laakso, M.; Puigserver, P.; Auwerx, J. Resveratrol improves mitochondrial function and protects against metabolic disease by activation SIRT1 and PGC-1alpha. Cell 2006, 127, 1108–1122. [Google Scholar]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, W.; Allard, J.S.; Lopez-Llunch, G.; Lewis, K.; Pistell, P.J.; Poosala, S.; Becker, K.G.; Boss, O.; Gwinn, D.; Wang, M.; Ramaswamy, S.; Fishbein, K.W.; Spencer, R.G.; Lakatta, E.G.; Le Couteur, D.; Shaw, R.J.; Navas, P.; Puigserver, P.; Ingram, D.K.; de Cabo, R.; Sinclair, D.A. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar]
- Tang, B.L. Sirt1’s complex roles in neuroprotection. Cell. Mol. Neurobiol. 2009, 29, 1093–1103. [Google Scholar] [CrossRef]
- Pfister, J.A.; Ma, C.; Morrison, B.E.; D'Mello, S.R. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One 2008, 3, e4090. [Google Scholar]
- Li, Y.; Xu, W.; McBurney, M.W.; Longo, V.D. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008, 8, 38–48. [Google Scholar] [CrossRef]
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular. Med. 2009, 11, 28–42. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; Bedalov, A.; Kennedy, B.K. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 17038–17045. [Google Scholar]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.C.; Atangan, L; Wang, M. Resveratrol is not a direct activator of SIRT1 enzyme activity. Chem. Biol. Drug Des. 2009, 74, 619–624. [Google Scholar] [CrossRef]
- Kuhnle, G.; Spencer, J.P.; Chowrimootoo, G.; Schroeter, H.; Debnam, E.S.; Srai, S.K.; Rice-Evans, C.; Hahn, U. Resveratrol is absorbed in the small intestine as resveratrol glucuronide. Biochem. Biophys. Res. Commun. 2000, 272, 212–217. [Google Scholar] [CrossRef]
- El Mohsen, M.A.; Bayele, H.; Kuhnle, G.; Gibson, G.; Debnam, E.; Srai, S.K.; Rice-Evans, C.; Spencer, J.P. Distribution of [3H]trans-resveratrol in rat tissues following oral administration. Br. J. Nutrit. 2006, 96, 62–70. [Google Scholar] [CrossRef]
- Goldberg, D.M.; Yan, J.; Soleas, G.J. Absorption of three wine-related polyphenols in three different matrices by health subjects. Clin. Biochem. 2003, 36, 79–87. [Google Scholar] [CrossRef]
- Marier, J.F.; Vachon, P.; Gritsas, A.; Zhang, J.; Moreau, J.P.; Ducharme, M.P. Metabolism and disposition of resveratrol in rats: extend of absorption, glucuronidation, nand enterohepatic recirculation evidenced by a linked-rat model. J. Pharmacol. Exp. Ther. 2002, 302, 369–373. [Google Scholar] [CrossRef]
- Sale, S.; Verschoyle, R.D.; Boocock, D.; Jones, D.J.; Wilsher, N.; Ruparelia, K.C.; Potter, G.A.; Farmer, P.B.; Steward, W.P.; Gescher, A.J. Pharmacokinetics in mice and growth-inhibitory properties of the putative cancer chemopreventative agent resveratrol and the synthetic analogue trans 3,4,5,4’-tetramethoxystilbene. Br. J. Cancer 2004, 90, 736–744. [Google Scholar] [CrossRef]
- Juan, M.E.; Maijó, M.; Planas, J.M. Quantification of trans-resveratrol and its metabolites in rat plasma and tissues by HPLC. J. Pharm. Biomed. Anal. 2010, 51, 391–398. [Google Scholar] [CrossRef]
- Vaz-da-Silva, M.; Loureiro, A.L.; Falcao, A.; Nunes, T.; Rocha, J.F.; Fernandes-Lopes, C.; Soares, E.; Wright, L.; Almeida, L.; Soares-da-Silva, P. Effect of food on the pharmacokinetic profile of trans-resveratrol. Int. J. Clin. Pharmacol. Ther. 2008, 46, 564–570. [Google Scholar]
- Stone, W.L.; Smith, M. Therapeutic use of antioxidant liposomes. Mol. Biotechnol. 2004, 27, 217–230. [Google Scholar] [CrossRef]
- Kristl, J.; Teskac, K.; Caddeo, C.; Abramović, A.; Sentjurc, M. Improvements of cellular stress response on resveratrol in liposomes. Eur. J. Pharm. Biopharm. 2009, 72, 253–259. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Robb, E.L.; Stuart, J.A. trans-Resveratrol as A Neuroprotectant. Molecules 2010, 15, 1196-1212. https://doi.org/10.3390/molecules15031196
Robb EL, Stuart JA. trans-Resveratrol as A Neuroprotectant. Molecules. 2010; 15(3):1196-1212. https://doi.org/10.3390/molecules15031196
Chicago/Turabian StyleRobb, Ellen L., and Jeffrey A. Stuart. 2010. "trans-Resveratrol as A Neuroprotectant" Molecules 15, no. 3: 1196-1212. https://doi.org/10.3390/molecules15031196
APA StyleRobb, E. L., & Stuart, J. A. (2010). trans-Resveratrol as A Neuroprotectant. Molecules, 15(3), 1196-1212. https://doi.org/10.3390/molecules15031196