Density Functional Theory Study of the Trans-Trans-Cis (TTC)→Trans-Trans-Trans (TTT) Isomerization of a Photochromic Spiropyran Merocyanine

Abstract
:Introduction

Computational Methods
Results and Discussion
Isomerization energy

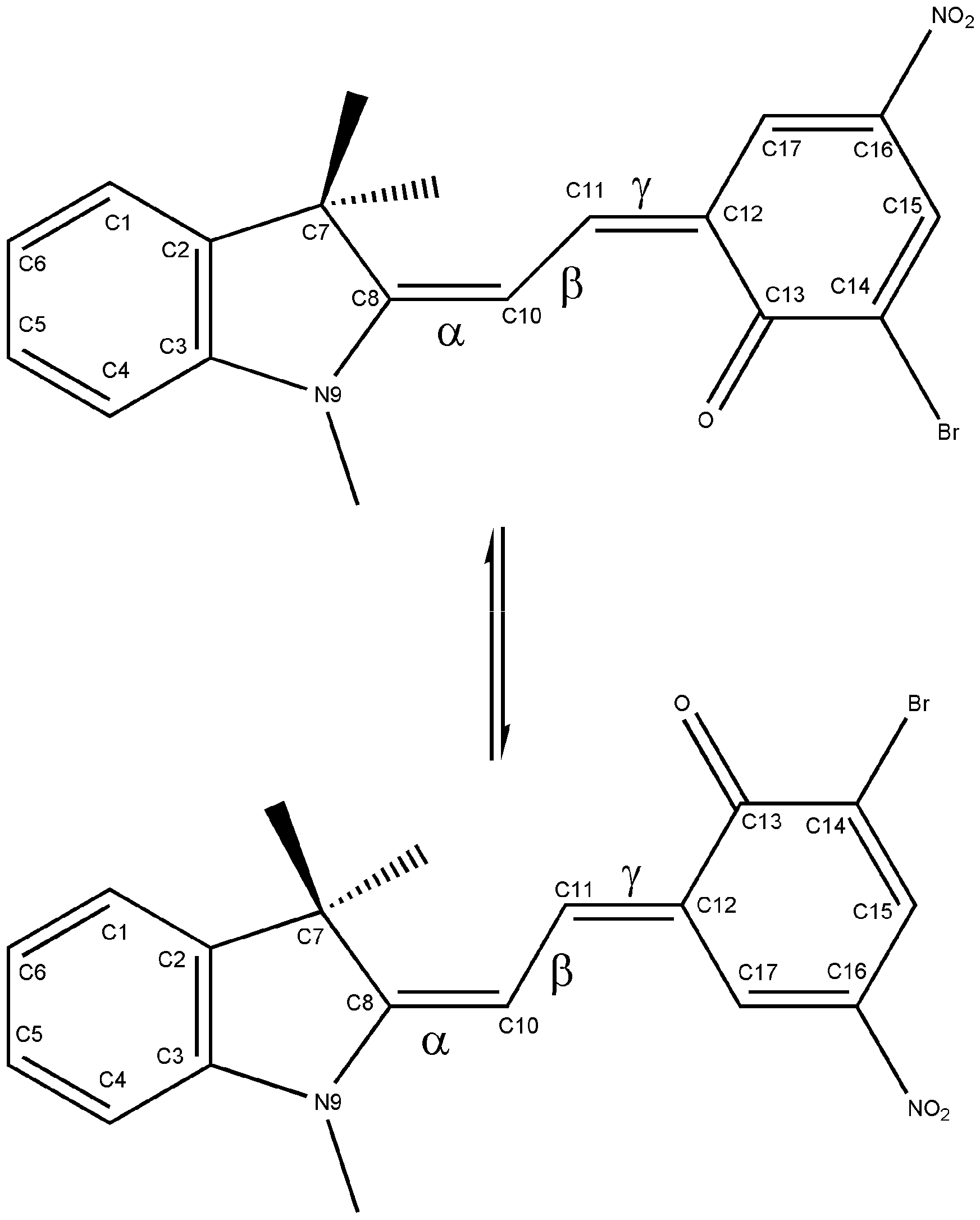{kind=link}
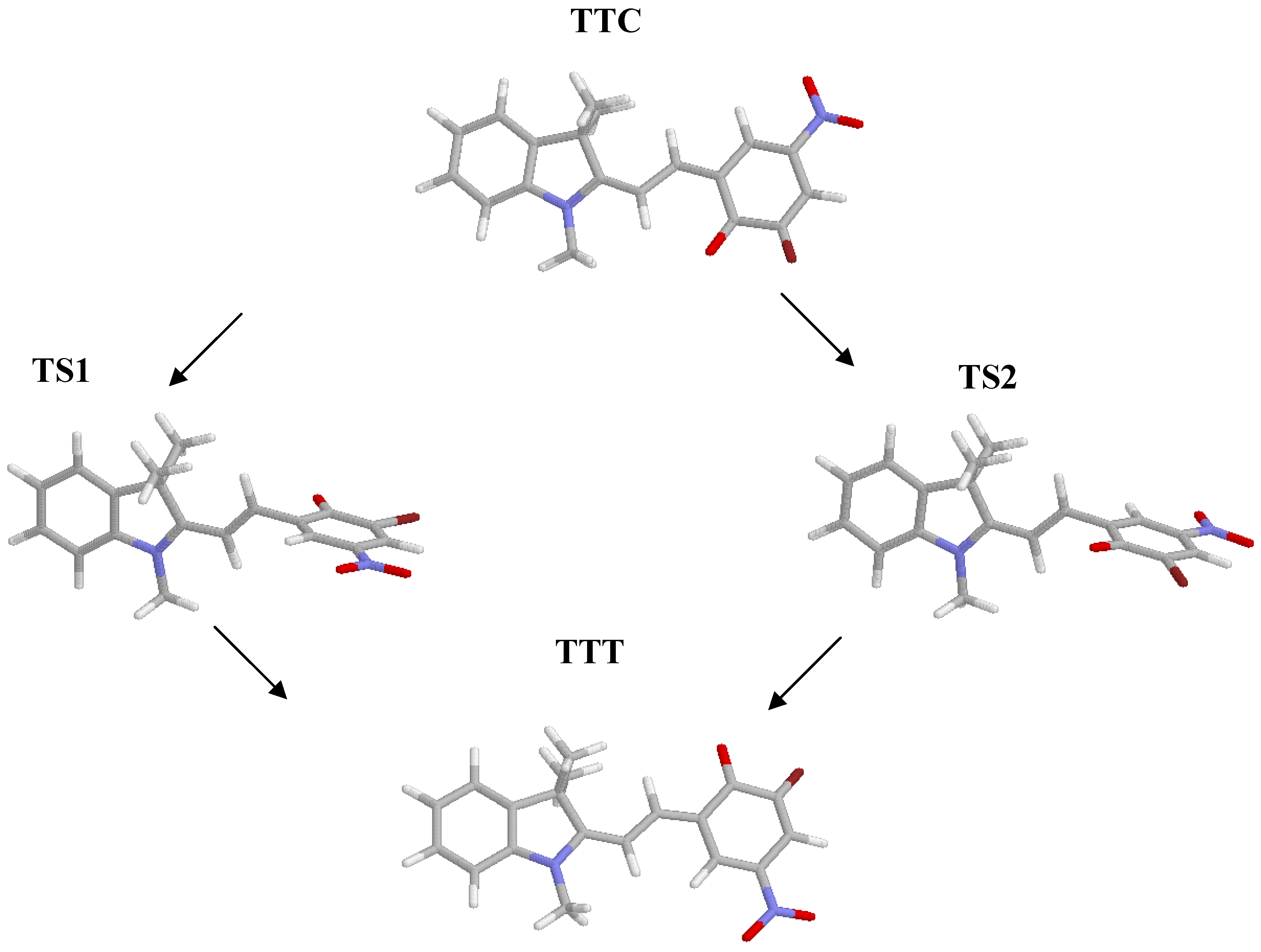{kind=link}
| 8-Br-6-NO2-BIPS in vacuo | 8-Br-6-NO2-BIPS in CH2Cl2 solution | |||
|---|---|---|---|---|
| TTC | TTT | TTC | TTT | |
| E | 0. | 6.8 | 0. | 5.8 |
| H190 | 0. | 6.3 | 0. | 6.0 |
| G190 | 0. | 5.8 | 0. | 5.3 |
| H298 | 0. | 6.6 | 0. | 6.0 |
| G298 | 0. | 5.8 | 0. | 4.5 |
| α | 180. | 180. | 180. | 180. |
| β | 180. | 180. | -179.9 | 180. |
| γ | 0. | 180. | 0.1 | 180. |
| C8-C10 | 1.390 | 1.388 | 1.407 | 1.405 |
| C10-C11 | 1.398 | 1.400 | 1. 384 | 1.383 |
| C11-C12 | 1.406 | 1.398 | 1.424 | 1.418 |
Transition states structures and activation energy
| 6-NO2-8-Br-BIPS in vacuo | 6-NO2-8-Br-BIPS in CH2Cl2 solution | |||
|---|---|---|---|---|
| TS1 | TS2 | TS1 | TS2 | |
| E | 125.0 | 126.1 | 70.9 | 70.9 |
| H190 | 120.3 | 119.5 | 66.2 | 65.9 |
| G190 | 120.6 | 122.9 | 72.5 | 72.8 |
| H298 | 119.8 | 118.5 | 64.6 | 64.6 |
| G298 | 120.8 | 125.3 | 76.4 | 76.7 |
| α | -174.5 | 178.6 | -178.1 | -178.0 |
| β | -170.1 | 173.5 | -179.5 | 179.5 |
| γ | 86.3 | -87.1 | 85.9 | -85.4 |
| C8-C10 | 1.420 | 1.417 | 1.436 | 1.436 |
| C10-C11 | 1.363 | 1.364 | 1.354 | 1.354 |
| C11-C12 | 1.473 | 1.476 | 1.482 | 1.482 |

Acknowledgements
References
- Kawata, S.; Kawata, J. Three-dimensional optical data storage using photochromic materials. Chem. Rev. 2000, 100, 1777–1788. [Google Scholar] [CrossRef]
- Pisignano, D.; Mele, E.; Persano, L.; Athanassiou, A.; Fotakis., C.; Cingolani, R. Optical gain from the open form of a photochromic molecule in the solid state. J. Phys. Chem. B 2006, 110, 4506–4509. [Google Scholar]
