Supramolecular Self-Assembled Chaos: Polyphenolic Lignin’s Barrier to Cost-Effective Lignocellulosic Biofuels

Abstract
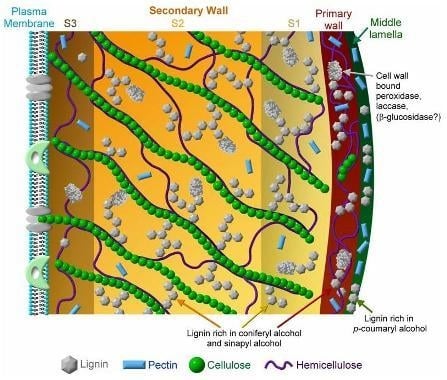
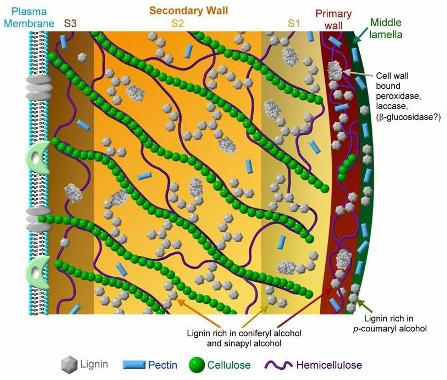
:
1. Introduction
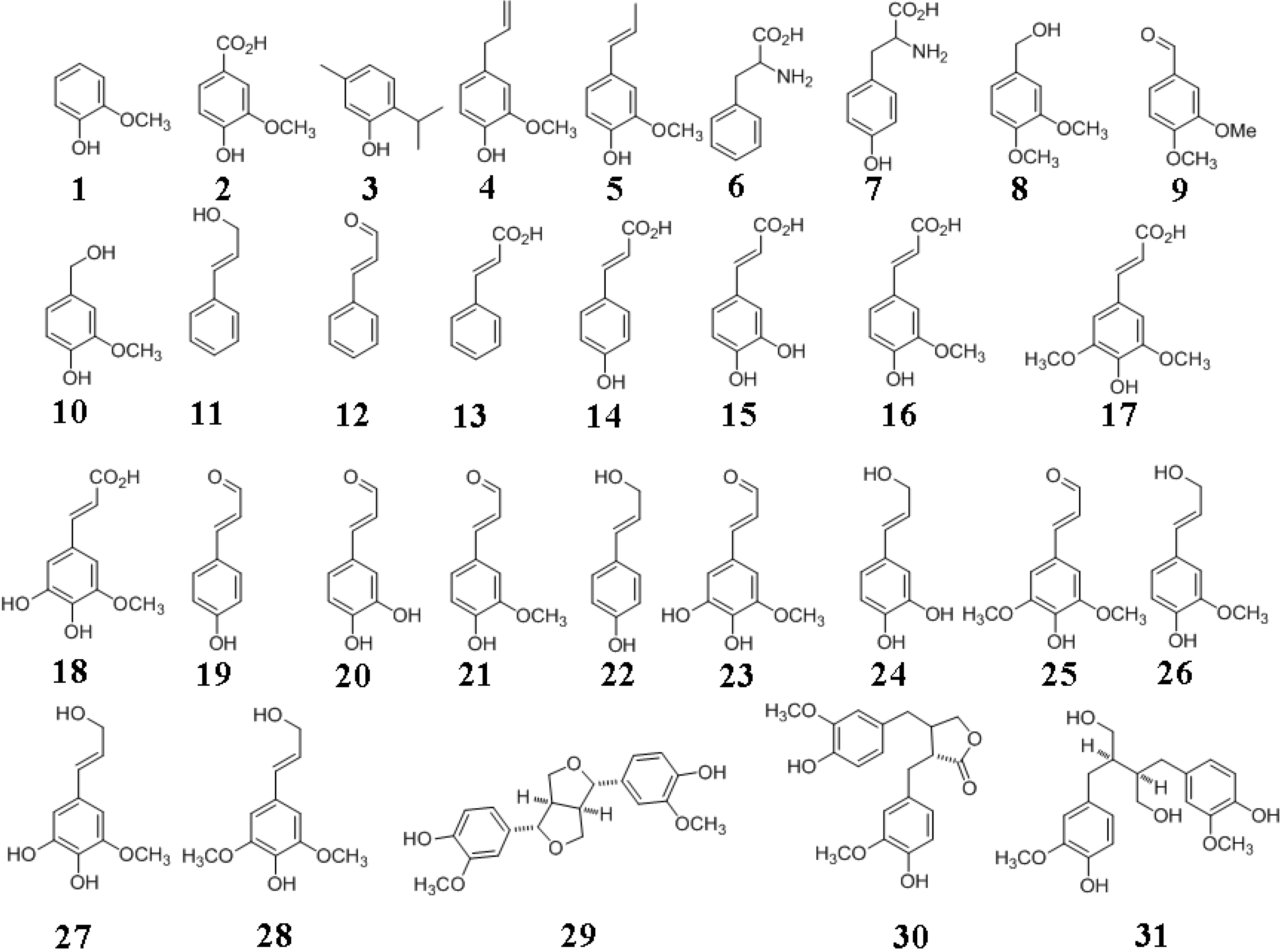
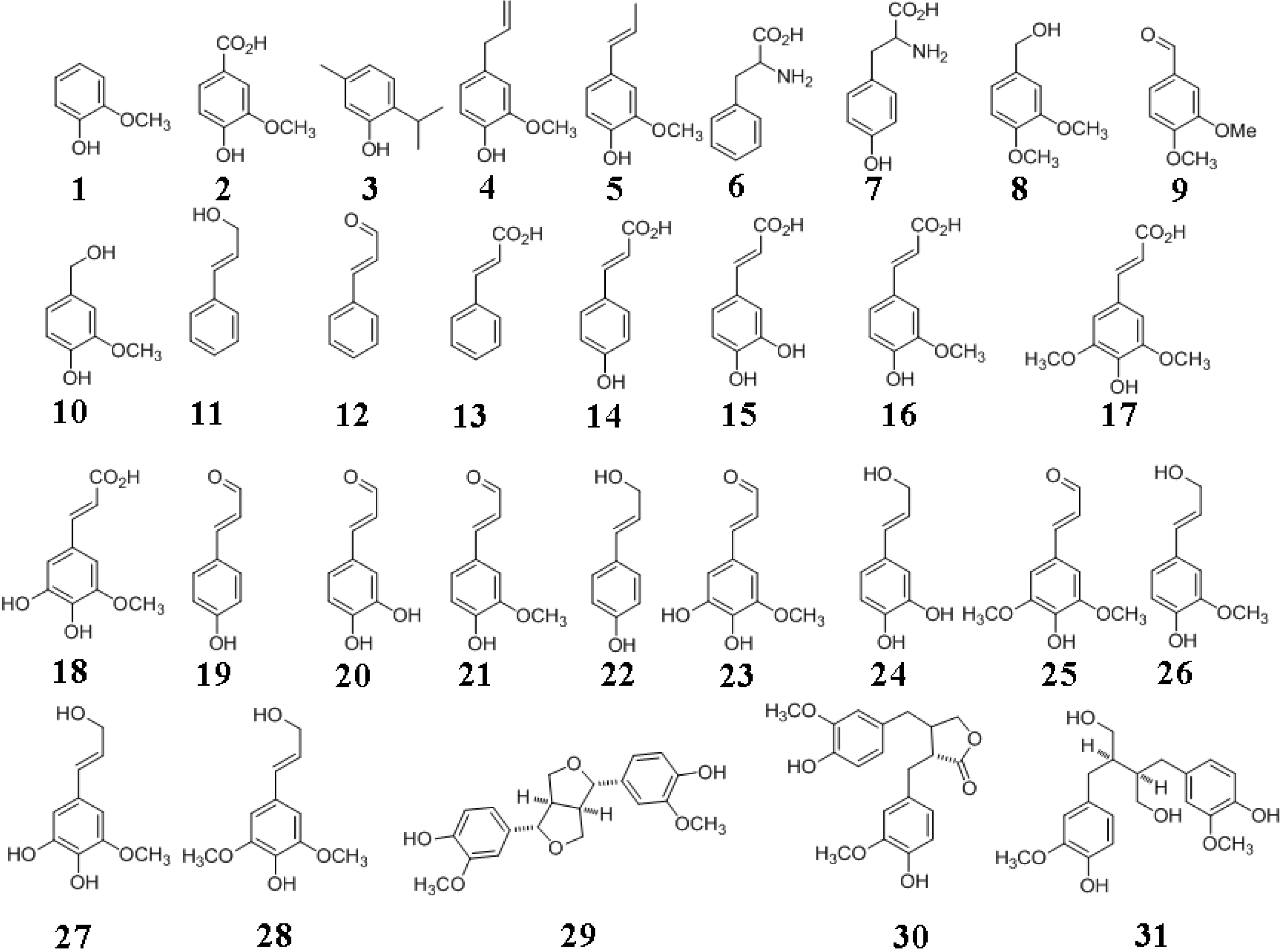
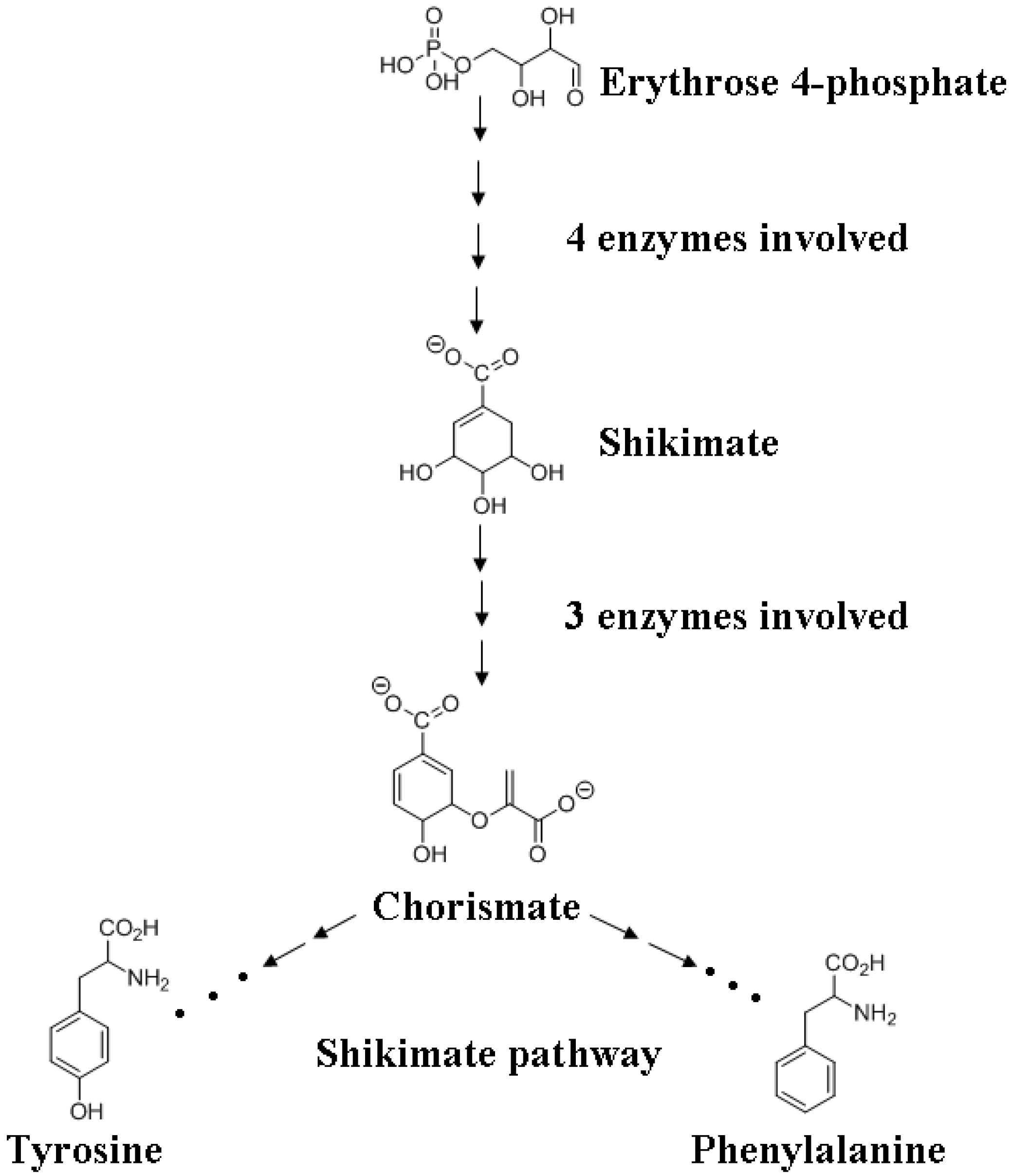
2. Plants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gymnosperms | Angiosperms |
|---|---|
| Softwoods | Hardwoods |
| Non-flowering (some exceptions) | Flowering monocotyledons (eg., corn) and dicotyledons (eg., beans) |
| Non-fruiting trees | Fruit trees |
| Coniferous “ever greens” (some conifers are deciduous) – conifers are major gymnosperms | All woody angiosperms are dicotyledons (not all dicotyledons are woody) |
| “Naked” seeds; bear cones | Seeds covered in fruit or nut |
| Retain/shed leaves throughout the year | Shed leaves at one particular time of year |
| Needle shaped leaves mostly | Well formed leaf structure |
| Cedar, Fir, Pine, Spruce, Redwood, Juniper, Cypress, Giant Sequoia, etc. | Ash, Mahogany, Oak, Aspen, Walnut, Balsa, Elm, Birch, Maple, etc. |
| Temperate growth regions | Temperate/tropical growth regions |
| Lower density wood | Higher density wood |
| Less expensive | More expensive |
| Smaller group (~20% of plant kingdom) | Largest group (~80% of plant kingdom) |
| Evolutionarily “primitive” | Evolutionarily “advanced” |
| Evolutionarily first seed-bearing plants | Seed plants evolved later |
| Oldest and largest trees (eg., giant sequoias) | More recent and smaller trees mostly |
| Reaction wood is mostly compression wood | Reaction wood is mostly tension wood |
3. General Properties of Lignin
| Softwood Lignin | Hardwood Lignin |
|---|---|
| Lignin content is ~ 28% | Lignin content is ~ 20% |
| Lignin dissociates faster in solution | Lignin dissociates slower in solution |
| Lignin self-associates greater in solution | Lignin self-associates less in solution |
| Harder to breakdown lignocellulosic biomass | Easier to breakdown lignocellulosic biomass |
| Coniferyl alchol primarily (~80%) | Coniferyl (~56%) and Sinapyl (~40%) alcohols |
| Guaiacyl (coniferyl alcohol derived) G-lignin | Guaiacyl-Syringyl (G-S) lignin; Syringyl is sinapyl alcohol derived lignin |
| Gymnosperms | Angiosperms, Dicotyledons |
| Molecular mass is larger than hardwood lignin | Molecular mass is lower than softwood lignin |
| Branching is higher | Branching is lower; Lignin is more linear |
| Cross-links are greater | Cross-links are fewer |
| C-C bonds are greater | C-C bonds are fewer |
| 5' Linkages more common | 5' Linkages less common |
| –OCH3 content is ~20% | –OCH3 content is ~14% |
| β-O-4 ether bonds are lower | β-O-4 ether bonds are higher |
| β-β and β-5 bonds are higher | β-β and β-5 bonds are fewer |
| Deconstruction is harder | Deconstruction is easier |
| Lignin is condensed | - - - - - |
4. Lignin Formation
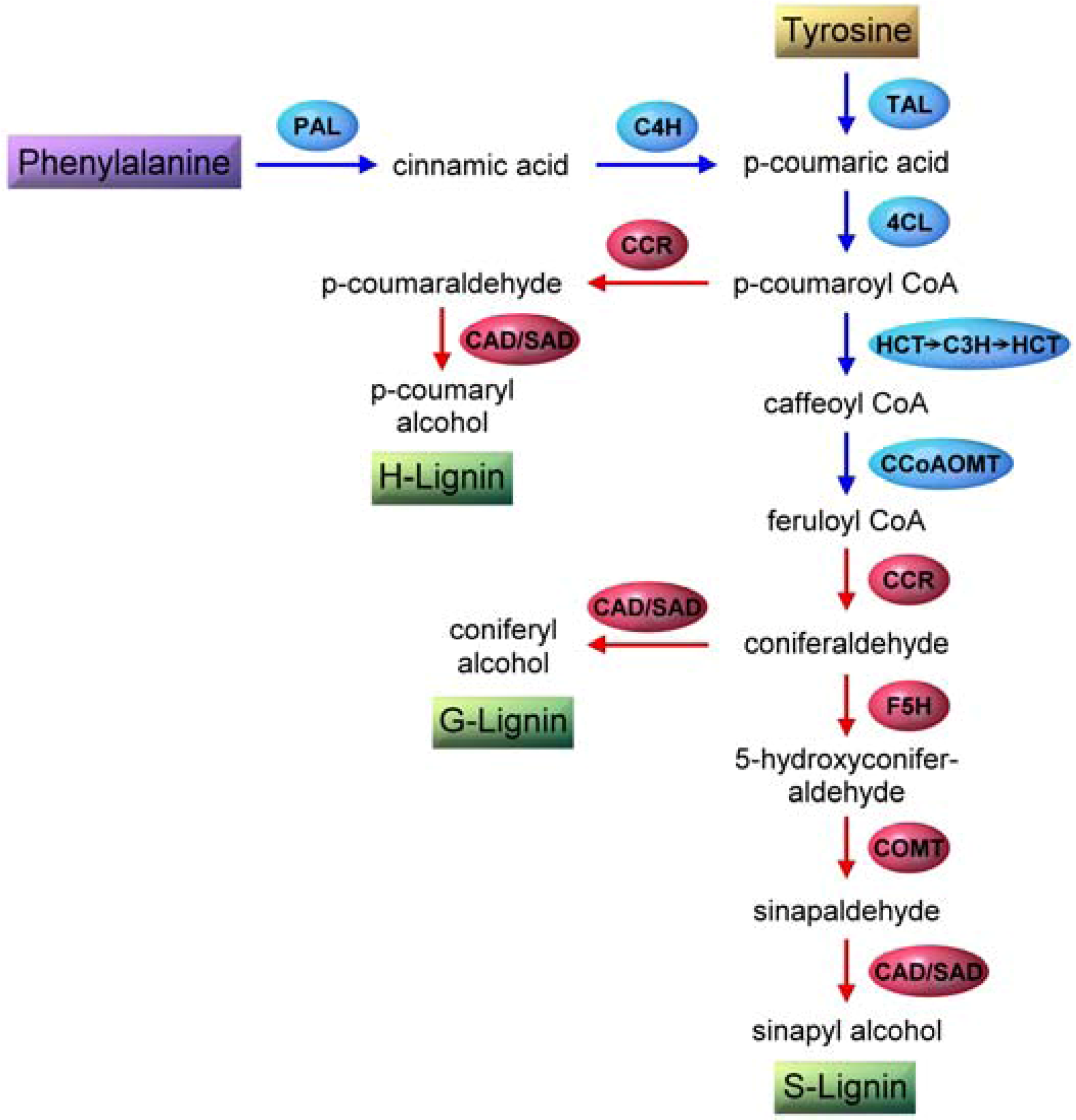
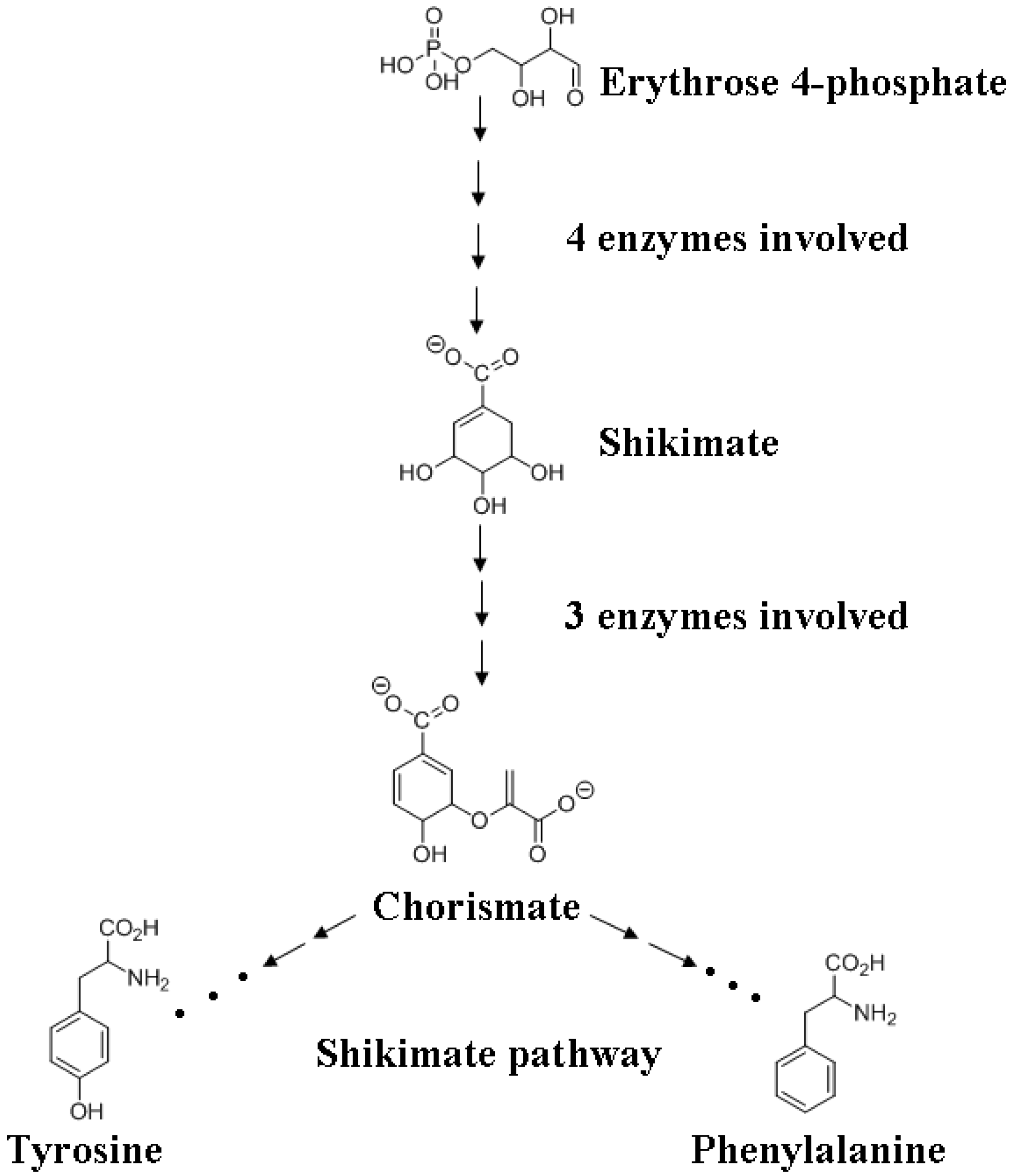
4.1. Monolignol Biosynthesis

4.2. Monolignol Transport
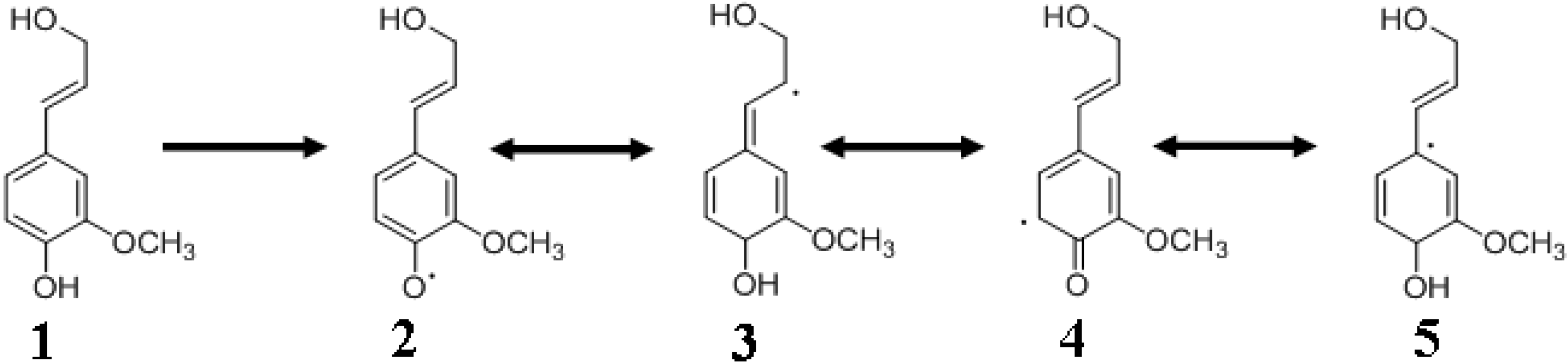
4.3. Monolignol Radical Formation

4.4. Monolignol Polymerization
4.5. Quinone Methides
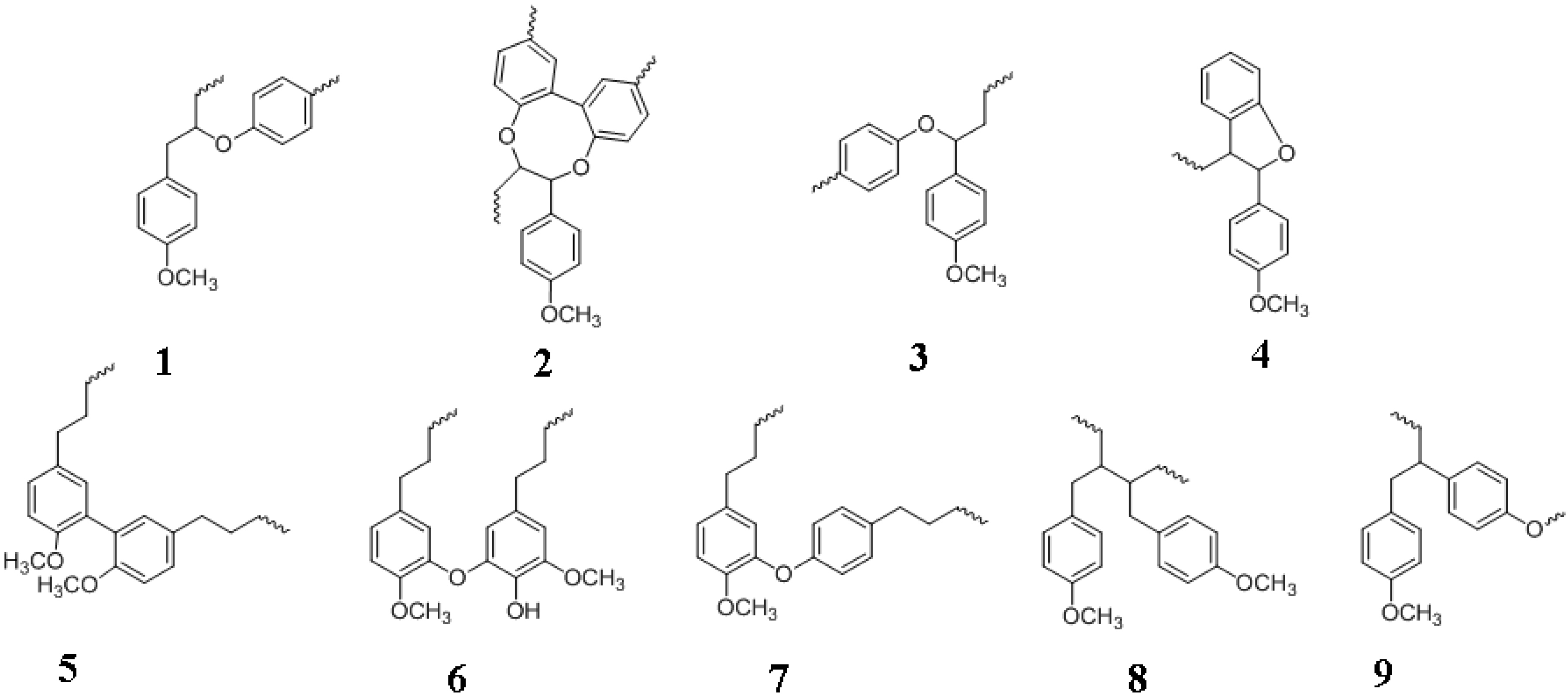
4.6. Lignin Formation and Structure
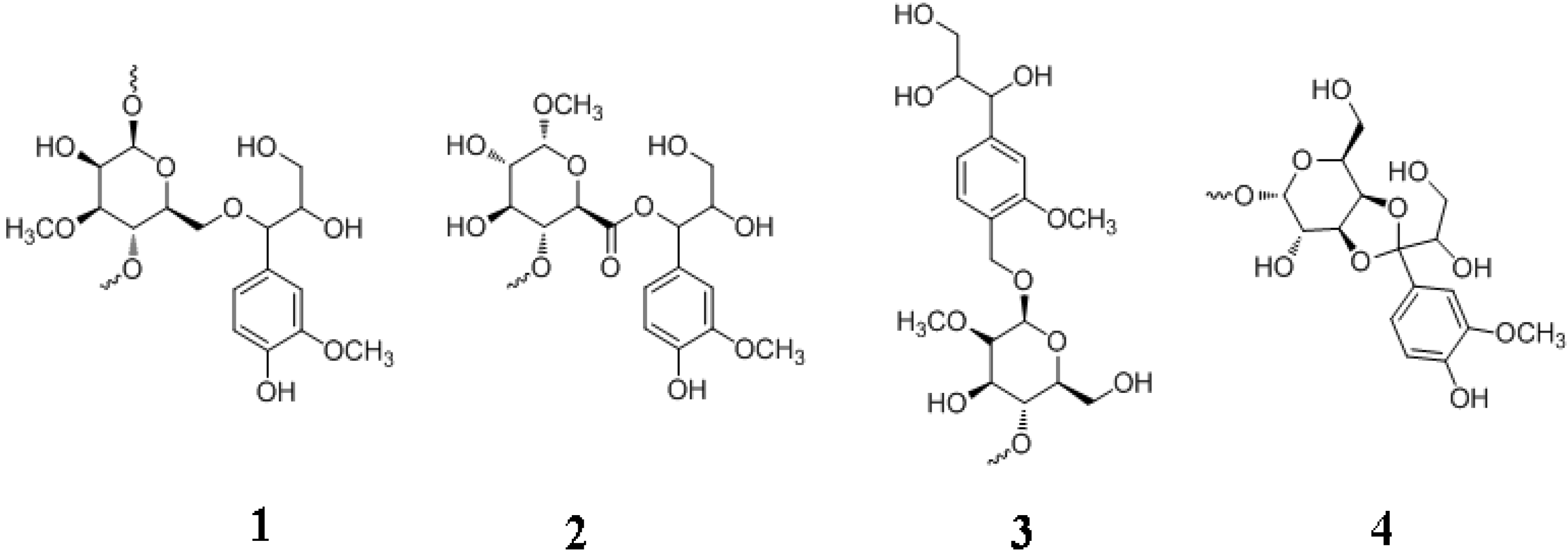
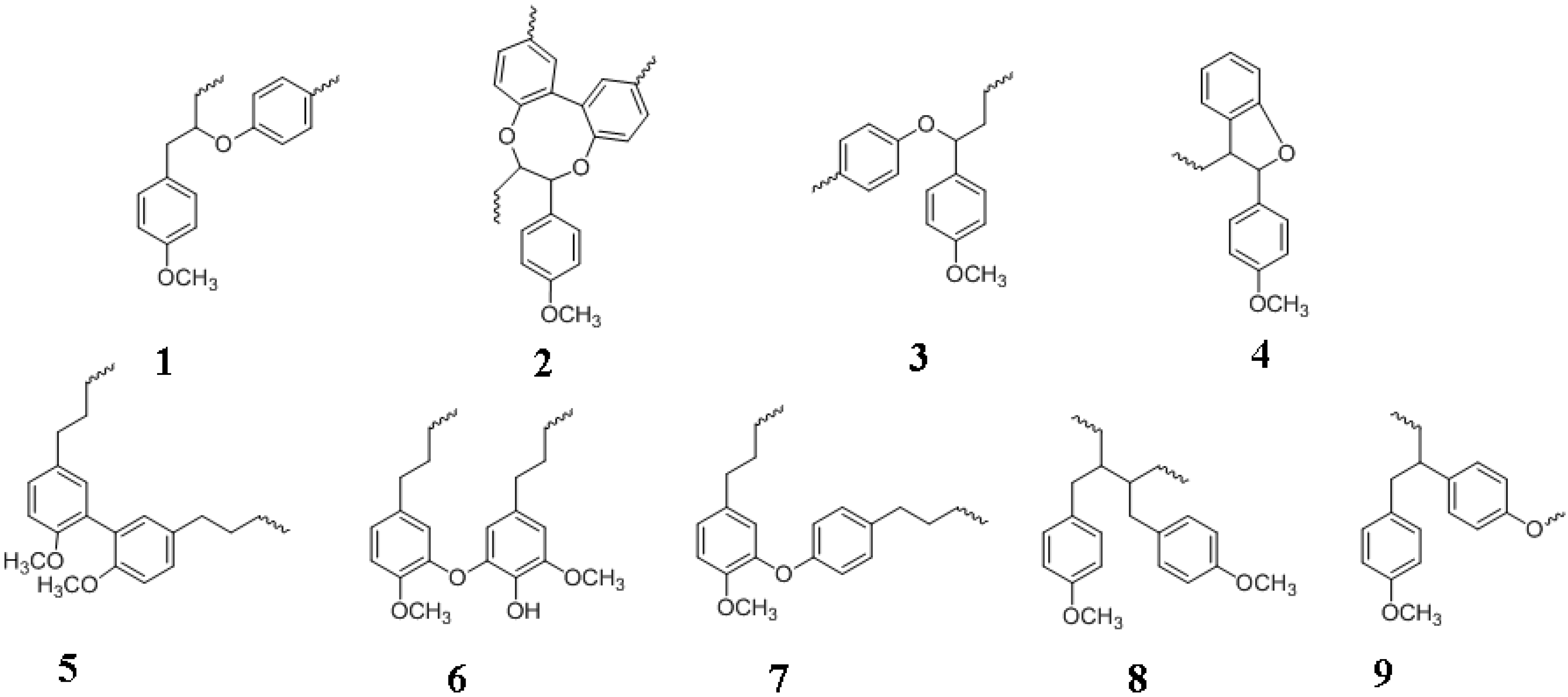
5. Lignans and Other Compounds
6. Lignin-Protein Interactions?
7. Lignin-Carbohydrate Complex (LCC)
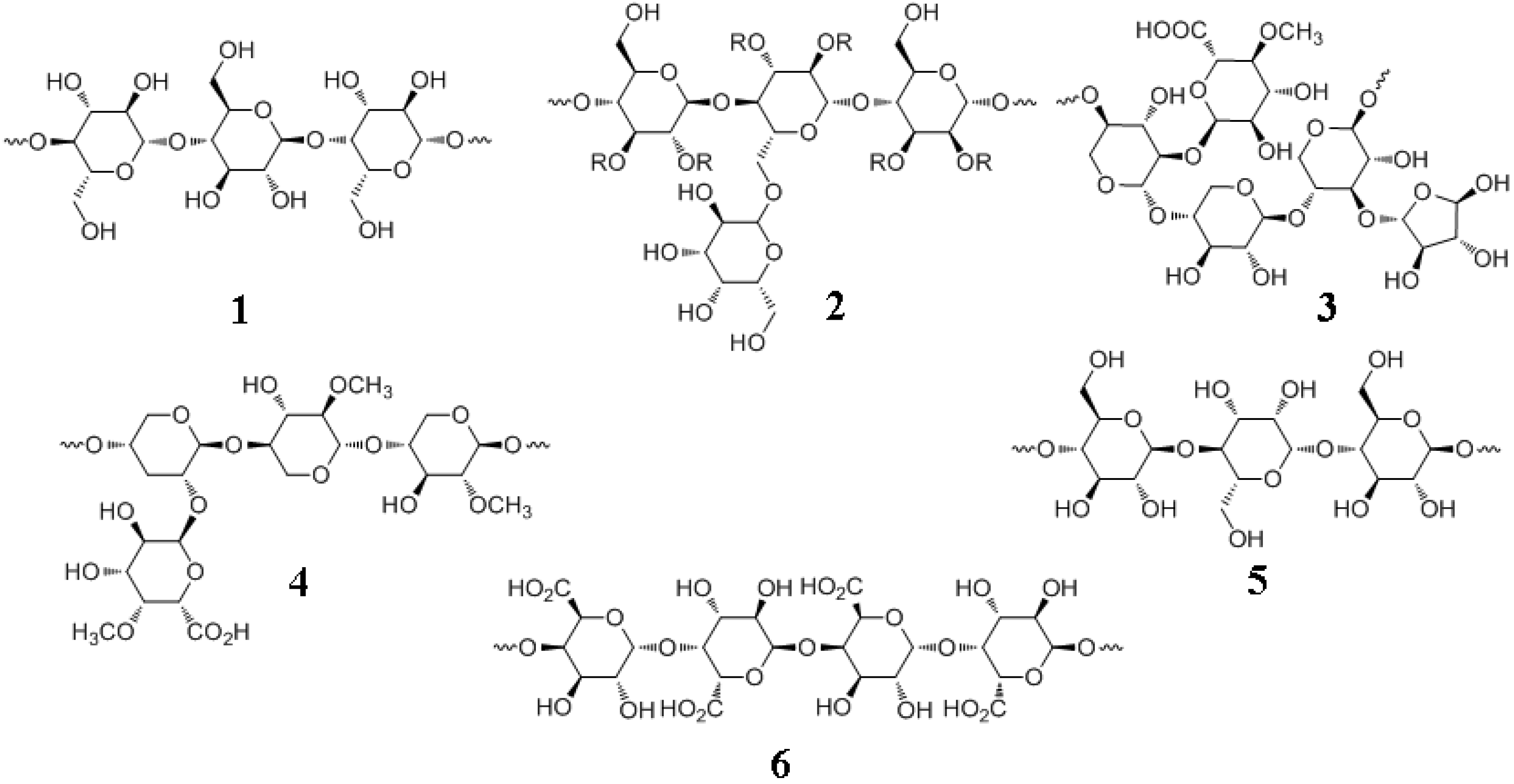
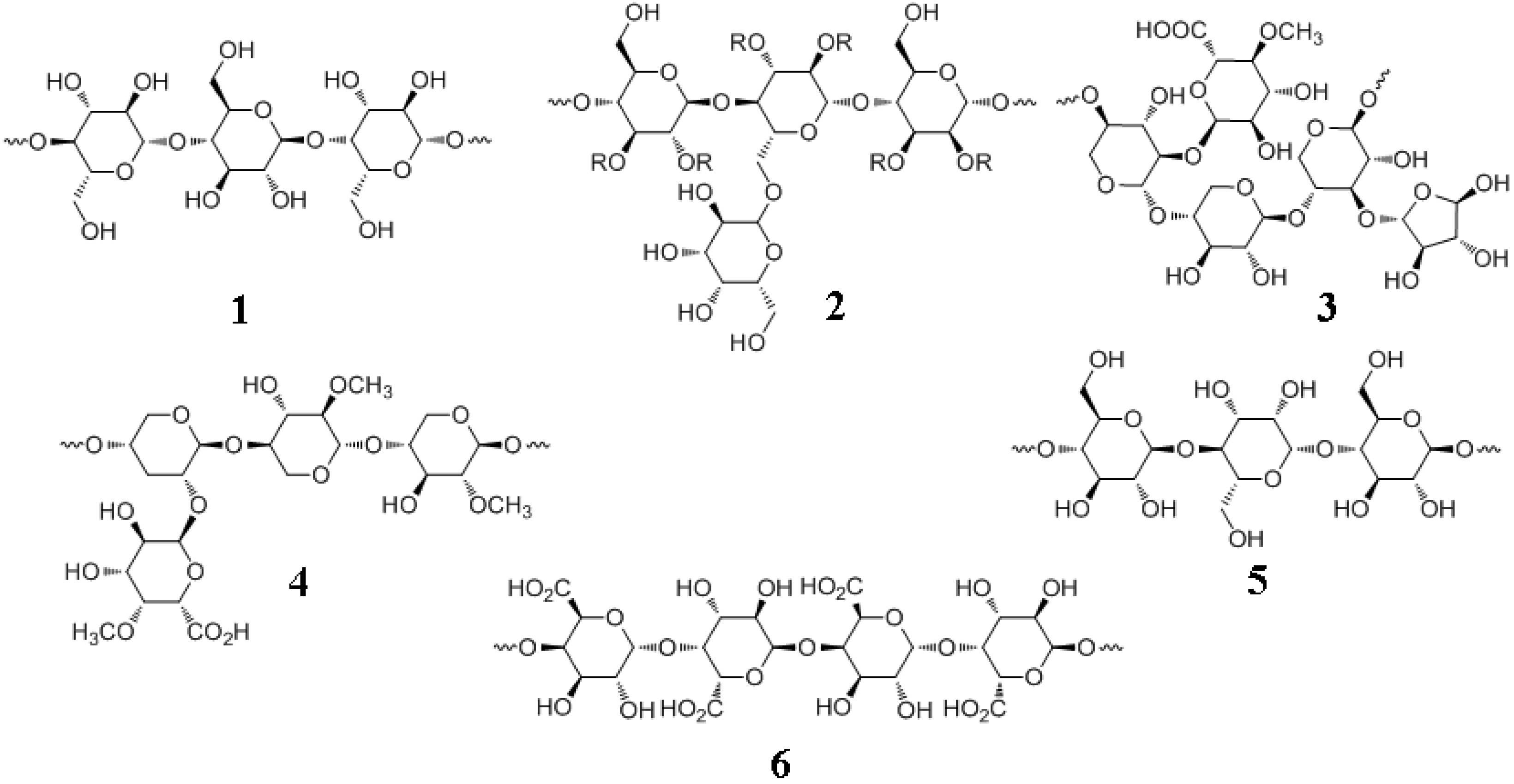
7.1. Major Plant Polysaccharides
7.2. Mechanisms for Lignin-Carbohydrate Complex (LCC) Formation

7.3. Mimicking LCCs
8. Supramolecular Self Assembly
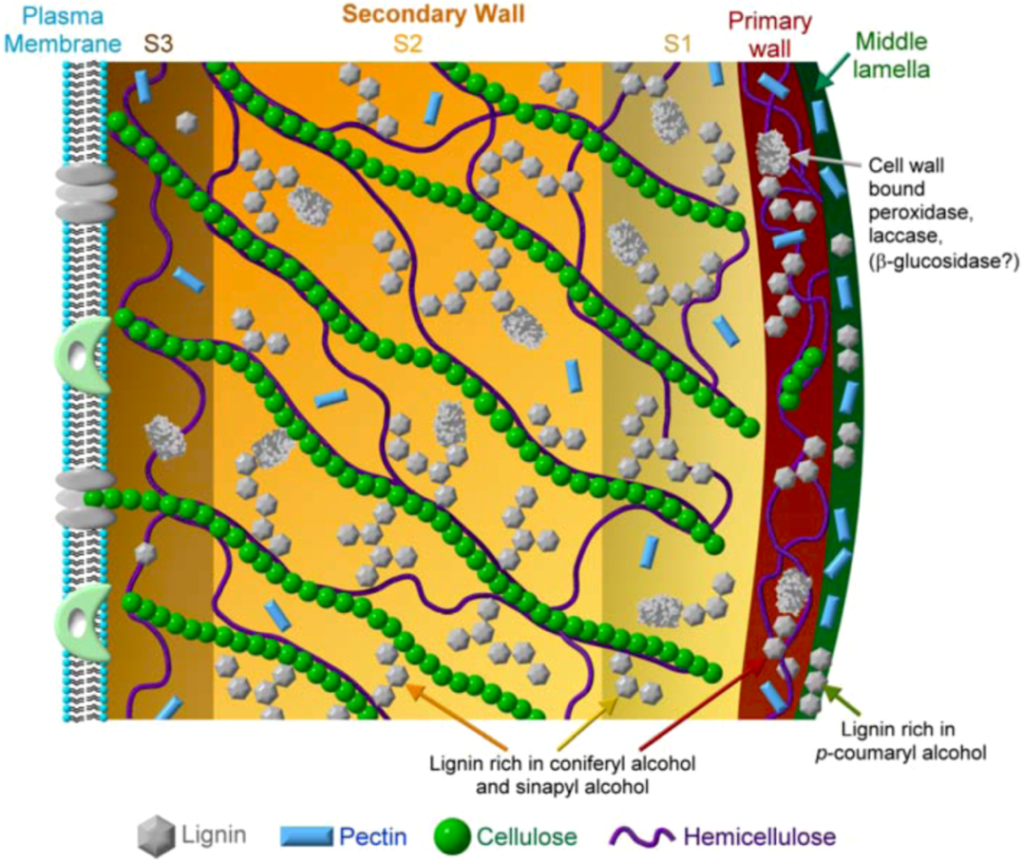
8.1. Xylogenesis
8.2. Cell Wall Formation
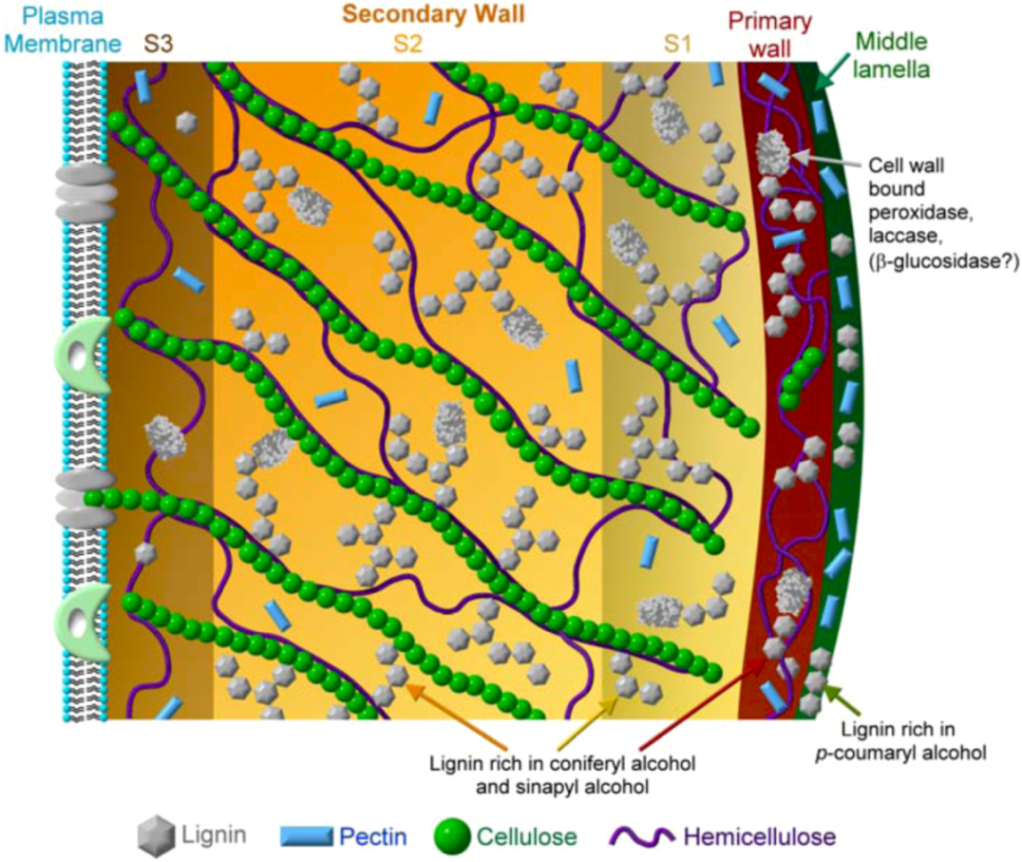
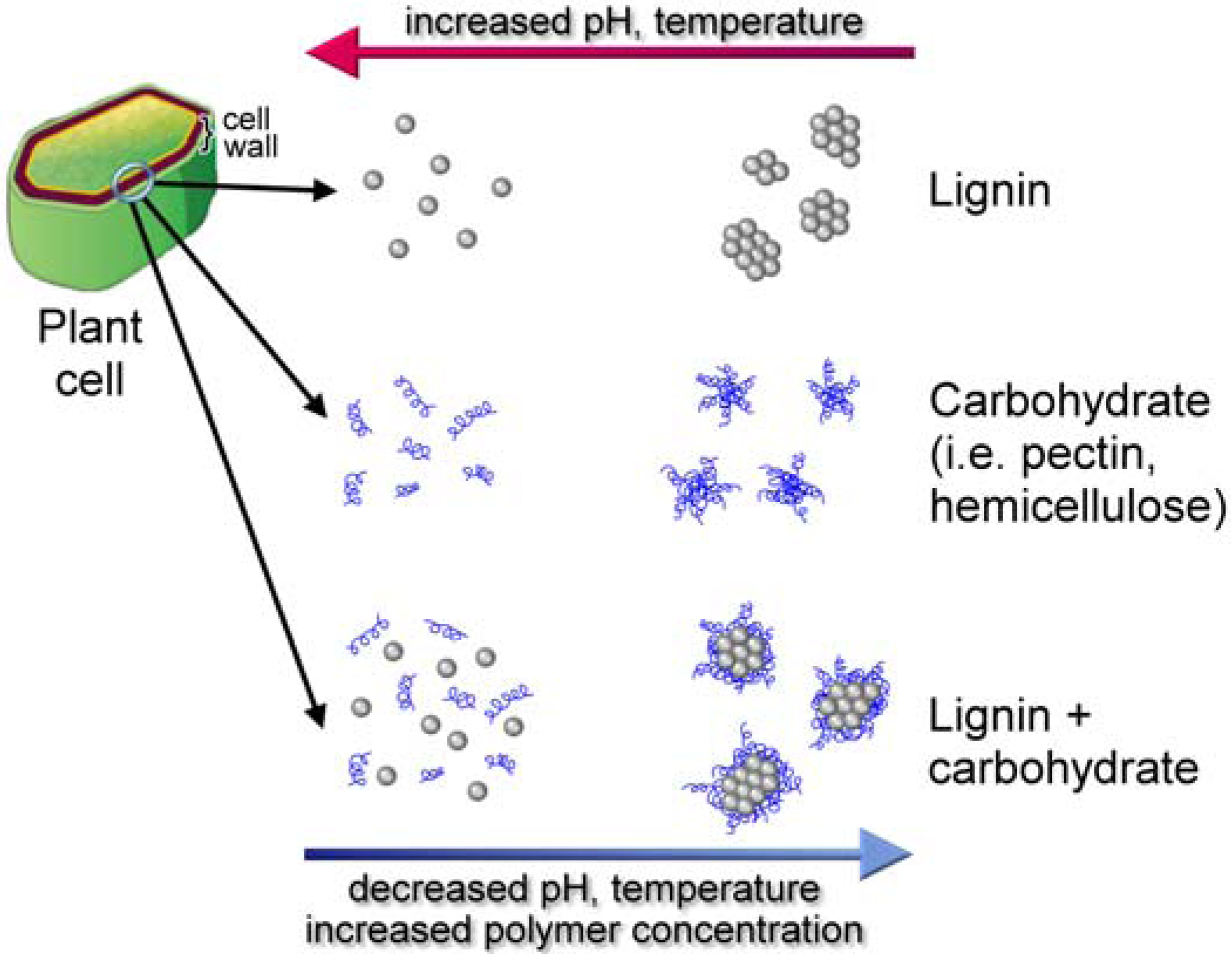
8.3. Supramolecular Lignin
8.4. Substratum Effects on Supramolecular Self-Assembly
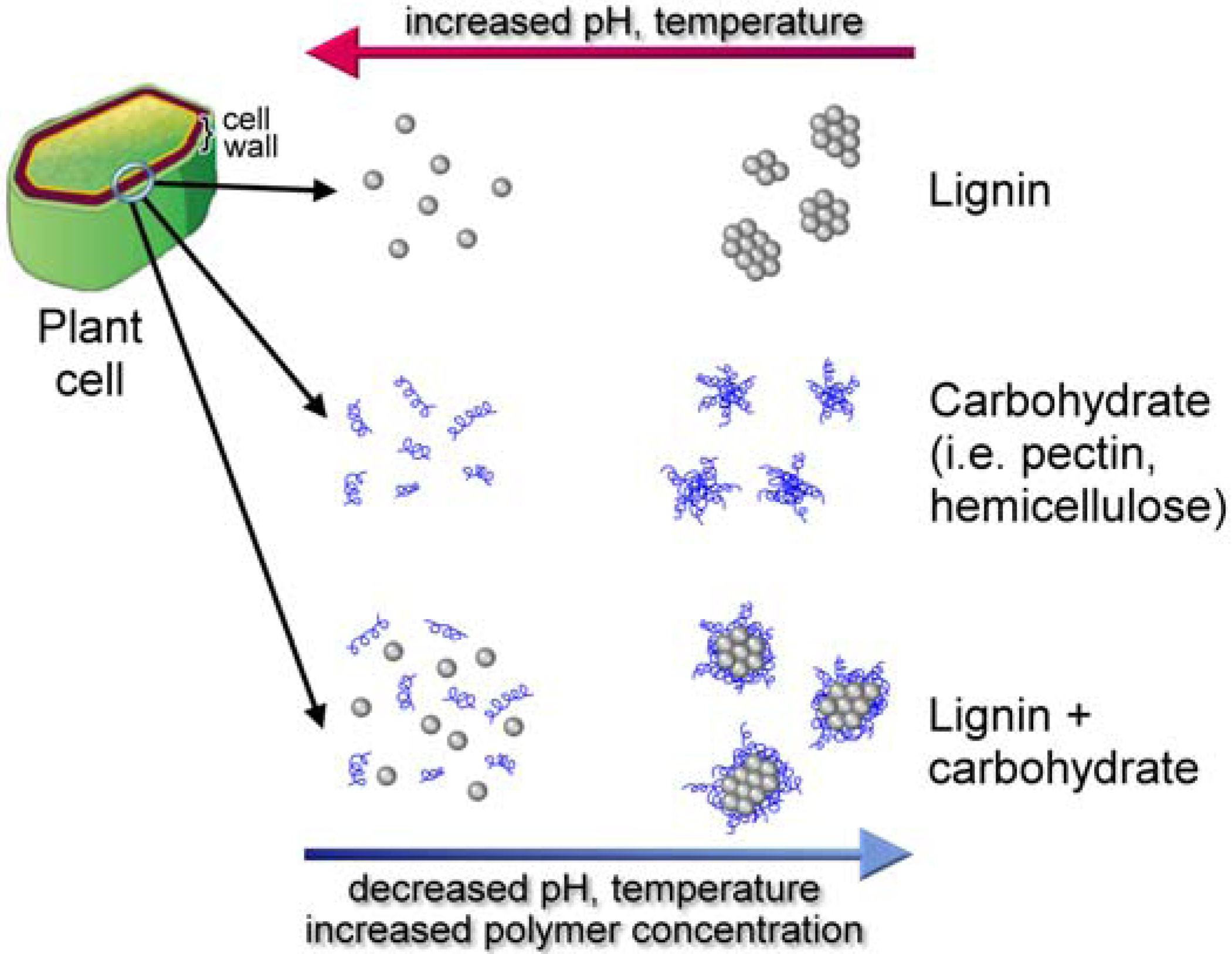
8.5. Lignin Conformation
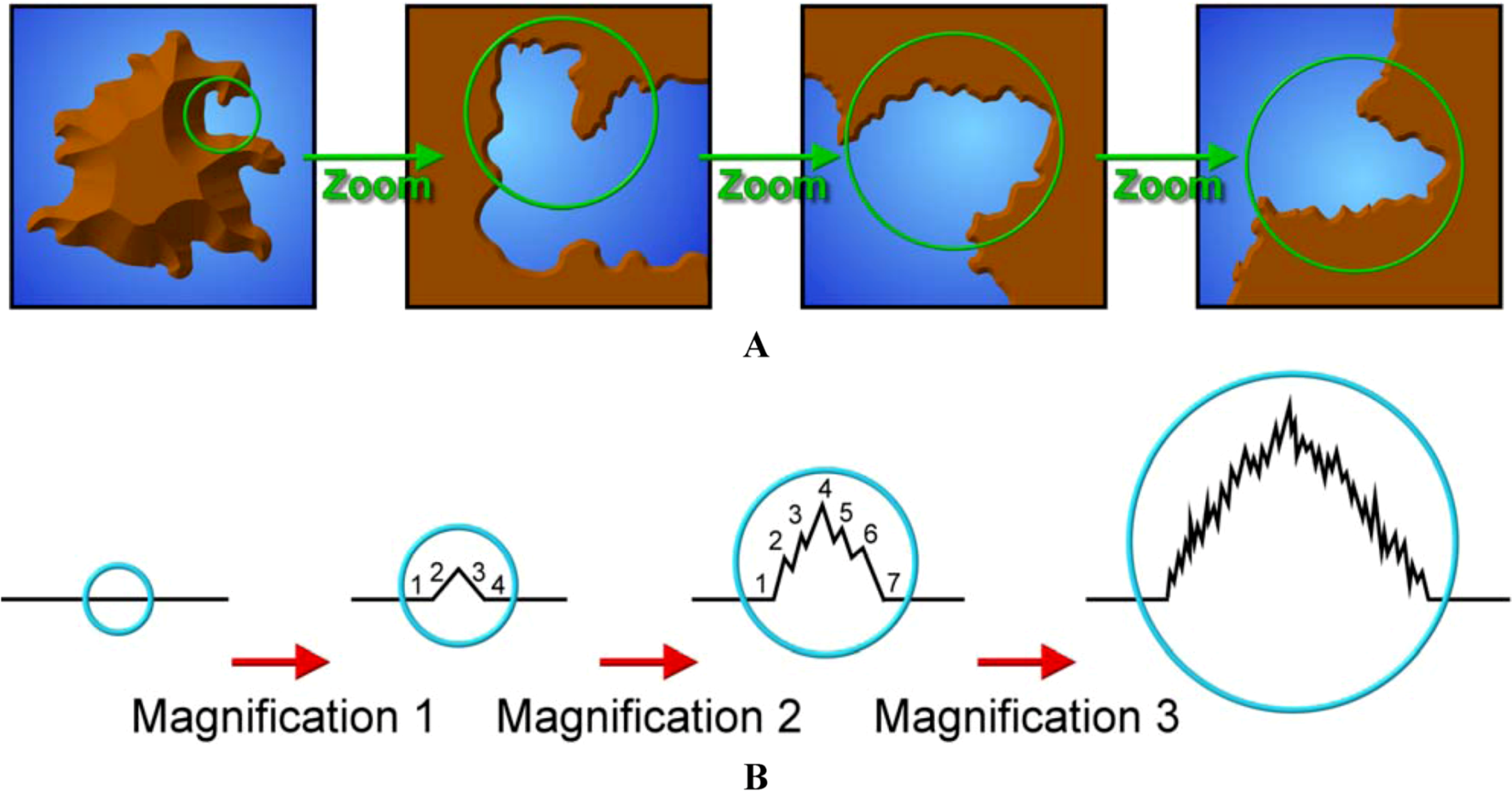
8.6. Fractal Properties of Lignin

8.7. Wood Attributes
9. Tools and Techniques
9.1. Raman Spectroscopy
9.2. Molecular Spectroscopy
9.3. Scanning Probe Microscopy (SPM)
9.4. NMR
9.5. Mechanical Properties
9.6. Computational Tools
10. Lignin Deconstruction—Nature’s Instructions
10.1. Lignin Barrier
10.2. Altering Lignin
10.3. Our Microbial Teachers
10.4. Delignifying Enzymes
11. Conclusions
Acknowledgements
- Sample Availability: Not available.
References and Notes
- Vacek, J.; Ulrichova, J.; Klejdus, B.; Simanek, V. Analytical methods and strategies in the study of plant polyphenolics in clinical samples. Anal. Meth. 2010, 2, 604–613. [Google Scholar] [CrossRef]
- Boudet, A.M.; Kajita, S.; Grima-Pettenati, J.; Goffner, D. Lignins and lignocellulosics: A better control of synthesis for new and improved uses. Trends Plant Sci. 2003, 8, 576–581. [Google Scholar] [CrossRef]
- Kenrick, P.; Crane, P.R. The origin and early evolution of plants on land. Nature 1997, 389, 33–39. [Google Scholar] [CrossRef]
- Stocker, M. Biofuels and biomass-to-liquid fuels in the biorefinery: Catalytic conversion of lignocellulosic biomass using porous materials. Angew. Chem. Int. Ed. 2008, 47, 9200–9211. [Google Scholar] [CrossRef]
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; Boerjan, W. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 2004, 3, 29–60. [Google Scholar] [CrossRef]
- Barcelo, A.R.; Ros, L.V.G.; Gabaldon, C.; Lopez-Serrono, M.; Pomar, F.; Carrion, J.S.; Pedreno, M.A. Basic peroxidases: the gateway for lignin evolution. Phytochem. Rev. 2004, 3, 61–78. [Google Scholar] [CrossRef]
