Novel Prodrugs for Targeting Diagnostic and Therapeutic Radionuclides to Solid Tumors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction

Data Mining
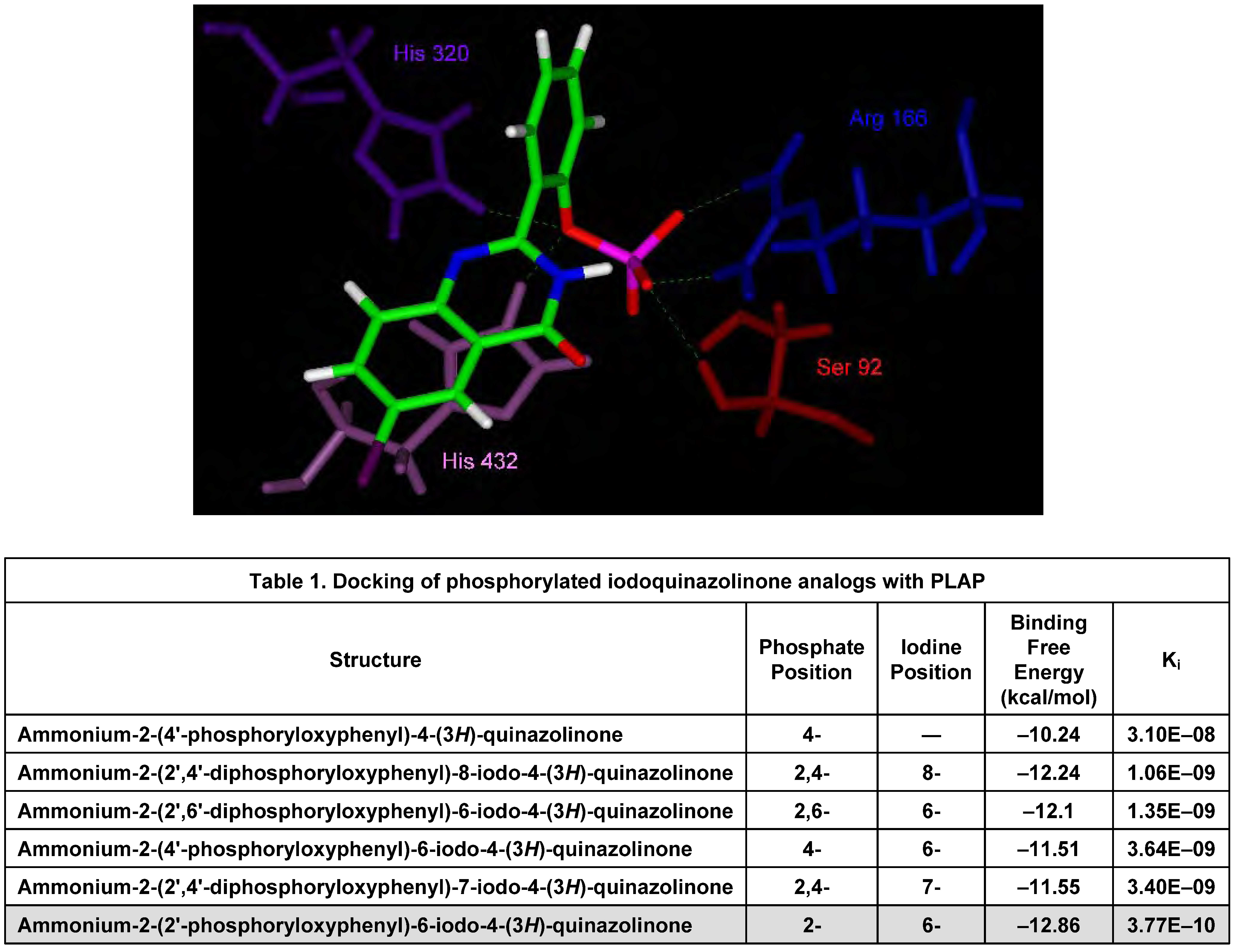
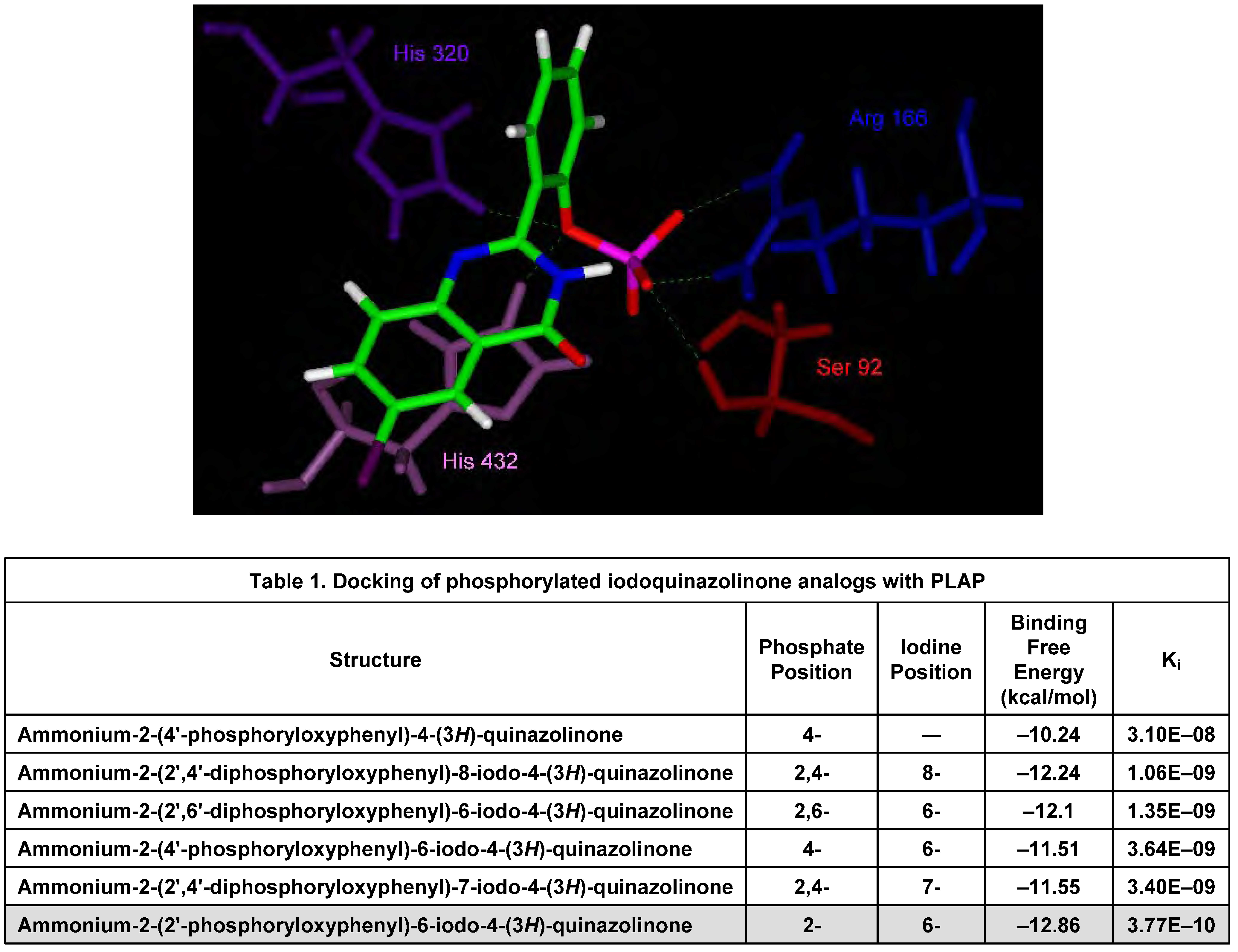
In Silico Molecular Modeling Studies
Synthesis, Radioiodination, and Characterization of Q2–P Derivatives [5,6,7]
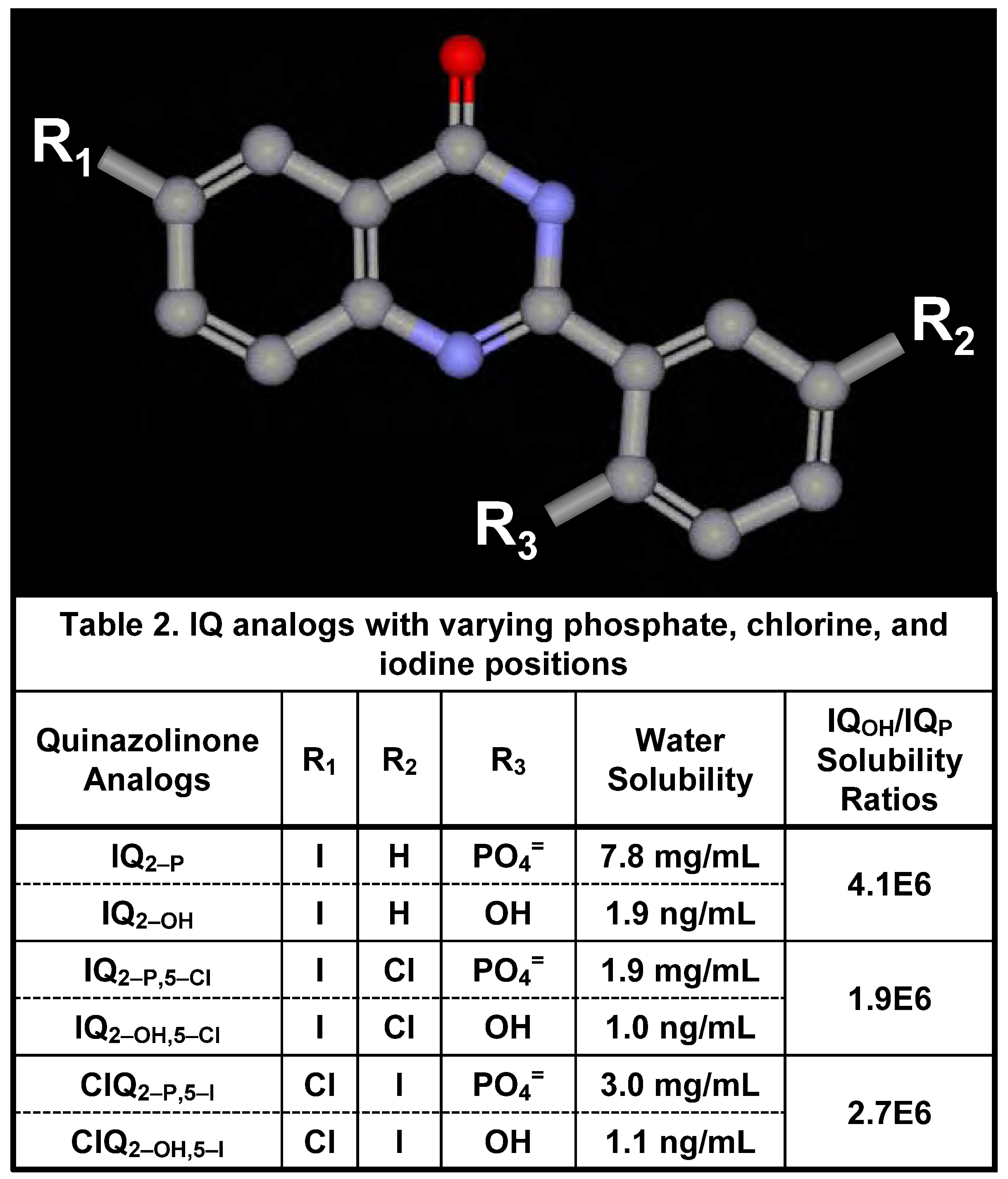
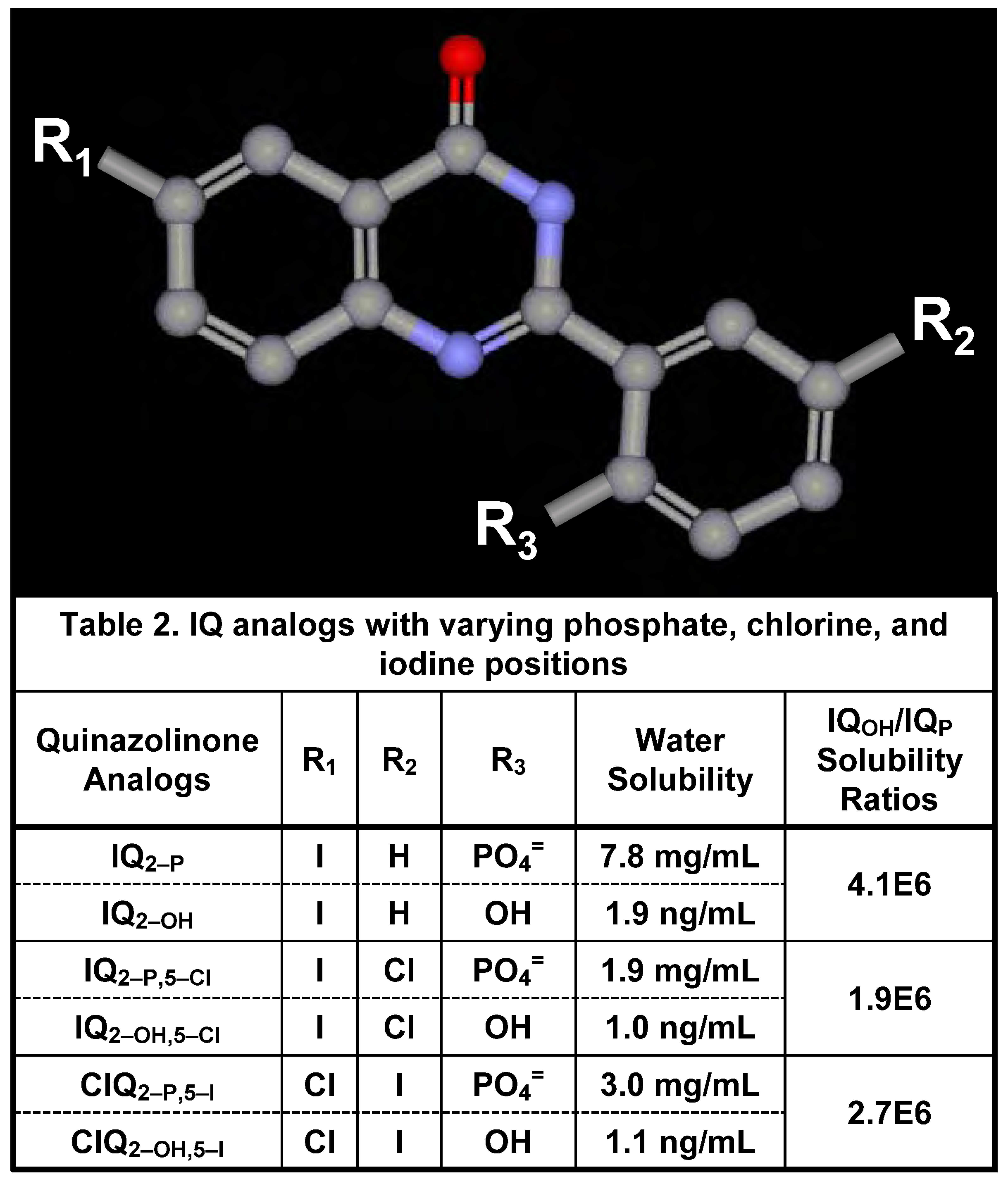
Water Solubility of Iodinated Q2–P and Q2–OH Derivatives
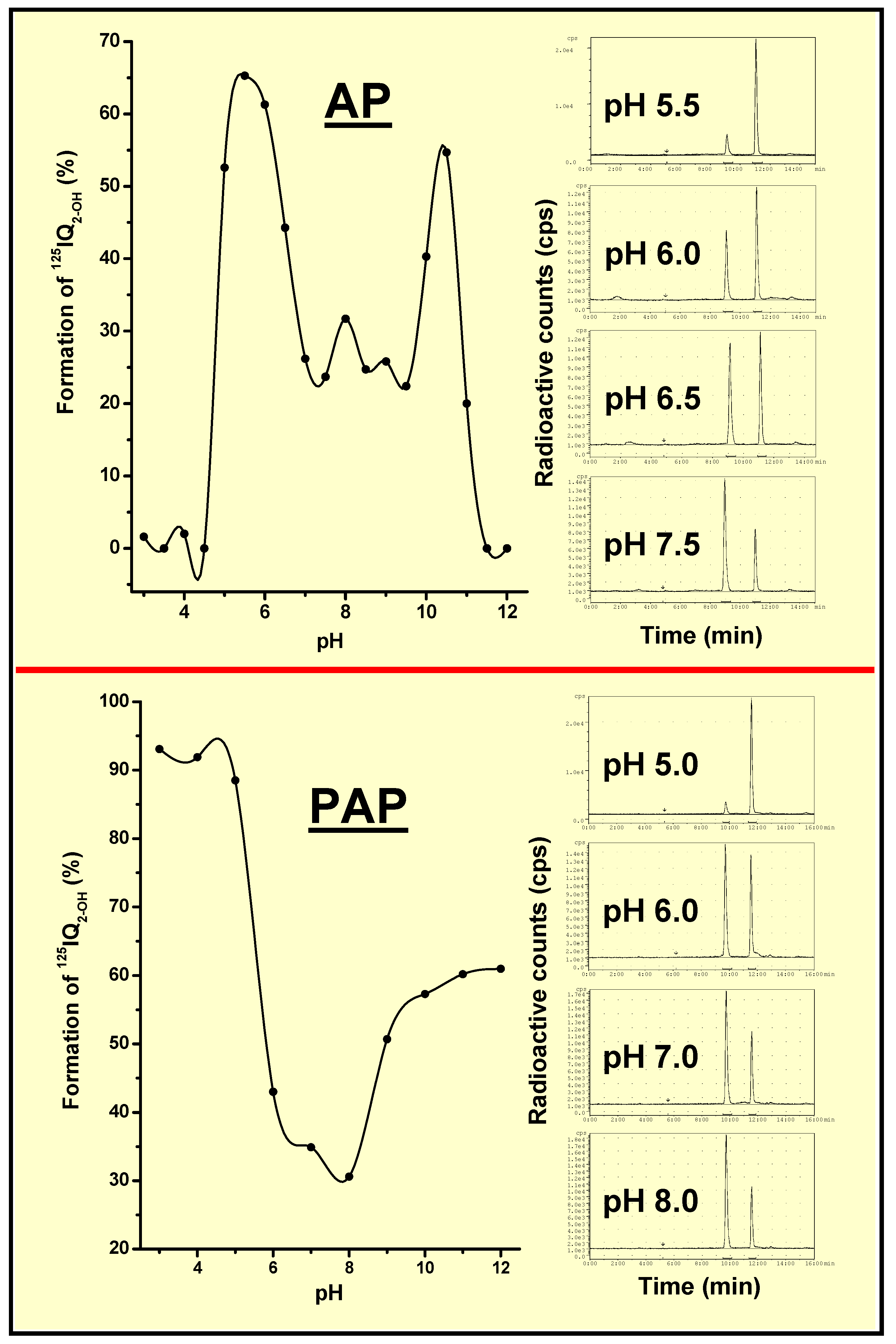
