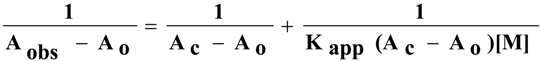Introduction
Conformationally flexible medium-sized cyclophanes are macrocycles of great importance for supramolecular chemistry and thus the attention of numerous chemists has focused on synthetic approaches to such molecules [
1,
2,
3]. In particular, great interest has been created in the synthesis and characterization of constrained macrocycles for the use as specific receptors for the selective complexation of neutral guests [
3,
4,
5] with the emphasis on the binding of a wide variety of biologically relavant molecules. Moreover, chemically robust aromatic macrocycles able to complex cations, in particular lanthanides, are needed as luminescent sensors and for diagnostic bioassays in medicine [
6,
7], not least for use in powerful screening techniques such as Magnetic Resonance Imaging (MRI), Proton Emission Computed Assisted Tomography (SPECT) or Proton Emission Tomography (PET) once complexed with radioactive lanthanides and actinides.
The search for rigid receptor molecules for organic dicationic guests is a very fertile field in chemistry. In particular, there exists a need to develop three-dimensional building blocks containing selected functional groups in order to introduce binding sites suitable for biologically relevant molecules into small stereochemically rigid cyclophanes [
1,
2,
3,
4,
5].
In fact, the selective recognition of biologically relevant molecules, as well as cations and especially anions, by synthetic receptors is a very fertile and interesting field of investigation in Supramolecular Chemistry [
8]. In particular, high emphasis is placed on the synthesis of conformationally preorganized building blocks from which specific hosts with desired properties can be prepared by appropriate and selective functionalization reactions.
In our approach, among the various possible functionalization procedures possible, the introduction of a phosphonic group into the molecule, directly linked to the aromatic rings of the phanes or as pendant arm, appears to be the most promising route. In fact, for constructing an effective artificial receptor for amino alcohols and dicationic amino acids molecules the host must possess not only negative charges arising from the phosphonate moieties but also a hydrophobic binding site like electron rich aromatic residues.
Furthermore, because it would be of great interest for biochemistry as well as for pharmacological studies to dispose of preorganized rigid chiral hosts for biorelevant molecules we designed
inter alia, some new chiral macrocycles capable of a triple binding mode,
i.e., exert attraction for the guests by electrostatic, hydrogen-bond and π-cation interactions. For the electrostatic interactions we resorted to the use of phosphonate dialkyl ester moiety, which can bear a negative charge when it is hydrolyzed to the mono-ester functionality and the ionization occurs at almost neutral pH values a prerequisite for molecular recognition at physiological conditions [
9].
Considering that the spirobisindanol moiety 1 is an easily available preorganized chiral molecule which exists as a pair of enantiomers, and can be used as a chiral template for building chiral polycondensates or inducing chirality in replicant strands, we resorted to its use for constructing macrocycles that could be of interest also for chiral recognition and chiral separations.
Thus, in this paper we shall review the salient aspects of some macrocycles synthetized in our laboratory, all possessing the spirobisindanol phosphonate moiety and able to act as complexing agents for cations and organic substrates. In particular, we shall describe their NMR characterization, their stereochemistry in solution and in the solid state, and the use of some of them as chiral receptors for biorelevant molecules.
[1+1] Cyclophanes
It is well known [
10] that dialkyl aryl phosphates can be rearranged by treatment with lithium diisopropylamide (LDA) or butyllithium to dialkyl aryl phosphonates, which, in turn, can be easily hydrolyzed to
o-hydroxyaryl phosphonic acids. Thus the bis-hydroxy aryl diphosphonate of the spirobisindanol (
1) was prepared [
11] and we used it for the synthesis of novel macrocyclic polyethers possessing ancillary groups (the phosphonic ones) which improve their water solubility or their complexing properties towards particular cations [
12], such as the lanthanides, extensively used in diagnostic medicine [
6,
13].
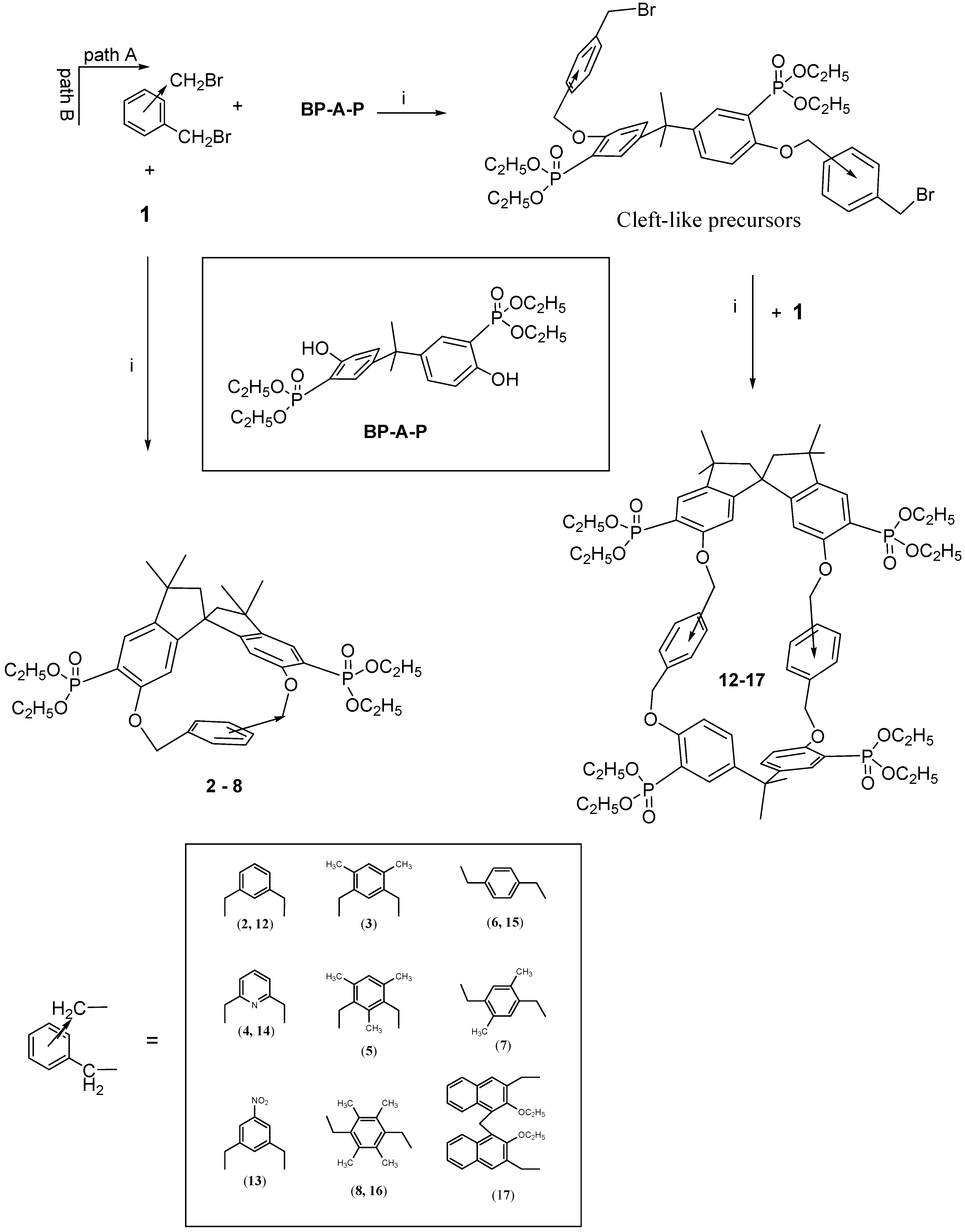
The Williamson synthetic condensation using the stereochemically constrained spirobisindanol monomer
1 and 1,3- or 1,4-aromatic dialkyl halides produced in high yield the [1+1] rigid cyclophanes as depicted in
Scheme 1, path B.
Characterization in solution was performed by proton, carbon and phosphorus NMR [
14]. By NMR analyses, it was shown that cycles
2-4 (path B) show mobility of the aryl rings, which by π radians rotation, average the benzylic bridging protons as well as the spirobisindane phosphonic groups.
Interestingly enough, the inner aromatic proton positioned between the two benzylic bridging groups is strongly upfield shifted, ca. 0.6 ppm, by the aromatic ring current effect of the cavity, indicating that it is pointing inside the cage. On the contrary, macrocycle 5, as well as those obtained by condensing the dihydroxy monomer 1 with 1,4-bis-chloromethyl aryl derivatives, i.e., compounds 6-8, are all stereochemically rigid on the NMR time scale. In particular, for 5, restricted rotation of the 1,3 bridged-mesityl ring renders the molecule asymmetric, as evidenced by the fact that all nuclei are chemically and magnetically different and this is confirmed by 1H-, 13C- and 31P-NMR observations. The inner methyl group of the mesitylene ring resonates at δ 1.59 ppm, much more up-field compared with the values of the other mesityl methyl protons (δ 2.54 and 2.43 ppm). The two bridging benzylic groups are not anymore homotopic and thus their Ha and Hb hydrogens (as well as Ha’ and Hb’) which are diastereotopic give rise, in the 1H-NMR spectrum, to two different doublets of doublets centered at δ 5.20 and 5.33 ppm, respectively.
Scheme 1.
i: Refluxing acetonitrile, K2CO3, 24 h.
Cycles 6-8 have a chiral structure (point group symmetry C2) and for all of them kinetic restricted rotation of the 1,4-bridged aryl ring was observed, as evidenced by the presence of two sets of signals for the nuclei in positions 2 and 3.
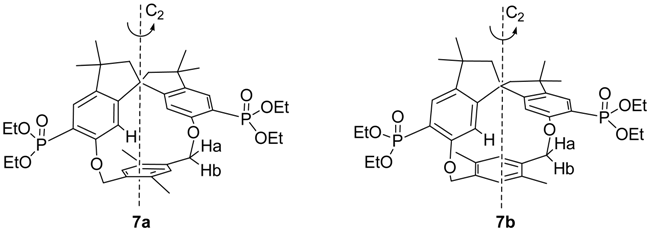
The stereochemical pattern in our macrocycles is quite intriguing due to the presence in the same molecule of a C2-symmetrical spirobisindane unit (which is in itself chiral) and of a xylylene bridging moiety, which could impart planar chirality. It follows that choosing the appropriate unit (i.e., the 2,5-disubstituted 1,4-xylylene bridge) under conditions of restricted rotation of this unit, two different diastereomers are possible for macrocycle 7. In fact for our series only in 7 the xylylene bridging unit is lacking a local C2 axis passing through the CH2 bridges.
The two structures
7a and
7b are depicted above. In
7a the two homotopic methyl groups of the 1,4-substituted
p-xylene ring are pointing out of the molecular cavity and are far from the ring shielding cone of the spirobisindane moiety; the opposite situation arises for
7b. After work-up of the reaction mixture, only one single diastereomer was isolated with a sharp melting point (m.p. 158-159 °C) and which showed only one set of signals for all the diastereotopic nuclei [
14].