- Ralph, J.; Brunow, G.; Harris, P.; Dixon, R.A.; Schatz, P.F.; Boerjan, W. Lignification: Are lignins biosynthesized via simple combinatorial chemistry or via proteinaceous control and template replication? In Recent Advances in Polyphenols Research; Daayf, F., El Hadrami, A., Adam, L., Ballance, G.M., Eds.; Wiley-Blackwell: Oxford, UK, 2008; pp. 36–66. [Google Scholar]
- Morreel, K.; Dima, O.; Kim, H.; Lu, F.; Niculaes, C.; Vanholme, R.; Dauwe, R.; Goeminne, G.; Inze, D.; Messens, E.; Ralph, J.; Boerjan, W. Mass spectrometry-based sequencing of lignin oligomers. Plant Physiol. 2010, 152, 1464–1478. [Google Scholar]
- Davin, L.B.; Lewis, N.G. Lignin primary structures and dirigent sites. Curr. Opin. Biotechnol. 2005, 16, 407–415. [Google Scholar] [CrossRef]
- Janshekar, H.; Haltmeier, T.; Brown, C. Fungal degradation of pine and straw alkali lignins. Eur. J. Appl. Microbiol. Biotechnol. 1982, 14, 174–181. [Google Scholar] [CrossRef]
- Ralph, J. What makes a good monolignol substitute? In The Science and Lore of the Plant Cell Wall; Hayashi, T., Ed.; Brown Walker Press: Boca Raton, FL, USA, 2006; pp. 285–293. [Google Scholar]
- Mansfield, S.D. Solutions for dissolution – engineering cell walls for deconstruction. Curr. Opin. Biotechnol. 2009, 20, 286–294. [Google Scholar] [CrossRef]
- Karmanov, A.P.; Monakov, Y.B. Lignin structural organization and fractal properties. Russ. Chem. Rev. 2003, 72, 715–734. [Google Scholar] [CrossRef]
- Vainio, U.; Maximova, N.; Hortling, B.; Laine, J.; Stenius, P.; Simola, L.K.; Gravitis, J.; Serimaa, R. Morphology of dry lignins and size and shape of dissolved kraft lignin particles by X-ray scattering. Langmuir 2004, 20, 9736–9744. [Google Scholar] [CrossRef]
- Wong, D.W.S. Stucture and action mechanisms of ligninolytic enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef]
- Karkonen, A.; Koutaniemi, S. Lignin biosynthesis studies in plant tissue cultures. J. Integr. Plant Biol. 2010, 52, 176–185. [Google Scholar] [CrossRef]
- Humphreys, J.M.; Chapple, C. Rewriting the lignin roadmap. Curr. Opin. Plant Biol. 2002, 5, 224–229. [Google Scholar] [CrossRef]
- Martinez, A.T.; Ruiz-Duenas, F.J.; Martinez, M.J.; del Rio, J.C.; Gutierrez, A. Enzymatic delignification of plant cell wall: from nature to mill. Curr. Opin. Biotechnol. 2009, 20, 348–357. [Google Scholar] [CrossRef]
- Battle, M.; Bender, M.L.; Tans, P.P.; White, J.W.C.; Ellis, J.T.; Conway, T.; Francey, R.J. Global carbon sinks and their variability inferred from atmospheric O2 and δ13C. Science 2000, 287, 2467–2470. [Google Scholar]
- Lewis, N.G.; Yamamoto, E. Lignin: Occurrence, biogenesis and biodegradation. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1990, 41, 455–496. [Google Scholar] [CrossRef]
- Ralph, J.; Hatfield, R.D.; Sederoff, R.R.; MacKay, J.J. Order and randomness in lignin and lignification: Is a new paradigm for lignification required? Research Summaries USDFRC 1998, 39–41. [Google Scholar]
- Sederoff, R.R.; MacKay, J.J.; Ralph, J.; Hatfield, R.D. Unexpected variation in lignin. Curr. Opin. Plant Biol. 1999, 2, 145–152. [Google Scholar] [CrossRef]
- Weng, J.K.; Li, X.; Bonawitz, N.D.; Chapple, C. Emerging strategies of lignin engineering and degradation for cellulosic biofuel production. Curr. Opin. Biotechnol. 2008, 19, 166–172. [Google Scholar] [CrossRef]
- Amthor, J.S. Efficiency of lignin biosynthesis: A quantitative analysis. Ann. Bot. 2003, 91, 673–695. [Google Scholar] [CrossRef]
- Patzlaff, A.; McInnis, S.; Courtenay, A.; Surman, C.; Newman, L.J.; Smith, C.; Bevan, M.W.; Mansfield, S.; Whetten, R.W.; Sederoff, R.R.; Campbell, M.M. Characterisation of a pine MYB that regulates lignification. Plant J. 2003, 36, 743–754. [Google Scholar] [CrossRef]
- Martone, P.T.; Estevez, J.M.; Lu, F.; Ruel, K.; Denny, M.W.; Somerville, C.; Ralph, J. Discovery of lignin in seaweed reveals convergent evolution of cell-wall architecture. Curr. Biol. 2009, 19, 169–175. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Dixon, R.A.; Chen, F.; Guo, D.; Parvathi, K. The biosynthesis of monolignols: A “metabolic grid”, or independent pathways to guaiacyl and syringyl units. Phytochemistry 2001, 57, 1069–1084. [Google Scholar] [CrossRef]
- Moura, J.C.M.S.; Bonnie, C.A.V.; Viana, J.O.F.; Dornelas, M.C.; Mazzafera, P. Abiotic and biotic stresses and changes in lignin content and composition in plants. J. Integr. Plant Biol. 2010, 52, 360–376. [Google Scholar] [CrossRef]
- Hatfield, R.; Vermerris, W. Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol. 2001, 126, 1351–1357. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Austin, M.B.; Stewart, C., Jr.; Noel, J.P. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol. Biochem. 2008, 46, 356–370. [Google Scholar] [CrossRef]
- Arcuri, H.A.; Zafalon, G.F.D.; Marucci, E.A.; Bonalumi, C.E.; da Silveira, N.J.F.; Machado, J.M.; de Azevedo, W.F., Jr.; Palma, M.S. SKPDB: A structural database of shikimate pathway enzymes. BMC Bioinformatics 2010, 11, 1–7. [Google Scholar]
- Chiang, V.L. Monolignol biosynthesis and genetic engineering of lignin in trees, a review. Environ. Chem. Lett. 2006, 4, 143–146. [Google Scholar] [CrossRef]
- Vanholme, R.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin engineering. Curr. Opin. Plant Biol. 2008, 11, 278–285. [Google Scholar] [CrossRef]
- Sticklen, M.B. Plant genetic engineering for biofuel production: Towards affordable cellulosic ethanol. Nature Rev. Genet. 2008, 9, 433–443. [Google Scholar] [CrossRef]