AP- and PAP-Dependent Conversion of Iodoquinazolinone Derivatives
In Vitro and In Vivo Studies
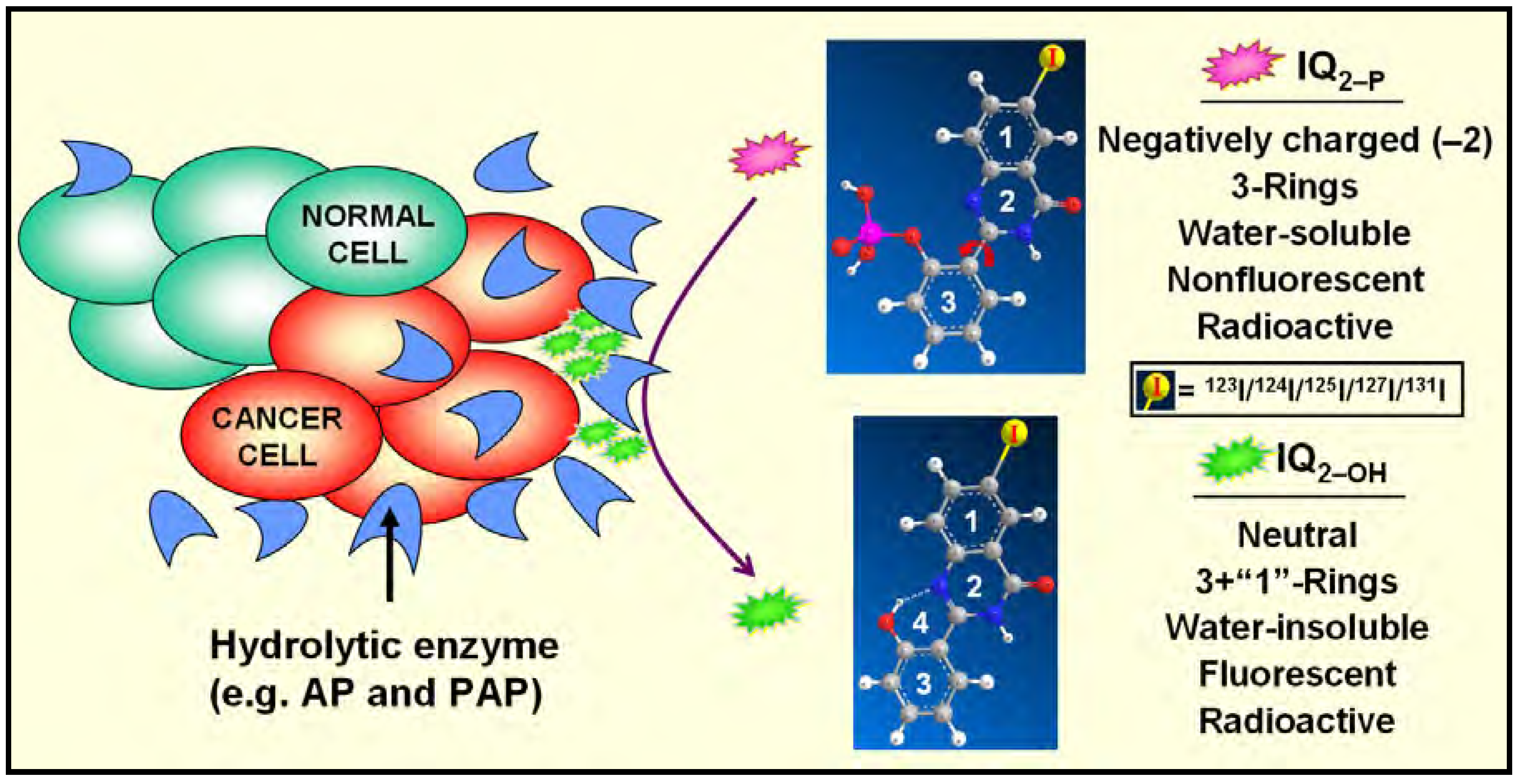
Hydrolysis of IQ2–P by Mammalian Cells [3,5,6,7]
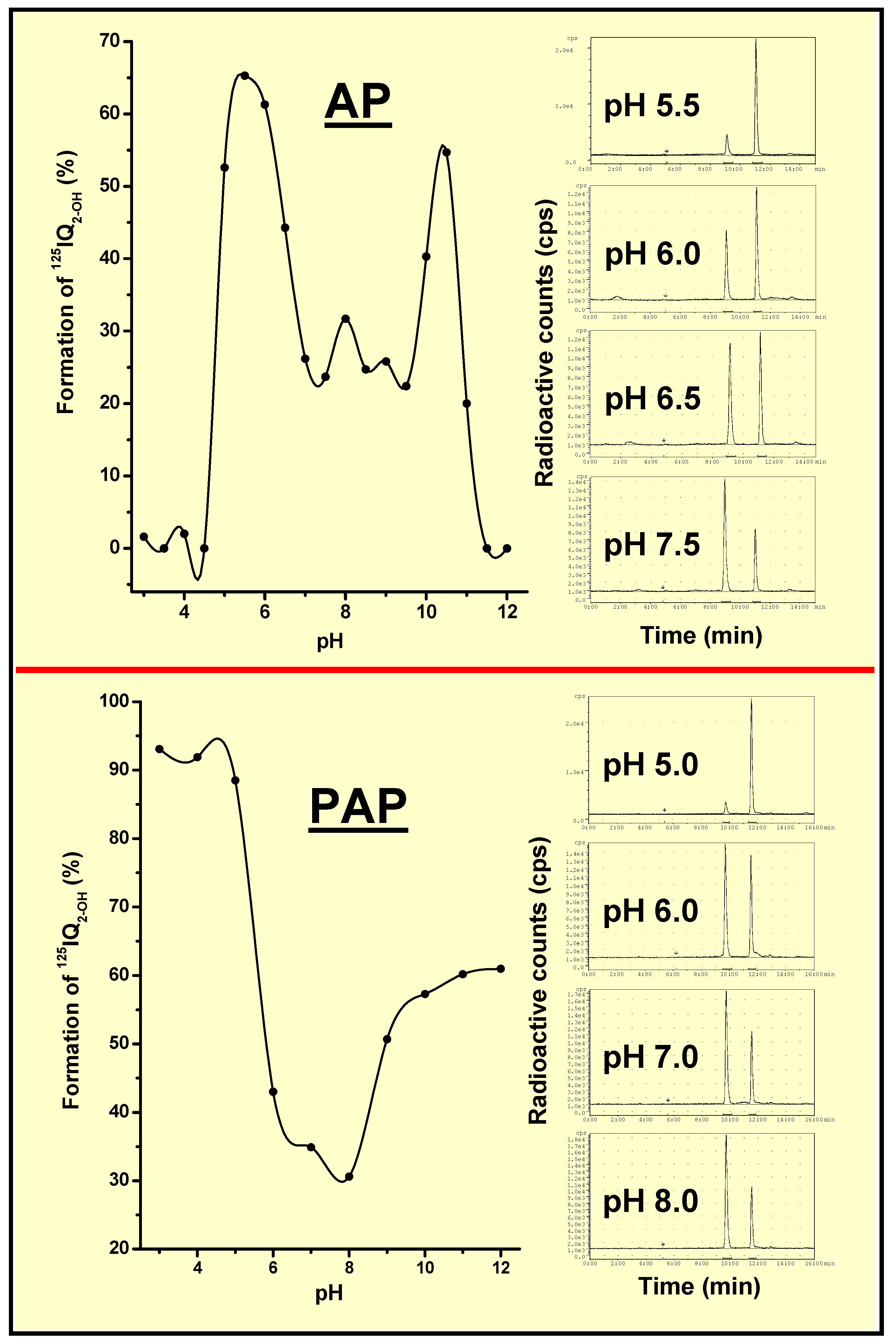

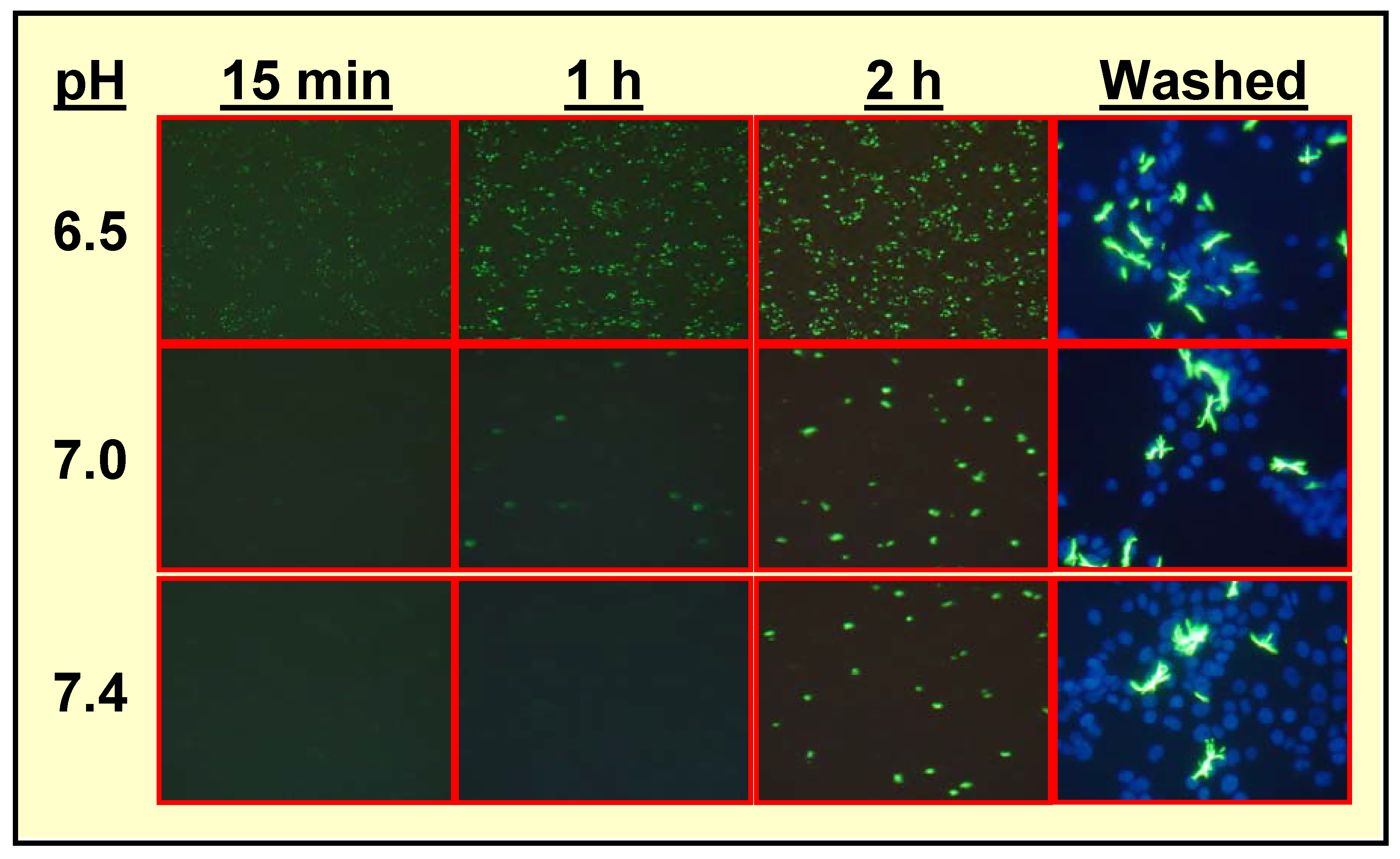
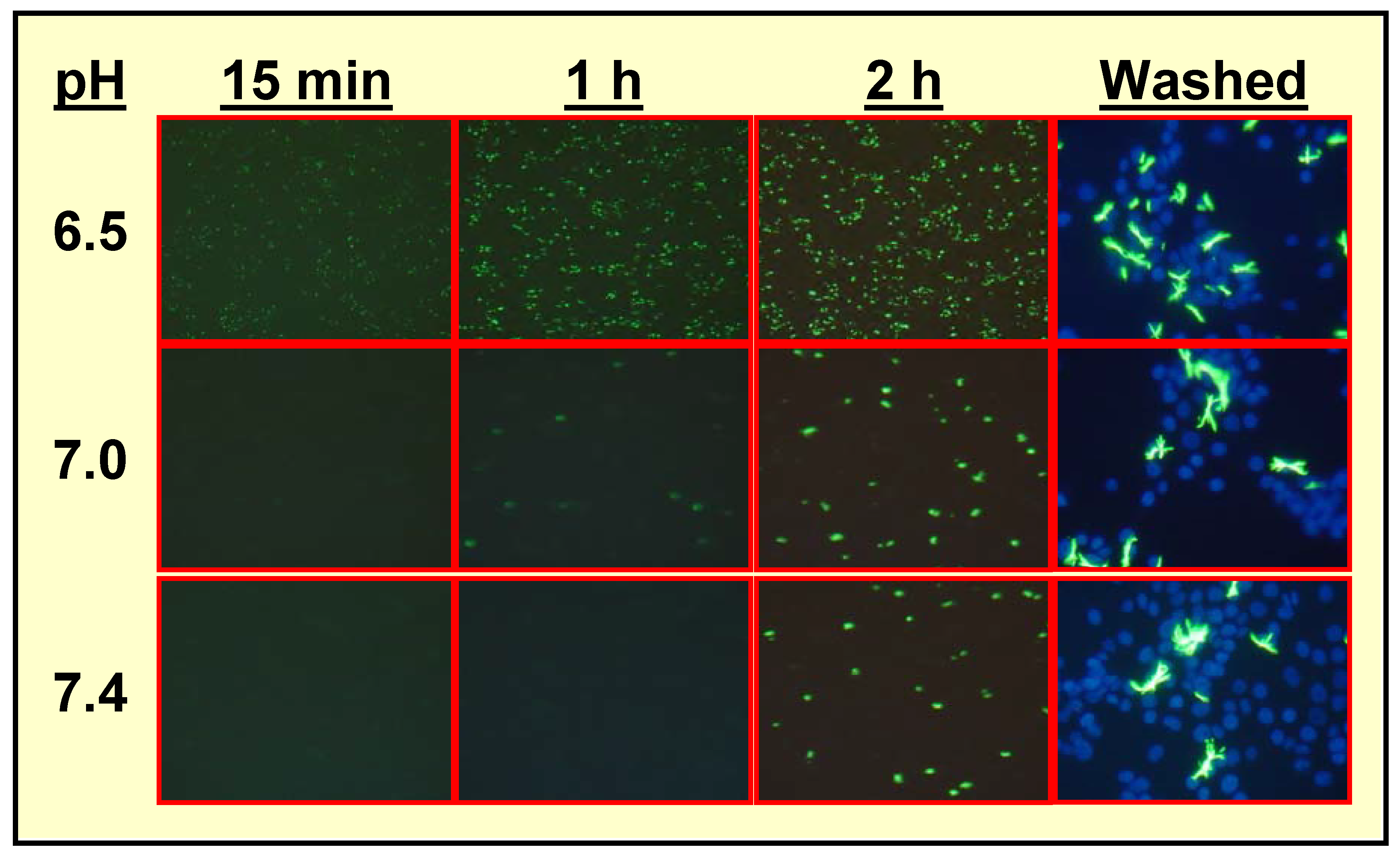
Specific Localization of IQ2–P in AP-Rich Regions and Entrapment of IQ2–OH within Tissues
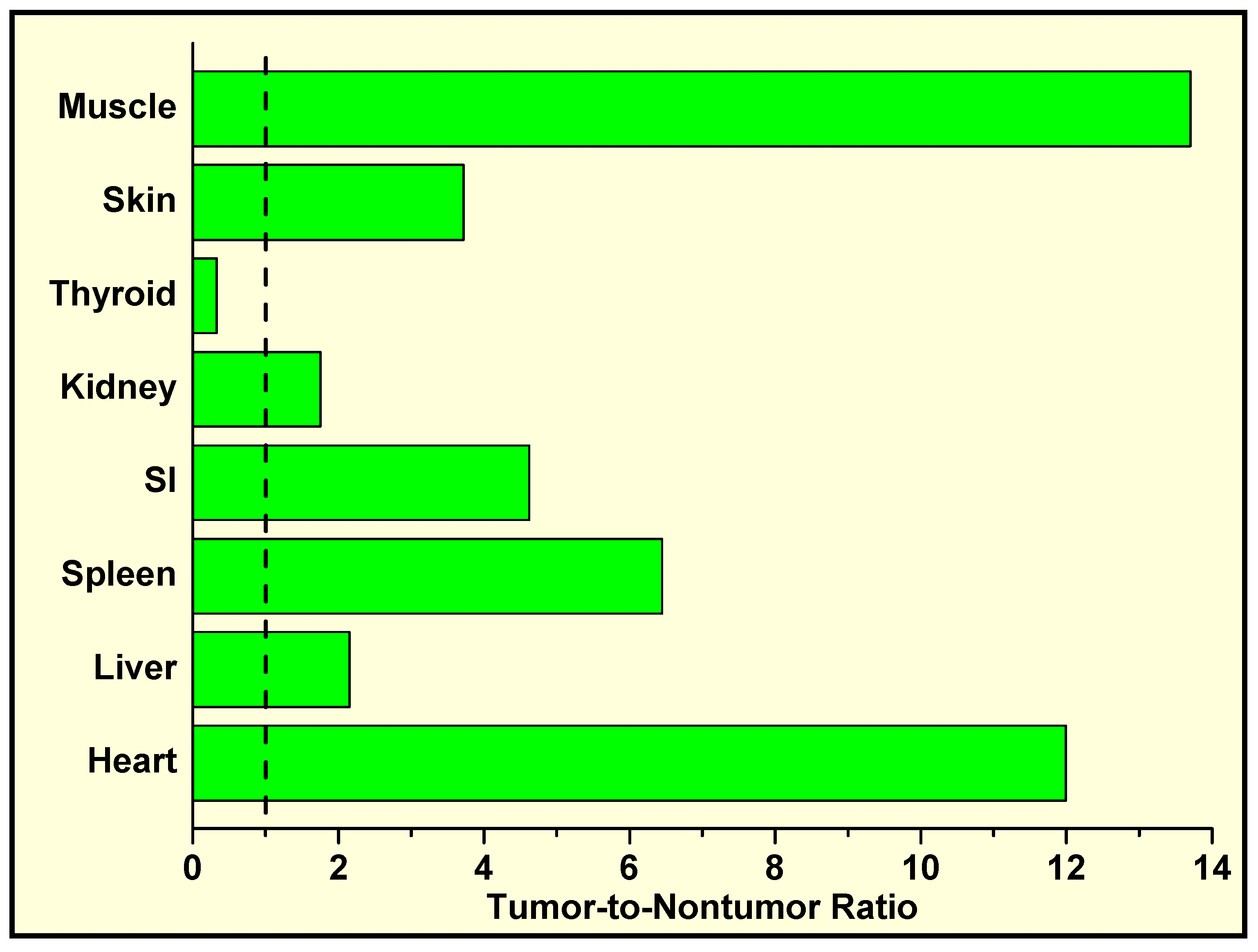
In Vivo Localization of 125I-Labeled Q2–P in Tumor-Bearing Mice and High Tumor-to-Normal Tissue (T/NT) Ratios