From chemical shifts considerations,
i.e., the aryl methyl groups resonate at normal values and do not show any up-field shift due to the aromatic ring current effect, but on the contrary such an effect is seen for the aryl ring protons, and from NOE experiments, which reveals that the aryl methyl protons are spatially close to the spirobisindane hydrogens, ortho to the oxygen bridge, we tentatively concluded that the isolated diastereomer has structure
7a, which is the most prevalent one (° 95%) formed in our cyclization reaction [
14].
With the aim of elucidating the preferred geometry in the solid state, an X-ray structure of macrocycles
5, complexed with two moles of cyclohexane, and of
7 was thus undertaken [
14].
The overall view of the molecular conformation of the host 5 reveals that the molecule is asymmetric (C1) and the inner mesityl methyl group is pointing inside the cavity under the shielding cone of the spirobisindane aromatic ring. Thus the solution geometry detected by NMR spectroscopy was consistent with that found in the solid state.
From X-ray data we measured the cavity dimensions in 5 and we found that the cavity is large, but not big enough to include small organic solvent molecules such as cyclohexane. This explains why, the two molecules of cyclohexane, enclathrated by host 5, are present in the lattice, exterior to 5. Once we realized that host 5 includes cyclohexane, we checked in some more details the ability of our cyclophanes to enclathrate small organic molecules. Only macrocycle 3 was found to form inclusion complexes once again with one mole of cyclohexane, as judged by 1H-NMR integrations. Some other solvents (benzene, chloroform, ethyl acetate, etc.) were tested but NMR analyses did not detect the presence of any of them as guests in 3 and 5.
We previously postulated that for macrocycle
7 only diastereomer
7a was expected to be formed through a stereospecific coupling reaction; the X-ray structure of the isolated diastereomer with mp. 158-159°C reveals that the structure is consistent with that one deduced by NMR observations in solution [
14]. A comparison of the geometry of cycles
5 and
7 reveals that in
7, for example, the
para substituted phanes are quite hindered with respect to π rotation of the 1,4-substituted aryl ring, which flattens the structure and in turn reduces the cavity size of the cycle. Thus, the
meta substituted phanes of this series generally present a higher degree of rotational freedom and a large cavity size, as found experimentally.
We resolved by HPLC using chiral columns our macrocycles and we found that compounds
2 and
3, which showed internal mobility of the aryl rings on the NMR time scale, are not separated into their enantiomers. Compound
4, although possessing also a mobile aryl ring, shows a good separation; this can be due to greater interaction of the 2,5 disubstituted pyridine ring with the 3,5-dimethylphenyl end groups of the carbammate moieties of the chiral stationary phase. Compounds
5-
8, which are stereochemically rigid, are well separated and an increase in the polarity of the mobile phase has a beneficial effect on their separation factor [
14].
The good resolution factor (1.5) and low elution times of the enantiomeric pair for compound
5 afforded a quantitative separation of the enantiomers and collection of the eluates. The CD spectra of both eluates were measured and they were almost mirror images of each other, indicating the enantiomeric relationship between the two eluates. Their specific rotations were also measured, [α] = ‑189.8° and 190.3°, for the first and second eluted enantiomers, respectively [
14]. Analogously, a similar isolation procedure was applied to the compound
1 and the specific rotation measured gave [α] = 25.9° and -24.0° for the first and second eluted enantiomers, respectively. Analytical HPLC reruns of the eluates indicated enantiomeric purity (100 %) for the both peaks of compound
5 and 98 % (ee) and 88 % (ee) for the first and second eluted peak of compound
1.
The CD spectrum of compound
1 showed remarkable Cotton effects (CE). In fact the CD split shows a positive CE (Δε +60 at 215 nm, Δε -78 at 205 nm) for the less retained enantiomer. A negative CE is observed for the most retained enantiomer. A similar behavior can be envisaged for compound
5, although the CD splitting cannot be observed due to the shorter wavelengths of the CD bands [
14].
To improve the complexing properties of our macrocycles, especially for longer biologically relevant α,ω-diamine dications, we wanted to introduce an additional binding site into one of the congested cyclophanes in a particularly strategic position, i.e., we synthesized a novel sterically congested cleft-like receptor molecule 9 according to the reaction reported below:
The NMR data confirmed that macrocycle
9 is stereochemically rigid on the NMR time scale. In particular, the restricted rotation of the 1,3-bridged mesityl ring renders the molecule asymmetric, as evidenced by the fact that all nuclei are chemically and magnetically different. The two bridging benzylic groups, as well as the two phosphorus atoms attached to the
C2-symmetrical spirobisindane unit, are no longer homotopic. Thus, their hydrogens, which are diastereotopic, give rise to two different doublets of doublets, while the phosphorus atoms give rise to two sharp singlets at δ 18.97 and 17.74 ppm, respectively [
15].
The molecular structure of compound
9 was solved by X-ray diffraction [
15] techniques and it almost completely coincides with that one of bis-(5,5
'-diethyloxyphosphonyl)-6,6
'-(2,4,5-trimethyl-1,3-benzyloxy)-3,3,3
',3
'-tetramethyl-1,1
'-spirobisindane,
i.e. compound
5. Although the molecule of
9 is chiral, it is regularly alternated with its optical antipode in the centrosymmetric
P21/
c space group, giving a racemic crystal. The macrocycle shows its usual heart-like shape and the steric hindrance of the ring atoms does not allow guest molecules to be enclathrated so that no solvent molecules are found in the structure. Moreover, some degree of tension is present in the macrocycle that slightly twists the mesityl group, which is not exactly planar.
Chiral Receptors
Considering that the effective and selective molecular recognition of amino acids in water by synthetic receptors is of great interest [
16] we prepared the anions of the phosphonate spiro-bisindanol
1 and of macrocycle
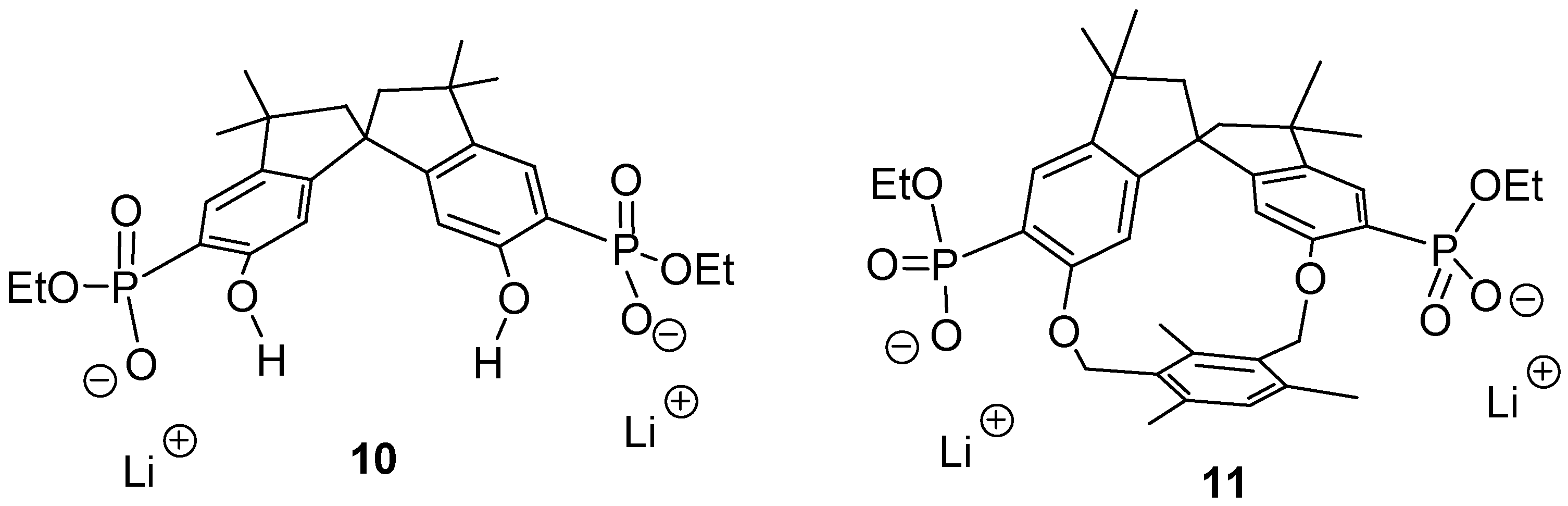
5, which according to X-ray analysis and molecular modeling adopt a favorable conformation for a highly effective chelate complex with the cationic group of biologically relevant amino acids.
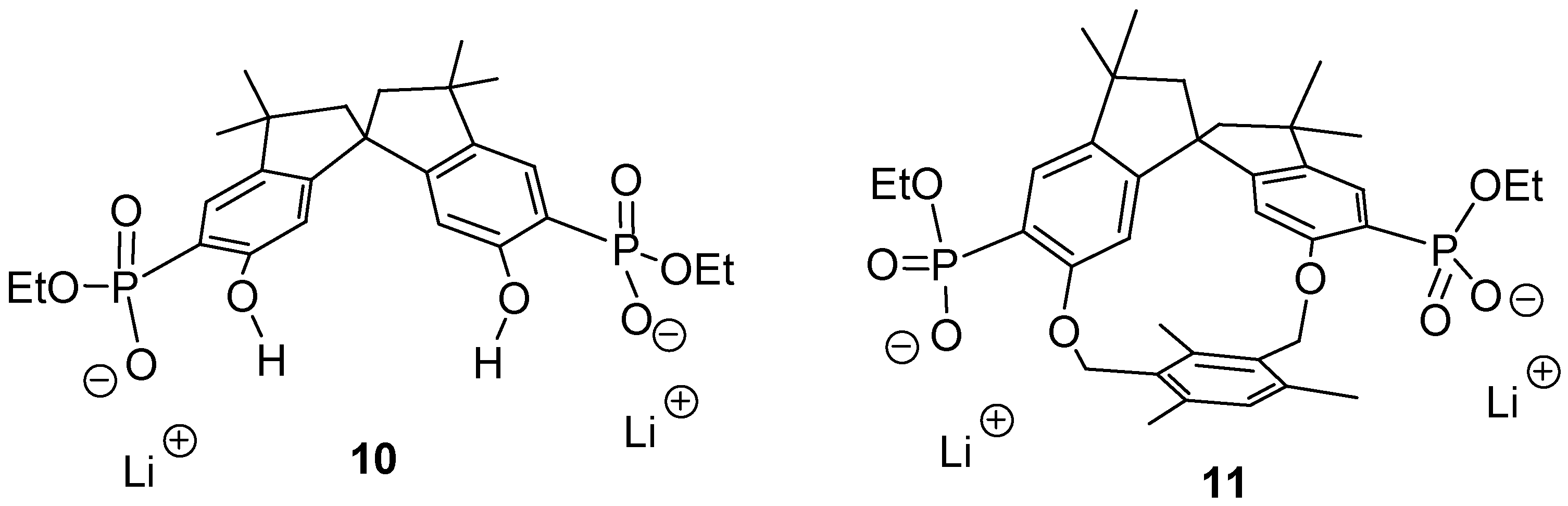
The respective anions
10 and
11 (
Figure 1) should be able to perform an induced fit on approach of a dication with the correct spacer. Thus, a highly stable 1:1-complex could be formed, possibly even in water. Heating of a 2-hexanone solution of the tetraethyl ester precursors with 2 equivalents of dry lithium bromide for 1 week afforded, after recrystallization and dialysis, a new class of cleftlike receptor molecules
10 and
11 based on bisphosphonates and fully equipped for multipoint binding of their dicationic substrate [
17]. These compounds were well soluble in polar solvents such as methanol, DMSO and water.
