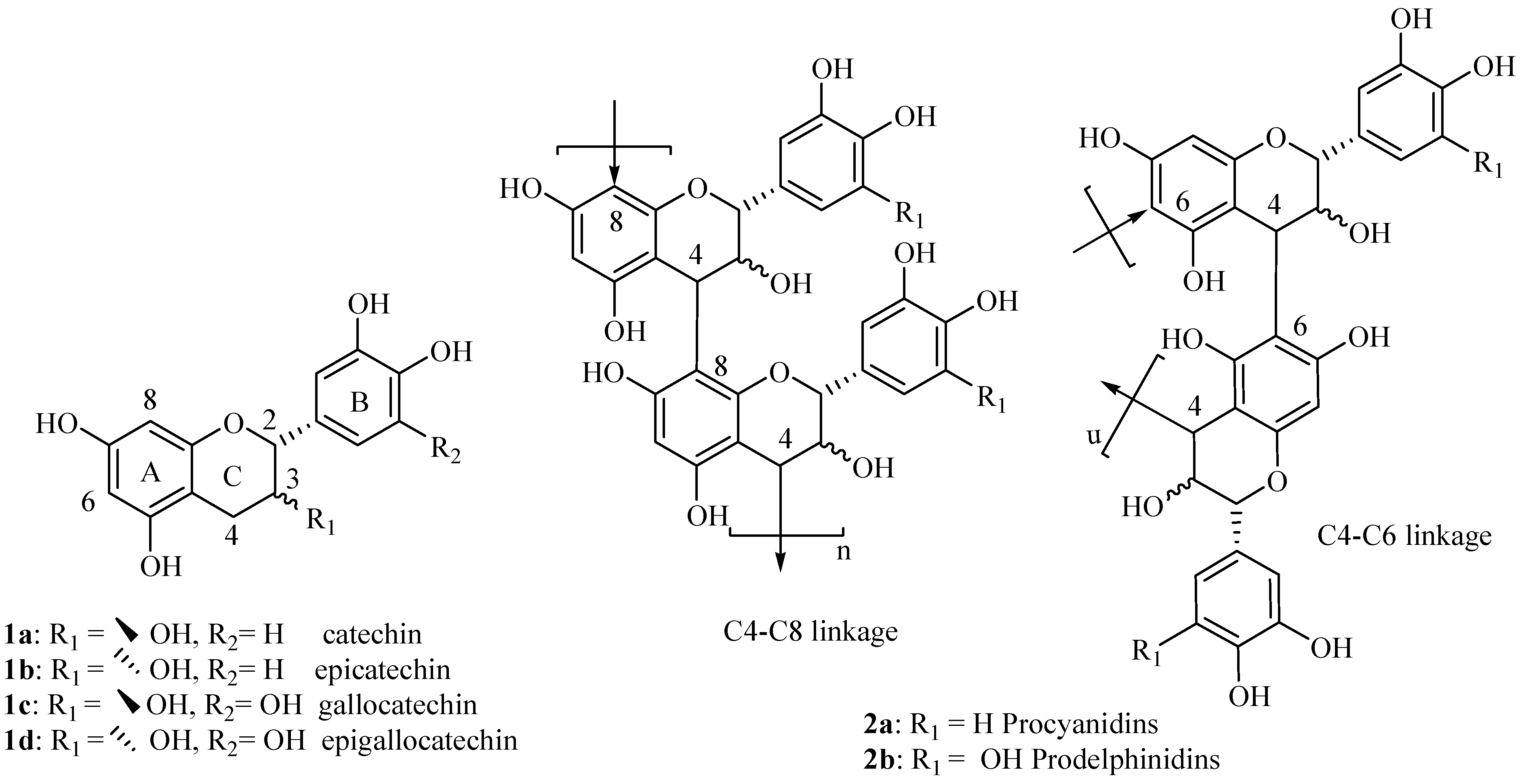
Synthesis of modified (+)-catechin derivatives
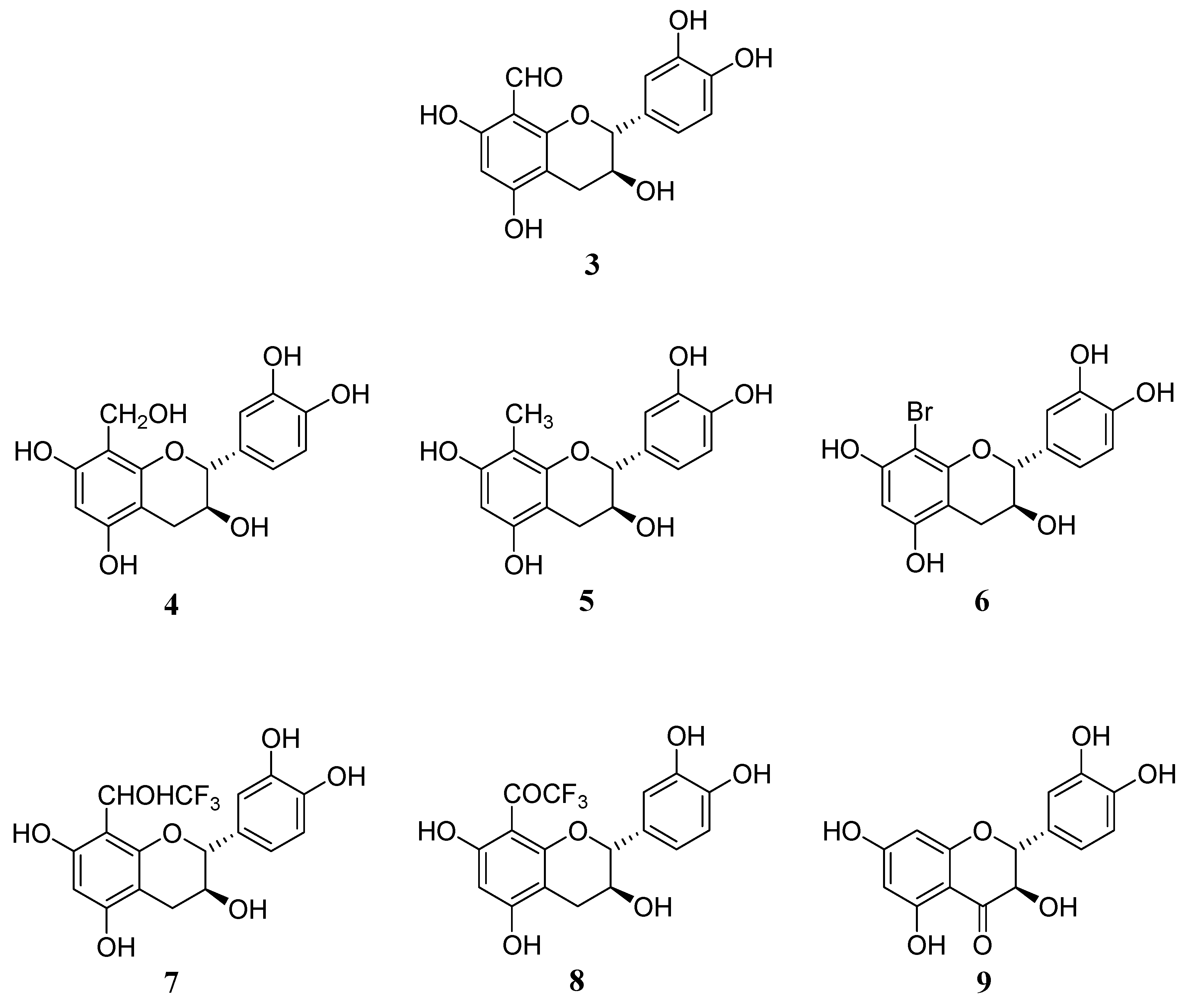
We started investigations on the synthesis of new modified monomeric units derived from catechin with the objective of exploring the impact of the A ring substitution on their biological properties. For this study, the six 8-substituted derivatives of flavan-3-ols,
3-
8, in addition to taxifolin (
9) were synthesized and their antioxidative activity investigated. The modified flavan-3-ols monomer derivatives described in this work are shown in
Figure 2and their synthesis pathways are depicted in
Scheme 1. As can be noticed, the studied compounds were synthesized in their benzylated forms
11,
12,
14-
16 and
23. Their free forms were obtained through deprotection in a MeOH/THF medium, under H
2 and in presence of Pd as catalyst.
Figure 2.
Structures of the studied modified flavan-3-ol monomers 3-9.
Figure 2.
Structures of the studied modified flavan-3-ol monomers 3-9.
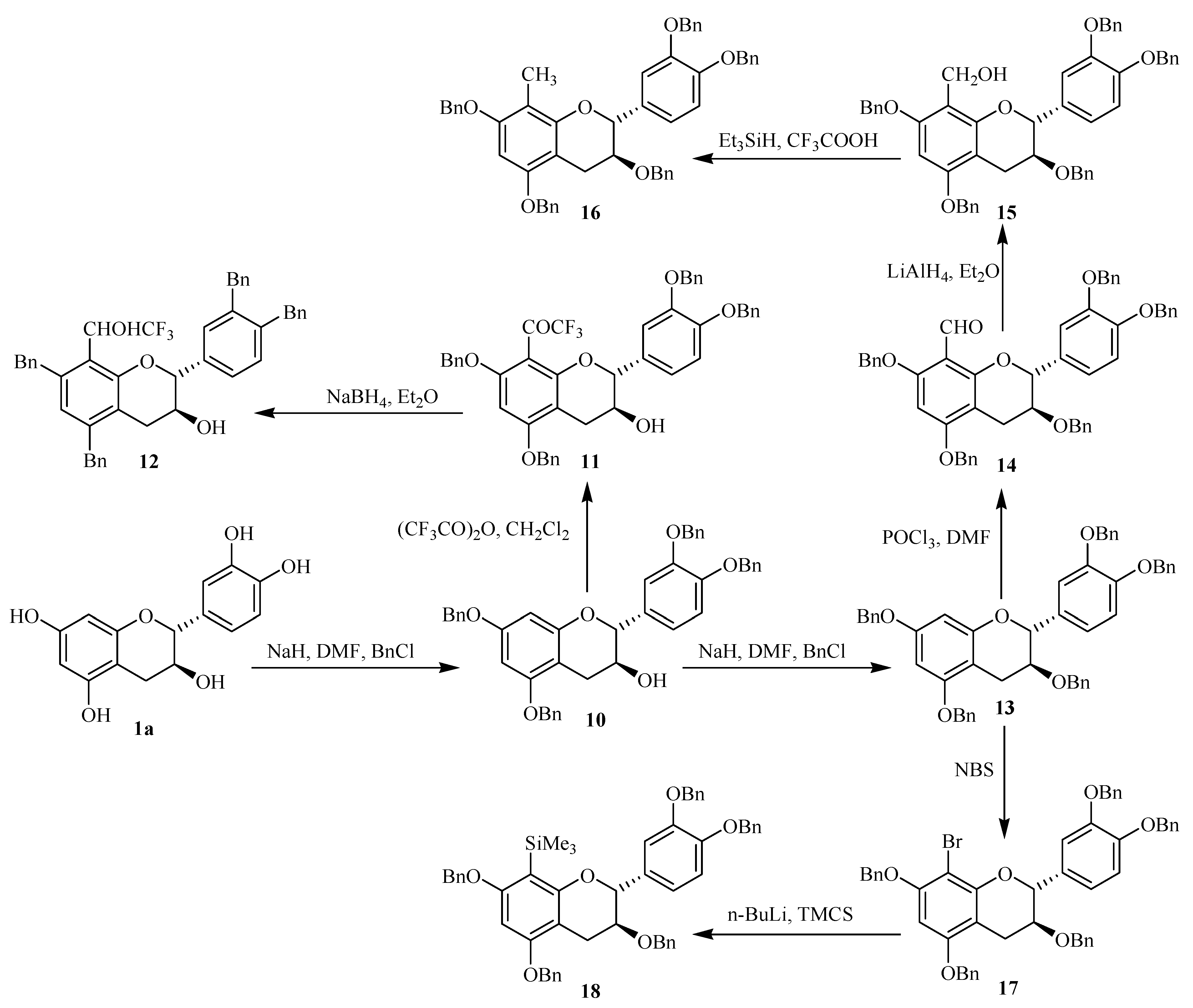
Compound
11 was prepared by action of trifluoacetic anhydride on tetrabenzylated catechin
10 following a Friedel-Craft’s reaction on the nucleophilic C-8 position, as previously described [
14,
15]. Further reduction by NaBH
4 afforded compound
12. Compound
14 was prepared starting from the pentabenzylated catechin
13. After formylation through the classical Vilsmeier reaction, the obtained compound
14 was reduced by LiAlH
4 giving the hydroxymethyl derivative
15. Further reduction of the latter gave the target product
16 with a good yield. The bromided adduct
17 was obtained from
13 by action of NBS. Gram-scale amounts of taxifolin (
9) were prepared from (+)-catechin through reactions involving oxidation processes as previously described [
17].
The structures of these modified catechin derivatives were determined through UV, MS and NMR spectroscopy. Structure elucidation of compound
16 will be detailed as example. The ESI-MS spectrum recorded in the positive ion mode showed signals located at
m/z 755, 772 and 777 corresponding to [M+H]
+, [M+NH
4]
+ and [M+Na]
+ ions respectively and indicating a molecular weight of 754 amu in agreement with the structure of compound
16. The usual characteristic flavan-3-ol RDA fragmentation was also observed at
m/z 423, [M+H-332]
+ ion, corresponding to the protonated A moiety (
Figure 3).
Scheme 1.
Synthesis pathways of the studied modified flavanol monomers.
Scheme 1.
Synthesis pathways of the studied modified flavanol monomers.
Figure 3.
Main fragmentations observed in compounds 11, 16 and 18 and main 1H-13C long range correlations observed for compound 16.
Figure 3.
Main fragmentations observed in compounds 11, 16 and 18 and main 1H-13C long range correlations observed for compound 16.
The remaining outstanding question that needed to be resolved was related to the position of the methyl group on the flavanol A ring. This constitutes the most encountered problem in flavanol structural characterization. Since the two positions 6 and 8 are almost magnetically equivalent, they could not be distinguished on the basis of their chemical shift. In our case, assuming that the substitution occurs at the more nucleophilic positions of the flavanol skeleton, i.e 6 or 8, as confirmed through ES-MS spectrometry, determination of the residual proton (H6 or H8) could not be achieved based only on its chemical shift but would rather requires the use of 2D NMR analysis. The position of the CH3 group on the aromatic A ring was elucidated by long range distance carbon-proton correlations established by 2D NMR HMBC experiments through the following reasoning. The usual pyran ring protons H4 [(2.7 ppm, dd, J= 16.7 and 5.6 Hz) and 3.0 ppm (dd, J= 16.7 and 8.7 Hz)], H3 (6.6 ppm, m) and H2 (7.9 ppm, d, J= 8.0 Hz) were easily assigned by 1H-NMR analysis. The three B ring protons were observed between 6.9 and 7.0 ppm. For the aromatic A ring, only one proton signal appearing as a singlet at 6.2 ppm was present indicating a monosubstitution. The presence of the CH3 group was confirmed through 1H-NMR analysis showing a singulet at 2.1 ppm. The protonated carbon chemical shifts were assigned through NMR HSQC analysis.
The definitive structure elucidation of compound
16 was achieved by HMBC experiment which allowed assignment of all hydrogen and carbon atoms. In addition to their correlations with C2 (79.8 ppm) and C3 (75.0 ppm), H4 protons (2.8 and 3.0 ppm) correlated with 3 carbons located at 102.7, 153.1 and 154.8 ppm. Carbons C4a, C8a and C5 are in a favorable position to give such correlations (
Figure 3). The signal observed at 102.7 ppm was attributed to C4a due to its chemical shift position compared to C8a and C5 which are linked to an oxygen atom. The carbon signal located at 153.1 ppm also gave a correlation with H2, which pointed to the C8a carbon and thus the remaining signal observed at 154.8 ppm was attributed to C5. The C8a signal thus attributed did not show any correlation with the residual A ring aromatic protons which is thus H6. This was also confirmed by the presence of a correlation between C5 and the residual aromatic proton and between the methyl protons and the C8a (
Figure 3). The position of the methyl group on the A ring 8 position was thus demonstrated.