- Ralph, J.; Brunow, G.; Boerjan, W. Lignins. In Encyclopedia of Life Sciences; John Wiley & Sons Ltd: Hoboken, NJ, USA, 2007; pp. 1–10. [Google Scholar]
- Steeves, V.; Forster, H.; Pommer, U.; Savidge, R. Coniferyl alcohol metabolism in conifers – I. Glucosidic turnover of cinnamyl aldehydes by UDPG: Coniferyl alcohol glucosyltransferase from pine cambium. Phytochemistry 2001, 57, 1085–1093. [Google Scholar] [CrossRef]
- Terashima, N.; Ralph, S.A.; Landucci, L.L. New facile syntheses of monolignol glucosides; p-glucocoumaryl alcohol, coniferin and syringin. Holzforschung 1995, 50, 151–155. [Google Scholar]
- Dharmawardhana, D.P.; Ellis, B.E.; Carlson, J.E. A β-glucosidase from lodgepole pine xylem specific for the lignin precursor coniferin. Plant Physiol. 1995, 107, 331–339. [Google Scholar]
- Samuels, A.L.; Rensing, K.H.; Douglas, C.J.; Mansfield, S.D.; Dharmawardhana, D.P.; Ellis, B.E. Cellular machinery of wood production: Differentiation of secondary xylem in Pinus contorta var. latifolia. Planta 2002, 216, 72–82. [Google Scholar] [CrossRef]
- Tsuji, Y.; Chen, F.; Yasuda, S.; Fukushima, K. Unexpected behavior of coniferin in lignin biosynthesis of Ginkgo biloba L. Planta 2005, 222, 58–69. [Google Scholar] [CrossRef]
- Kaneda, M.; Rensing, K.H.; Wong, J.C.; Banno, B.; Mansfield, S.D.; Samuels, A.L. Tracking monolignols during wood development in lodgepole pine. Plant Physiol. 2008, 147, 1750–1760. [Google Scholar] [CrossRef]
- Galvez, M.; Martin-Cordero, C.; Ayuso, M.J. Pharmacological activities of phenylpropanoids glycosides. Studies Nat. Prod. Chem. 2006, 33, 675–718. [Google Scholar] [CrossRef]
- Onnerud, H.; Zhang, L.; Gellerstedt, G.; Henriksson, G. Polymerization of monolignols by redox shuttle–mediated enzymatic oxidation. A new model in lignin biosynthesis I. Plant Cell 2002, 14, 1953–1962. [Google Scholar] [CrossRef]
- Fourand, D.; Cathala, B.; Lapierre, C. Initial steps of the peroxidase-catalyzed polymerization of coniferyl alcohol and/or sinapyl aldehyde: Capillary zone electrophoresis study of pH effect. Phytochemistry 2003, 62, 139–146. [Google Scholar]
- Harvey, P.J.; Schoemaker, H.E.; Palmer, J.M. Veratryl alcohol as a mediator and the role of radical cations in lignin biodegradation by Phanerochaete chrysosporium. FEBS Lett. 1986, 195, 242–246. [Google Scholar] [CrossRef]
- Caparros-Ruiz, D.; Fornale, S.; Civardi, L.; Puigdomenech, P.; Rigau, J. Isolation and characterisation of a family of laccases in maize. Plant Sci. 2006, 171, 217–225. [Google Scholar] [CrossRef]
- McCaig, B.C.; Meagher, R.B.; Dean, J.F.D. Gene structure and molecular analysis of the laccase-like multicopper oxidase (LMCO) gene family in Arabidopsis thaliana. Planta 2005, 221, 619–636. [Google Scholar] [CrossRef]
- Barcelo, A.R.; Gomez Ros, L.V.; Carrasco, A.E. Looking for syringyl peroxidases. Trends Plant Sci. 2007, 12, 486–491. [Google Scholar] [CrossRef]
- Walling, C.; El-Taliawi, G.; Amarnath, K. Oxidation of styrene derivatives by peroxydisulfate(2-) ion-copper(II) in acetic acid and acetonitrile. Reaction paths in oxidations via radical cations. J. Am. Chem. Soc. 1984, 106, 7573–7578. [Google Scholar] [CrossRef]
- Rouhi, M. Only facts will end lignin war. Chem. Engg. News 2001, 79, 52–56. [Google Scholar] [CrossRef]
- Davin, L.B.; Lewis, N.G. Dirigent phenoxy radical coupling: Advances and challenges. Curr. Opin. Biotechnol. 2005, 16, 398–406. [Google Scholar] [CrossRef]
- Chen, Y.; Sarkanen, S. Macromolecular replication during lignin biosynthesis. Phytochemistry 2010, 71, 453–462. [Google Scholar] [CrossRef]
- Ralph, J.; Peng, J.; Lu, F.; Hatfield, R.D.; Helm, R.F. Are lignins optically active? J. Agric. Food Chem. 1999, 47, 2991–2996. [Google Scholar] [CrossRef]
- Lu, F.C.; Ralph, J. Detection and determination of p-coumaroylated units in lignins. J. Agric. Food Chem. 1999, 47, 1988–1992. [Google Scholar] [CrossRef]
- Facchini, P.J.; Hagel, J.; Zulak, K.G. Hydroxycinnamic acid amide metabolism: Physiology and biochemistry. Can. J. Bot. 2002, 80, 577–589. [Google Scholar] [CrossRef]
- Morreel, K.; Ralph, J.; Kim, H.; Lu, F.; Goeminne, G.; Ralph, S.; Messens, E.; Boerjan, W. Profiling of oligolignols reveals monolignol coupling conditions in lignifying poplar xylem. Plant Physiol. 2004, 136, 3537–3549. [Google Scholar] [CrossRef]
- Lu, F.C.; Ralph, J.; Morreel, K.; Messens, E.; Boerjan, W. Preparation and relevance of a cross-coupling product between sinapyl alcohol and sinapyl p-hydroxybenzoate. Org. Biomol. Chem. 2004, 2, 2888–2890. [Google Scholar] [CrossRef]
- Alber, E. Lignin chemistry – past, present and future. Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar] [CrossRef]
- Freudenberg, K. Beitrage zur erforschung des lignins (“Contributions to study the lignins”). Angew. Chem. 1956, 68, 508–512. [Google Scholar] [CrossRef]
- Grabber, J.H. How do lignin composition, structure, and cross-linking affect degradability? A review of cell wall model studies. Crop Sci. 2005, 45, 820–831. [Google Scholar] [CrossRef]
- van Parijs, F.R.D.; Morreel, K.; Ralph, J.; Boerjan, W.; Merks, R.M.H. Modeling lignin polymerization. I. Simulation model of dehydrogenation polymers. Plant Physiol. 2010, 153, 1332–1344. [Google Scholar] [CrossRef]
- Leary, G.J. Quinone methides and the structure of lignin. Wood Sci. Technol. 1980, 14, 21–34. [Google Scholar] [CrossRef]
- Scaiano, J.C.; Berinstain, A.B.; Whittlesey, M.K.; Malenfant, P.R.L.; Bensimon, C. Lignin-like molecules: Structure and photophysics of crystalline α-guaiacoxyacetoveratrone. Chem. Mater. 1993, 5, 700–704. [Google Scholar] [CrossRef]