Figure 1.
Two new host molecule: the cleft 10 has C2- whereas macrocycle 11 has C1-symmetry.
Figure 1.
Two new host molecule: the cleft 10 has C2- whereas macrocycle 11 has C1-symmetry.
The spirobisindane skeleton in 10 and 11 guarantees a high degree of rigidity, which should lead to a pronounced preorganization of the different binding sites. A series of NMR binding experiments revealed a remarkably different behavior of the two new host molecules with respect to their chemo selectivity as well as their enantioselectivity.
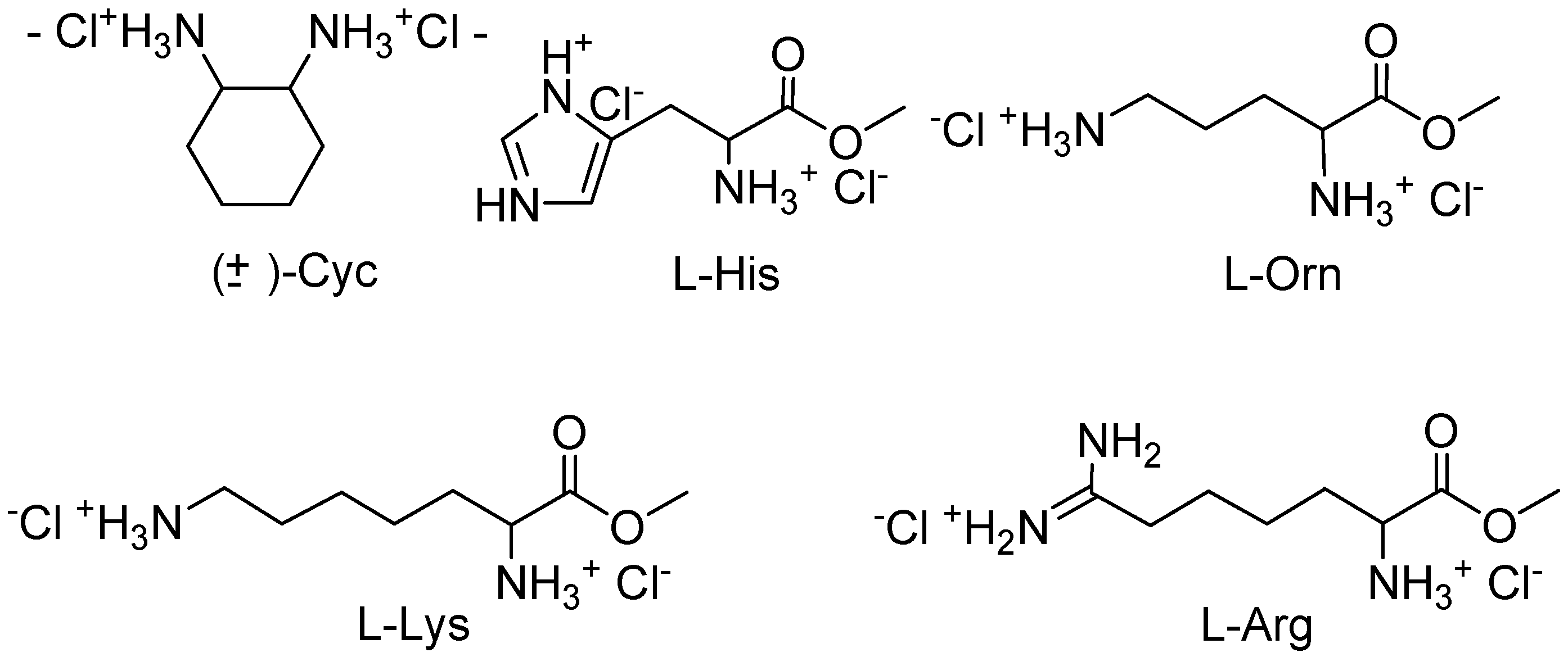
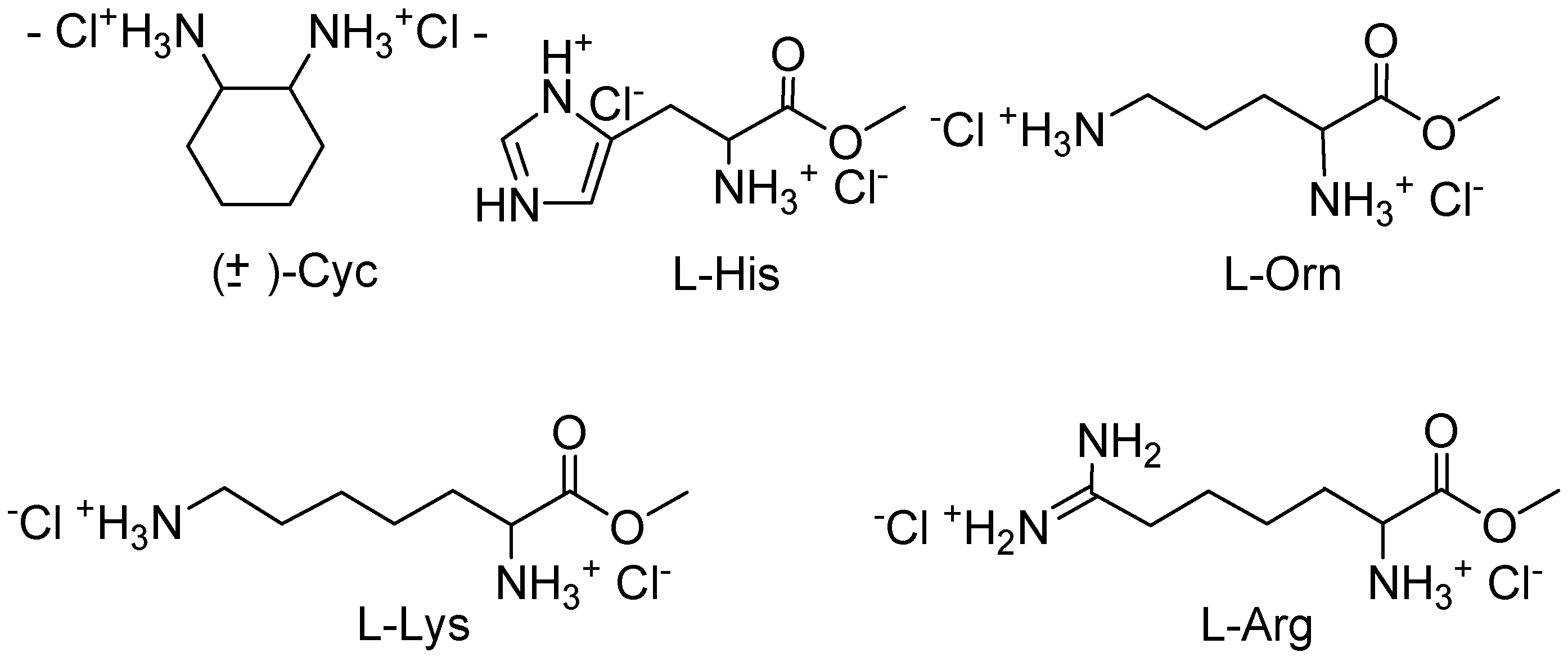
Job plots with
10,
11, and several α,ω-diammonium compounds (
Figure 2) proved that
10 forms 1:1 complexes in all cases, whereas
11 binds two guest molecules as long as they are shorter than lysine or arginine [
17]. This divergent behaviour could be explained with the different sterical accessibility of the cleft in
10 and
11. In
10 there is ample room for both cationic groups to approach the phosphonate anions from within the cleft, with the effect that small α,ω-diammonium compounds should lead to the highest binding enthalpy, governed mainly by optimal electrostatic interactions. In
11, on the contrary, the cleft is closed by the rigid mesitylene bridge, so that only long α,ω-dicationic guests can reach from one phosphonate group to the other; at the same time their carbon chain comes into close contact with the mesitylene bridge, so that molecular recognition can be expected to become more sensitive. We performed NMR titrations of various dicationic guests with a successively increasing distance between their positively charged nitrogen atoms.
Figure 2.
Investigated chiral guest molecules.
Figure 2.
Investigated chiral guest molecules.
The dependence of the order of total binding energies from the guest structure differed enormously between host 10 and 11: the open chain host 10 was selective for short rigid α,ω-diamines (Ka up to 8 x 103 M-1); macrocycle 11, however, preferred longer dications (Ka up to 1.2 x 104 M-1).
No enantiodiscrimination wss found for the open chain host
10, and even
11 could only distinguish between enantiomeric guests with an N
+•••N
+ distance of more than five bonds. Force field calculations suggested that only lysine and arginine are long enough to span the bridged cleft of
11, so that two salt bridges could be formed between their cationic groups and the respective phosphonates. This indicated once more that for an efficient chiral recognition the two binding sites within the
C2-symmetrical open chain host molecule
10 are not sufficient. Only the additional third binding site offered in
C1-symmetrical macrocycle
11 by the chiral surface of the mesitylene bridge leads to a detectable enantiomeric discrimination. Although the ee’s were not very high (
10:L
-Arg, 17%;
11:L
-Lys, 33%), well-resolved NMR signals were produced during the NMR titration, so that optically pure
11 may be used as a shift reagent for the quantitative determination of the enantiomeric purity of arginine and lysine derivatives [
17].
In summary, we have presented a new class of rigid receptor molecules for organic dicationic guests. Depending on the accessibility of their cleft, these hosts are selective for short or long α,ω-diammonium and guanidinium compounds. Because of their inherent chirality, we could examine their potential for chiral discrimination and found that only those host-guest combinations which allow a three-point interaction were effective. Thus, the bridged macrocyle 11 represents a chiral shift reagent for arginine and lysine derivatives.
Lanthanides Complexation
Lanthanide ions, particularly Tb
3+ and Eu
3+ are popular luminescent probes for the development of fluoroimmunoassays [
18,
19,
20,
21,
22,
23]. Therefore some of our phosphonate-spirobisindanol cyclophanes have been used for studying the efficiency of excited-states interactions between lanthanide ions (Tb
3+ and Eu
3+) and the cyclophane moiety. We used as substrates for our complexing studies [
24] macrocycles
2,
4 and
8.
The apparent association constant (K
app) of (Tb
3+ and Eu
3+ with
2,
4 and
8) complexes in DMSO were obtained [
24] by using Benesi-Hildebrand -type plot equation
1.
where A
o, A
c and A
obs are the absorbance of the ligand of the complex and of the ligand at various concentrations of the metal ions, respectively. K
app and [M] are the formation constant and the concentration of the metal ion, respectively.