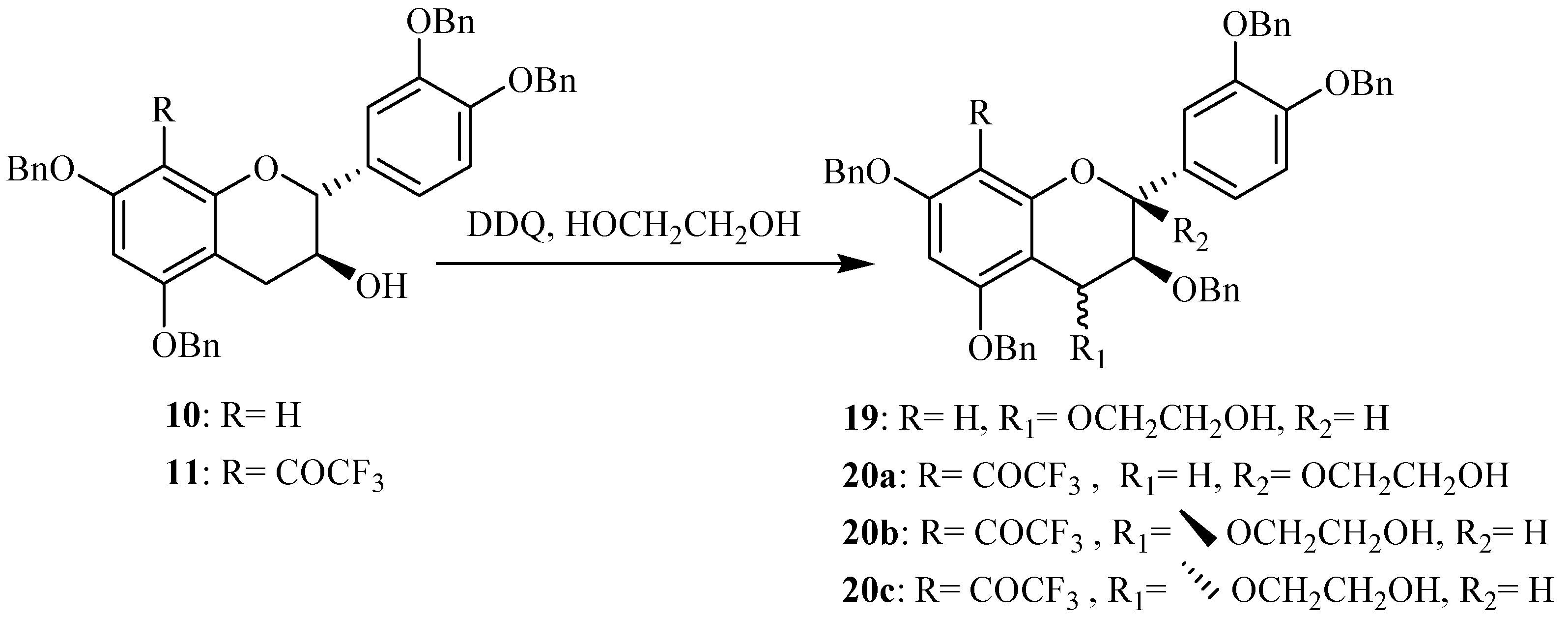
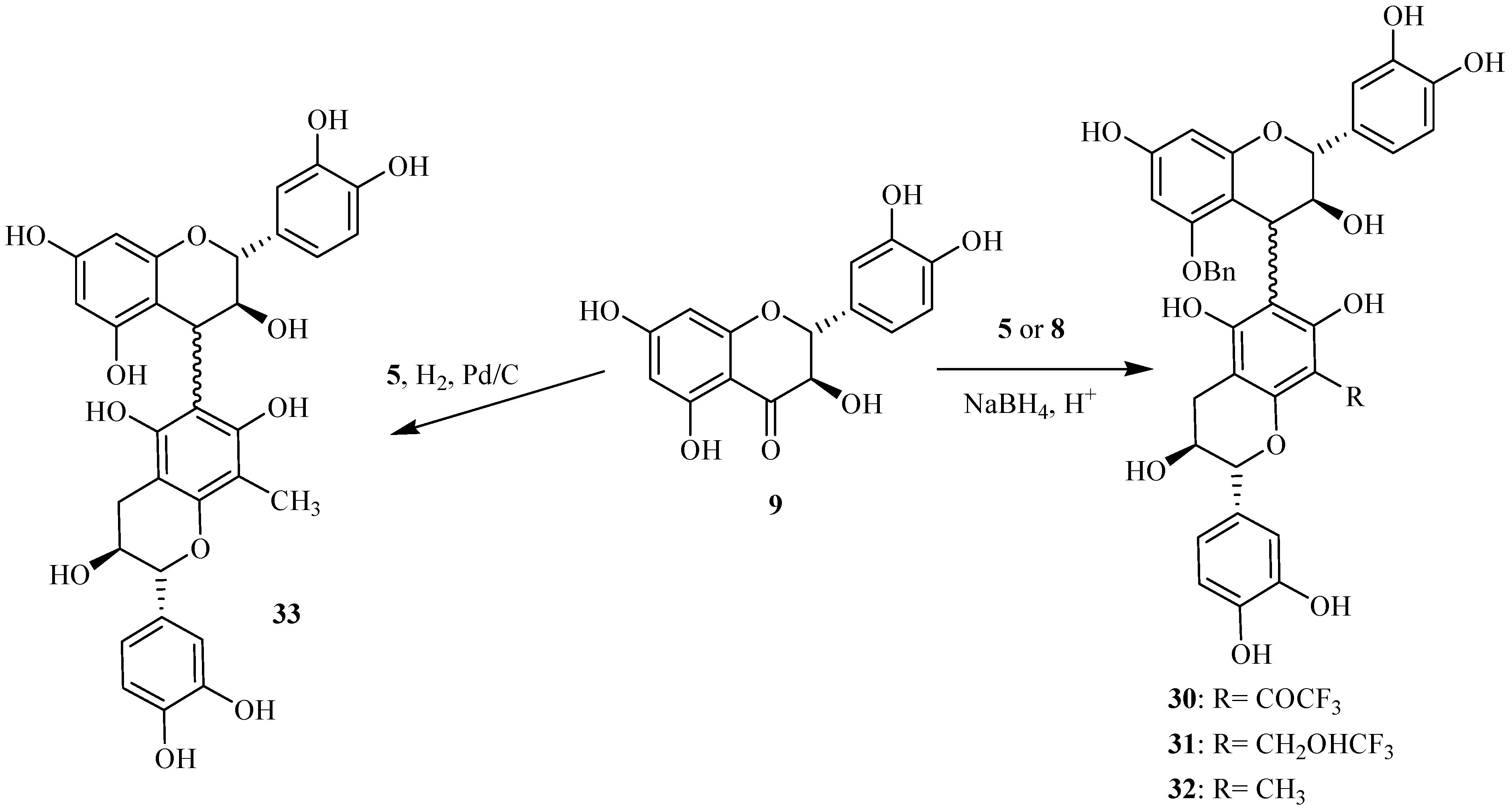
Synthesis of activated flavanols
Oxidation at the benzylic C-4 position of flavanols constitutes a fundamental step in proanthocyanidin synthesis giving
O-alkylated adducts. The use of DDQ [
15,
16] as oxidant of tetrabenzylated catechin
10 in presence of ethylene glycol gave the corresponding 4-
O-alkylated catechin
19 with good yields attaining 70 %, as indicated in our previous work [
14] (
Scheme 2).
Scheme 2.
Synthesis of the activated catechin derivatives.
Scheme 2.
Synthesis of the activated catechin derivatives.
In order to investigate the use of other electrophiles in the proanthocyanidin synthesis, the oxidation of pentabenzylated catechin
13 with DDQ/CH
2Cl
2 was conducted in presence of water (
Scheme 3).
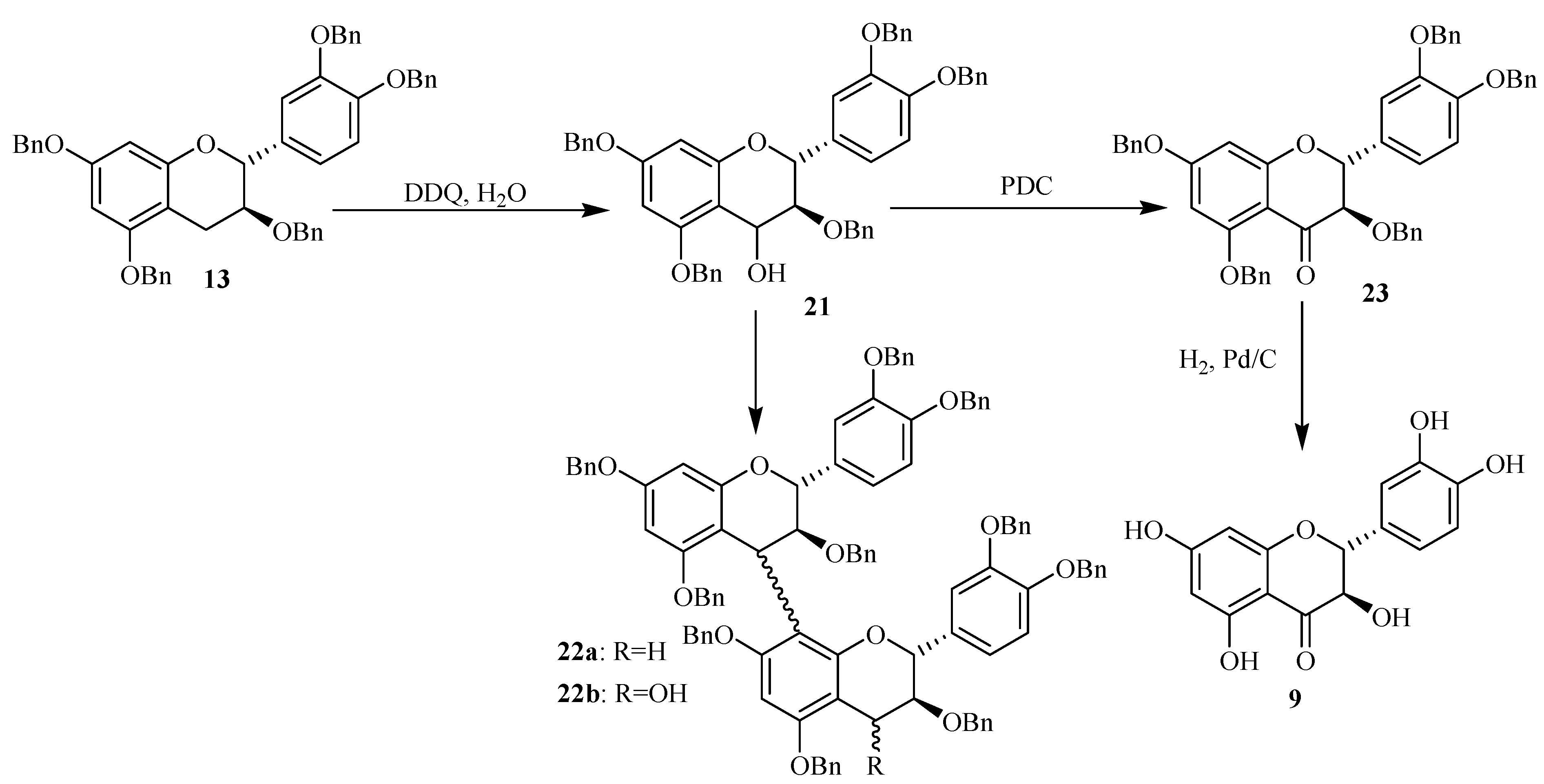
Scheme 3.
Synthesis of 4-activated catechin through DDQ oxidation and in presence of H2O.
Scheme 3.
Synthesis of 4-activated catechin through DDQ oxidation and in presence of H2O.
The flavan-3,4-diol
21 and the flavan-4-one
23 were obtained respectively in 66 and 15% yield and their structures were confirmed by MS and NMR spectroscopy [
17]. All the obtained data were in agreement with the structures proposed for these compounds. Moreover, the coupling constant for the AMX spin system of the C-ring protons (
J3,4 = 3.6 Hz) of compound
21 indicated a 3,4-
cis relative configuration for this ring, i.e a 4β-bond.
The flavan-4-one was probably formed by oxidation of the first formed flavan-3,4-diol 21. In addition, the formation of the dimeric compounds 22a (10 %) and 22b (7 %) was also detected through LC/ESI-MS analysis conducted in the positive ion mode. Compound 22a was detected by its [M+H]+ ion located at m/z 1479.7, corresponding to a dimeric structure consisting of two catechin units. Its formation involved probably the formation of a carbocation from compound 21 and its further reaction on a free benzylated catechin 13. Furthermore the 4-hydroxy oxidized form of compound 22a was also detected through LC/ESI-MS analysis in the positive ion mode. The mass spectrum of this compound noted 22b showed signals located at m/z 1496, 1513 and 1518 corresponding respectively to [M+H]+, [MNH4]+ and [MNa]+ in agreement with the (+)-catechin dimeric structure with an additional hydroxy group.
The mass spectrum of compound 22b showed several signals corresponding to the flavanol characteristic fragmentations. Its structure was finally confirmed through NMR analysis. The formation of this compound could be achieved through oxidation of compound 22a or by action of the carbocation generated from compound 21 and its further coupling with an already formed 4-hydroxybenzylated catechin 21. The formation of such dimeric precursor showed that the oxidation through DDQ could also occur on more long chains and then could be considered as a pathway for the preparation of more oligomerized flavanol precursors.
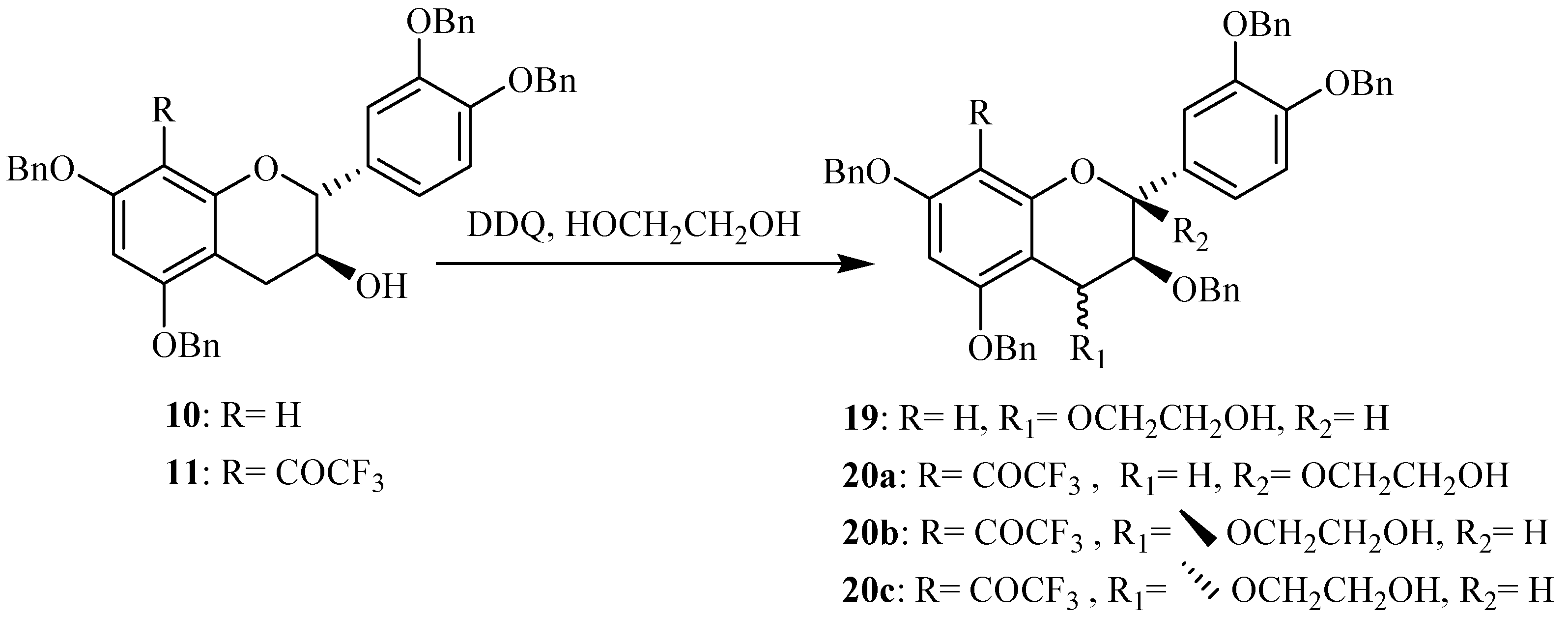
The preparation of these 4-activated (+)-catechin derivatives was achieved in order to use them as nucleophiles for the synthesis of modified proanthocyanidins having a modified catechin as terminal units and a free (+)-catechin as upper unit. Within the scope of preparing proanthocyanidins with a modified catechin as upper units, we were interested in investigating the oxidation of the previously prepared 8-substituted catechins. The 8-trifluoroacetyl derivative of tetrabenzylated catechin
11 was used as starting material in this first assay and using DDQ as oxidation agent in presence of ethylene glycol (
Scheme 2). No oxidation was observed in the conditions previously used on tetrabenzylated catechin. After optimization of the reaction conditions, the formation of three new compounds was observed. Their isolation was achieved through column chromatography and their structure elucidation was initiated through MS and NMR spectroscopy.
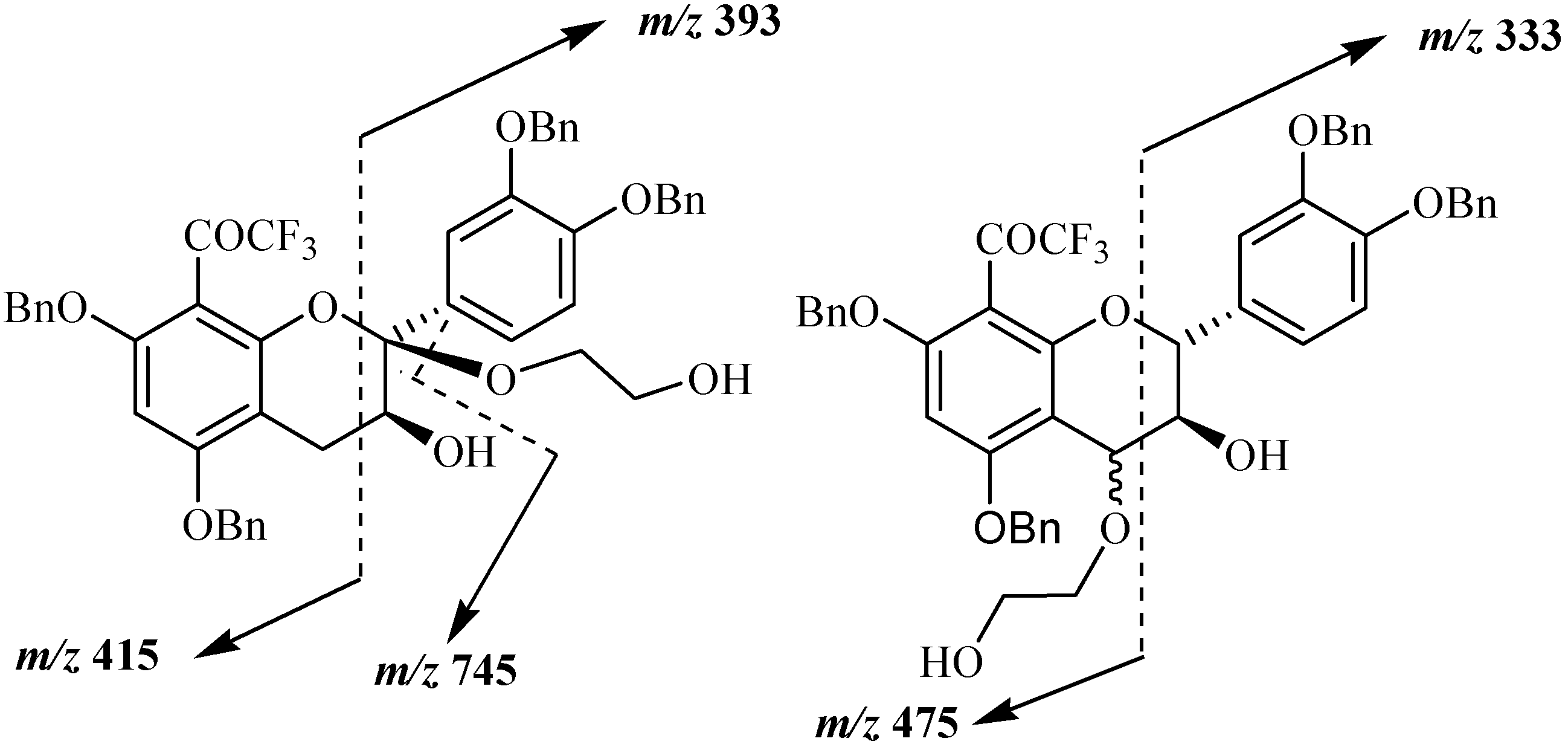
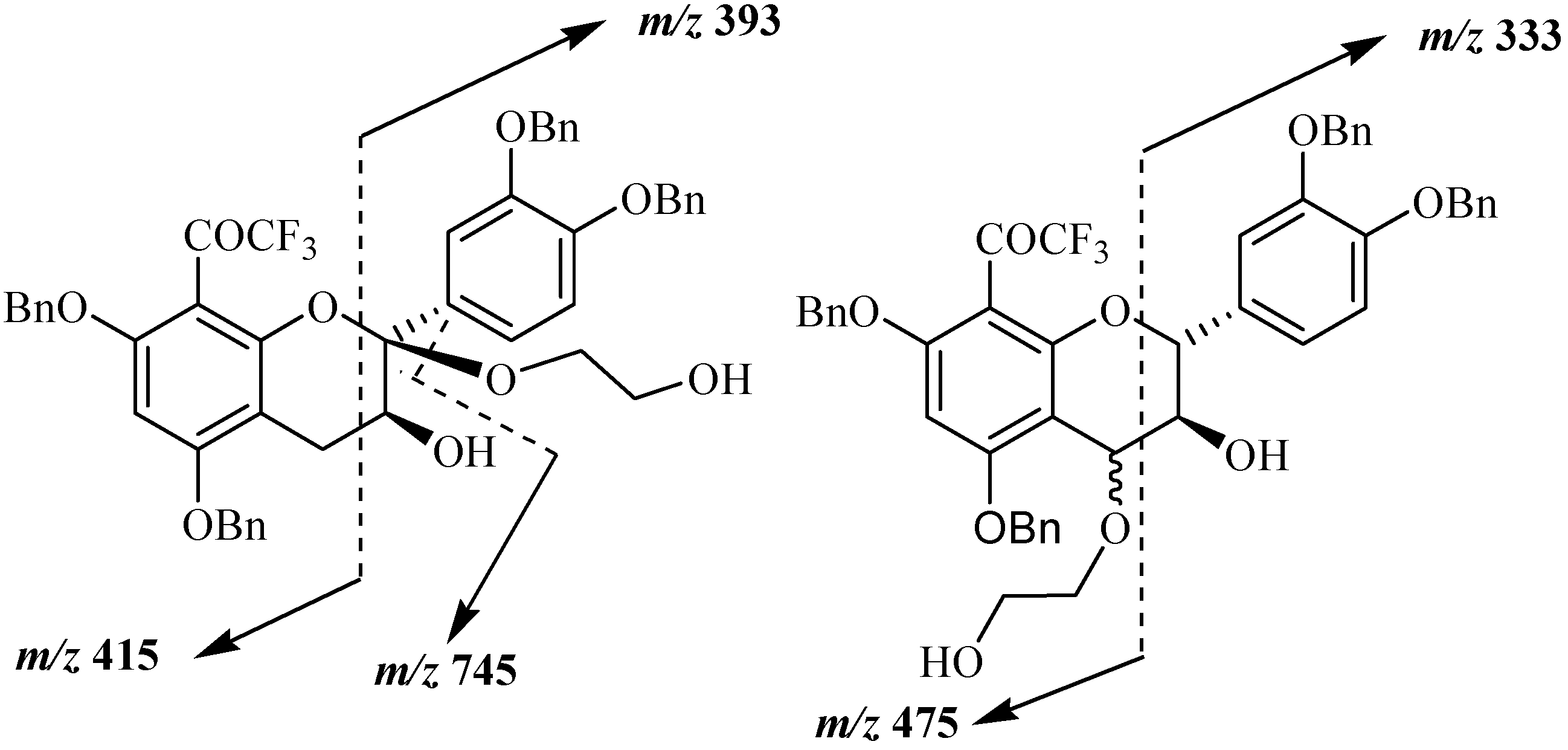
Mass spectral analysis of the three isolated compounds, conducted in the positive ion mode, showed signals located at
m/z 807, 824, 829 and 845. These signals corresponded to [M+H]
+, [M+NH
4]
+, [M+Na]
+ and [M+K]
+, respectively, and indicating a molecular weight of 806 amu. This result is in agreement with structures consisting of a tetrabenzylated moiety linked to a hydroxyethoxy group meaning that oxidation reaction has occurred in presence of DDQ. Since the three MS and UV data supported the conclusion that the three structures consisted of flavan-3-ol units linked to a HOCH
2CH
2O group, the primary problem was the establishment of the alkoxy location. Even if the mass spectra of the three compounds indicated the same molecular ions, some differences were observed in their fragmentation pattern. Thus signal peaks at
m/z 393 and 415 were observed for compound
20a, while
20b and
20c showed signals at
m/z 333 and 475 suggesting different alkoxyl group locations for these compounds shown in
Figure 4.