- Fukushima, K.; Terashima, N. Heterogeneity in formation of lignin. Wood Sci. Technol. 1991, 25, 371–381. [Google Scholar]
- Campbell, M.M.; Sederoff, R.R. Variation in lignin content and composition: Mechanisms of control and implications for the genetic improvements of plants. Plant Physiol. 1995, 110, 3–13. [Google Scholar]
- Monties, B. A look back upon some ‘non-conventional’ biochemical mechanisms of supramolecular structures during peroxidase-oxydase catalyzed polymerization of lignins and the related dehydropolymers (DHP) formation. Cellulose Chem. Technol. 2007, 41, 495–504. [Google Scholar]
- Saleem, M.; Kim, H.J.; Ali, M.S.; Lee, Y.S. A update on bioactive plant lignans. Nat. Prod. Rep. 2005, 22, 696–716. [Google Scholar] [CrossRef]
- Pan, J.Y.; Chen, S.L.; Yang, M.H.; Wu, J.; Sinkkonen, J.; Zou, K. An update on lignans: Natural products and synthesis. Nat. Prod. Rep. 2009, 26, 1251–1292. [Google Scholar] [CrossRef]
- Koeduka, T.; Fridman, E.; Gang, D.R.; Vassa, D.G.; Jackson, B.L.; Kish, C.M.; Orlova, I.; Spassova, S.M.; Lewis, N.G.; Noel, J.P.; Baiga, T.J.; Dudareva, N.; Pichersky, E. Eugenol and isoeugenol, characteristic aromatic constituents of spices, are biosynthesized via reduction of a coniferyl alcohol ester. Proc. Natl. Acad. Sci. USA 2006, 103, 10128–10133. [Google Scholar]
- Cassab, G.I. Plant cell wall proteins. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 281–309. [Google Scholar] [CrossRef]
- Showalter, A.M. Introduction: plant cell wall proteins. Cell Mol. Life Sci. 2001, 58, 1361–132. [Google Scholar]
- Estermann, E.V.; Peterson, G.H.; McLaben, A.D. Digestion of clay-protein, lignin-protein, and silica-protein complexes by enzymes and bacteria. Soil Sci. Soc. Am. J. 1959, 23, 31–36. [Google Scholar] [CrossRef]
- Chen, M.; Sommer, A.J.; McClure, J.W. Fourier transform IR determination of protein contamination in thioglycolic acid lignin from radish seedlings, and improved methods for extractive-free cell wall preparation. Phytochem. Anal. 2000, 11, 153–159. [Google Scholar] [CrossRef]
- Henderson, M.E.K. Fungal metabolism of certain aromatic compounds related to lignin. Pure Appl. Chem. 1963, 7, 589–602. [Google Scholar] [CrossRef]
- Sutcliffe, R.; Sadler, J.N. The role of lignin in the adsorption of cellulases during enzymatic treatment of lignocellulosic material. Biotechnol. Bioeng. 1986, 17, 749–762. [Google Scholar]
- Tatsumoto, K.; Baker, J.O.; Tucker, M.P.; Oh, K.K.; Mohagheghi, A.; Grohmann, K.; Himmel, M.E. Digestion of pretreated aspen substrates: hydrolysis rates and adsorptive loss of cellulase enzymes. Appl. Biochem. Biotechnol. 1988, 18, 159–174. [Google Scholar] [CrossRef]
- Rajeev, K.; Wyman, C.E. Access of cellulose to cellulose and lignin or poplar solids produced by leading pretreatment technologies. Biotechnol. Progr. 2009, 25, 807–819. [Google Scholar] [CrossRef]
- Tu, M.; Pan, X.; Saddler, J.N. Adsorption of cellulase on cellulolytic enzyme lignin from Lodgepole pine. J. Agric. Food Chem. 2009, 57, 7771–7778. [Google Scholar] [CrossRef]
- Berlin, A.; Gilkes, N.; Kurabi, A.; Bura, R.; Tu, M.; Kilburn, D.; Saddler, J.N. Weak lignin-binding enzymes: A novel approach to improve activity of cellulases for hydrolysis of lignocellulosics. Appl. Biochem. Biotechnol. 2005, 121-124, 163–170. [Google Scholar]
- Berlin, A.; Balakshin, M.; Gilkes, N.; Kadla, J.; Maximenko, V.; Kubo, S.; Saddler, J. Inhibition of cellulase, xylanase and β-glucosidase activities by softwood lignin preparations. J. Biotechnol. 2006, 125, 198–209. [Google Scholar]
- Berlin, A.; Maximenko, V.; Gilkes, N.; Saddler, J. Optimization of enzyme complexes for lignocellulose hydrolysis. Biotechnol. Bioeng. 2007, 97, 287–296. [Google Scholar] [CrossRef]
- Chandra, R.P.; Bura, R.; Mabee, W.E.; Berlin, A.; Pan, X.; Saddler, J.N. Substrate pretreatment: the key to effective enzymatic hydrolysis of lignocellulosics? Adv. Biochem. Eng. Biotechnol. 2007, 108, 67–93. [Google Scholar]
- Kaewtatip, K.; Menut, P.; Auvergne, R.; Tanrattanakul, V.; Morel, M.H.; Guilbrt, S. Interactions of Kraft lignin and wheat gluten during biomaterial processing: evidence for the role of phenolic groups. J. Agric. Food Chem. 2010, 58, 4185–4192. [Google Scholar] [CrossRef]
- Yang, B.; Wyman, C.E. BSA treatment to enhance enzymatic hydrolysis of cellulose in lignin containing substrates. Biotechnol. Bioeng. 2006, 94, 611–617. [Google Scholar] [CrossRef]
- Yang, B.; Wyman, C.E. Lignin blockers and uses thereof. US Patent 2006/0088922A1, 27 April 2006. [Google Scholar]
- Yang, B.; Wyman, C.E. Ligin-blocking treatment of biomass and uses thereof. US Patent US7,604,967B2, 20 October 2009. [Google Scholar]
- Zahedifar, M.; Castro, F.B.; Orskov, E.R. Effect of hydrolytic lignin on formation of protein-lignin complexes and protein degradation by rumen microbes. Anim. Feed Sci. Technol. 2002, 95, 83–92. [Google Scholar] [CrossRef]
- Waffenschmidt, S.; Woessner, J.P.; Beer, K.; Goodenough, U.W. Isodityrosine cross-linking mediates insolubilization of cell walls in Chlamydomonas. Plant Cell 1993, 5, 809–820. [Google Scholar]
- Bao, W.; O’Malley, D.M.; Sederoff, R.R. Wood contains a cell-wall structural protein. Proc. Natl. Acad. Sci. USA 1992, 89, 6604–6608. [Google Scholar] [CrossRef]
- Bradley, D.J.; Kjellbom, P.; Lamb, C.J. Elicitor- and wound-induced oxidative crosslinking of proline-rich plant cell wall protein: a novel, rapid defense response. Cell 1992, 70, 21–30. [Google Scholar] [CrossRef]