The absorption spectra of 2 x10
-5 M for the ligands (
2,
4 and
8) in methanol showed broad bands in the UV at 290, 289 and 284 nm, with molar absorptivity coefficient (ε =11850, 11500 and 8600 M
-1cm
-1), respectively. These bands were attributed to π – π* transitions in pyridine, benzene and o-xylene moieties, respectively. Upon complexation to lanthanide cation no red shift in (π–π*) transition wss observed indicating a very weak ion-dipole interaction in this solvent between lanthanide cation and the oxygen atoms in the ring of cyclophanes [
25].
Moreover lanthanide ions do not contribute to the spectra of their complexes since f-f transition is Laporte-Forbidden with a very weak (ε), (0.5-3.0 M
-1 cm
-1). On the other hand, charge-transfer bands involving lanthanide orbitals are also typically not observed in the near-UV and spectral regions
. Hence, the absorption bands of lanthanide complexes were completely attributed to the ligand-centered (LC) transitions and some perturbation was observable upon complexation [
26].
The unusual stability of lanthanide ions with diethoxyphosphonate cyclophanes that have one or two binding sites was dominated by two primary factors: (1) ion-dipole interaction between metal ion and cyclophanes donating oxygens and (2) long-range interaction between metal ion and ethoxy group [
27].
Absorption at λ= 290 nm as a measure of the complex concentration, increased with metal ion concentration, and a high K
formation values in case of Tb
3+ (small size = 1.0 Å) compared with Eu
3+ (large size = 1.066 Å) and [M
n+] = 8X 10
-5 M, are obtained [
27].
It has been found that the quantum yield values of Tb
3+ and Eu
3+ complexes were consistent with the observed K
app values, i.e. the luminescence quantum yield followed the order Me-(
8) > Me-(
2) > Me-(
4). This behaviour can be explained on the fact that the largest heavy atom effect (Tb
3+> Eu
3+) results when the metal ion perturber is located along the out-of-plane axis originating from the center of the benzene chromophore which applies in the case of benzene crown ether metal ion (Tb
3+ and Eu
3+) [
28]. This in turn was attributed to the symmetry restrictions which enter the spin-orbit matrix elements via the overlap integrals between π electrons of benzene and p orbitals of the heavy atom [
29,
30,
31,
32]. According to this effect the triplet (T
1) population and radiative decay of the T
1 state should be a maximum in
8 (among
4,
2 and
8) due to the heavy atom perturbation [
29,
30,
31,
32].
Furthermore, the enhanced emission of Tb3+-cyclophanes and Eu3+- cyclophanes in DMSO, DMF and CH3CN was caused by the formation of anhydrous solvates, Tb3+-cyclophanes.n (solvent) and Eu3+- cyclophanes.n (solvent). Introducing solvent molecules in the first coordination sphere of Tb3+-cyclophanes and Eu3+- cyclophanes leads to the enhancement of the intensity of all transitions and especially 5D0 → 7F2 and 5D4 → 7F5 transitions have been increased significantly in Tb3+ and Eu3+ and consequently the radiative rate of the complexed ion.
By increasing the radiative rate, Tb
3+ and Eu
3+ excited states will become less sensitive to deactivation processes, ultimately resulting in a more efficiently emissive Tb
3+ and Eu
3+ complexes [
33]. It is also observed that the luminescence intensities for the complexes in DMSO, DMF and CH
3CN solutions were stronger than those in hydroxy solvents (such as methanol and ethanol) solutions. This was attributed to vibrational energy transfer to the solvent molecules. It is well know that the excited state of the lanthanide ions is efficiently quenched by interactions with high-energy vibrations like O-H groups thereby the luminescence of these complexes in –OH containing solvents can be quenched easily because of the O-H oscillators [
34].
Furthermore, the luminescence intensity and the quantum yield values of terbium complexes were higher than those of europium complexes in all solvents. The luminescence of Ln
3+-chelates is related to the efficiency of the intramolecular energy transfer between the triplet level of ligand and the emitting level of the ions, which depends on the energy gap between the two levels. In the organic solvents, the energy gap between the ligand triplet levels and the emitting level of the terbium favors the energy transfer process for terbium [
24].
In conclusion, terbium and europium ions formed stable complexes with three cyclophanes containing spirobisindanol phosphonates. Energy gap value between the T1 of the ligands and the emitting states of the lanthanide ions is possible significant factor affecting the extent of energy transfer from the ligand and consequently the quantum yield values. In the organic solvents, the energy gap between the ligand triplet levels and the emitting level of the terbium favored the energy transfer process for terbium over europium. Moreover, the luminescence intensities for the complexes in DMSO, DMF and CH3CN solutions were stronger than those in hydroxy solvents. The enhancement of the luminescence intensity observed in PMMA matrix for both Tb and Eu ions reflected the higher energy transfer from the ligand to the metal ion due to restricted free volume effect.
[2+2] Chiral Macrocycles
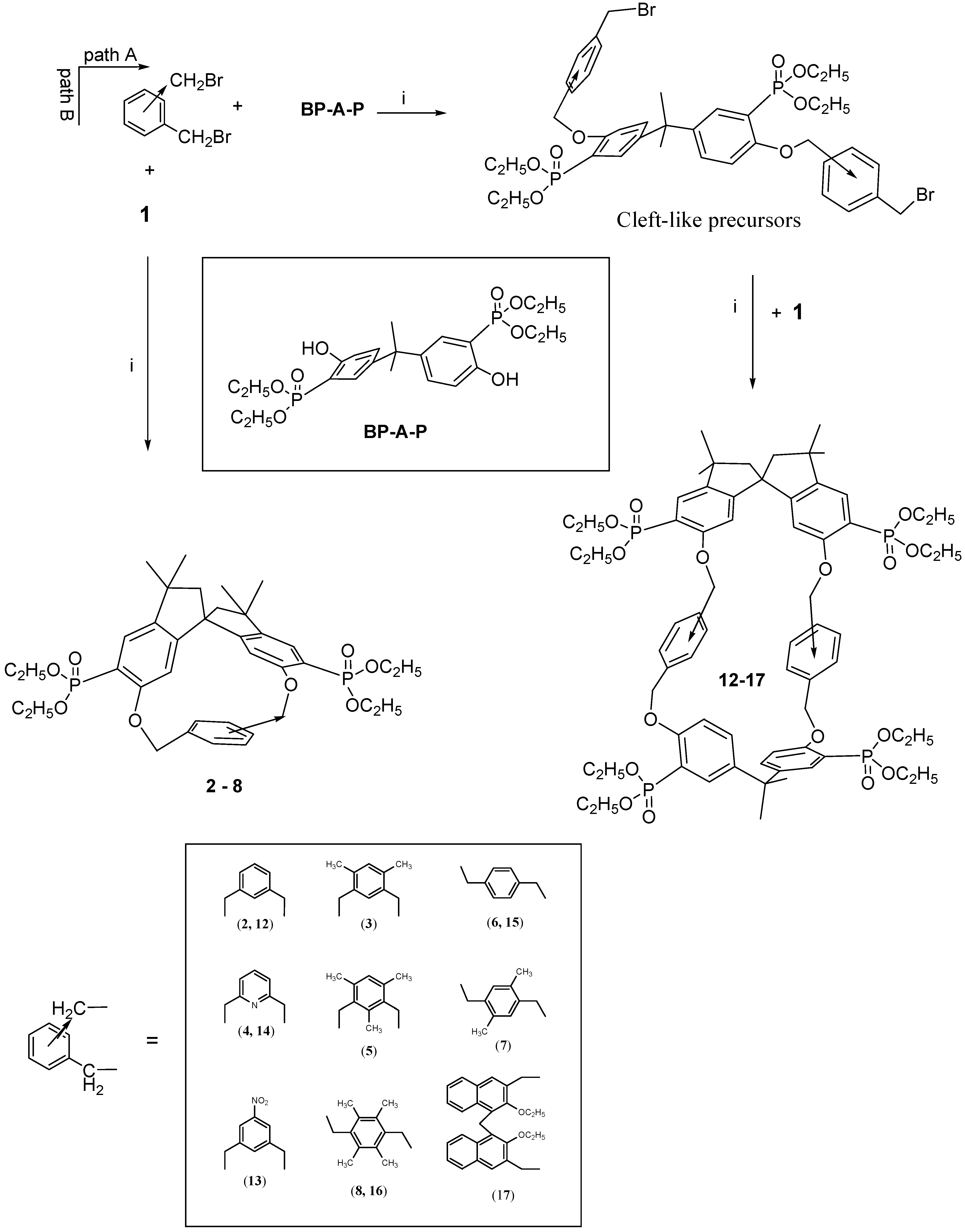
The survey of the macrocycles so far described revealed that although the [1 + 1] macrocycles 2-8 are chiral, their cavity is not large enough to include bulky guests.Thus, with the idea of synthetizing chiral bis-chelating phosphonate macrocycles, which can include neutral guests and, after hydrolysis to tetra-monoester could become water-soluble receptors for positive charged molecules, we adopted a new strategy for the synthesis of different kinds of robust inherently chiral cyclophanes, able to act as potential resolving agent for antipods of biologically relevant molecules.
We noticed that by condensing the spirobisindane monomer
1 with 1,3- or 1,4-aryldibenzylhalides, only [1+1] cyclization products were obtained in high yield [
14]; whereas when the 2,2-bis(3-diethylphosphono-4-hydroxy-phenyl)propane (
BP-A-P) was used as condensing agent, the main isolated products were a [2+2] macrocycle [
35]. It follows that for the synthesis of novel macrocycles containing both units (
BP-A-P) and
1 in the structure, the one-pot method is not a possible synthetic route. A two step synthesis was then used and, in principle, two different synthetic paths could be envisioned for the preparation of such novel inherently chiral macrocycles, but only one was the effective route followed.
Scheme 1 gives a schematic description of the synthetic strategy used for the preparation of our [2+2] macrocycles. The idea was to synthesize at first a bis-halomethyled derivative containing the moiety (
BP-A-P) and then cyclize it to the desidered cyclophane
12-17.
Thus ten equivalents of
meta- or
para-bis(halomethyl)benzene and one equivalent of phosphorylated bisphenol A (
BP-A-P) in acetonitrile in the presence of solid K
2CO
3 as a base, according to the Williams condensation gave with high yield (60-80%) the cleft-shaped precursor molecules, which were isolated from the reaction mixture and fully characterized [
36].
Each of the cleft-like precursors was then allowed to react under high dilution conditions with
1 (the phosphorus-modified spirobisindanol) to give macrocycles
12-17. They are all inherently chiral and they differ on size and polarity of their cavity. Proton, carbon and phosphorus NMR spectroscopy, a FAB-MS and X-ray analysis data supported the formulation of the synthesized compounds [
36].
The 500 MHz 1H-NMR spectrum of macrocycle 15 at room temperature in CDCl3 solution showed two triplets (δ = 1.31 and 1.36) and a multiplet (δ = 4.10-4.28) for the ethoxy groups linked to the phosphorus atoms, three sharp singlets for the methyl groups attached to the bridgehead quaternary carbons, a doublet of doublet centered at (δ = 2.16 and δ = 2.45, respectively) for the methyne hydrogens of the spirobisindane unit, a couple of two different doublets of doublets centered at (δ = 4.80 and δ = 4.93; δ = 5.21 and δ = 5.28, respectively) for the benzylic protons linked to the p-xylyl nucleus and the expected multiplicity for the aromatic hydrogens. This pattern, coupled with the 13C- and 31P-NMR spectra, clearly indicated a chiral structure (point group symmetry C2), and a great mobility of macrocycle 15, which interconverts itself in solution among various possible conformations.