Figure 4.
Main fragmentations observed in compounds 20a, 20b and 20c.
Figure 4.
Main fragmentations observed in compounds 20a, 20b and 20c.
The position of the alkoxyl group was finally achieved through NMR analysis which easily indicated that the aromatic A proton in addition to those of the B ring were still present for the three compounds meaning that the oxidation occurred on the pyranic C ring. In the case of compound
20a (10 %) the presence of the two double doublets characteristic of the two H4 protons was observed meaning that the oxidation occurred at the 2 position. This was confirmed through
13C and DEPT NMR analysis, where only one singularily protonated carbon corresponding to C3 was observed. It must be noted that similar C2 oxidation product was already observed in the reaction involving DDQ oxidation of tetrabenzylated epicatechin in presence of methanol [
29]. In the case of compounds
20b (55 %) and
20c (20 %) similar NMR spectra were obtained. In the
1H spectrum the absence of the AMX characteristic signal of the two H4 protons was observed. This was also confirmed through
13C and DEPT analysis showing that the two compounds were stereoisomers
R- and
S-4-(2-hydroxyethoxy) derivatives of the 8-trifluoroacetyl adduct of tetrabenzylated catechin. These compounds could then serve as monomer precursors for the preparation of modified proanthocyanidins with modified catechin upper unit.
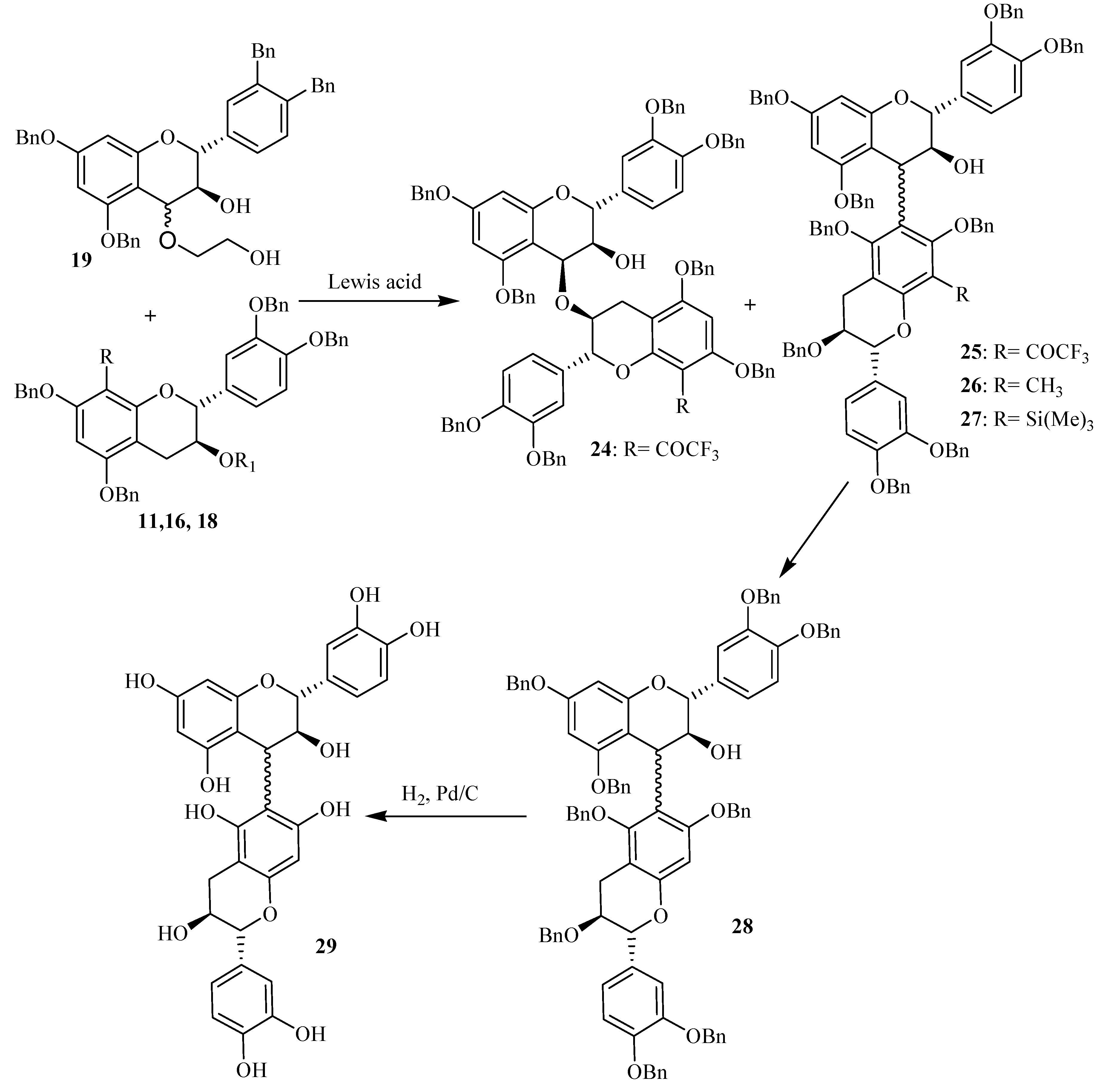
Lewis acid induced flavanol couplings
Lewis acids have been employed in literature to synthesize proanthocyanidins. Thus TiCl
4, AgBF
4, SnCl
4, TMSOTf have all been used to synthesize dimeric and oligomeric procyanidins of (+)-catechin and (-)-epicatechin units [
29,
30,
31,
32,
33,
34,
35,
36]. In these reactions, the role of Lewis acids is to promote the formation of the benzylic carbocation at C4 of a flavanol subunit starting from a C4 hetero substituted flavanol, which thereafter undergoes a Friedel-Craft-like addition on a second flavanol subunit. For this study, the Lewis acid TiCl
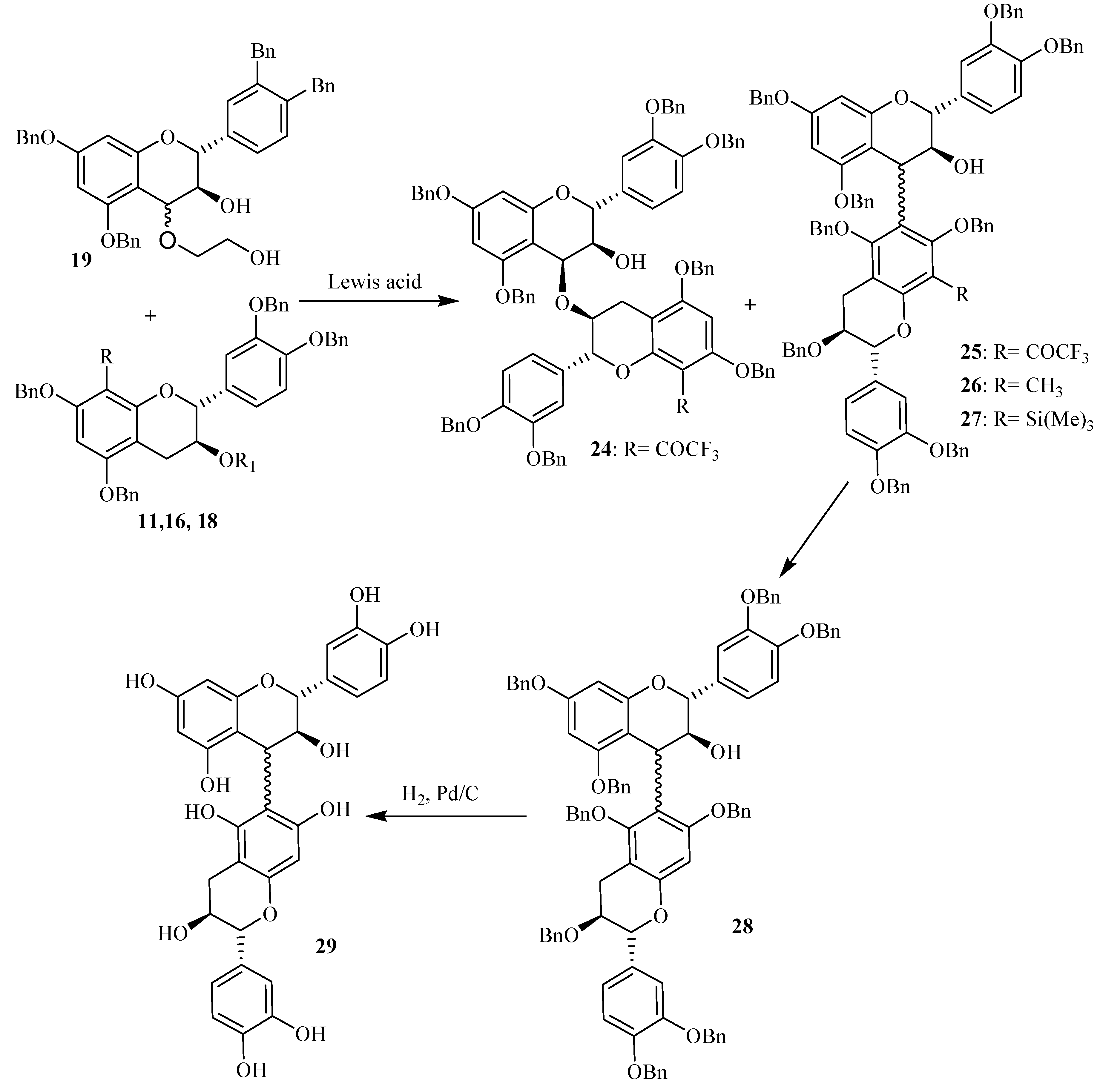
4 was used as a carbocation promoting agent from the 4-(2-hydroxyethyloxy) flavan-3-ol
19. Coupling reaction between compound
11 and
19 in a 6/1 molar ratio was investigated in CH
2Cl
2 according to
Scheme 4. The reaction was monitored by TLC and HPLC and showed the disappearance of compound
11 and appearance of new compounds. Among the products formed, compound
24 was obtained in sufficient amounts to allow its structure investigation. This was achieved through UV, LC/ESMS, ES CAD MS/MS and NMR analysis.
The UV spectrum of compound 24 exhibited similar maxima (285 and 305 nm) to that of compound 11, indicating that the original flavan structure with the COCF3 group was retained. The mass spectrum obtained in the positive ion mode showed signals at m/z 1395, 1412, 1417 and 1433 corresponding respectively to [M+H]+, [M+NH4]+, [M+Na]+ and [M+K]+ indicating a molecular weight of 1394 amu in agreement with a dimeric structure consisting of tetrabenzylated (+)-catechin 10 linked to its trifluoroacylated derivative 11. However, the remaining problem was the establishment of the position of linkage to compound 11, as the tetrabenzylated catechin moiety is linked through its 4 position.
Scheme 4.
TiCl4-catalyzed flavanols coupling reactions.
Scheme 4.
TiCl4-catalyzed flavanols coupling reactions.