- Berkovic, G.; Krongauz, V.; Weiss, V. Spiropyrans and spirooxazines for memories and switches. Chem. Rev. 2000, 100, 1741–1753. [Google Scholar] [CrossRef]
- Wojtyk, J.T.C.; Kazmaier, P.M.; Buncel, E. Modulation of the spiropyran-merocyanine reversion via metal-ion selective complexation : Trapping of the “transient” cis-merocyanine. Chem. Mater. 2001, 13, 2547–2551. [Google Scholar] [CrossRef]
- Wojtyk, J.T.C.; Wasey, A.; Kazmaier, P.M.; Hoz., S.; Buncel, E. Thermal reversion mechanism of N-functionalized merocyanines to spiropyrans: A solvatochromic, solvatokinetic, and semi-empirical study. J. Phys. Chem. A 2000, 104, 9046–9055. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Molecular switches controlled by light. Chem. Comm. 2006, 1169–1178. [Google Scholar] [CrossRef]
- Bose, M.; Groff, D.; Xie, J.; Brustad, E.; Schultz, P.G. The incorporation of a photoisomerizable amino acid into proteins in E.coli. J. Am. Chem. Soc. 2006, 128, 388–389. [Google Scholar] [CrossRef]
- Koçer, A.; Walko, M.; Meijberg, W.; Feringa, B.L. A light-actuated nanovalve derived from a channel protein. Science 2005, 309, 755–758. [Google Scholar] [CrossRef]
- Cottone, G.; Noto, R.; La Manna, G. Theoretical study of spiropyran-merocyanine thermal isomerization. Chem. Phys. Lett. 2004, 388, 218–222. [Google Scholar] [CrossRef]
- Cottone, G.; Noto, R.; La Manna, G.; Fornili, S.L. Ab initio study on the photoisomers of a nitro-substituted spiropyran. Chem. Phys. Lett. 2000, 319, 51–59. [Google Scholar] [CrossRef]
- Görner, H. Photochromism of nitrospiropyrans: Effects of structure, solvent and temperature. Phys. Chem. Chem. Phys. 2001, 3, 416–423. [Google Scholar] [CrossRef]