Interestingly enough, crystals of
15, obtained from a cyclohexane/ethyl acetate solution showed, in the NMR spectrum, the presence of the saturated hydrocarbon enclathrated in a molar ratio 1:1; the same observation was also found by thermogravimetric analysis which shows total loss of one mol of cycloexane at 120°C; furthermore TGA shows total loss of two moles of water in a large range of temperature (30÷90°C). The molecular structure of
15 was solved by X-ray diffraction techniques [
36] and due to the presence of the
spiro carbon C(18) and of the great number of
sp2 carbons, the macrocycle has not a plane ring structure, but is heavily puckered and its cavity is rather closed.
Moreover, by recrystallization of cycles 12-17 from a mixture of cyclohexane and other organic solvents (ethyl acetate, chloroform, dichloromethane, benzene, etc.), was found that all macrocycles are able to form inclusion complexes only with cyclohexane.
Molecular “Hamburger”
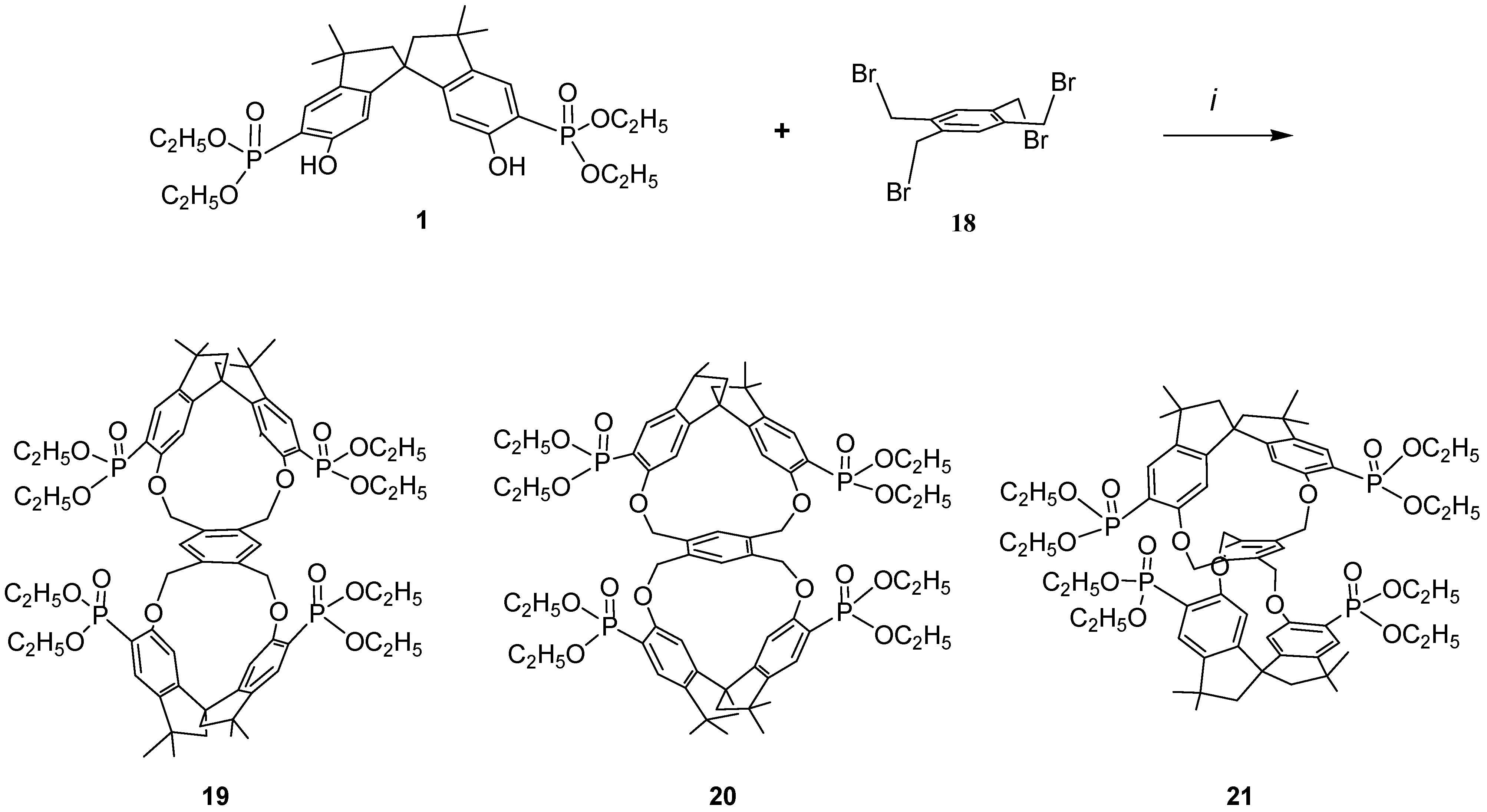
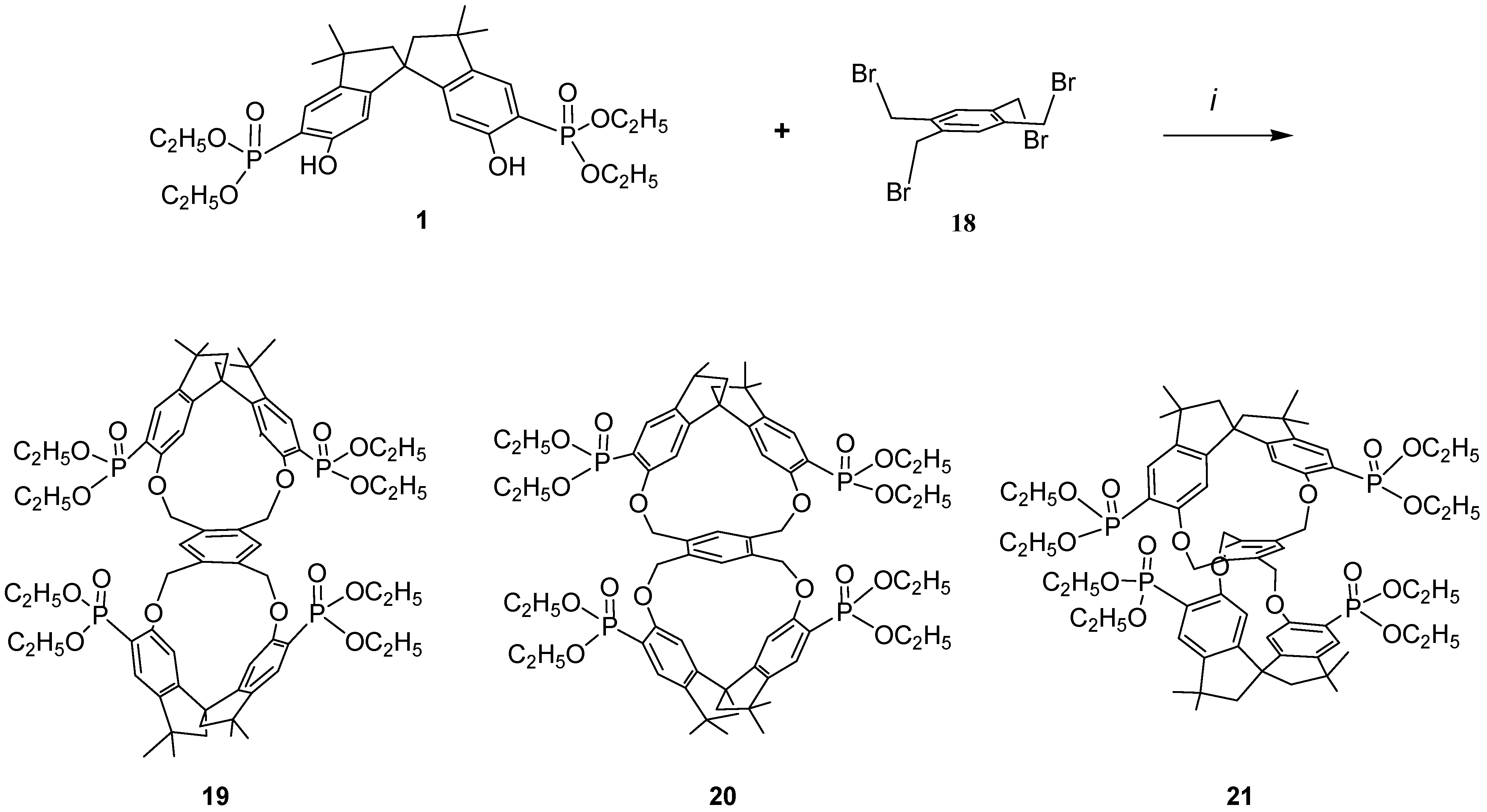
Because it would be of great interest for biochemistry and for pharmacological studies to dispose of bulky rigid hosts we designed some new chiral bismacrocycles. In order to induce chirality in our macrobicycles, we used the spirobisindanol phosphonate unit 1, which possesses a C2 rigid conformation and a geometric constrained structure in which the two hydroxyl groups are fixed in a favorable position for yielding by a template effect the [1+1] cyclization process, as described before. Thus, we reacted 1 with 1,2,4,5-tetrakisbromethylbenzene (18) according to the Williamson condensation in order to produce a new chiral macrobicyclic compound, in which a benzene ring is positioned in the centre of a pair of extended chiral cyclophane frames in a molecular equivalent of a ‘hamburger’; the phosphonate groups are in external strategic positions for complexation.
Scheme 2 shows the synthetic strategy employed for the preparation of our cyclophanes. The Williamson ether synthetic procedure proved to be very satisfactory; all reagents were converted almost quantitatively and only [2+1] cyclization products were obtained in very high yield. Reaction of the racemic monomer phosphonate
1 with 1,2,4,5-tetrakis(bromomethyl)benzene (
18) in acetonitrile containing solid potassium carbonate afforded a bromine free product having m/z = 1287.5 (MH
+), thus confirming the 2:1 stoichiometry of the reaction [
37].
The incorporation of the common tetra substituted benzene ring between the rigid bisphosphonate spirobisindane units gives rise theoretically to three different isomers i.e. ortho–ortho 19, meta–meta 20 and para–para 21.
Scheme 2.
i, Refluxing acetonitrile, K2CO3
Moreover, considering that the spirobisindane phosphonate monomer
1 is a geometrically constrained chiral molecule, which exists as a pair of enantiomers, for each of them a pair of diastereoisomers (i.e. meso and racemic forms) could be obtainable. Comparison of the OH—OH separation of the phenolic groups in
1 (Hyperchem, 6.93 Å; by x-ray [
14], 6.58 Å) with the bromomethyl C—C distances in
18 (AM1, ranging from 2.91 to 5.77 Å) supports the preferential formation of the dual para-substituted product
21, because less deformation of the molecular frame is required to achieve cyclization.