Conclusions
Acknowledgments
References
- Kassis, A.I.; Harapanhalli, R.S. Methods for enzyme-mediated tumor diagnosis and therapy. U.S. Patent Pending application no. 2001-839779, 2001. [Google Scholar]
- Ho, N.; Harapanhalli, R.S.; Dahman, B.A.; Chen, K.; Wang, K.; Adelstein, S.J.; Kassis, A.I. Synthesis and biologic evaluation of a radioiodinated quinazolinone derivative for enzyme-mediated insolubilization therapy. Bioconj. Chem. 2002, 13, 357–364. [Google Scholar] [CrossRef]
- Chen, K.; Wang, K.; Kirichian, A.M.; Al Aowad, A.F.; Iyer, L.K.; Adelstein, S.J.; Kassis, A.I. In silico design, synthesis, and biological evaluation of radioiodinated quinazolinone derivatives for alkaline phosphatase–mediated cancer diagnosis and therapy. Mol. Cancer Ther. 2006, 5, 3001–3013. [Google Scholar] [CrossRef]
- Pospisil, P.; Iyer, L.K.; Adelstein, S.J.; Kassis, A.I. A combined approach to data mining of textual and structured data to identify cancer-related targets. BMC Bioinformatics 2006, 7, 354. [Google Scholar]
