
Pathological AT1R-B2R Protein Aggregation and Preeclampsia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Preeclampsia: Risk Factors, Symptoms and Diagnosis
2.1. Risk Factors of Preeclampsia
2.2. Major Symptoms of Preeclampsia
2.3. Diagnosis of Preeclampsia
2.4. Prevention Therapy of Preeclampsia with Aspirin
3. Pathomechanisms of Preeclampsia Leading to Hypertension
3.1. The Interrelationship between Impaired Arterial Function, Dysfunctional Placentation, and Hypertension
3.2. The Angiotensin II AT1 Receptor Hypersensitivity of Preeclampsia
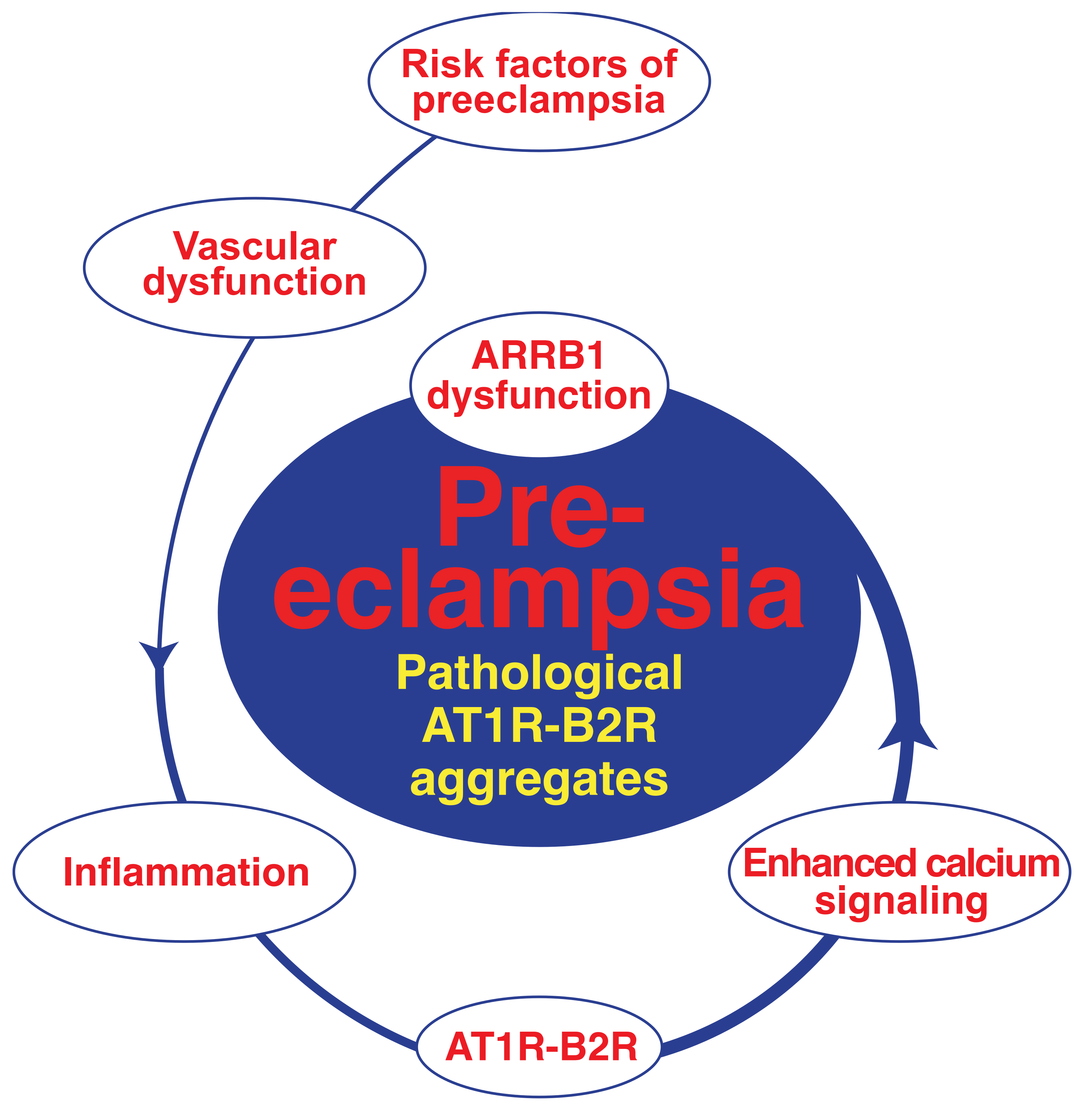
4. Pathologic GPCR Protein Aggregation and Preeclampsia
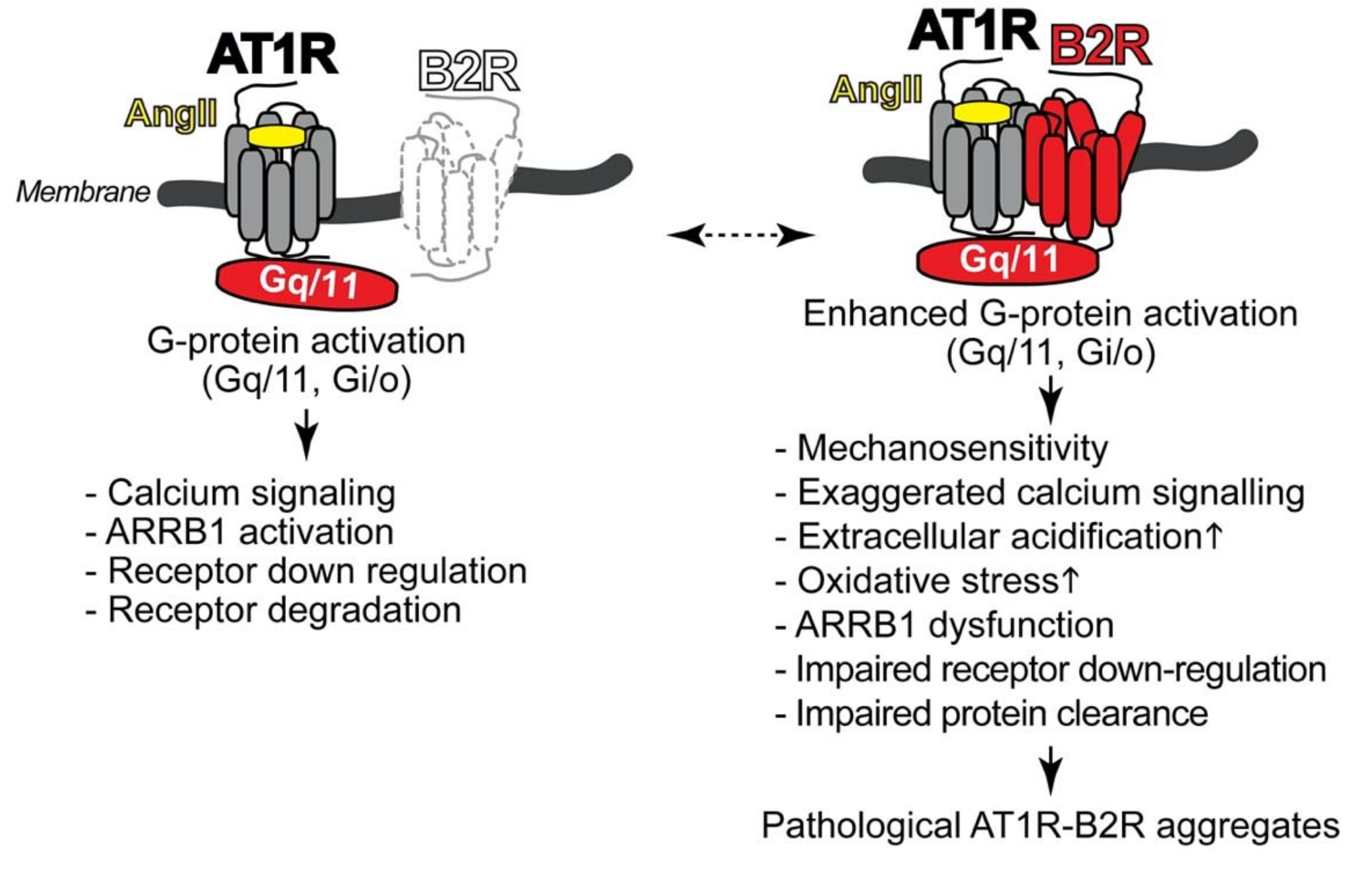