The 500 MHz
1H-NMR spectrum of the isolated crude powder at room temperature in CDCl
3 solution, obtained from the reaction outlined in
Scheme 1, indicated that a complex mixture of cyclic products was formed. To confirm that our synthesis leads to cyclophanes, we used the
31P-NMR technique. The spectrum revealed the presence of peaks at δ 18.38 ppm (30%), 18.11 ppm (30%), 17.79 ppm (5%) and 17.53 ppm (35%), which were shifted upfield with respect to the corresponding moieties of spirobisindane
1 which resonate at δ 21.24 ppm.
If we assume that the two peaks with the same percentage come from the same structure, then the three different sets of signals, which integrate in the ratio 60% (hereafter called major product), 35% (hereafter called minor product) and 5%, suggest that a mixture of three isomers was obtained from the cyclization reaction [
37].
From chemical and steric factors, we could exclude the presence of 19 in the reaction products. In fact, when under the same conditions the spirobisindane compound 1 was reacted with an equimolar amount of the bifunctional α,α’-dibromo-o-xylene, no evidence of [1+1] cyclic products was observed and only polymeric compounds were isolated.
Preliminary separation of the crude powder was made on silica gel and after cromatography we recovered 70% of a material, in which the 5% isomer is absent. The other two isomers, possessing the same Rf, were then separated by successive crystallizations; the major isomer had m.p. 145–147°C and the minor isomer 241–243°C.
Characterization was performed by FAB-MS, which confirmed that the major and the minor products possess the same molecular weight and they are isomers. In solution the characterization was performed by
1H-,
13C- and
31P-NMR spectroscopy; further, the chemical shifts of the 1-D spectra were assigned to specific atomic positions by COSY and TROESY [
37].
NMR spectroscopy
The 500 MHz 1H-NMR spectrum of the minor product at room temperature in CDCl3 solution showed a doublet (δ 7.44, 4,4’-H) and a doublet (δ 5.24, 7,7’-H) for the aromatic hydrogens of the spirobisindane units, a sharp singlet (δ 6.38) for the two aromatic hydrogens of the tetrakismethylbenzene unit, a doublet of doublets centred at (δ 5.40, Δδ = 0.36, 2JHH =12.5 Hz) for the four bridging benzylic groups and the expected multiplicity for the aliphatic hydrogens. This pattern of 1H-NMR resonances, combined with the 13C and 31P-NMR spectra, induced us to suppose that the minor product possess a high degree of symmetry (e.g. D2) according to the fact that the hydrogens of the two spirobisindane units and those of the four CH2 groups are equivalent.
In contrast, the major product showed more complex
1H-,
13C- and
31P-NMR spectra. The two spirobisindane units were no longer homotopic, hence their 4,4’-H
a and 4,4’-H
b hydrogens give rise, in the
1H-NMR spectrum, to two different doublets centred at δ 7.52 and 7.49, and the 7,7’-H
a and 7,7’-H
b hydrogens give rise to two different doublets centred at δ 5.39 and 5.13. Moreover, the four bridging benzylic groups are now only pair wise homotopic. In a pair of diastereotopic methylene groups, the hydrogens of each gave rise to two doublets of doublets centred at δ 5.26, Δδ = 0.91 and δ 5.14, Δδ = 0.26, respectively. The expected signals with the appropriate multiplicity were found also for the aliphatic hydrogens. From the 2-D spectra it was also possible to assign the relative chemical shifts to the methyl groups of the phosphonic groups and to the methylene groups of the spirobisindane fragment. Thus the major product has a chiral structure (point group symmetry
C2), as evidenced by the presence of two sets of signals, in the
1H-,
13C- and
31P-NMR spectra, for the nuclei of the two spirobisindane units [
37].
ROESY spectra were measured in order to investigate further the conformation of the macrocycles. The ROESY spectrum of the isolated major product showed the expected NOE correlation between the aromatic hydrogens (δ 6.89) of the tetrakismethylbenzene and the two doublets of doublets of the bridging benzylic hydrogens; more interesting the aromatic hydrogens at δ 6.89 showed also NOE with the 7,7’-Ha and 7,7’-Hb hydrogens of the spirobisindane units, which indicated that the macrocycle is congested and stereochemically rigid on the NMR time-scale.
Having gained such information and having already excluded the formation of macrobicycle 19, the questions that arise now are the following: are the isolated products constitutional or stereoisomers, and if stereoisomers, what is their relative stereochemistry? In order to answer these questions, the following considerations can be of help.
The chemical shifts of the 7,7’-H hydrogens of the spirobisindane units in these macrocycles were very diagnostic for distinguishing between the para and meta connections within the xylyl unit. In fact, while the 4,4’-H chemical shifts were not affected by the connection, the 7,7’-H chemical shifts were strongly dependent on the connection with the xylyl unit. In the para connection they were shifted strongly upfield by ca. 0.8 ppm compared with the meta connection, indicating that they were pointing inside the cage experiencing the deshielding effect of the aromatic nuclei. Thus from chemical shift considerations and NOE experiments, which revealed that the aryl methyl protons were spatially close to the spirobisindane hydrogens, we concluded, invoking Occam’s razor, that the two isolated macrocycles were the two possible diastereoisomers of structure
21 in
Scheme 2 [
37].
Stereochemistry
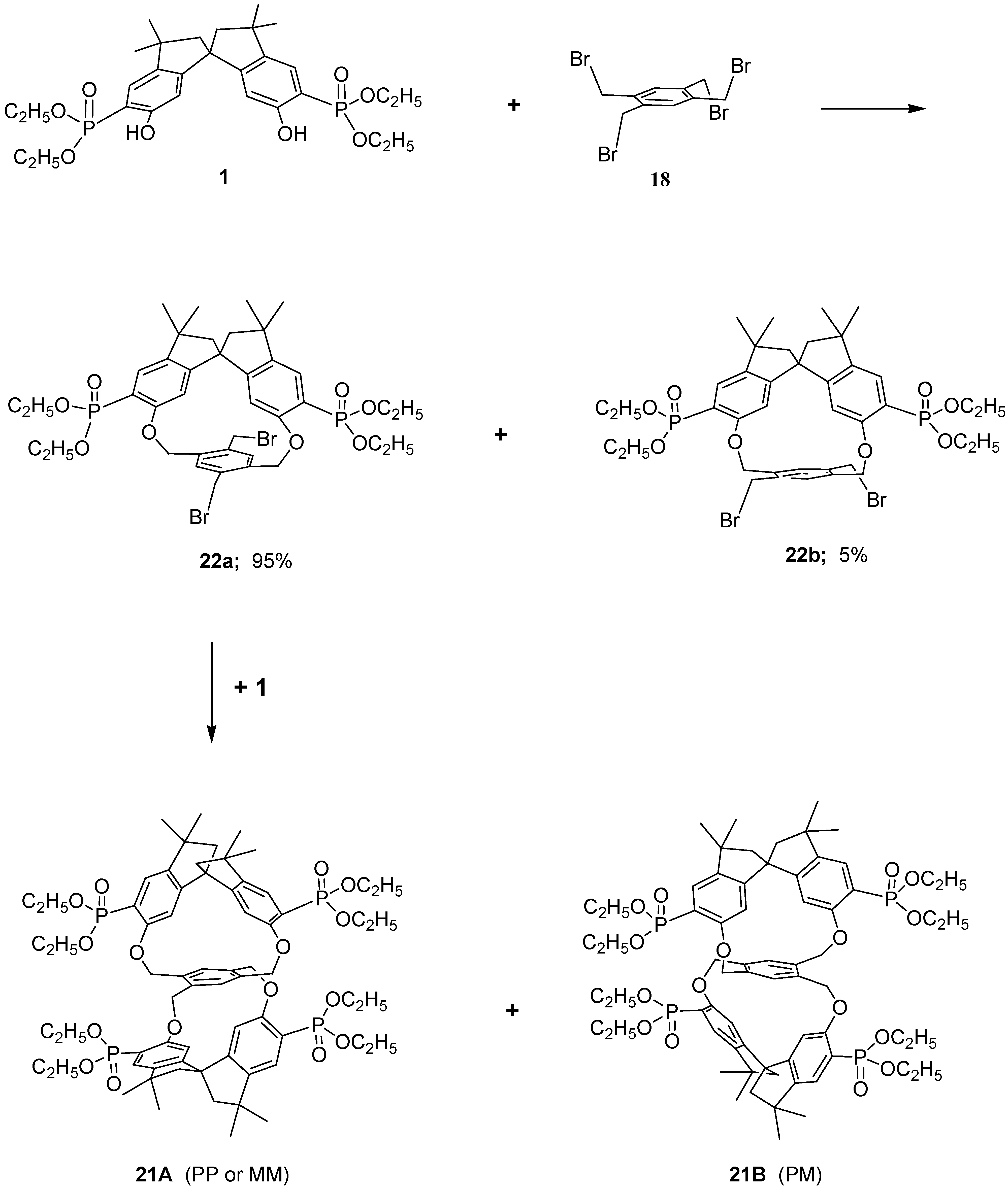
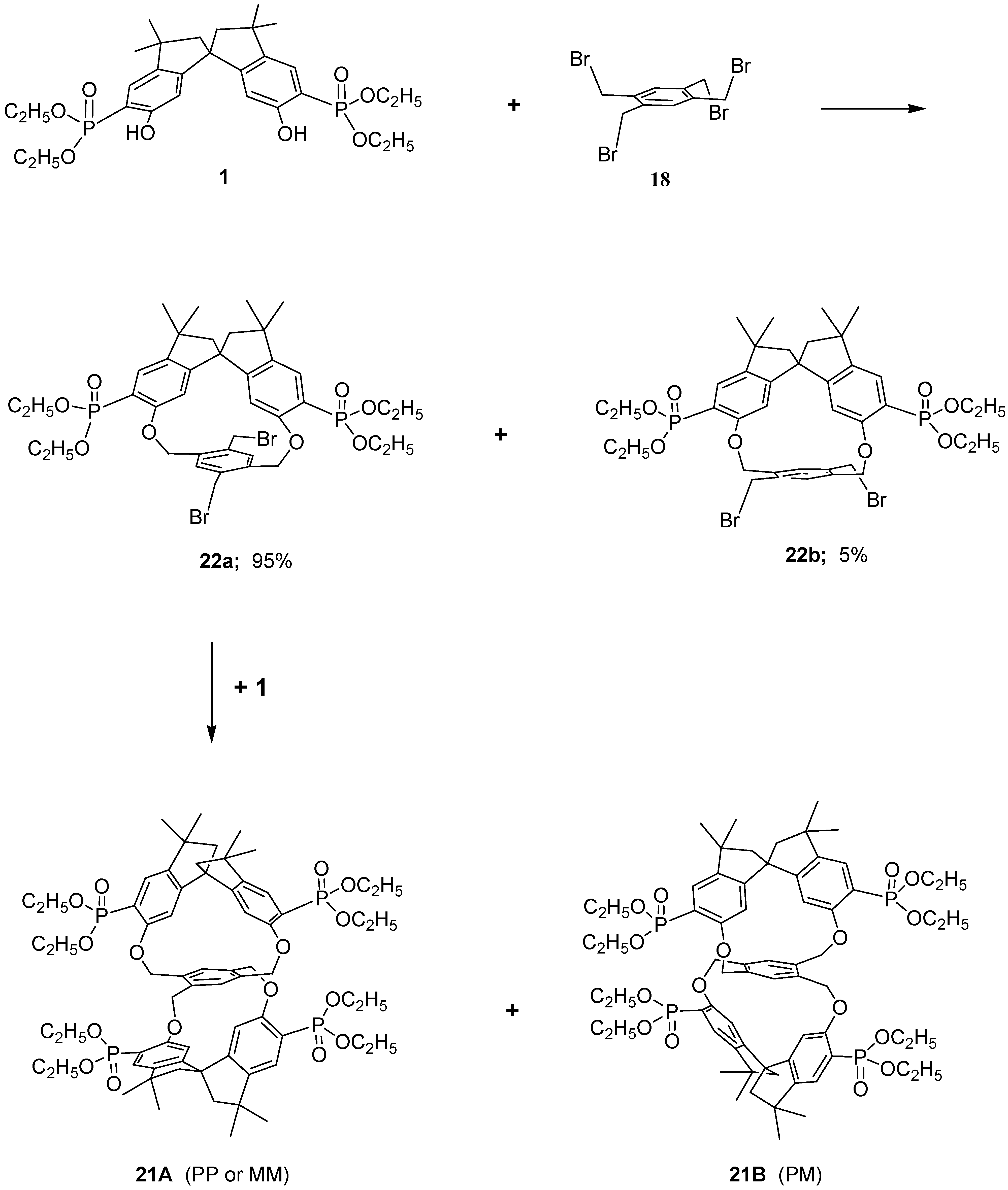
The stereochemical pattern in the macrocycles is intriguing owing to the presence, in the same molecule, of a
C2-symmetrical spirobisindane unit (which is itself chiral) and of a tetrakis-methylbenzene bridging moiety which can adopt a chiral conformation after the connection. (Ortho–ortho and meta–meta connections give rise to macrobicycles which are conformational mobile and possess symmetry elements of second order, then they are achiral). It follows that under conditions of restricted rotation of this unit, only two different diastereoisomers are possible in the first step of the macrocyclization reaction (
Scheme 3). In
22a the two homotopic methylene groups of the 1,4-substituted tetrakismethylbenzene ring are pointing out of the molecular cavity and are far from the ring shielding cone of the spirobisindane moiety; the opposite situation arises for
22b. Evidence of the formation of structures
22a and
22b came from the model reaction of 2,5-dibromomethyl-1,4-xylylene and the spirobisindane phosphonates
1. From this reaction, as noticed before, two diastereoisomers were formed in a ratio of 95:5 as judged by
31P-NMR and the predominant product of such [1+1] cyclization process was the one diastereomers in which the two xylyl methyl groups are pointing out the molecular cavity of the cycle.