- Chen, K.; Al Aowad, A.F.; Adelstein, S.J.; Kassis, A.I. Molecular-docking-guided design, synthesis, and biologic evaluation of radioiodinated quinazolinone prodrugs. J. Med. Chem. 2007, 50, 663–673. [Google Scholar] [CrossRef]
- Pospisil, P.; Wang, K.; Al Aowad, A.F.; Iyer, L.K.; Adelstein, S.J.; Kassis, A.I. Computational modeling and experimental evaluation of a novel prodrug for targeting the extracellular space of prostate tumors. Cancer Res. 2007, 67, 2197–2205. [Google Scholar] [CrossRef]
- Wang, K.; Kirichian, A.M.; Al Aowad, A.F.; Adelstein, S.J.; Kassis, A.I. Evaluation of chemical, physical, and biologic properties of tumor-targeting radioiodinated quinazolinone derivative. Bioconjugate Chem. 2007, 18, 754–764. [Google Scholar] [CrossRef]
- Kassis, A.I. Compounds and methods for enzyme-mediated tumor imaging and therapy. U.S. provisional patent pending 2007. [Google Scholar]
- Kassis, A.I. Radiotargeting agents for cancer therapy. Expert Opin. Drug Deliv. 2005, 2, 981–991. [Google Scholar] [CrossRef]
- Blaschke, C.; Yeh, A.; Camon, E.; Colosimo, M.; Apweiler, R.; Hirschman, L.; Valencia, A. Do you do text? Bioinformatics 2005, 21, 4199–4200. [Google Scholar]
- NCBI PubMed. Available online: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi.
- UniProt, the Universal Protein Resource. Available online: http://www.pir.uniprot.org/.
- NCBI Entrez Gene (supercedes LocusLink). Available online: http://www.ncbi.nlm.nih.gov/projects/LocusLink/.
- IT.Omics LSGraph®. Available online: http://lsgraph.it-omics.com/.
- Ingenuity® Systems. Available online: http://www.ingenuity.com/products/pathways_analysis.html.