- Hobley, J.; Malatesta, V. Energy barrier to TTC-TTT isomerisation for the merocyanine of a photochromic spiropyran. Phys. Chem. Chem. Phys. 2000, 2, 57–59. [Google Scholar] [CrossRef]
- Gaussian 03. Revision B.04,; Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. (Eds.) Gaussian, Inc.: Wallingford, CT, 2004.
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Sheng, Y.; Leszczynski, J.; Garcia, A.A.; Rosario, R.; Gust, D.; Springer, J. Comprehensive Theoretical Study of the Conversion Reactions of Spiropyrans: Substituent and Solvent Effects. J. Phys. Chem. B 2004, 108, 16233–16243. [Google Scholar] [CrossRef]
- Sicinska, D.; Truhlar, D.G.; Paneth, P. Solvent-Dependent Transition States for Decarboxylations. J. Am. Chem. Soc. 2001, 123, 7683–7686. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cottone, G.; Noto, R.; La Manna, G. Density Functional Theory Study of the Trans-Trans-Cis (TTC)→Trans-Trans-Trans (TTT) Isomerization of a Photochromic Spiropyran Merocyanine. Molecules 2008, 13, 1246-1252. https://doi.org/10.3390/molecules13061246
Cottone G, Noto R, La Manna G. Density Functional Theory Study of the Trans-Trans-Cis (TTC)→Trans-Trans-Trans (TTT) Isomerization of a Photochromic Spiropyran Merocyanine. Molecules. 2008; 13(6):1246-1252. https://doi.org/10.3390/molecules13061246
Chicago/Turabian StyleCottone, Grazia, Rosina Noto, and Gianfranco La Manna. 2008. "Density Functional Theory Study of the Trans-Trans-Cis (TTC)→Trans-Trans-Trans (TTT) Isomerization of a Photochromic Spiropyran Merocyanine" Molecules 13, no. 6: 1246-1252. https://doi.org/10.3390/molecules13061246
APA StyleCottone, G., Noto, R., & La Manna, G. (2008). Density Functional Theory Study of the Trans-Trans-Cis (TTC)→Trans-Trans-Trans (TTT) Isomerization of a Photochromic Spiropyran Merocyanine. Molecules, 13(6), 1246-1252. https://doi.org/10.3390/molecules13061246