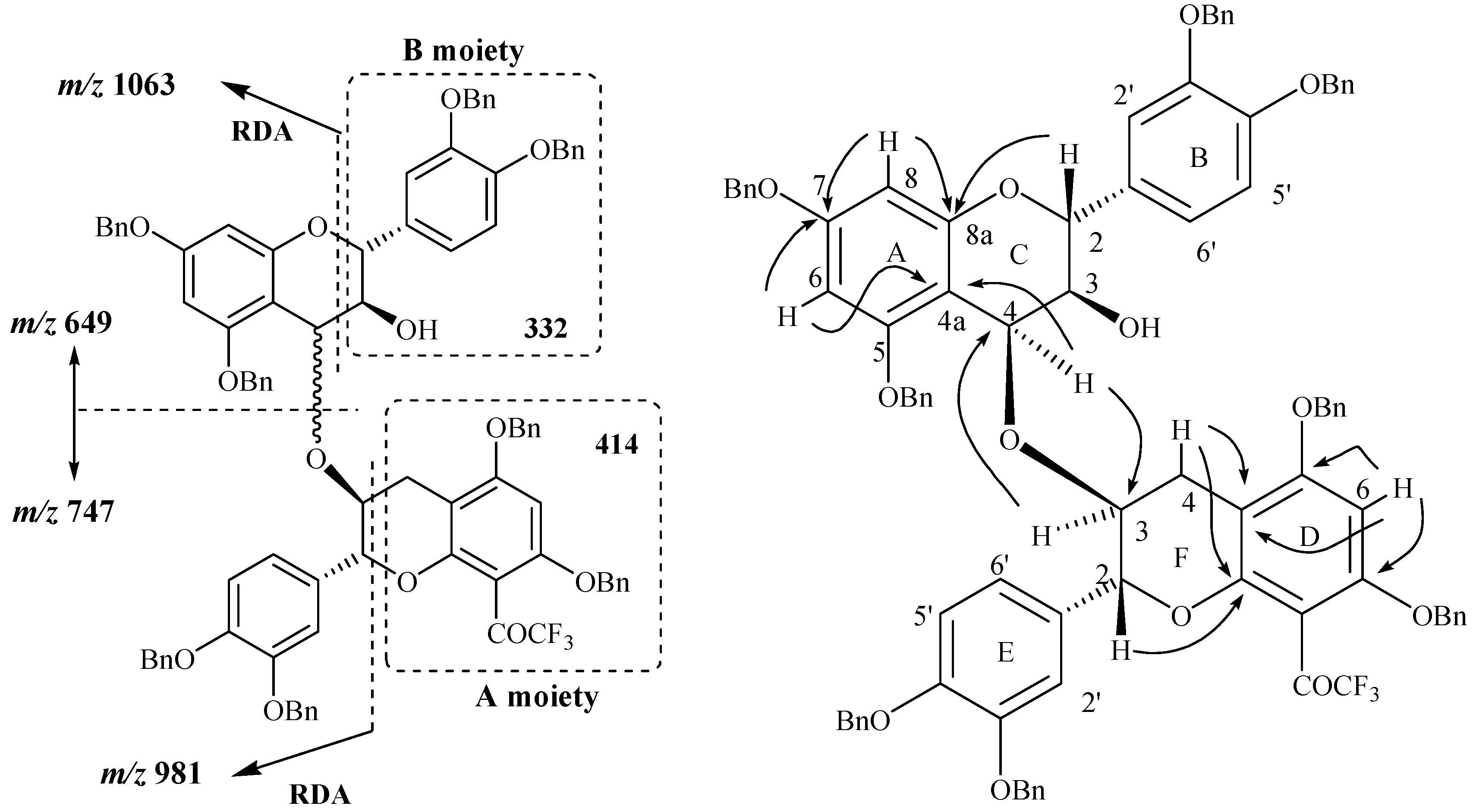
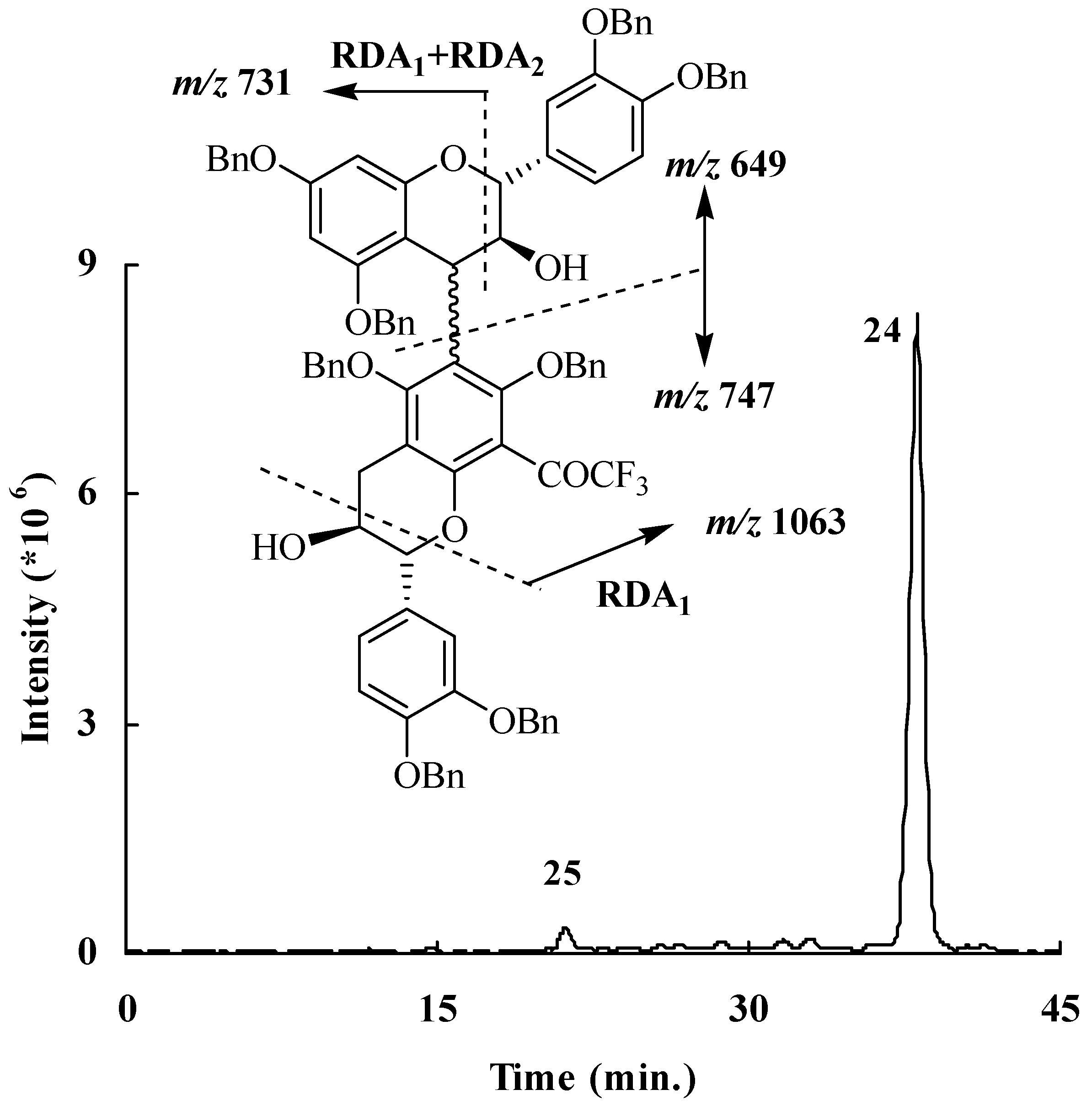
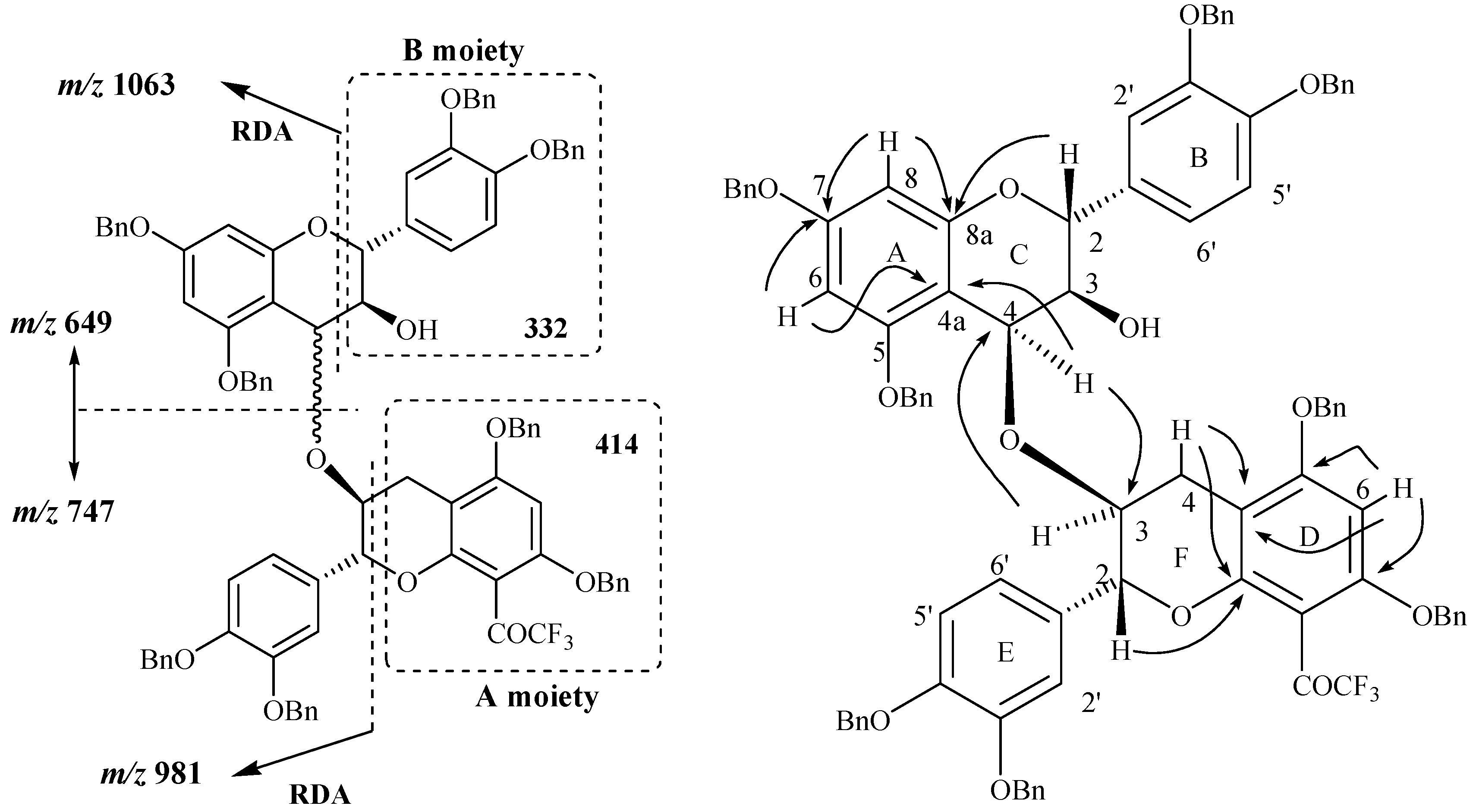
In addition to the signals indicated above, the mass spectrum of compound
24 also showed signals at
m/z 747 and 649 corresponding to the fission of the bond between the two constitutive units. Among the other observed signals two of them were located at
m/z 1063 and 981 and were also observed in the spectrum obtained through positive ES CAD MS/MS fragmentation of the signal located at
m/z 1395 ([M+H]
+ ion). The signal observed at
m/z 1063 was attributed to the characteristic RDA fragmentation corresponding to the [M+H-332]
+ ion as what was observed for compound
11 through a loss of the B moiety. The second fragmentation observed at
m/z 981 correspond in fact to the [M+H-414]
+ ion, meaning a loss of the A moiety of compound
11 unit (
Figure 5) and corresponding to another RDA fragmentation. The occurrence of this fission indicated the presence of the A moiety in the structure of compound
24. In other words, this means that the isolated compound is not a C4→C6 dimer since only one RDA fragmentation corresponding to the [M+H-332]
+ ion could be possible in this case. The possible linkage is thus expected to occur
via the 2 or 3 position of the F ring or possibly the 2’, 5’ or 6’ positions of the ring E.
Figure 5.
Main fragmentations observed and main 1H-13C long range correlations observed in compound 24.
Figure 5.
Main fragmentations observed and main 1H-13C long range correlations observed in compound 24.
Through NMR analysis and in conjunction with the absence of a doubly benzylic methylene proton characteristic of a C4→C6 linkage and taking into account the dimeric structure of the compound as supported by MS analysis, the NMR data collectively indicated a dimeric structure with an interflavanyl ether bond connecting the two heterocyclic C and F rings. Taking into account the fact that the linkage did not involve the H2, H3, H4 F ring protons since they were all evidenced through NMR analysis, a (4-
O-3) mode of linkage was thus concluded to occur between the two flavan-3-ols units. This was also confirmed by comparison of the chemical shifts of the H4 and H3 resonances of both the C and the F rings with those of their precursors. Finally the structure of compound was univocally confirmed through HMBC analysis where several long range correlations were observed. In particular correlations involving proton and carbon of both C and F rings via the oxygen atom were observed and confirmed thus the ether linkage involved in compound
24. Full assignment of the protons and carbon chemical shifts was achieved through HMBC analysis (
Figure 5) where the main correlations involving H/C of the C and F rings were showed in agreement with the proposed structure.
It was concluded that a (4-
O-3) linkage was occurred between the two flavan-3-ol units. Moreover, coupling constants for the AMX spin system of the C-ring protons (
J3,4 = 3.2 Hz) indicated a 3,4-
cis relative configuration for this ring, which was determined through homonuclear decoupling experiment. The complete stereoselectivity of the reaction remains, however, to be explained and should presumably be due to a participation of the hydroxy group at C3 of
11. However, its involvement in the stereochemical course of the reaction cannot be, in our case, related to the formation of a protonated epoxide similar to that reported by Bennie
et al. [
37] in a work dealing with the dimerization of epioritin-4-ol derivatives.
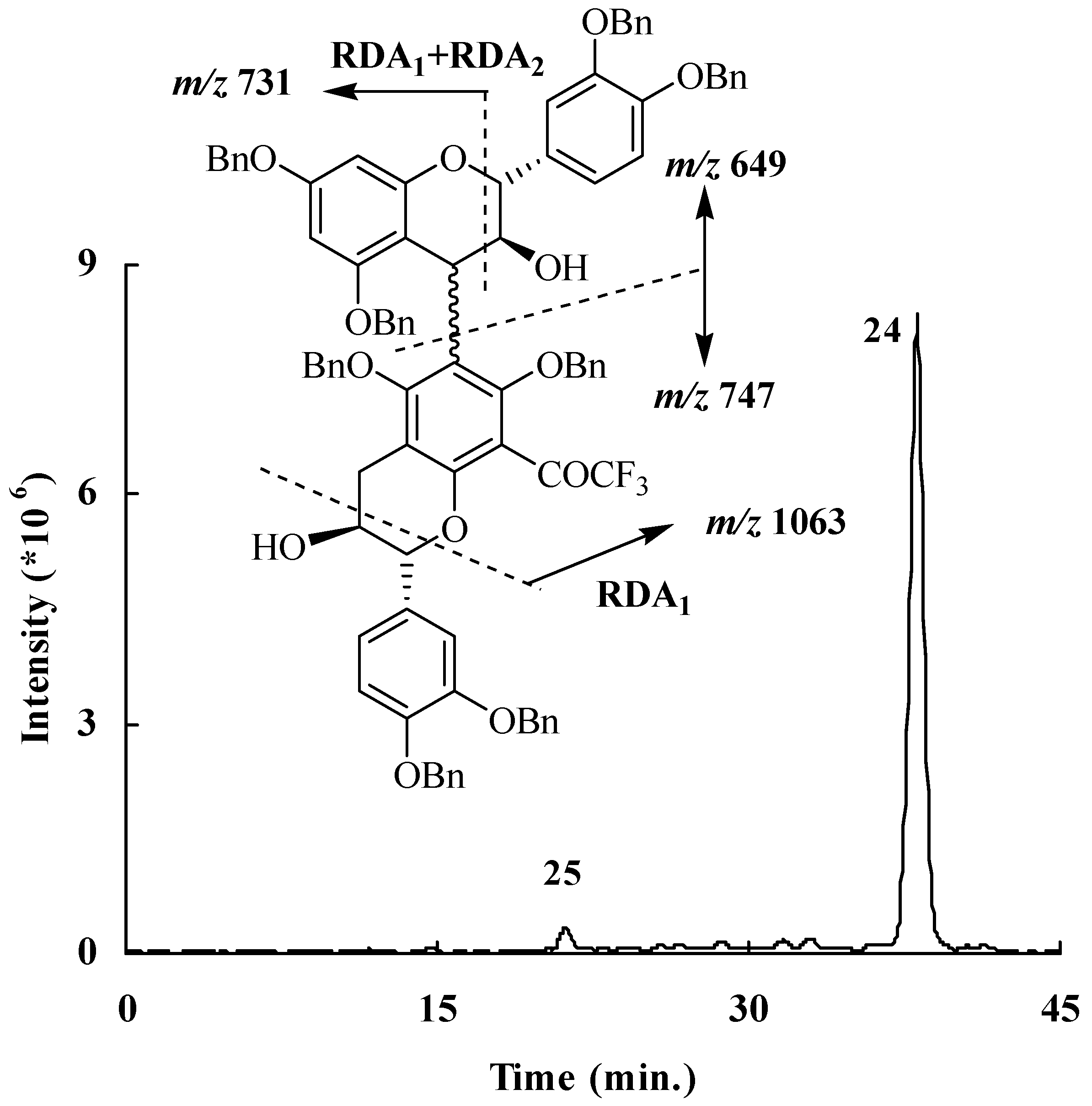
Figure 6.
Extracted ion chromatogram recorded at m/z 1395 and main fragmentations observed in compound 25.
Figure 6.
Extracted ion chromatogram recorded at m/z 1395 and main fragmentations observed in compound 25.
In order to verify the presence of other dimeric structures the mixture was explored by HPLC coupled to a mass spectrometry detection operating in the positive ion mode. An extracted ion current chromatogram recorded at
m/z 1395 and 1412 (
Figure 6) and corresponding to a dimeric structure molecular weight showed the presence, in addition to compound
24, of a minor compound, which is possibly the carbon-carbon coupled dimer
25. The fragmentations observed for compound
25 were in agreement with the proposed dimeric structure consisting of tetrabenzylated (+)-catechin
10 coupled to its trifluroacylated derivative
11 through a C4→C6 linkage (
Figure 6).
The almost exclusive, high yielding formation under these conditions of the novel ether-linked procyanidins as main compound rather than its carbon-carbon C4→C6 coupled analogue reflects the importance of electronic features in the formation of flavan-3-ol dimers. The poor nucleophilicity of the A ring monomeric precursor, caused by the presence of the COCF3 group, permits alternative centers to participate in the interflavanyl bond formation.
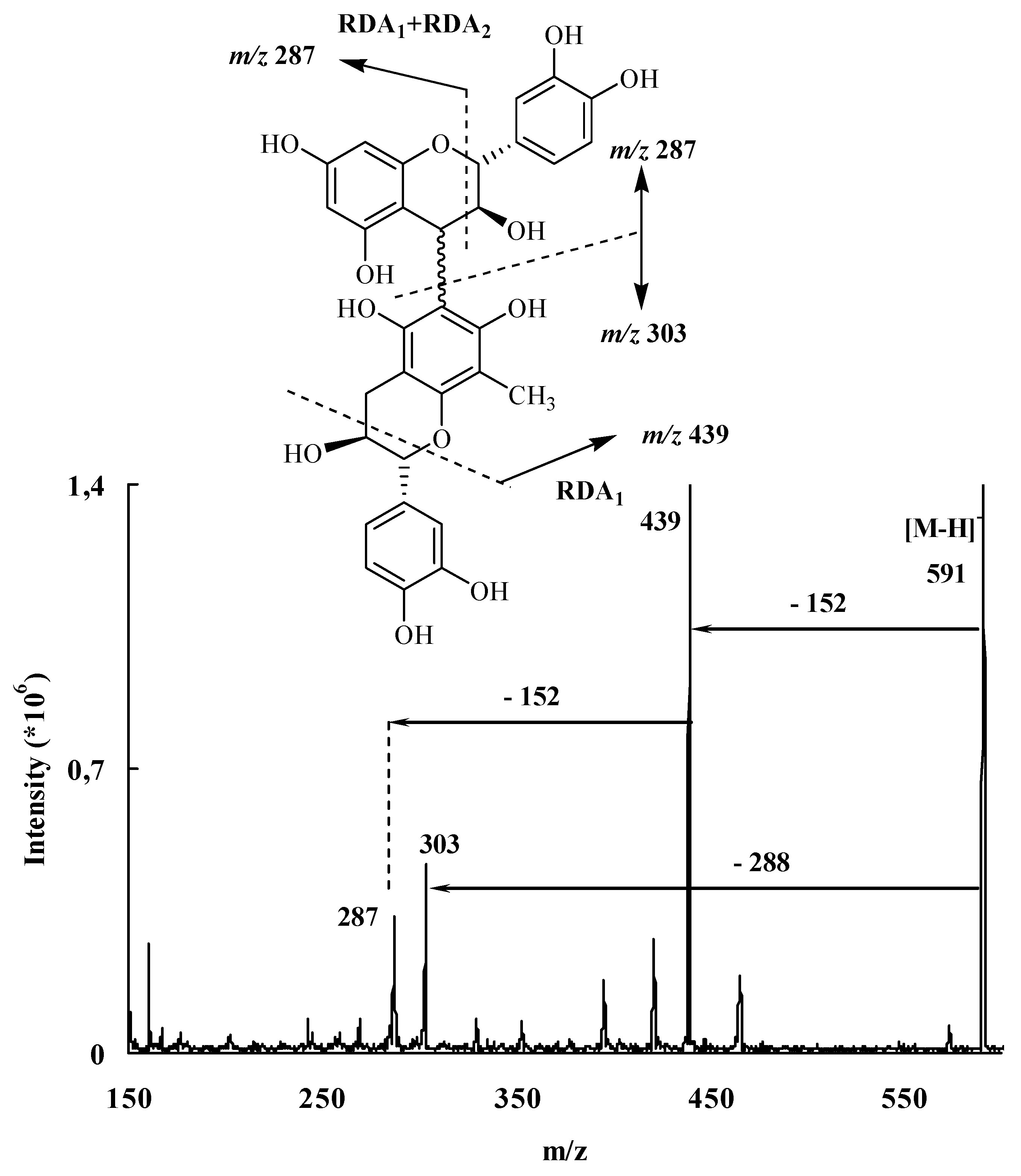
The obtained results discussed above showed that the interflavanic carbon-carbon linkage formation was largely inhibited during Lewis acid induced flavanols coupling reactions by the presence of an electron-withdrawing group, while this was not the case with an electron-donor group like the methyl substituent. This could constitute thus a setereoselective method for the formation of C4→C6 procyanidin dimers which are usually obtained as minor compounds the C4→C8 ones. In order to apply this technique to the stereoselective preparation of natural dimeric procyanidin of the C4→C6 type, we unsuccefully tried to remove the methyl group from the modified derivatives 26. Then we used the trimethylsilyl group which is easily removable by hydrolysis. Indeed, during purification of compound 18 on silicagel chromatography column, we saw that this compound was easily transformed to 10 by loss of the 8 substituent group. This prompted us to use this compound as nucleophile acceptor unit in the Lewis acid coupling reaction. The reaction was monitored through TLC and LC-MS, showing the presence of compound with a molecular mass of 1460 amu corresponding to the dimeric compound 27. After hydrolysis and hydrogenolysis, the corresponding product 29 was separated and analyzed through mass spectroscopy. The obtained results indicated a molecular mass of 578 amu in agreement with the dimeric structure of compounds 29. Indeed, the mass spectrum of compound 29 showed procyanidin characteristic RDA fragmentation giving an ion fragment by a loss of 152 mass units. We tried to proof the interfalvanic linkage of the obtained compound through 2D NMR technique, but due to degradation of the obtained compound, our attempt was unsuccessful.