- Ringli, C.; Hauf, G.; Keller, B. Hydrophobic interactions of the structural protein GRP1.8 in the cell wall of protoxylem elements. Plant Physiol. 2001, 125, 673–682. [Google Scholar] [CrossRef]
- Iiyama, K.; Lam, T.B.T.; Stone, B.A. Covalent cross-links in the cell wall. Plant Physiol. 1994, 104, 315–320. [Google Scholar]
- Cannon, M.C.; Terneus, K.; Hall, Q.; Tan, L.; Wang, Y.; Wegenhart, B.L.; Chen, L.; Lamport, D.T.A.; Chen, Y.; Kieliszewski, M.J. Self-assembly of the plant cell wall requires an extensin scaffold. Proc. Natl. Acad. Sci. USA 2008, 105, 2226–2231. [Google Scholar]
- Dill, I.; Salnikow, J.; Kraepelin, G. Hydroxyproline-rich protein material in wood and lignin of Fagus sylvatica. Appl. Environ. Microbiol. 1984, 48, 1259–1261. [Google Scholar]
- Bacic, A.; Harris, P.J.; Stone, B.A. Structure and function of plant cell walls. In The Biochemistry of Plants: a comprehensive treatise; Stumpf, P.K., Conn, E.E., Eds.; Carbohydrates, Acad. Press: New York, NY, USA, 1988; Volume 14, pp. 297–371. [Google Scholar]
- Iiyama, K.; Lam, T.B.T.; Meikle, P.J.; Ng, K.; Rhodes, D.; Stone, B.A. Cell wall biosynthesis and its regulation. In Forage cell wall structure and digestibility; Jung, H., Buxton, D., Hatfield, R., Ralph, J., Eds.; Am. Soc. Agron.: Madison, WI, USA, 1993; pp. 621–683. [Google Scholar]
- Whitmore, F.W. Lignin-protein complex catalyzed by peroxidase. Plant Sci. Lett. 1978, 13, 241–245. [Google Scholar] [CrossRef]
- Whitmore, F.W. Lignin carbohydrate complex formed in isolated cell walls of callus. Phytochemistry 1978, 17, 421–425. [Google Scholar] [CrossRef]
- Whitmore, F.W. Lignin-protein complex in cell walls of Pinus elliotti: Amino acid constituents. Phytochemistry 1982, 21, 315–318. [Google Scholar] [CrossRef]
- Evans, J.J.; Himmelsbach, D.S. Incorporation of peroxidase into synthetic lignin. J. Agric. Food Chem. 1991, 39, 830–832. [Google Scholar] [CrossRef]
- Morimoto, S.; Tateishi, N.; Inuyama, H.; Tara, F.; Tanaka, H.; Shoyama, Y. Identification and molecular characterization of novel peroxidase with structural protein-like properties. J. Biol. Chem. 1999, 274, 26192–26198. [Google Scholar]
- McDougall, G.J.; Stewart, D.; Morrison, I.M. Tyrosine residues enhance cross-linking of synthetic proteins into lignin-like dehydrogenation products. Phytochemistry 1996, 41, 43–47. [Google Scholar]
- Liang, H.; Frost, C.J.; Wei, X.; Brown, N.R.; Carlson, J.E.; Tien, M. Improved sugar release from lignocellulosic material by introducing a tyrosine-rich cell wall peptide gene in poplar. Clean Soil Air Water 2008, 36, 662–668. [Google Scholar] [CrossRef]
- Boukari, I.; Putaux, J.L.; Cathala, B.; Barakat, A.; Saake, B.; Remond, C.; O’Donohue, M.; Chabbert, L.B. In vitro model assemblies to study the impact of lignin-carbohydrate interactions on the enzymatic conversion of xylan. Biomacromolecules 2009, 10, 2489–2498. [Google Scholar] [CrossRef]
- Grant Reid, J.S. Carbohydrate metabolism: Structural carbohydrates. In Plant Biochemistry; Dey, P.M., Harborne, J.B., Eds.; Academic Press: San Diego, CA, USA, 1997; pp. 205–236. [Google Scholar]
- Perez, J.; Munoz-Dorado, J.; de la Rubia, T.; Martinez, J. Biodegradation and biological treatments of cellulose, hemicellulose and lignin: An overview. Int. Microbiol. 2002, 5, 53–63. [Google Scholar] [CrossRef]
- Westbye, P.; Kohnke, T.; Glasser, W.; Gatenholm, P. The influence of lignin on the self-assembly behavior of xylan rich fractions from birch. Cellulose 2007, 14, 603–613. [Google Scholar] [CrossRef]
- Jeffries, T.W. Biodegradation of lignin-carbohydrate complexes. Biodegradation 1990, 1, 163–176. [Google Scholar] [CrossRef]
- Koshijima, T.; Watanabe, T. Association between lignin and carbohydrates in wood and other plant tissues. In Springer Series in Wood Science; Timell, T.E., Ed.; Springer-Verlag: New York, NY, USA, 2003; p. 329. [Google Scholar]
- Habrant, A.; Gaillard, C.; Ralet, M.-C.; Laivez, D.; Cathala, B. Relation between chemical structure and supramolecular organization of synthetic lignin-pectin particles. Biomacromolecules 2009, 10, 3151–3156. [Google Scholar] [CrossRef]
- Barakat, A.; Putaux, J.-L.; Saulnier, L.; Chabbert, B.; Cathala, B. Characterization of arabinoxylan-dehydrogenation polymer (synthetic lignin polymer) nanoparticles. Biomacromolecules 2007, 8, 1236–1245. [Google Scholar] [CrossRef]
- Barakat, A.; Gaillard, C.; Lairez, D.; Saulnier, L.; Chabbert, B.; Cathala, B. Supramolecular organization of heteroxylan-dehydrogenation polymers (synthetic lignin) nanoparticles. Biomacromolecules 2008, 9, 487–493. [Google Scholar] [CrossRef]
- Barakat, A.; Winter, H.; Rondeau-Mouro, C.; Saake, B.; Chabbert, B.; Cathala, B. Studies of xylan interactions and cross-linking to synthetic lignins formed by bulk and end-wise polymerization: A model study of lignin carbohydrate complex formation. Planta 2007, 226, 267–281. [Google Scholar] [CrossRef]
- Uraki, Y.; Nakamura, A.; Kshimoto, T.; Ubakata, M. Interaction of hemicelluloses with monolignols. J. Wood. Chem. Technol. 2007, 27, 9–21. [Google Scholar] [CrossRef]
- Lairez, D.; Cathala, B.; Monties, B.; Bedos-Behal, F.; Duran, H.; Gorrichon, L. Aggregation during coniferyl alcohol polymerization in pectin solution: A biomimetic approach of the first steps of lignification. Biomacromolecules 2005, 6, 763–774. [Google Scholar] [CrossRef]
- Lehn, J.M. Supramolecular chemistry. Science 1993, 260, 1762–1763. [Google Scholar]