Therefore, reasoning along these lines the racemic compound 22a (the only one present in our reaction in considerable amount) in the second step reacts again with 1, which is also a racemic mixture, giving rise to a pair of enantiomers (PP, MM, 21A) and the meso form (PM, 21B), in a ratio of 35:60, respectively.
Examination of structure 21A (PP or MM, i.e. racemic form) revealed that this molecule is chiral, having a chiral structure (point group symmetry D2), then all constitutionally equivalent atoms of the spirobisindane units and those of the central tetrakismethylbenzene unit were related by symmetry. It follows that the 1H-, 13C- and 31P-NMR spectra of this diastereoisomer should feature one set of signals, with the expected spin–spin coupling, for the 4,4’-H, for the 7,7’-H, for the four diastereotopic CH2 bridged hydrogens, etc.
In contrast, examination of structure 21B (PM, i.e. meso form) reveals that this molecule has point group symmetry C2, then also this structure is chiral but the constitutional equivalent atoms of the spirobisindane units and those of the central tetrakismethylbenzene unit are therefore related pair wise by symmetry. It follows that the 1H-, 13C- and 31P-NMR spectra of the structure 21B should feature, in the absence of accidental isochronies, two sets of signals, with the expected spin–spin coupling, for the 4,4’-H, for the 7,7’-H, for the four diastereotopic CH2 bridged hydrogens, etc.
The 1H-NMR spectrum of the major product at room temperature in CDCl3 solution as said before, revealed two doublets for the 4,4’-H, two doublets for the 7,7’-H of the aromatic spirobisindane units, two doublets of doublets for the CH2 bridging hydrogens and also in the aliphatic region two sets of signals were present. Moreover, two sets of peaks were present in the 13C-NMR spectra and two sharp peaks with equal intensity were present in the 31P{1H}-NMR spectra. These results strongly suggested that the major product isolated from our macrocyclization reaction has structure 21B and the minor product has structure 21A.
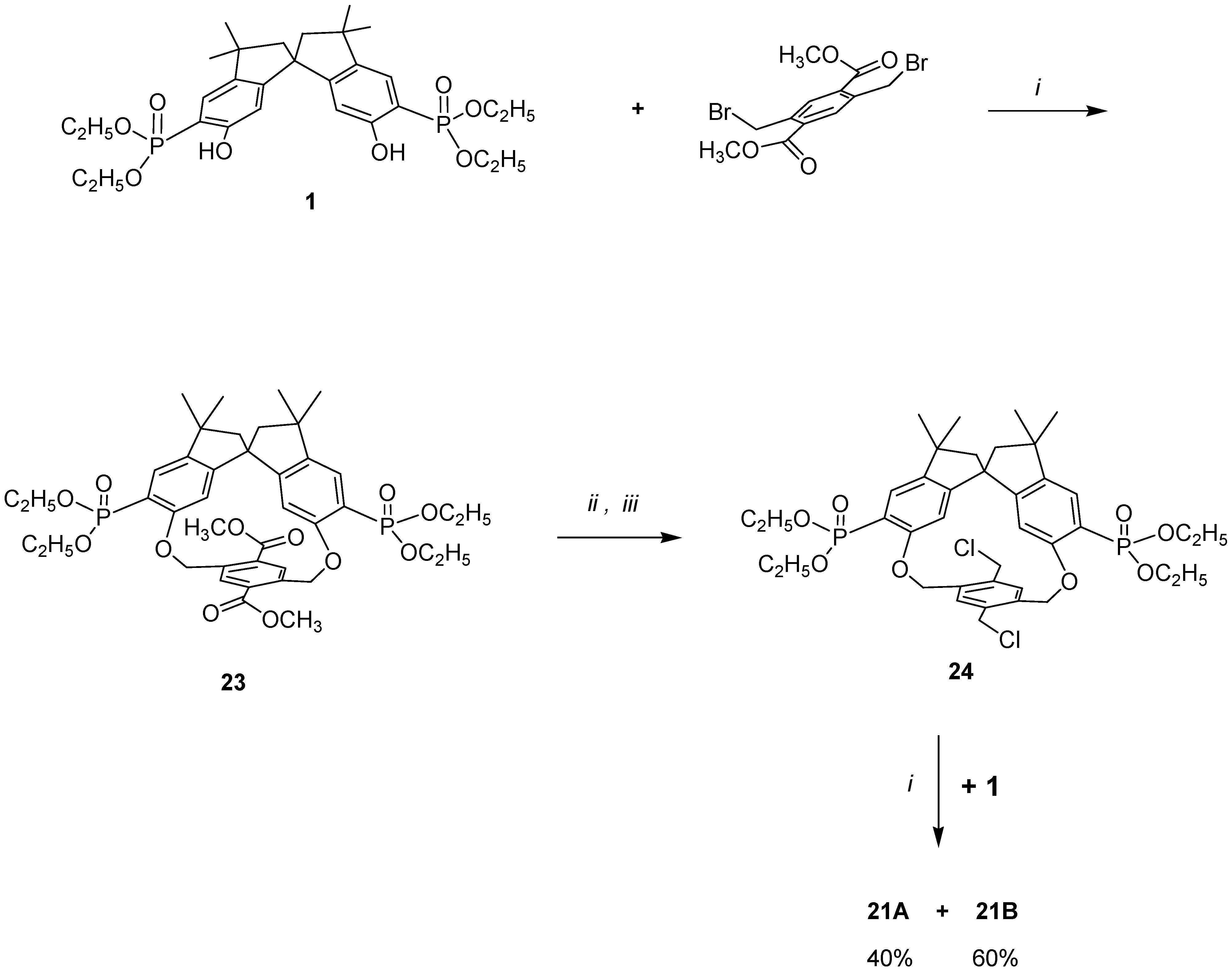
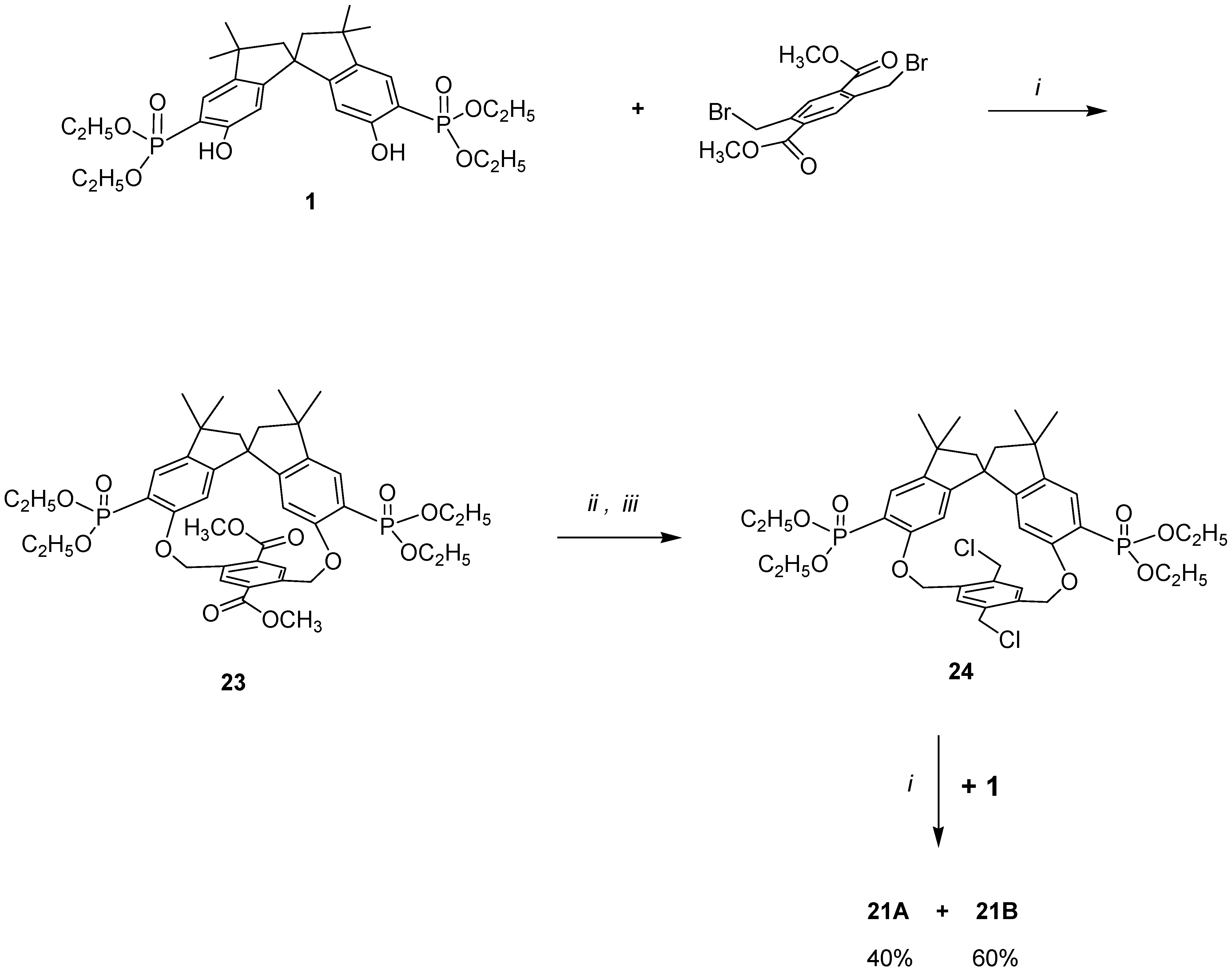
In order to demonstrate that in our cyclization reaction only a step-by-step para–para condensation was occurring, we resorted to a stepwise synthesis of macrobicycles
21A and
21B as outlined in
Scheme 4, using an alternative chemical route. Thus, condensing
1 with 2,5-dibromomethyl-1,4-carbomethoxybenzene the major diastereomer
23 (formed in ≥ 90% yield) was isolated.