- Davies, J.O.; Davies, E.R.; Howe, K.; Jackson, P.C.; Pitcher, E.M.; Sadowski, C.S.; Stirrat, G.M.; Sunderland, C.A. Radionuclide imaging of ovarian tumours with 123I-labelled monoclonal antibody (NDOG2) directed against placental alkaline phosphatase. Br. J. Obstet. Gynaecol. 1985, 92, 277–286. [Google Scholar] [CrossRef]
- Critchley, M.; Brownless, S.; Patten, M.; McLaughlin, P.J.; Tromans, P.M.; McDicken, I.W.; Johnson, P.M. Radionuclide imaging of epithelial ovarian tumours with 123I-labelled monoclonal antibody (H317) specific for placental-type alkaline phosphatase. Clin. Radiol. 1986, 37, 107–112. [Google Scholar] [CrossRef]
- Stigbrand, T.; Hietala, S.-O.; Johansson, B.; Makiya, R.; Riklund, K.; Ekelund, L. Tumour radioimmunolocalization in nude mice by use of antiplacental alkaline phosphatase monoclonal antibodies. Tumor Biol. 1989, 10, 243–251. [Google Scholar] [CrossRef]
- Kosmas, C.; Kalofonos, H.P.; Hird, V.; Epenetos, A.A. Monoclonal antibody targeting of ovarian carcinoma. Oncology 1998, 55, 435–446. [Google Scholar] [CrossRef]
- Le Du, M.H.; Stigbrand, T.; Taussig, M.J.; Ménez, A.; Stura, E.A. Crystal structure of alkaline phosphatase from human placenta at 1.8 Å resolution: implication for a substrate specificity. J. Biol. Chem. 2001, 276, 9158–9165. [Google Scholar]
- Ho, D.H.W. Distribution of kinase and deaminase of 1-β-D-arabinofuranosylcytosine in tissues of man and mouse. Cancer Res. 1973, 33, 2816–2820. [Google Scholar]
- Ortlund, E.; LaCount, M.W.; Lebioda, L. Crystal structures of human prostatic acid phosphatase in complex with a phosphate ion and α-benzylaminobenzylphosphonic acid update the mechanistic picture and offer new insights into inhibitor design. Biochemistry (Mosc) 2003, 42, 383–389. [Google Scholar] [CrossRef]
- Schneider, G.; Lindqvist, Y.; Vihko, P. Three-dimensional structure of rat acid phosphatase. EMBO J. 1993, 12, 2609–2615. [Google Scholar]
- Porvari, K.S.; Herrala, A.M.; Kurkela, R.M.; Taavitsainen, P.A.; Lindqvist, Y.; Schneider, G.; Vihko, P.T. Site-directed mutagenesis of prostatic acid phosphatase: catalytically important aspartic acid 258, substrate specificity, and oligomerization. J. Biol. Chem. 1994, 269, 22642–22646. [Google Scholar]
- Lindqvist, Y.; Schneider, G.; Vihko, P. Crystal structures of rat acid phosphatase complexed with the transition-state analogs vanadate and molybdate: implications for the reaction mechanism. Eur. J. Biochem. 1994, 221, 139–142. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Kim, S.-G.; Evelhoch, J.L.; Ackerman, J.J.H. Nonglycolytic acidification of murine radiation-induced fibrosarcoma 1 tumor via 3-O-methyl-D-glucose monitored by 1H, 2H, 13C, and 31P nuclear magnetic resonance spectroscopy. Cancer Res. 1992, 52, 1259–1266. [Google Scholar]
- Raghunand, N.; Altbach, M.I.; van Sluis, R.; Baggett, B.; Taylor, C.W.; Bhujwalla, Z.M.; Gillies, R.J. Plasmalemmal pH-gradients in drug-sensitive and drug-resistant MCF-7 human breast carcinoma xenografts measured by 31P magnetic resonance spectroscopy. Biochem. Pharmacol. 1999, 57, 309–312. [Google Scholar] [CrossRef]
- Gillies, R.J.; Liu, Z.; Bhujwalla, Z. 31P-MRS measurements of extracellular pH of tumors using 3-aminopropylphosphonate. Am. J. Physiol. 1994, 267, C195–C203. [Google Scholar]
- Timperley, W.R. Alkaline-phosphatase-secreting tumour of lung. Lancet 1968, 2, 356. [Google Scholar] [CrossRef]
- Suzuki, H.; Iino, S.; Endo, Y.; Torii, M.; Miki, K.; Oda, T. Tumor-specific alkaline phosphatase in hepatoma. Ann. N. Y. Acad. Sci. 1975, 259, 307–320. [Google Scholar] [CrossRef]
- Higashino, K.; Kudo, S.; Ohtani, R.; Yamamura, Y. Further observation on Kasahara isoenzyme in patients with malignant diseases. Gann 1976, 67, 909–911. [Google Scholar]
- Benham, F.J.; Povey, M.S.; Harris, H. Placental-like alkaline phosphatase in malignant and benign ovarian tumors. Clin. Chim. Acta 1978, 86, 201–215. [Google Scholar] [CrossRef]
- Nadji, M.; Tabei, Z.; Castro, A.; Morales, A.R. Immunohistological demonstration of prostatic origin of malignant neoplasms. Lancet 1979, 1, 671–672. [Google Scholar]
- Loor, R.; Wang, M.C.; Valenzuela, L.; Chu, T.M. Expression of prostatic acid phosphatase in human prostate cancer. Cancer Lett. 1981, 14, 63–69. [Google Scholar] [CrossRef]
- Wick, M.R.; Swanson, P.E.; Manivel, J.C. Placental-like alkaline phosphatase reactivity in human tumors: an immunohistochemical study of 520 cases. Hum. Pathol. 1987, 18, 946–954. [Google Scholar] [CrossRef]
- Azumi, N.; Traweek, S.T.; Battifora, H. Prostatic acid phosphatase in carcinoid tumors: immunohistochemical and immunoblot studies. Am. J. Surg. Pathol. 1991, 15, 785–790. [Google Scholar] [CrossRef]
- Dabare, A.A.N.P.M.; Nouri, A.M.E.; Cannell, H.; Moss, T.; Nigam, A.K.; Oliver, R.T.D. Profile of placental alkaline phosphatase expression in human malignancies: effect of tumour cell activation on alkaline phosphatase expression. Urol. Int. 1999, 63, 168–174. [Google Scholar] [CrossRef]
- Samples Availability: Not available.
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Kassis, A.I.; Korideck, H.; Wang, K.; Pospisil, P.; Adelstein, S.J. Novel Prodrugs for Targeting Diagnostic and Therapeutic Radionuclides to Solid Tumors. Molecules 2008, 13, 391-404. https://doi.org/10.3390/molecules13020391
Kassis AI, Korideck H, Wang K, Pospisil P, Adelstein SJ. Novel Prodrugs for Targeting Diagnostic and Therapeutic Radionuclides to Solid Tumors. Molecules. 2008; 13(2):391-404. https://doi.org/10.3390/molecules13020391
Chicago/Turabian StyleKassis, Amin I., Houari Korideck, Ketai Wang, Pavel Pospisil, and S. James Adelstein. 2008. "Novel Prodrugs for Targeting Diagnostic and Therapeutic Radionuclides to Solid Tumors" Molecules 13, no. 2: 391-404. https://doi.org/10.3390/molecules13020391
APA StyleKassis, A. I., Korideck, H., Wang, K., Pospisil, P., & Adelstein, S. J. (2008). Novel Prodrugs for Targeting Diagnostic and Therapeutic Radionuclides to Solid Tumors. Molecules, 13(2), 391-404. https://doi.org/10.3390/molecules13020391