- Ariga, K.; Hill, J.P.; Lee, M.V.; Vinu, A.; Chrvet, R.; Acharya, S. Challenges and breakthroughs in recent research on self-assembly. Sci. Technol. Adv. Mater. 2008, 9, 1–96. [Google Scholar]
- Inomata, F.; Takabe, K.; Saiki, H. Cell wall formation of conifer tracheid as revealed by rapid-freeze and substitution method. J. Electron Microsc. (Tokyo) 1992, 41, 369–374. [Google Scholar]
- Ralph, J.; Grabber, J.H.; Hatfield, R.D. Lignin-ferulate cross-links in grasses: Active incorporation of ferulate polysaccharide esters into ryegrass lignins. Carbohydrate Res. 1995, 275, 167–178. [Google Scholar] [CrossRef]
- Donaldson, L.A. Lignification and lignin topochemistry – an ultrastructural view. Phytochemistry 2001, 57, 859–873. [Google Scholar] [CrossRef]
- Gorshkova, T.A.; Mikshina, P.V.; Gurjanov, O.P.; Chemikosova, S.B. Formation of plant cell wall supramolecular structure. Biochemistry (Moscow) 2010, 75, 159–172. [Google Scholar] [CrossRef]
- Boudet, A.M. Towards an understanding of the supramolecular organization of the lignified cell wall. In The Plant Cell Wall; Rose, J.K.C., Ed.; 2003; pp. 1555–182. [Google Scholar]
- Connors, W.J.; Sarkanen, S.; McCarthy, J.L. Gel chromatography and association complexes of lignin. Holzforschung 1980, 34, 80–85. [Google Scholar] [CrossRef]
- Sarkanen, S.; Teller, D.C.; Stevens, C.R.; McCarthy, J.L. Lignins. 20. Associative interactions between Kraft lignin components. Macromolecules 1984, 17, 2588–2597. [Google Scholar] [CrossRef]
- Woerner, D.L.; McCarthy, J.L. Lignin. 24. Ultrafiltration and light-scattering evidence for association of Kraft lignins in aqueous solutions. Macromolecules 1988, 21, 2160–2166. [Google Scholar] [CrossRef]
- Gilardi, G.; Cass, A.E.G. Associative and colloidal behavior of lignin and implications for its biodegradation in vitro. Langmuir 1993, 9, 1721–1726. [Google Scholar] [CrossRef]
- Guerra, A.; Gaspar, A.R.; Contreras, S.; Lucia, L.A.; Crestini, C.; Argyropoulos, D.S. On the propensity of lignin to associate: A size exclusion chromatography study with lignin derivatives isolated from different plant species. Phytochemistry 2007, 68, 2570–2583. [Google Scholar] [CrossRef]
- Contreras, S.; Gaspar, A.R.; Guerra, A.; Lucia, L.A.; Argyropoulos, D.S. Propensity of lignin to associate: Light scattering photometry study with native lignins. Biomacromolecules 2008, 9, 3362–3369. [Google Scholar] [CrossRef]
- Micic, Ml.; Jeremic, M.; Radotic, K.; Leblanc, R.M. A comparative study of enzymatically and photochemically polymerized artificial lignin supramolecular structures using environmental scanning electron microscopy. J. Colloid Interface Sci. 2000, 231, 190–194. [Google Scholar] [CrossRef]
- Micic, M.; Jeremic, M.; Radotic, K.; Mavers, M.; Leblanc, R.M. Visualization of artificial lignin supramolecular structures. Scanning 2000, 22, 288–294. [Google Scholar]
- Micic, M.; Radotic, K.; Benitez, I.; Ruano, M.; Jeremic, M.; Moy, V.; Mabrouki, M.; Leblanc, R.M. Topographical characterization and surface force spectroscopy of the photochemical lignin model compound. Biophys. Chem. 2001, 94, 257–263. [Google Scholar] [CrossRef]
- Micic, M.; Orbulescu, J.; Radotic, K.; Jeremic, M.; Sui, G.; Zheng, Y.; Leblanc, R.M. ZL-DHP lignin model compound at the air-water interface. Biophys. Chem. 2002, 99, 55–62. [Google Scholar] [CrossRef]
- Micic, M.; Radotic, K.; Jeremic, M.; Djikanovic, D.; Kammer, S.B. Study of the lignin model compound supramolecular structure by combination of near-field scanning optical microscopy and atomic force microscopy. Colloids Surf. B: Biointerfaces 2004, 34, 33–40. [Google Scholar] [CrossRef]
- Micic, M.; Benitez, I.; Ruano, M.; Mavers, M.; Jeremic, M.; Radotic, K.; Moy, V.; Leblanc, R.M. Probing the lignin nanomechanical properties and lignin-lignin interactions using the atomic force microscopy. Chem. Phys. Lett. 2007, 347, 41–45. [Google Scholar]
- Terashima, N.; Kitano, K.; Kojima, M.; Yoshida, M.; Yamamoto, H.; Westermark, U. Nanostructural assembly of cellulose, hemicellulose, and lignin in the middle layer of secondary wall of ginkgo tracheid. J. Wood Sci. 2009, 55, 409–416. [Google Scholar] [CrossRef]
- Micic, M.; Radotic, K.; Jeremic, M.; Leblanc, R.M. Study of self-assembly of the lignin model compound on cellulose model substrate. Macromol. Biosci. 2003, 3, 100–106. [Google Scholar] [CrossRef]
- Abreu, H.S.; Latorraca, J.V.F.; Pereira, R.P.W.; Monteiro, M.B.O.; Abreu, F.A.; Amparado, K.F. A superamolecular proposal of lignin structure and its relation with the wood properties. Ann. Brazilian Acad. Sci. 2009, 81, 137–142. [Google Scholar] [CrossRef]
- Norgren, M.; Notley, S.M.; Majtnerova, A.; Gellerstedt, G. Smooth model surfaces from lignin derivatives. I. Preparation and characterization. Langmuir 2006, 22, 1209–1214. [Google Scholar] [CrossRef]
- Djikanovic, A.; Kalauzi, M.; Jeremic, M.; Micic, M.; Radotic, K. Deconvolution of fluorescence spectra: contribution to the structural analysis of complex molecules. Colloids Surf. B Biointerfaces 2007, 54, 188–192. [Google Scholar] [CrossRef]
- Kocheva, L.S.; Karmanov, A.P.; Mironov, M.V.; Belyi, V.A.; Belyaev, V.Y.; Monakov, Y.B. Straw lignins: Hydrodynamic and conformational properties of the macromolecules. Russ. J. Appl. Chem. 2008, 81, 2033–2039. [Google Scholar] [CrossRef]
- Liebovitch, L.S. Fractals and chaos simplified for the life sciences; Oxford University Press: New York, NY, USA, 1998; p. 268. [Google Scholar]
- Karmanov, A.P.; Monakov, Y.B. Hydrodynamic properties and structure of lignin. Int. J. Polym. Mater. 2000, 48, 151–175. [Google Scholar] [CrossRef]