Trametes versicolor laccase induced oxidation of flavonoids
Although a number of papers in the literature have already discussed the enzymatic oxidation of flavonols, most of them only reported the kinetic features of the reaction measured through the changes of UV absorption. Few have tried to look at the precise structure of the oxidized products and the mechanistic pathway(s) of their formation. Moreover, the main reports have focused only on the enzymatic oxidation of quercetin and quercetin glycosides. In order to allow chemical mechanism determination for this type of reaction, we decided to test our enzymatic system on several commercially available flavonols regarding their substitution either on B or C rings. Indeed, most of the already published studies on flavonol have underlined the predominance of B ring oxidation, as laccases are essentially reported to have a phenol or catechol oxidase activity giving rise in most cases either to oxidized monomeric species or oligomeric compounds obtained through radical-radical couplings [
40].
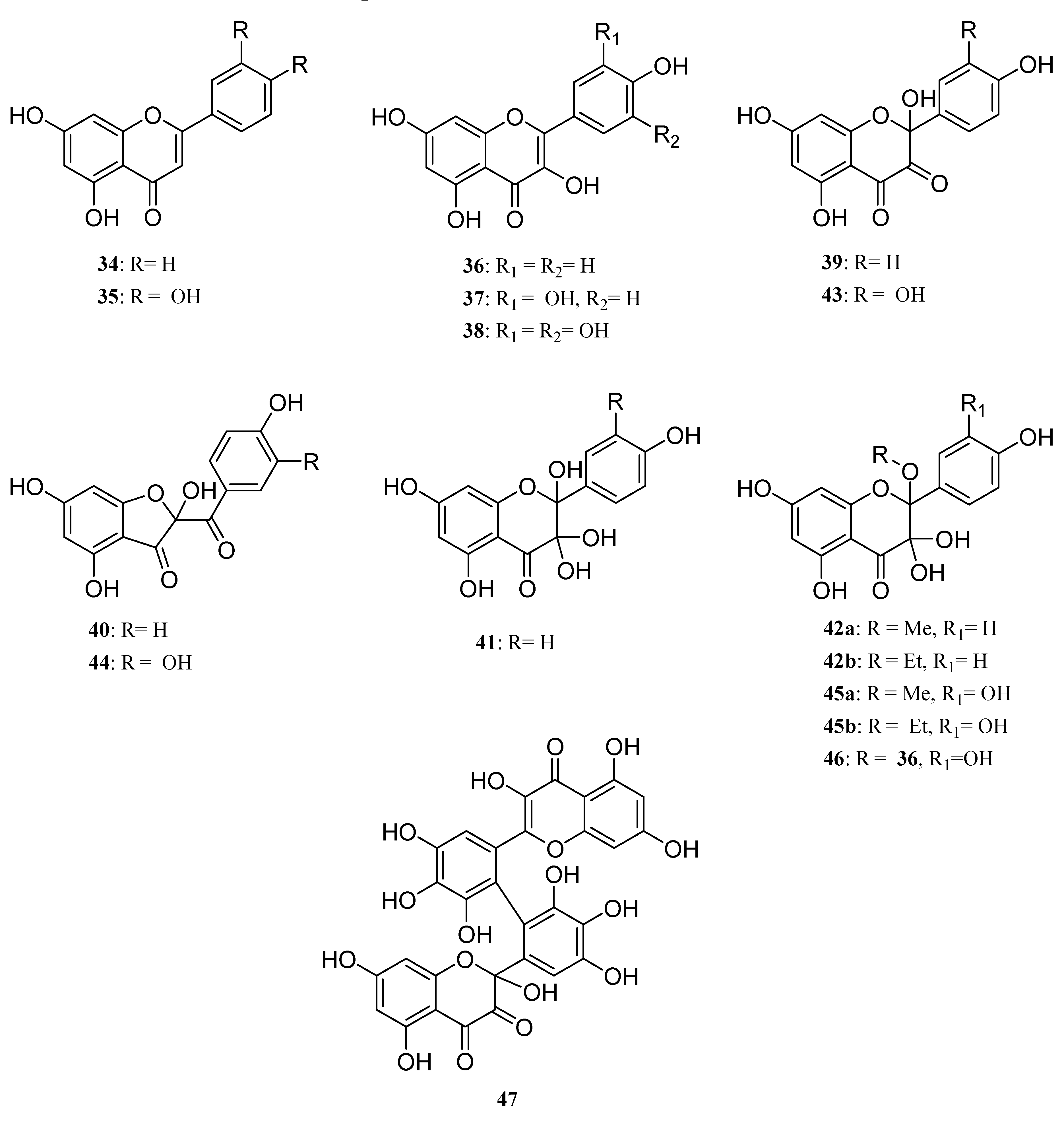
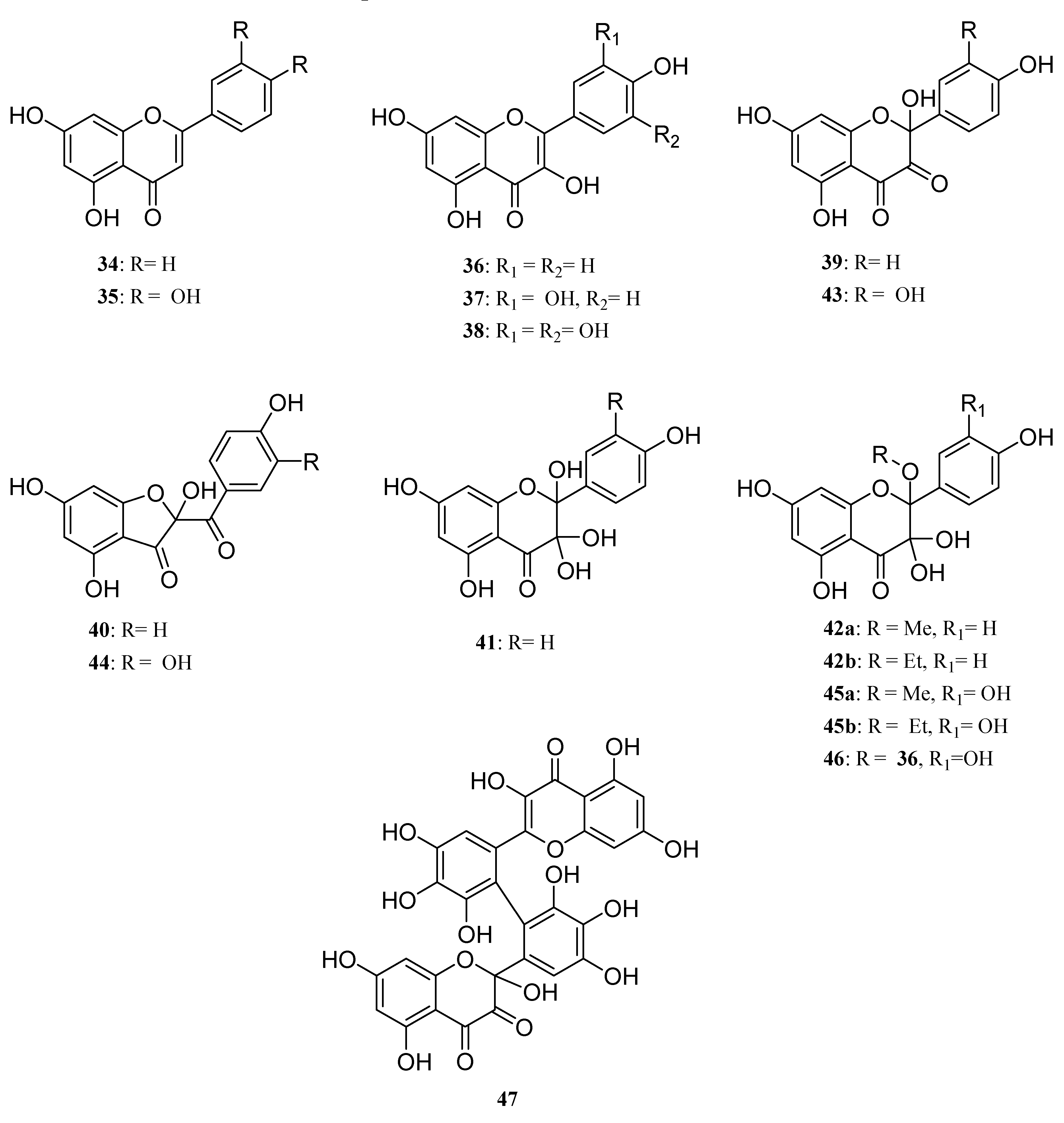
The choice of the starting materials (
Figure 8) was made on account of several results published in the literature on the structure-antioxidant activity relationships of flavonoids. It has indeed been clearly established that the B-ring is the most important site for H-transfer and consequently antioxidant capacity. In contrast, the A-ring seems to be less important. On the other hand, the 2-3 double bond should also contribute to the antioxidant activity, as it ensures π-electron delocalization between the B- and C-rings, which contributes to the stabilization of the radicals RO
• formed in the oxidation process after H atom abstract [
41]. Moreover, an important issue that is still under debate is the role of the 3-OH group.
In vitro studies demonstrated that this hydroxy group contributes to the antioxidant potential. Indeed, blocking the 3-OH group as in rutin or removing it as in luteolin significantly decreases the activity [
42]. The influence of the 3-OH group has also been proven during the metabolization of quercetin [
43], since quercetin is believed to coordinate to the copper containing active site of the 2,3-dioxygenase issued from human intestinal bacteria through the 3-OH and the carbonyl group at C4, allowing H abstraction and oxidation at C2 [
44].
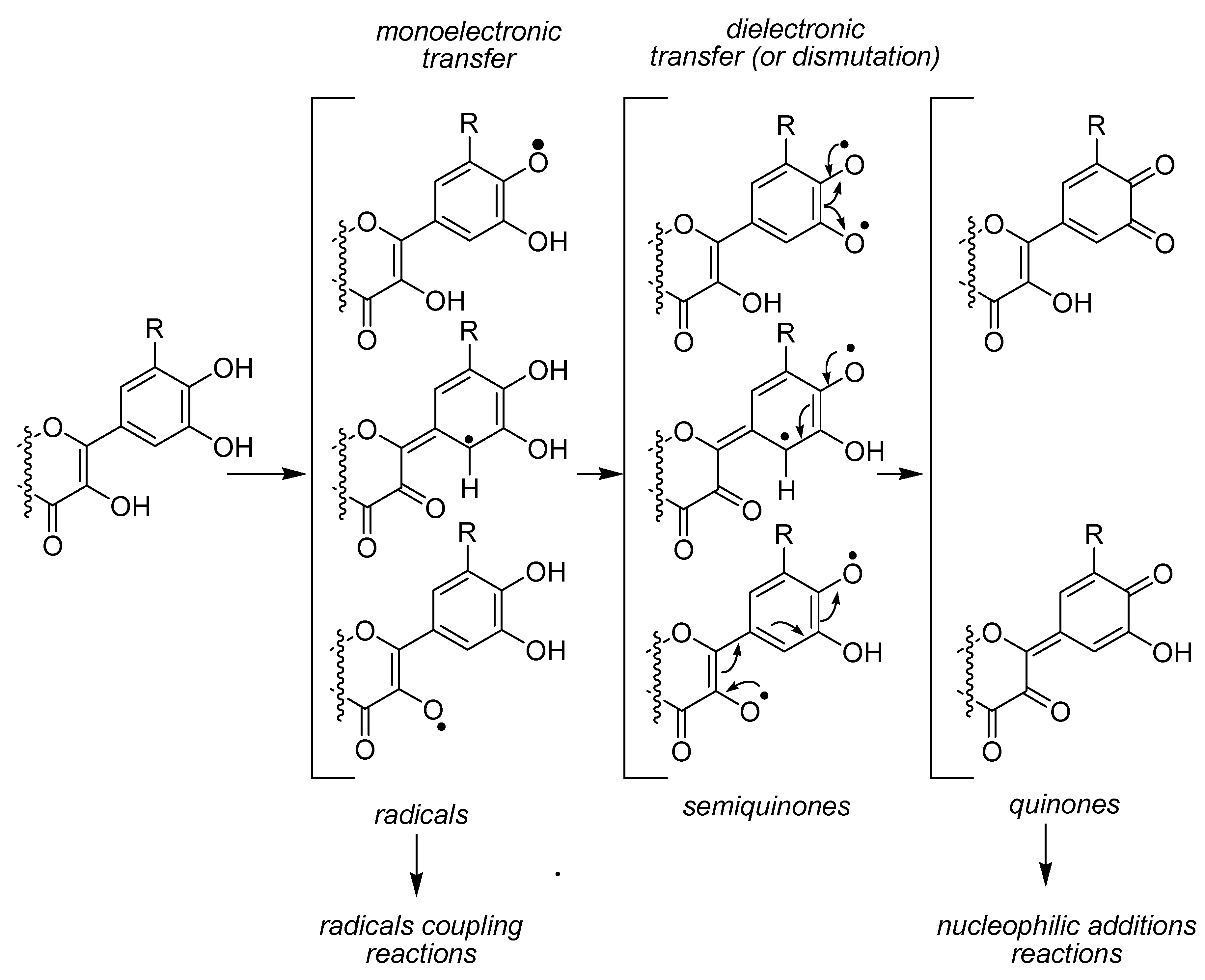
An important feature of our study compared to those already published is the use of a laccase as enzymatic oxidant. Indeed, other studies most often reported the oxidation of flavonoids either with chemical radical initiators like DPPH or enzymatic system like PPO (polyphenol oxidase; in this case, the flavonoids are not the primary substrate of the enzyme, the oxidation being mediated by the
oquinone from caffeoyl quinic acid). Therefore, in these two cases, oxidation of the flavonols may occur through either a mono- or a di-electronic transfer (
Figure 9).
The possibility of a di-electronic transfer may indeed favor the oxidation of diphenol through formation of semiquinones and quinones, and the thermodynamic of the reaction would thereby be directed by the stability of the potentially formed products rather than by the stability of the radicals formed through H-abstraction. In the case of a laccase, albeit the complete mechanisms of the reaction, and mainly the pathway leading to oxygen reduction is not clearly determined [
45], the monoelectronic transfer is the more privileged mechanism, therefore allowing to investigate the stability and the reactivity of the various RO
• potentially formed as primary product of the oxidation since quinones may only be the result of radical dismutation, kinetic of which would probably not be competitive with this of a radical coupling reaction. Indeed, upon the four copper atoms contained in the structure of the laccase, only one is involved in the oxidation of the substrates, the three other atoms being responsible for the oxygen reduction and oxidation of the Cu(I) atom of the active site of the enzyme.
Figure 8.
Structures of the flavonoids studied in the enzymatic oxidation. The structures of the obtained oxidized compounds are also shown.
Figure 8.
Structures of the flavonoids studied in the enzymatic oxidation. The structures of the obtained oxidized compounds are also shown.