Following the synthetic routes
ii and
iii, it was converted to the corresponding bischloromethyl derivative
24, which was then allowed to react with an equivalent amount of
1. Two diastereomers in a ratio of 40:60 were obtained, which possess the same melting-point and the same spectroscopic data for the major and minor products obtained from the one-pot reaction (structures in
Scheme 3). The products obtained,
21A and
21B, represent the first example of a chiral molecular ‘hambuger’ and thus open up new frontiers in the stereochemical use of such molecules [
37].
The highly selective construction of ‘hamburger’ products 21A and 21B in a para–para manner, rather than in either a meta–meta or an ortho–ortho manner, is a direct consequence of the geometry and of the spatial position of the phenolic OH of the spirobisindane unit.
Scheme 3.
Possible stepwise pathway for compounds 21A and 21B.
Scheme 3.
Possible stepwise pathway for compounds 21A and 21B.
Scheme 4.
i: Refluxing acetonitrile, K2CO3; ii: room temperature, THF, LiBH4;
iii: 0°C, DCM, K2CO3, SOCl2.
Chiral Resolution
Considering that both diastereomers
21A and
21B are chiral and taking into account our experience in the enantioseparation of spirobisindane phosphonate monomer
1 [
14] we tried to resolve them by enantioselective HPLC. We used two polysaccharide derived chiral stationary phases (CSPs) and several eluents in a normal-phase mode. A good enantioseparation of
21A was obtained by HPLC on Chiralpak AD. The best separation factor α was found to be 2.1 with the resolution factor
Rs = 1.4 [
38]. A slight increase in the polarity of the mobile phase (isooctane–ethanol, to 95:5) decreases both α and
Rs to 1.8 and 0.7, respectively[
39].
Based on the HPLC trace, we performed the isolation of the enantiomers of compound 21A. However, a ‘peak shaving’ was necessary to avoid the collection of impurities present as successive shoulders in the descending edge of the second peak.
The CD spectra of the eluted peaks were recorded after rotoevaporating the solutions from HPLC and dissolving again the residues in 1 ml of ethanol. They resulted in mirror images of each other, indicating the enantiomeric relationship between the two eluates [
39]. Interestingly, the CD curves were not bisignate and were formed by residual signals. The exciton coupling present in the CD spectra of the enantiomers of compound
1 is, in fact, absent in compound
21A. This is due to a partial ‘internal compensation’ between both chiral spirobisindane bis(phosphonate) moieties present in the macrocycle. We noticed that there was a distorsion in the reciprocal space orientation of these CD-active moieties present in the ‘upper’ and ‘lower’ parts of the molecule. Moreover, both moieties interact with the tetra-substituted benzene ring in the middle of the molecule, and a shoulder at 255–260 nm can be related to the strong UV absorbance of this chromophore at λmax = 226 and 260 nm, while compound
1 exhibits λmax = 206 nm and λmin = 260 nm.
Thus, the above results represent the first experimental proof of inherent chirality in a solution of compound 21A. We tried to resolve compound 21A using a polysaccharide CSP containing an additional stereogenic center, Chiralpak AS-H, and a similar eluent, but only broad peaks were observed.
Analogously, the enantioresolution of compound 21B was totally unsuccessful on both the CSPs described since several irreproducible peaks appeared in the HPLC experiments indicating either the lability of the compound or a severe contamination.
Miscellaneous Cyclophanes
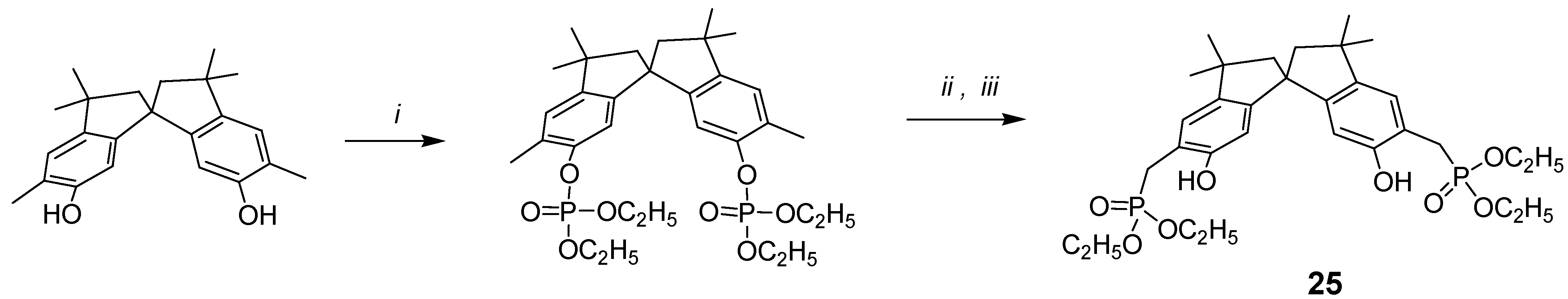
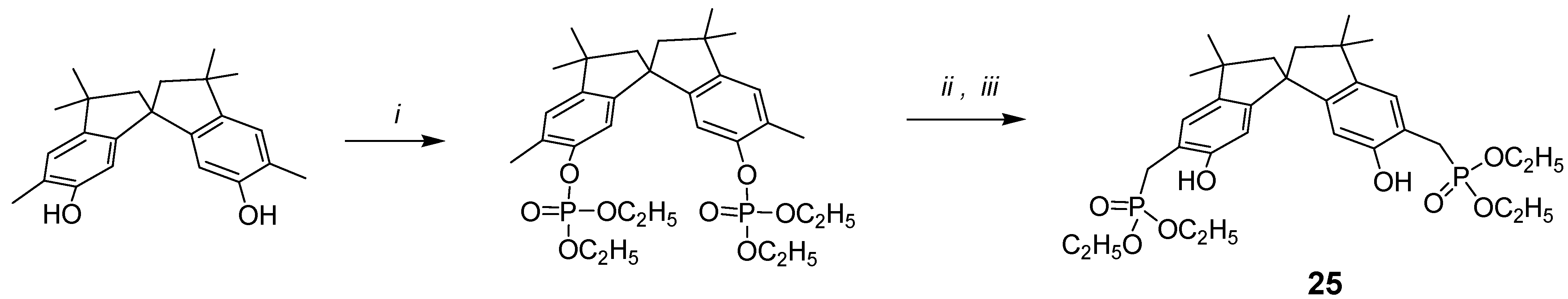
In search for novel building blocks to be incorporated into new types of molecular scaffolds, we have been attracted by compound 25, where the phosphonic groups are attached to the benzylic methylenes of the spirobisindane frame.
The new and general one-pot procedure for the synthesis of (o-hydroxyphenyl)methylphosphonic acid derivatives using ortho-disubstituted methyl phenol and lithium diisopropylamide (LDA) was undertaken, with the idea of synthetizing chiral bis-chelating phosphonates, which can include neutral guests and act as ligands for lanthanides. Furthermore, these molecules, after hydrolysis to the corresponding bis-monoesters, could become water-soluble receptors for positively charged molecules and thus act as potential resolving agents for antipods of biologically relevant molecules.
The synthesis is conceptually based on the phosphorilation of the OH phenolic group, which yields the phosphate derivative; the use of lithium salts at low temperature, in case of bis(orthomethyl)-substituted phenols rearranges the phosphate to the benzylic phosphate as reported in
Scheme 5.
Scheme 5.
i: HP(O)(OC2H5)2/CCl4, N(C2H5)3, 0°C; ii: LDA/THF, -78 °C; iii: saturated NH4Cl/H2O.
In other words, the treatment of phosphate esters with an excess of lithium diisopropylamide generates the methyl anion, which undergoes migration of the phosphorus from oxygen to carbon. Although O → C migrations of phosphorus are not without precedent [
10,
11], this new reaction represents the first example of a phosphate → phosphonate rearrangement involving the methyl group. Then, using the synthetic procedure outlined in
Scheme 5, compound
25 was easily synthetized starting from 5,5
'-dimethyl-6,6
'-spirobisindanol, which was readily converted into its diphosphate ester upon treatment with diethyl phosphite and CCl
4 in the presence of triethylamine, and then rearranged to crystalline product
25 by treatment of the diphosphate with a strong base (LDA). The possibility that the phosphonic group goes to the 7,7
'-position of the spirobisindanol skeleton by 1,3-rearrangement is very low and impossible because of the high steric congestion of such aromatic carbon atoms.
Phosphorus, carbon and proton NMR data provided convincing evidence for the structure assigned. The diphosphate compound showed a single phosphorus resonance at δ –5.76 ppm, while the rearrangement product
25 showed a single phosphorus resonance at δ 30.22 ppm, which is also so far from the resonance δ 22.12 ppm of compound
1, where the phosphorus atoms are directly attached to the aromatic carbons, and thus our new general synthetic procedure allows to produce
o-hydroxyphenyl methyl phosphonic acids in a very convenient and easy way [
40].
Macrocycle
26 is a novel chiral crown ether possessing phosphonate moieties in strategic position, which may enhance the possibility of hydrogen bonding formations in a three-dimensional chiral recognition. Therefore we prepared it by condensing under high dilution conditions the spirobisindane monomer
1 with pentaethylene glycol ditosylate under conditions of high dilution. The NMR investigations at room temperature of host
26 revealed that it possesses a
C2 symmetry in solution and the sets of signals due to the methylenes of the crown as well as that one’s of the spirobisindane moiety were in the expected range [
41].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}