- Radotic, K.; Simic-Krstic, J.; Jeremic, Ml.; Trifunovic, M. A study of lignin formation at the molecular level by scanning tunneling microscopy. Biophys. J. 1994, 66, 1763–1767. [Google Scholar] [CrossRef]
- Radotic, K.; Tasic, M.; Jeremic, M.; Budimlija, Z.; Simic-Krstic, J.; Polzovic, A.; Bozovic, Z. Fractal dimension of lignin structure at the molecular level. In UDC 541.64.BIBLID 0021-3225 Conference Paper; 1998; pp. 215–220. [Google Scholar]
- Radotic, K.; Tasic, M.; Jeremic, M.; Budimlija, J.; Simic-Krstic, J.; Polzovic, A.; Bozovic, Z. Fractal analysis of STM images of lignin polymer obtained by in vitro synthesis. Gen. Physiol. Biophys. 2000, 19, 171–180. [Google Scholar]
- Karmanov, A.P.; Matveev, D.V.; Monakov, Y.B. Polymerization dynamics of monomeric precursors of guaiacyl lignins. Doklady Chem. 2001, 380, 280–283. [Google Scholar] [CrossRef]
- Radotic, K.; Micic, M.; Jeremic, M. New insights into the structural organization of the plant polymer lignin. Ann. N.Y. Acad. Sci. 2005, 1048, 215–229. [Google Scholar] [CrossRef]
- Suchy, M.; Virtanen, J.; Kontturi, E.; Vuorinen, T. Impact of drying on wood ultrastructure by deuterium exchange and photoacoustic FT-IR spectroscopy. Biomacromolecules 2010, 11, 515–520. [Google Scholar] [CrossRef]
- Teeri, T.T.; Brumer, H.; Daniel, G.; Gatenholm, P. Biomimetic engineering of cellulose-based mateials. Trends Biotechnol. 2007, 25, 299–306. [Google Scholar] [CrossRef]
- Mellerowicz, E.J.; Sundberg, B. Wood cell walls: biosynthesis, developmental dynamics, and their implications for wood properties. Curr. Opin. Plant Biol. 2008, 11, 293–300. [Google Scholar] [CrossRef]
- Moreno, N.; Bougourd, S.; Haseloff, J.; Feijo, J.A. Imaging plant cells. In Handbook of Biological Confocal Microscopy, 3rd; Pawley, J.B., Ed.; Springer Science: New York, NY, USA, 2006; pp. 769–787, Chapter 44. [Google Scholar]
- Pedroso, M.C.; Sinclair, M.B.; Jones, H.D.T.; Haaland, D.M. Hyperspectral confocal fluorescence microscope: A new look into the cell. Micros. Today 2010, 18, 14–18. [Google Scholar]
- Abraham, V.C.; Taylor, D.L.; Haskins, J.R. High-content screening applied to large-scale cell biology. Trends Biotechnol. 2004, 22, 15–22. [Google Scholar] [CrossRef]
- Zhong, R.; Ripperger, A.; Ye, Z.H. Ectopic deposition of lignin in the pith of stems of two Arabidopsis mutants. Plant Physiol. 2000, 123, 59–69. [Google Scholar] [CrossRef]
- Heitner, C.; Dimmel, D.R.; Schmidt, J.A. Lignin and Lignans: Advances in chemistry; CRC Press: Boca Raton, FL, USA, 2010; p. 683. [Google Scholar]
- Raman, C.V.; Krishnan, K.S. A new type of secondary radiation. Nature 1928, 121, 501–502. [Google Scholar]
- Agarwal, U.P. Raman spectroscopic characterization of wood and pulp fibers. In Characterization of Lignocellulose Materials; Hu, T.Q., Ed.; Blackwell Publishing: Oxford, UK, 2008; pp. 17–35, Chapter 2. [Google Scholar]
- Schmidt, M.; Schwartzbg, A.M.; Perera, P.N.; Weber-Bargioni, W.; Carroll, A.; Sarkar, P.; Bosneaga, E.; Urban, J.J.; Song, J.; Balakshin, M.Y.; Caapanema, E.A.; Auer, M.; Adams, P.D.; Chiang, V. Label-free in situ imaging of lignification in the cell wall of low lignin transgenic Populus trichocarpa. Planta 2009, 230, 589–597. [Google Scholar] [CrossRef]
- Johnson, C.K.; Osborn, K.D.; Allen, M.W.; Slaughter, B.D. Single-molecule fluorescence spectroscopy: New probes of protein function and dynamics. Physiology 2005, 20, 10–14. [Google Scholar] [CrossRef]
- Berg, R.H. Evaluation of spectral imaging for plant cell analysis. J. Micros. 2004, 214, 174–181. [Google Scholar] [CrossRef]
- Singh, S.; Simmons, B.A.; Vogel, K.P. Visualization of biomass solubilization and cellulose regeneration during ionic liquid pretreatment of switchgrass. Biotechnol. Bioeng. 2009, 104, 68–74. [Google Scholar] [CrossRef]
- Achyuthan, K.E.; Adams, P.D.; Datta, S.; Simmons, B.A.; Singh, A.K. Hitherto unrecognized fluorescence properties of coniferyl alcohol. Molecules 2010, 15, 1645–1667. [Google Scholar] [CrossRef]
- Hatfield, R.; Fukushima, R.S. Can lignin be accurately measured? Crop Sci. 2005, 45, 832–839. [Google Scholar] [CrossRef]
- Nkansah, K.; Dawson-Andoh, B.; Slahor, J. Rapid characterization of biomass using near infrared spectroscopy coupled with multivariate data analysis: Part 1. Yellow-poplar (Liriodendron tulipifera L). Bioresource Technol. 2010, 101, 4570–4576. [Google Scholar] [CrossRef]
- Gidh, A.V.; Decker, S.R.; Vinzant, T.B.; Himmel, M.E.; Williford, C. Determination of lignin by size exclusion chromatography using multi angle laser light scattering. J. Chrom. A 2006, 1114, 102–110. [Google Scholar] [CrossRef]
- Koch, G.; Gunnar, G. Application of scanning UV microspectrophotometry to localize lignins and phenolic extractives in plant cell walls. Holzforschung 2001, 55, 563–567. [Google Scholar]
- Yarbrough, J.M.; Himmel, M.E.; Ding, S.Y. Plant cell wall characterization using scanning probe microscopy techniques. Biotechnol. Biofuels 2009, 2, 17. [Google Scholar] [CrossRef]
- Wimmer, R.; Lucas, B.N.; Tsui, T.Y.; Oliver, W.C. Longitudinal hardness and Young’s modulus of spruce tracheid secondary walls using nanoindentation technique. Wood Sci. Technol. 1997, 31, 131–141. [Google Scholar]
- Yelle, D.J.; Ralph, J.; Frihart, C.R. Characterization of nonderivatized plant cell wals using high-resolution solution-state NMR spectroscopy. Magn. Reson. Chem. 2008, 46, 508–517. [Google Scholar] [CrossRef]
- Shevchenko, S.M.; Bailey, G.W. The mystery of the lignin-carbohydrate complex: A computational approach. J. Mol. Struct. (Theochem.) 1996, 364, 197–208. [Google Scholar] [CrossRef]