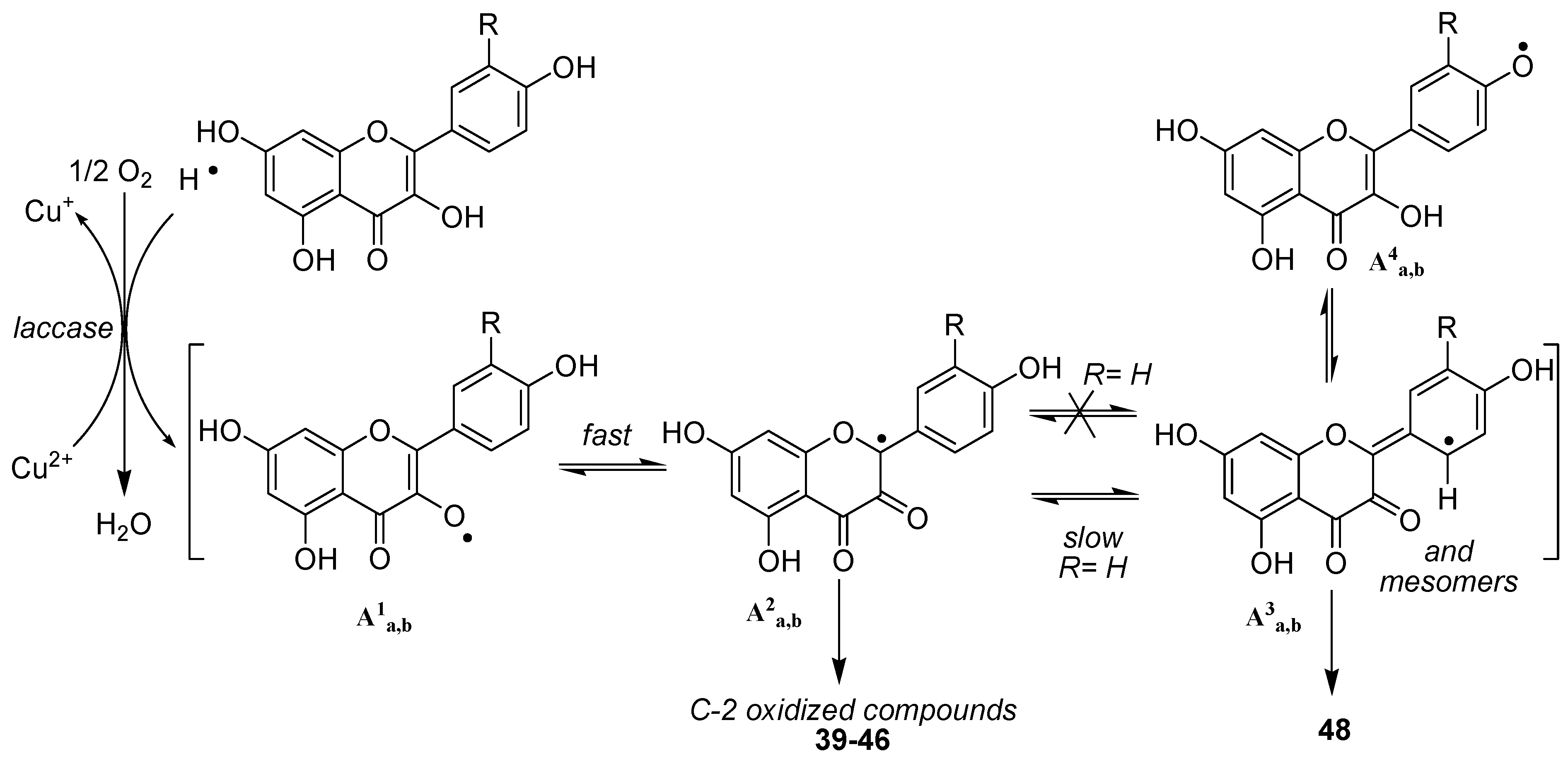
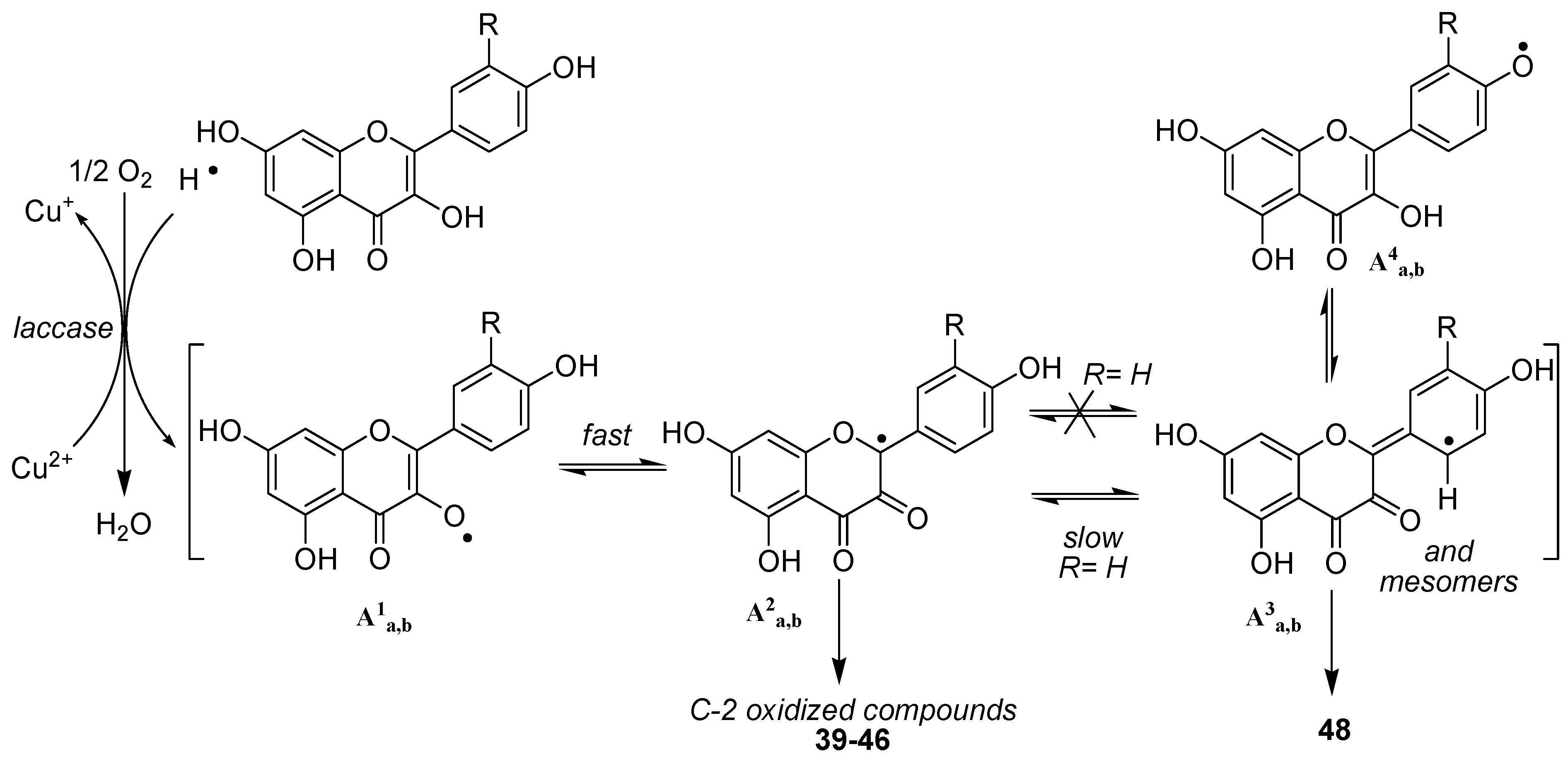
The first feature arising from our study was the non-oxidation of both
34 and
35, thereby confirming the crucial role of the 3-OH group in the oxidation process. Moreover, this first result also showed that the presence of an highly reactive catechol type B-ring is not sufficient to allow a one-electron oxidative process to occur, also reflecting the postulated important role (DFT study) [
46,
47] played by the hydroxy group at C3 in the first steps of oxidation of flavonols.
Turning thereafter to the enzymatic oxidation of flavonols 36 and 37, we were not surprised to observe that these two compounds, although not exhibiting the same hydroxylation pattern on the B-ring, presented similar behaviours in our oxidation conditions. Indeed, the reaction mixture was reflecting in both cases the presence of only few compounds, MS analysis of which showed that the major ones were isomeric products corresponding to the addition of one oxygen atom (M+16).
Figure 9.
General oxidation pathways for flavonols.
Figure 9.
General oxidation pathways for flavonols.
Depending on the solvent used for the reaction, the other products of the reactions were thereafter shown to be either dimeric compounds in aprotic conditions (acetonitrile) or products resulting from solvent addition in protic media (MeOH or EtOH). In this latter case, turning from methanol to ethanol was the key step allowing to unambiguously underline this solvent addition. Later, NMR analysis confirmed the structure of compounds
39-
42 and
42-
45 as oxidation products of kaempferol and quercetin respectively, structure of which have already been more or less described in the literature [
48,
49].
As an example, NMR spectra of
45a were used to determine its structure as follows. The
1H-NMR spectrum exhibited the same resonance pattern for the aromatic protons than quercetin but also showed a supplementary resonance at 3.1 ppm, integrating for 3 protons in accordance with the presence of a methyl group attached to an oxygen atom. The
13C-NMR spectrum was also quite similar to quercetin's, with two quaternary sp
2 carbon resonances less, which were replaced by two resonances at 91.7 and 100.9 ppm. HSQC and HMBC experiments were thereafter performed and allowed the complete assignment of all carbon resonances. Indeed, HMBC correlation were observed for the resonance at 100.9 ppm with protons of the methyl group as well as with protons H2', H5' and H6'. In the same time no HMBC correlations (
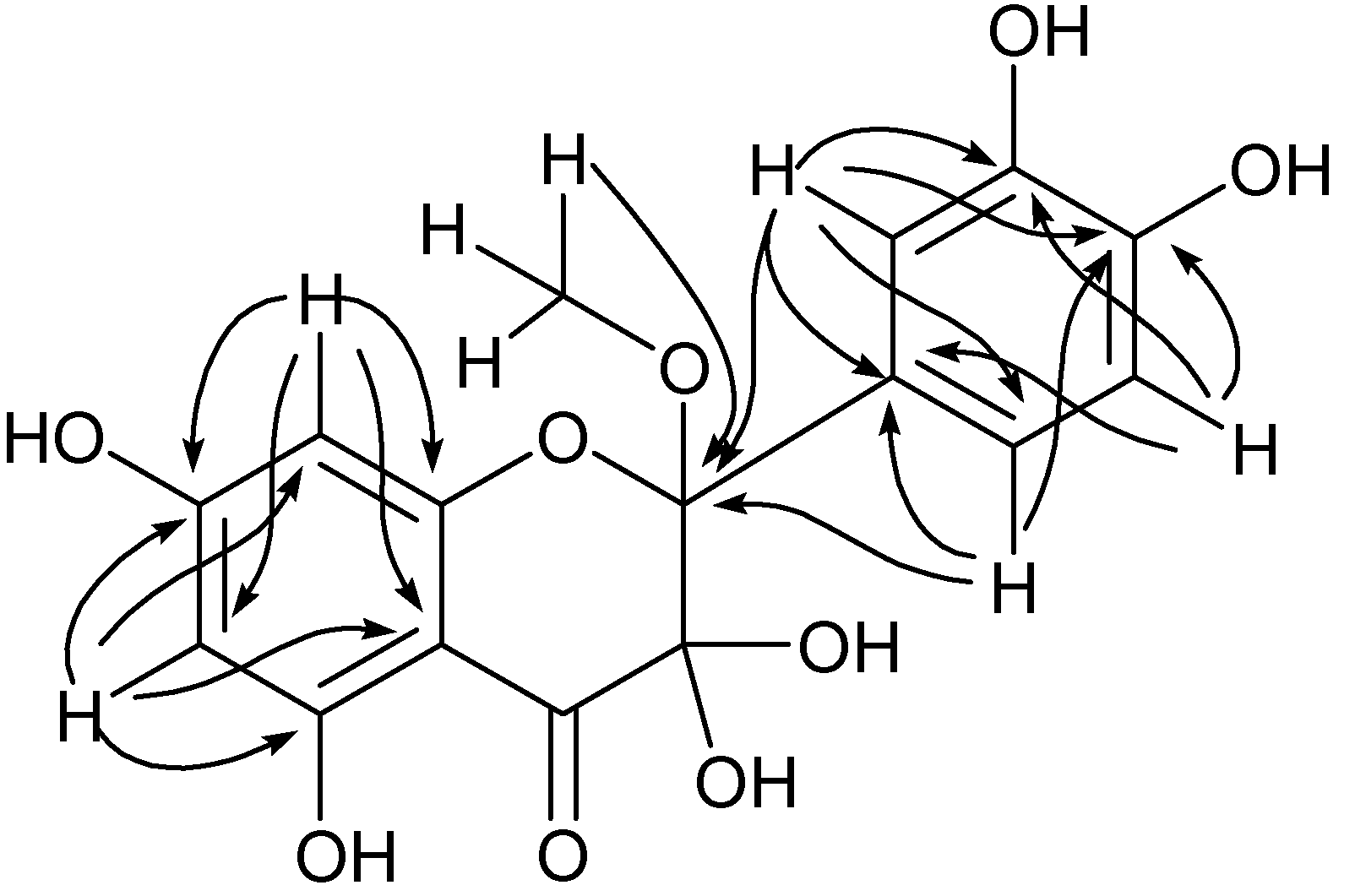
Figure 10) were observed for carbon resonance at 91.7, which thereby was attributed to C3 when the resonance at 100.9 ppm was assigned to C2, confirming the introduction of a methoxy group at C2 through trapping of the C2 radical. These results confirmed the possibility of enzymatic oxidation at C2 through activation of the 3-OH group in a monoelectronic transfer process.
Figure 10.
HMBC correlations observed for compound 45a.
Figure 10.
HMBC correlations observed for compound 45a.
The structure of the dimeric compounds obtained through oxidation of these flavonols in acetonitrile was however more difficult to establish because of their low abundance in the reaction mixture which did not allow us to get them in sufficient amount for NMR spectroscopy. However, kaempferol oxidation led to the formation of only one dimeric compound (
m/z 604) when quercetin allowed formation of two pseudo dimeric compounds at
m/z 498 and 618. Carefull examination of the MS spectra of compounds at
m/z 604 (
46) and 618 (
48), obtained from
36 and
37 respectively however reflected a behavioural discrepancy between these two flavonols. Indeed, when the formation of
48 is compatible with the formation of either a carbon-carbon or a carbon-oxygen single bond between the two monomeric subunits, formation of
46 is clearly not compatible with the formation of a carbon-carbon bond between two oxidized subunits but rather to the formation of a carbon-oxygen bond through intermolecular trapping of the C-2 radical formed in the first step of the oxidation by an hydroxy group of another flavanol molecule in a similar way as this observed for the formation of oxidized monomers
42a,b (
Scheme 6).
This assumption is also supported by the observation in the MS spectrum of 46 of a major fragment at m/z 302 also observed in compounds 42a,b, as a result of the easy cleavage of the OR bond. Unfortunately, we did not get sufficient data to determine which of the oxygen atom of kaempferol could be involved in this interflavanyl link. On the other hand, MS spectrum of 48 did not reveal similar fragmentations (no corresponding fragment at m/z 318) but rather those compatible with the classical fragmentations of the flavan skeleton. This may indicate a carbon-carbon single bond between both quercetin subunits, although we were unable to get sufficient data for a complete structure assignment. Compound at m/z 498, structure of which has also not been established, since it remains a very minor product of the reaction, should arise from a degradation of pseudo dimer 48.
Scheme 6.
Postulated oxidation mechanism for compounds 36 and 37.
Scheme 6.
Postulated oxidation mechanism for compounds 36 and 37.
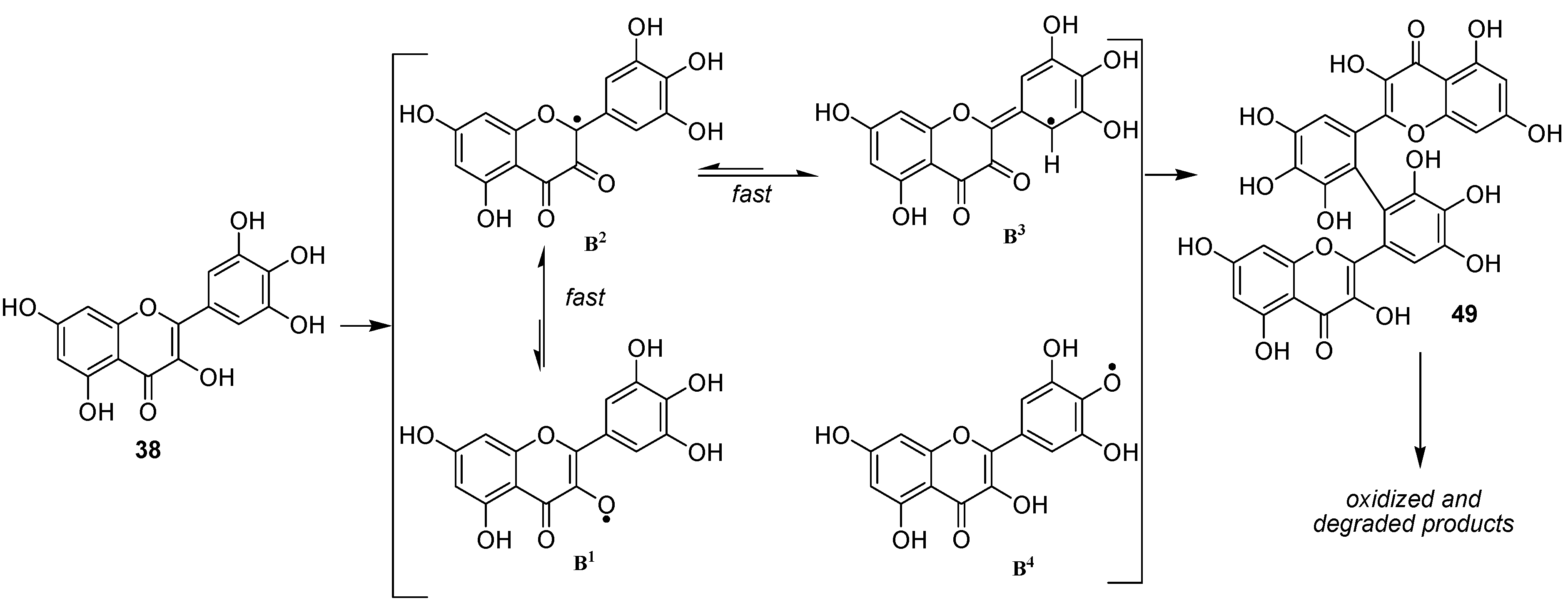
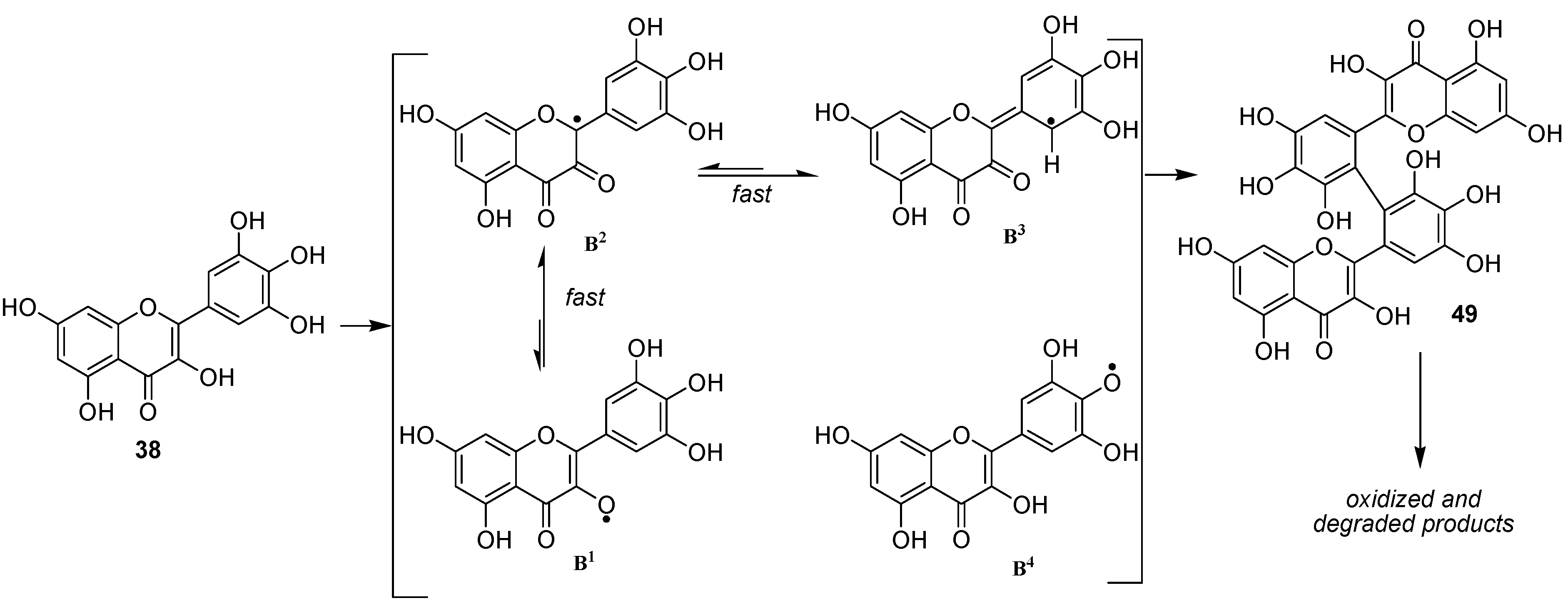
We thereafter turned to the investigation of the possible oxidation of myricetin (
38) by our enzymatic system. Albeit being already assumed as substrate of oxidase enzymes [
50], oxidation products of myricetin have, to our knowledge never been investigated. As for
36 and
37, three solvents (methanol, ethanol and acetonitrile) were used in order to see the influence of the solvent in this reaction and to be potentially able to trap the C2 radical in protic media. However in this case, we unexpectedly observed no formation of C2 oxidized monomers. Indeed, although not exhibiting exactly the same chromatographic profile, the reaction mixtures in these three solvents essentially showed the presence of dimers and oxidized dimers at
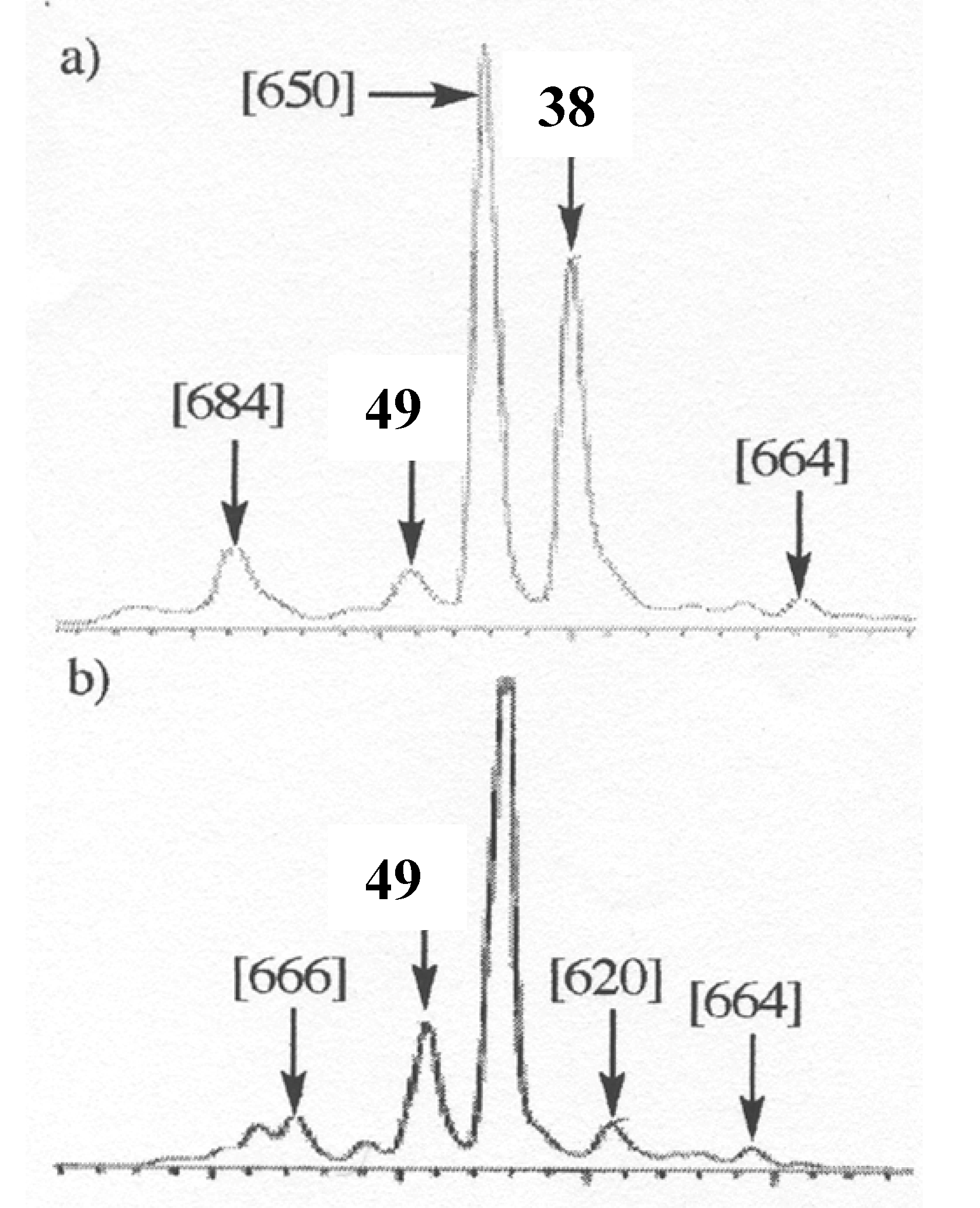
m/z 634 (2M-2H), 648, 650, 664, 666 and 684. Beside these compounds, oxidation conducted in protic media also exhibited some degraded dimers at
m/z 484 and 620. Focusing our interest on the "pure" dimer at
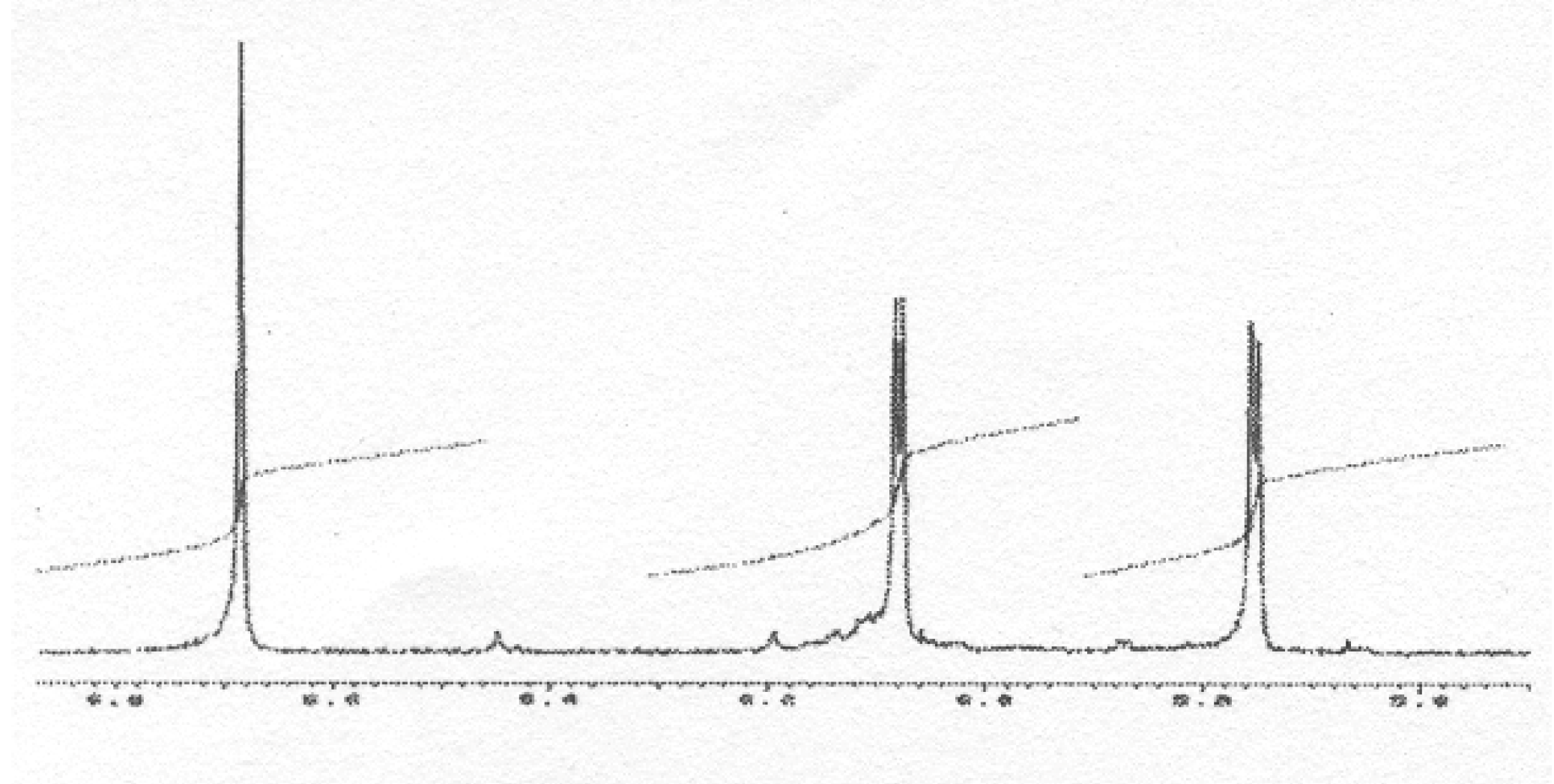
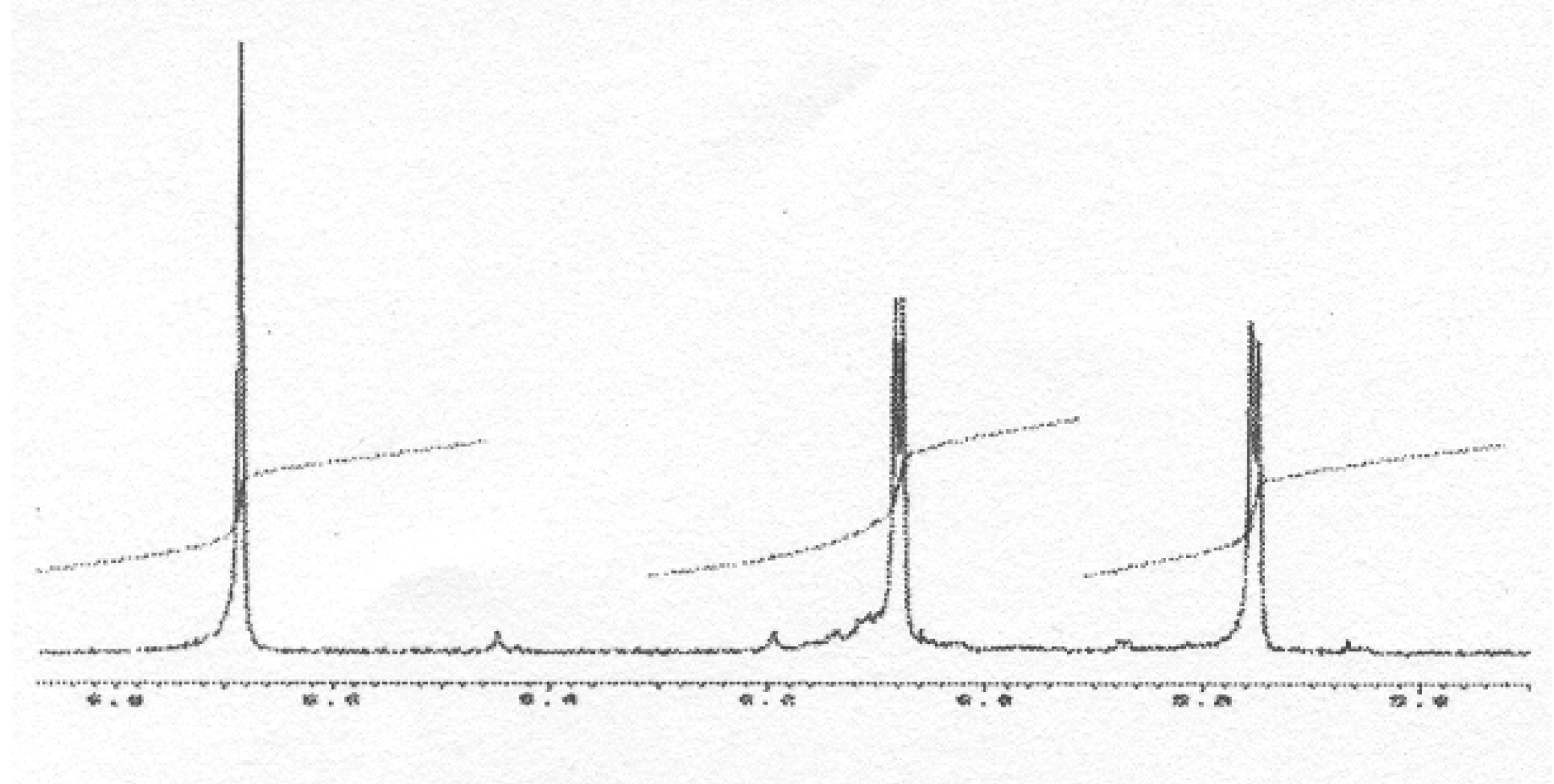
m/z 634, we were able to isolate it as a pure compound and its very simple
1H NMR spectrum, due to symmetry, (
Figure 11) allowed us to assign structure
49 to this compound, thus resulting from the coupling of two
B3 radicals (
Scheme 7).
Figure 11.
1H-NMR spectrum of 49 (exchangeable protons are not shown).
Figure 11.
1H-NMR spectrum of 49 (exchangeable protons are not shown).
Scheme 7.
Postulated oxidation mechanism for compound 38.
Scheme 7.
Postulated oxidation mechanism for compound 38.
The most important feature arising from this reaction is the fact that no other type of linkages than this occurring in
49 was observed in all the other dimeric oxidation products. Indeed, when performing the enzymatic oxidation with
49 as starting material, the chromatographic profile of the reaction (
Figure 12a), although not being completely similar from a quantitative point of view to that observed in the oxidation of
38 (
Figure 12b), qualitatively showed the presence of the same oxidized dimers.
Moreover, the evolution of the chromatographic profile of the enzymatic oxidation of 38 clearly showed that 49 is first accumulated in the reaction mixture and then transformed into the other compounds and mainly into the compound exhibiting a molecular weight of 650 amu corresponding to a further oxidation with incorporation of an oxygen atom. According to the structure of the oxidation products of quercetin, structure 47 could be putatively assigned to this compound.
Figure 12.
Chromatographic profiles of the oxidation reaction mixture at 3 hours of 38 (a) and 49 (b).
Figure 12.
Chromatographic profiles of the oxidation reaction mixture at 3 hours of 38 (a) and 49 (b).
From a mechanistic point of view, our results showed that the oxidation of flavonols occurs first at C2 through abstraction of the enolic proton at 3-OH and that, thereafter, the equilibrium between the possible isomeric radicals is directed by the substitution pattern of ring B. In the case of 36 and 37, radicals A3 and A4 are not sufficiently stabilized to allow the significative formation of dimeric compounds linked through a carbon-carbon biphenyl bond (A3-A3 radical coupling). On the other hand, in the case of myricetin 38, although the first formation of B4 radical could not be excluded, the most probable pathway remains the first formation of the B1 radical, which rapidly isomerizes into the B3 one, which is stabilized by the presence of the three phenolic groups, leading to the formation of 49.
However, the presence of compounds resulting of further oxidation of this first formed dimer as well as the no observation of higher order oligomers, indicates that in dimeric compounds, oxidation thereafter occurs mainly at C-2. Moreover, the observation of compounds at m/z 664, 666 and 684, clearly demonstrates that compound 49 may undergo at least three further consecutive oxidations, thereby underlining the high antioxidant capacity of 38.
Antioxidative activity of the synthesized compounds
Free radicals are known to be a major factor in biological damages, and DPPH
• has been used to evaluate the free radical-scavenging activity of natural antioxidants [
51]. DPPH
•, which is a molecule containing a stable free radical with a purple color, changes into a stable compound with a yellow color by reacting with an antioxidant which can donate an electron to DPPH
•. In such case, the purple color typical of the free DPPH
• radical decays, a change which can be followed either spectrophotometrically (517 nm) or by detecting changes in concentration of starting materials and/or end reaction products, using HPLC analysis [
52].
The fact that the extent of the reaction depends on the hydrogen donating ability of the antioxidant [
53] make of it an indication of the capacity of the tested products to scavenge free radicals. This simple test can provide information on the ability of a compound to donate an electron, the number of electrons a given molecule can donate and on the mechanism of antioxidant action. In cases where the structure of the electron donor is not known (e.g. a plant extract), this method can afford data on the reduction potential of unknown materials. The DPPH
• test is a very convenient method for screening small antioxidant molecules because the reaction can be observed visually using common TLC and also its intensity can be analysed by simple spectrophotometric assays [
54]. The DPPH
• radical is scavenged by antioxidants through the donation of hydrogen to form the stable reduced DPPH molecule. The antioxidant radicals formed are stabilized through the formation of non-radical products. This method has been also a useful and widely used to evaluate the free radical-scavenging effectiveness of various antioxidant substances in food systems [
55]. In order to investigate the antioxidant activities of the synthesized modified (+)-catechin derivatives the DPPH
• method was used and the results obtained are depicted in
Figure 13.
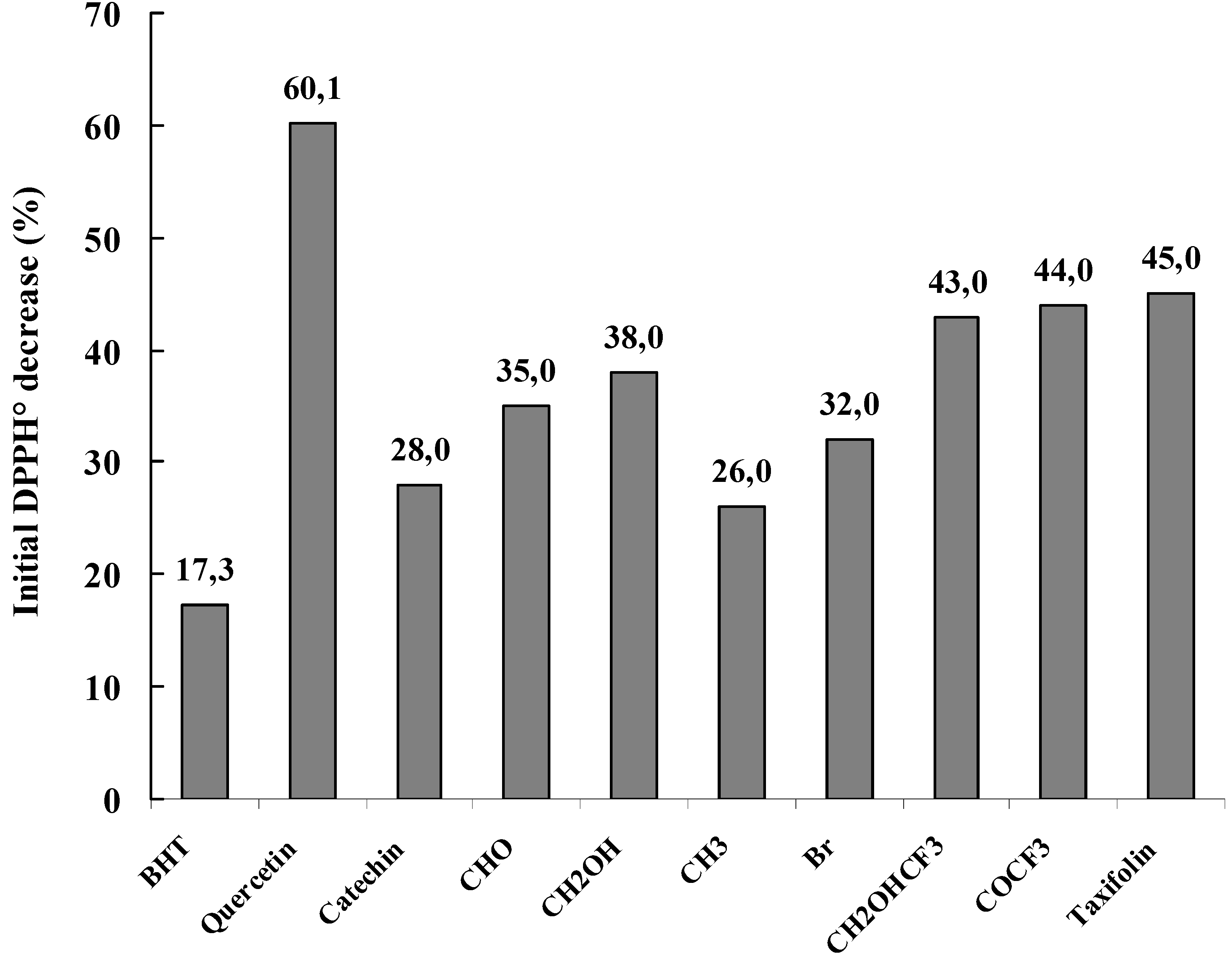
Figure 13.