- Faulon, J.L.; Hatcher, P.G. Is there any order in the structure of lignin. Energy Fuels 1994, 8, 402–407. [Google Scholar] [CrossRef]
- da Costa Sousa, L.; Chundawat, S.P.; Balan, V.; Dale, B.E. Cradle-to-grave’ assessment of existing lignocellulose pretreatment technologies. Curr. Opin. Biotechnol. 2009, 20, 339–347. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, Z.; Zhang, R. Overview of biomass pretreatment for cellulosic ethanol production. Int. J. Agric. Biol. Eng. 2009, 2, 51–68. [Google Scholar]
- Rastogi, S.; Dwivedi, U.N. Manipulation of lignin plants with special reference to O-methyltransferase. Plant Sci. 2008, 174, 264–277. [Google Scholar] [CrossRef]
- Leple, J.C.; Dauwe, R.; Morreel, K.; Storme, V.; Lapierre, C.; Pollet, B.; Naumann, A.; Kang, K.Y.; Kim, H.; Ruel, K.; Lefe’bvre, A.; Joseleau, J.P.; Grima-Pettenati, J.; De Rycke, R.; Andersson-Gunnera, S.; Erban, A.; Fehrle, I.; Petit-Conil, M.; Kopka, J.; Polle, A.; Messens, E.; Sundberg, B.; Mansfield, S.D.; Ralph, J.; Pilate, G.; Boerjan, W. Downregulation of cinnamoyl-coenzyme A reductase in poplar: multiple-level phenotyping reveals effects on cell wall polymer metabolism and structure. Plant Cell 2007, 19, 3669–3691. [Google Scholar] [CrossRef]
- Camarero, S.; Ibarra, D.; Martinez, M.J.; Martinez, A.T. Lignin-derived compounds as efficient laccase mediators for decolorization of different types of recalcitrant dyes. Appl. Environ. Microbiol. 2005, 71, 1775–1784. [Google Scholar] [CrossRef]
- Bourbonnais, R.; Paice, M.G.; Freiermuth, B.; Bodie, E.; Borneman, S. Reactivities of various mediators and laccases with Kraft pulp and lignin model compounds. Appl. Environ. Microbiol. 1997, 63, 4627–4632. [Google Scholar]
- Takahama, U. Oxidation of hydroxycinnamic acid and hydroxycinnamyl alcohol derivatives by laccase and peroxidase. Interactions among p-hydroxyphenyl, guaiacyl and syringyl groups during the oxidation reactions. Physiol. Plant. 1995, 93, 61–68. [Google Scholar]
- Takahama, U.; Oniki, T. Enhancement of peroxidase-dependent oxidation of sinapyl alcohol by an apoplstic component, 4-coumaric acid ester isolated from epicotyls of Vigna angularis L. Plant Cell Physiol. 1997, 38, 456–462. [Google Scholar] [CrossRef]
- Achyuthan, K.E.; Adams, P.D.; Simmons, B.A.; Singh, A.K. Spectroscopic analyses of the biofuels-critical phytochemical coniferyl alcohol and its enzyme-catalyzed oxidation products. Molecules 2009, 14, 4758–4778. [Google Scholar] [CrossRef]
- Martinez, D.; Larrondo, L.F.; Putnam, N.; Gelpke, M.D.S.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.; Coutinho, P.M.; Henrissat, B.; Berka, R.; Cullen, D.; Rokhsar, D. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef]
- Sonoki, T.; Otsuka, Y.; Ikeda, S.; Masai, E.; Kajita, S.; Katayama, Y. Close association between the enzymes involved in the lignin metabolic pathway of Sphingomonas paucimobilis SYK-6: Interaction of O-demethylase (LigX) and ring fission dioxygenase (LigZ). J. Wood Sci. 2002, 48, 250–252. [Google Scholar] [CrossRef]
- Kersten, P.; Cullen, D. Extracellular oxidative systems of the lignin-degrading basidomycete Phanerochaete chrysosporium. Fungal Genet. Biol. 2007, 44, 77–87. [Google Scholar] [CrossRef]
- Passardi, F.; Theiler, G.; Zamocky, M.; Cosio, C.; Rouhier, N.; Teixera, F.; Margis-Pinheiro, M.; Ioannidis, V.; Penel, C.; Falquet, L.; Dunand, C. PeroxiBase: The peroxidase database. Phytochemistry 2007, 68, 1605–1611. [Google Scholar]
- Sasaki, S.; Nishida, T.; Tsutsumi, Y.; Kondo, R. Lignin dehydrogenative polymerization mechanism: A poplar cell wall peroxidase directly oxidizes polymer lignin and produces in vitro dehydrogenative polymer rich in β-O-4 linkage. FEBS Lett. 2004, 562, 197–201. [Google Scholar] [CrossRef]
- Leonowicz, A.; Cho, N.S.; Luterek, J.; Wilkolazka, A.; Wojtaswasilewska, M.; Matuszewska, A.; Hofrichter, M.; Wesenberg, D.; Rogalski, J. Fungal laccase: Properties and activity on lignin. J. Basic Microbiol. 2001, 41, 183–225. [Google Scholar]
- Somerville, C. The billion-ton biofuels vision. Science 2006, 312, 1277. [Google Scholar] [CrossRef]
- Service, R.F. Is there a road ahead for cellulosic ethanol? Science 2010, 329, 784–785. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Achyuthan, K.E.; Achyuthan, A.M.; Adams, P.D.; Dirk, S.M.; Harper, J.C.; Simmons, B.A.; Singh, A.K. Supramolecular Self-Assembled Chaos: Polyphenolic Lignin’s Barrier to Cost-Effective Lignocellulosic Biofuels. Molecules 2010, 15, 8641-8688. https://doi.org/10.3390/molecules15118641
Achyuthan KE, Achyuthan AM, Adams PD, Dirk SM, Harper JC, Simmons BA, Singh AK. Supramolecular Self-Assembled Chaos: Polyphenolic Lignin’s Barrier to Cost-Effective Lignocellulosic Biofuels. Molecules. 2010; 15(12):8641-8688. https://doi.org/10.3390/molecules15118641
Chicago/Turabian StyleAchyuthan, Komandoor Elayavalli, Ann Mary Achyuthan, Paul David Adams, Shawn Matthew Dirk, Jason Carl Harper, Blake Alexander Simmons, and Anup Kumar Singh. 2010. "Supramolecular Self-Assembled Chaos: Polyphenolic Lignin’s Barrier to Cost-Effective Lignocellulosic Biofuels" Molecules 15, no. 12: 8641-8688. https://doi.org/10.3390/molecules15118641
APA StyleAchyuthan, K. E., Achyuthan, A. M., Adams, P. D., Dirk, S. M., Harper, J. C., Simmons, B. A., & Singh, A. K. (2010). Supramolecular Self-Assembled Chaos: Polyphenolic Lignin’s Barrier to Cost-Effective Lignocellulosic Biofuels. Molecules, 15(12), 8641-8688. https://doi.org/10.3390/molecules15118641