Free radical scavenging activity of the studied compounds. The results represent the decrease (%) of the initial DPPH• absorption at 517 nm.
Figure 13.
Free radical scavenging activity of the studied compounds. The results represent the decrease (%) of the initial DPPH• absorption at 517 nm.
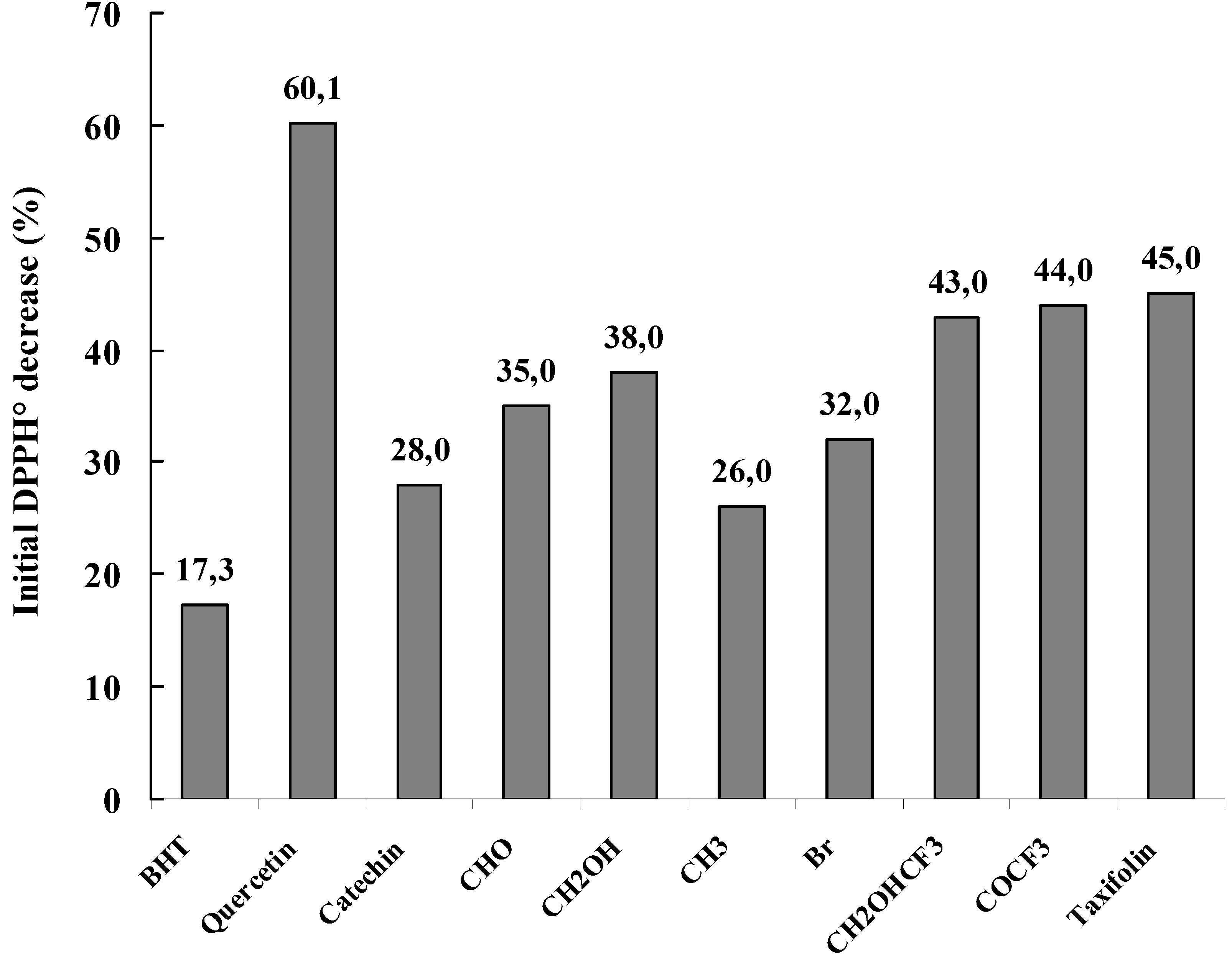
Figure 13 shows the DPPH
• free radical scavenging activity of each compound at a concentration of 10 µM where a general decrease in the absorbance at 517 nm was noticed. This indicated a DPPH
• free radical-scavenging activity of the tested compounds. It can be also noticed that most of the synthesized compounds showed obvious scavenging activity on DPPH
• radicals. All the modified flavan-3-ols displayed stronger activities than that of BHT and most of them showed better antioxidative activity than (+)-catechin. Among the tested modified flavan-3-ols, compounds
7 (CHOHCF
3),
8 (COCF
3) and
9 (taxifolin) displayed the strongest activity with an absorbance decrease of about 43, 44 and 45 % respectively. And the order of the activities of flavan-3-ol derivatives were showed to be as follow
9 (taxifolin) >
8 (COCF
3) >
7 (CHOHCF
3) >
4 (CH
2OH) >
3 (CHO) >
6 (Br) >
1a (catechin) >
5 (CH
3), that is, the modified flavan-3-ols with oxygenated and/or hydroxylated subsituents showed much higher antioxidant activities than that of (+)-catechin.
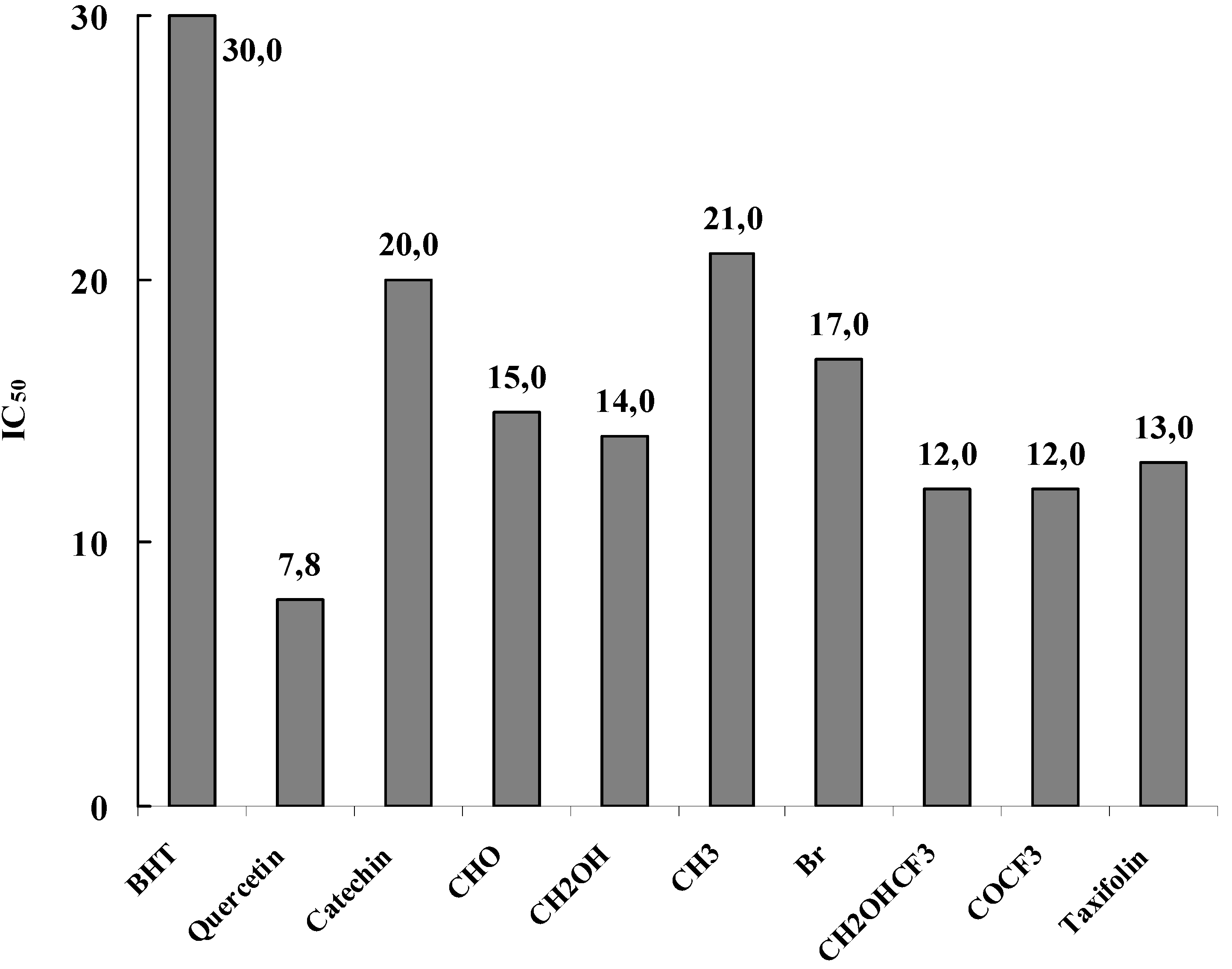
The free radical scavenging activity is usually expressed as percentage of DPPH• inhibition but also by the antioxidant concentration required for a 50 % DPPH• reduction (IC50). A dose-response curve was thus obtained for every product where all the tested compounds showed dose-dependant increase in activity and a spectrophotometric analysis was used in order to determine the inhibition concentration (IC50) of the studied samples, which is a widely used parameter for determining radical scavenging capacity of pure samples. IC50 is the amount of antioxidant necessary to determine the initial concentration of DPPH• radical by 50 %. IC50 value is considered to be a good measure of the antioxidant efficiency of pure compounds and extracts.
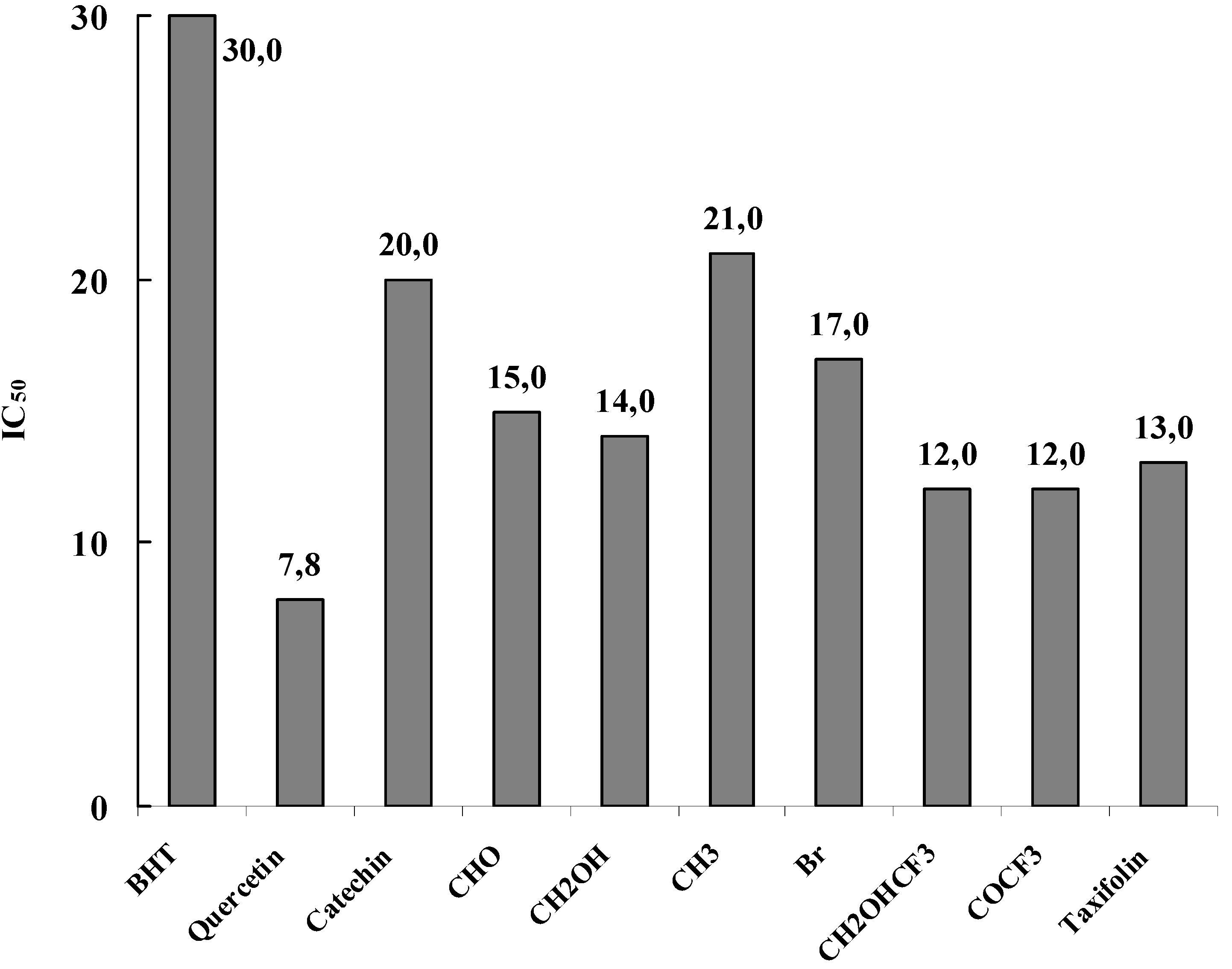
The obtained results, summarized in
Figure 14, showed that the new flavan-3-ol derivatives are potent free radical scavenging agents in the DPPH
• free radical assay. From IC
50 the ED
50 is determined. ED
50 represents the number of products (µmole) able to consume half the amount of free radical divided by µmole if initial DPPH
•. The inverse of ED
50 is a measure of the antiradical power (ARP). By multiplying the ED
50 by two, the stoichiometric value (theoretical concentration of antioxidant to reduce 100 % of the DPPH
•) is obtained. The inverse of this value represents the moles of DPPH
• reduced by one mole of antioxidant and gives an estimate of the number of hydrogen atoms involved in the process.
Table 1 summarizes the parameters obtained. It can be noticed that some of the tested compounds were more efficient than (+)-catechin
1a and BHT, being the order of antiradical power
9 (taxifolin) >
8 (COCF
3) >
7 (CHOHCF
3) >
4 (CH
2OH) >
3 (CHO) >
6 (Br) >
1a (catechin) >
5 (CH
3). The stoichiometric value obtained for (+)-catechin was 0.36 corresponding to the reduction of ca. three DPPH
•. Most of the modified tested flavan-3-ols were able to reduce roughly one more molecule of DPPH
• than the underivatised (+)-catechin. Compound
9 is clearly the most efficient of the new tested molecules. Among all the tested compounds, only compound
5 with a methyl group as substituent was less active than (+)-catechin. Quercetin, with five hydroxyl groups, donated an average of 4.8 electrons/molecule. Results shown in
Table 1 demonstrate that flavonoids which react with DPPH
• and donate an electron are those with an oxygenated substituent.
Figure 14.
Free radical scavenging activity of the studied compounds. The results represent the concentration IC50 needed to decrease by 50% the initial DPPH• absorption at 517 nm.
Figure 14.
Free radical scavenging activity of the studied compounds. The results represent the concentration IC50 needed to decrease by 50% the initial DPPH• absorption at 517 nm.
Table 1.
Antiradical power and stoichiometry from the DPPH assay.
Table 1.
Antiradical power and stoichiometry from the DPPH assay.
| Compound | ED50 | ARP | Stoichiometric value | H atoms per molecule |
|---|
| BHT | 0.33 | 3.00 | 0.67 | 1.50 |
| 37 : Quercetin | 0.10 | 9.60 | 0.21 | 4.80 |
| 1a : (+)-Catechin | 0.18 | 5.60 | 0.36 | 2.80 |
| 3: (CHO) | 0.14 | 7.00 | 0.29 | 3.50 |
| 4: (CH2OH) | 0.14 | 7.20 | 0.28 | 3.60 |
| 5: (CH3) | 0.19 | 5.20 | 0.38 | 2.60 |
| 6: (Br) | 0.15 | 6.60 | 0.30 | 3.30 |
| 7: (CHOHCF3) | 0.14 | 7.40 | 0.27 | 3.70 |
| 8: (COCF3) | 0.13 | 7.60 | 0.26 | 3.80 |
| 9: Taxifolin | 0.13 | 8.00 | 0.25 | 4.00 |
Structure-activity relationships
Many attempts at explaining the structure-activity relationships of some natural antioxidant compounds have been reported in the literature. It has been reported that the antioxidant activity of phenolic compounds may result from the neutralization of free radicals initiating oxidation processes, or from the termination of radical chain reactions, due to their hydrogen donating ability [
56]. It is also known that the antioxidant activity of polyphenolic compounds is closely associated with their structures, such as substitutions on the aromatic ring and side chain structure. Their accessibility to the radical centre of DPPH
• could also influence the order of the antioxidant power. Free radical-scavenging activity of polyphenolic compounds is believed to be influenced by the number and position of phenolic hydrogen in their molecules [
42]. It is also proposed that the higher antioxidant activity is related to the greater number of hydroxyl groups on the flavonoid nucleus [
57].
In overall agreement with structure-activity relationship studies of the free radical scavenging capacity of flavonoids [
42,
58], several structural features were shown to be important for the protective effect of flavonoids against glutamate-mediated programmed cell death [
59] including the presence of a hydroxyl group on C-3 and a 2-3 double bond in conjugation with a C-4 ketone function. The formation of hydrogen bonds between the ketonic oxygen and the hydroxyls at C-3 and C-5 may have some influence on the scavenging power as well [
58].
Catechins do not contain either the unsaturation or the ketone function on C-4 and this is the reason why compounds such as (+)-catechin
1a are less potent scavengers than flavonols such as quercetin. Some catechins such as galloylated or oligomeric derivatives compensate for this by the presence of more hydroxyl groups as well as the ester function at C-3 [
42,
60,
61].
While the antioxidative activity of (+)-catechin and its oligomeric forms (procyanidins) was investigated, little is known about the antioxidant activity of 8-modified flavan-3-ols. As shown in table 1, only one compounds 5 exhibited antioxidant activity lower than (+)-catechin, while the results obtained for all the tested compounds were higher than that of (+)-catechin and BHT. In the same conditions quercetin was the most potent antioxidative compound. The substituents on C-8 position of the aromatic A ring appear to have a clear influence of the antioxidant effect of (+)-catechin. The presence of a group on C-8, which allow for the formation of hydrogen bonds with the hydroxyls either C-7 may play a role in the ability of the (+)-catechin conjugates to scavenge free radicals in a manner similar to quercetin. Since the substituent moieties are attached to the side-chain hydroxyl group functionality of the flavanol skeleton, it is unlikely that they affect the aromatic hydroxyl group that is responsible for antioxidant activity.
Flavonoid hydrophobicity has also been proposed to play an important role the free radical scavenging activity. In view of the chemical structure of the compounds active, hydrophobicity may be most crucial. Instead, the 8-substitutents influence might also influence the polarity of the compounds becoming more or less hydrophobic, and thus might give negative/positive influences on their antioxidant activity when assayed in aqueous conditions. This may explain why compound 5 with a methyl group presents a lower free radical scavenging activity than (+)-catechin.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}