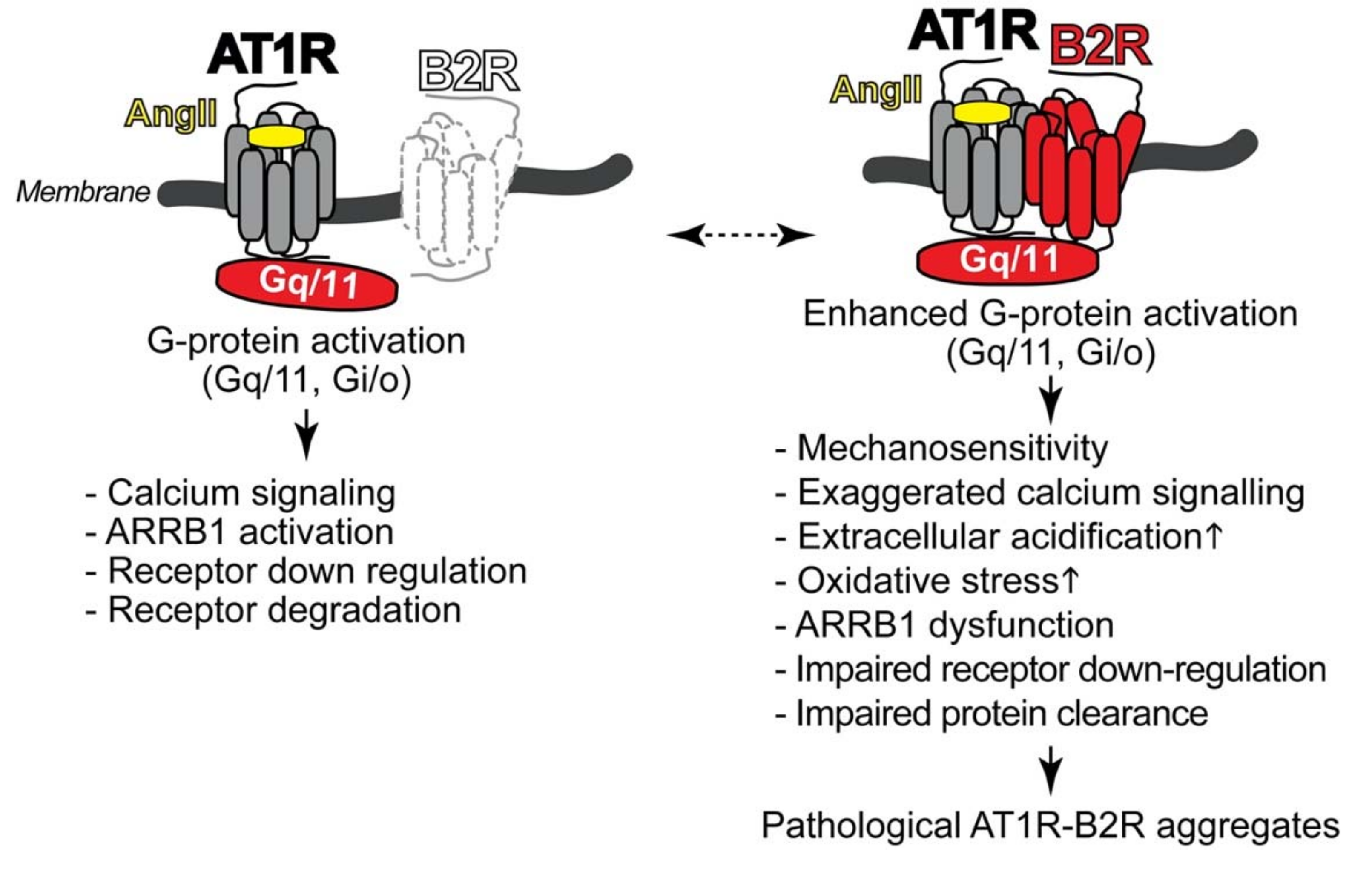
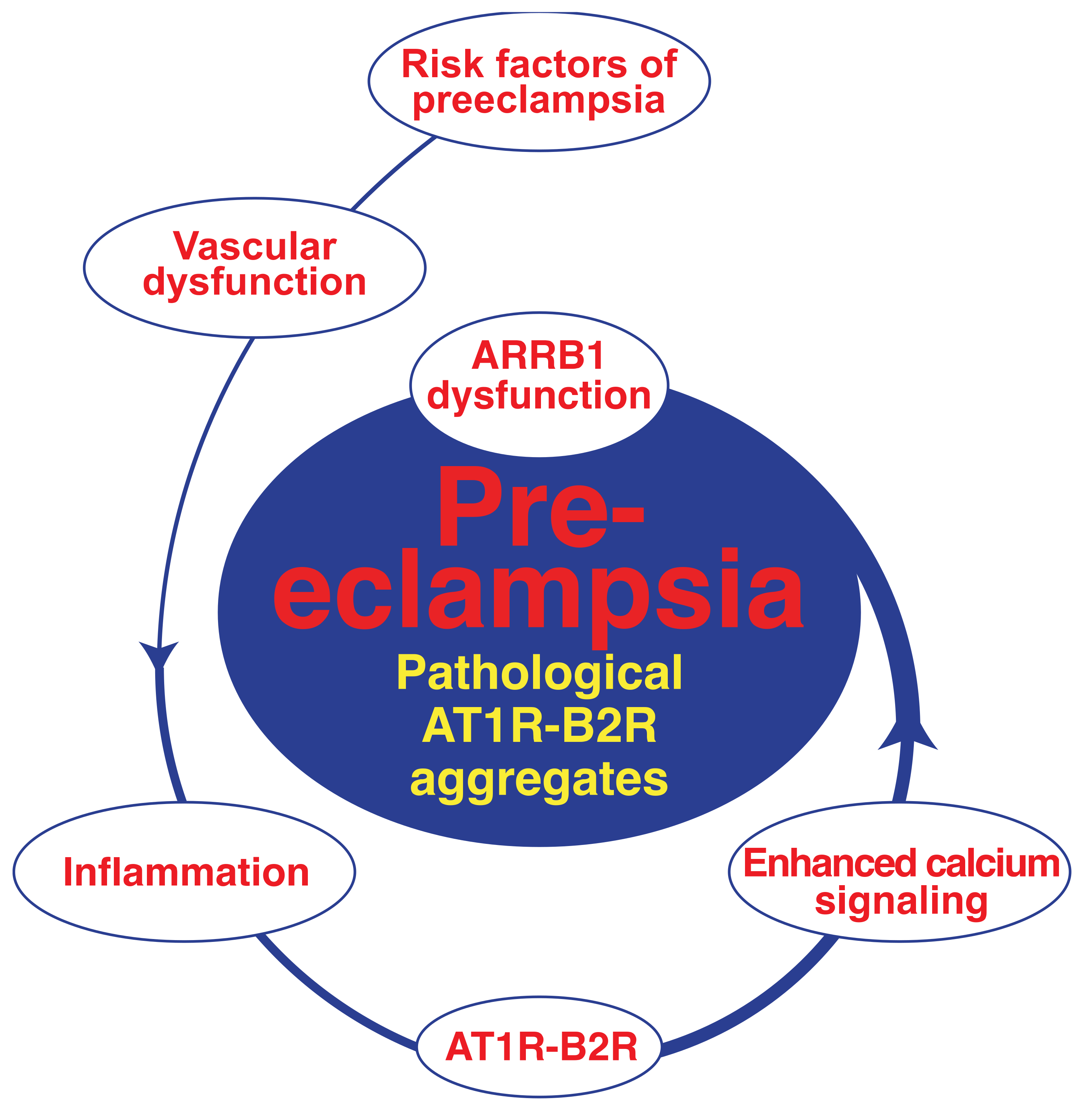
4.1. Pathologic Protein Complex Formation between the AT1 Receptor and the B2 Receptor Causes Angiotensin II AT1 Receptor Hypersensitivity of Preeclampsia
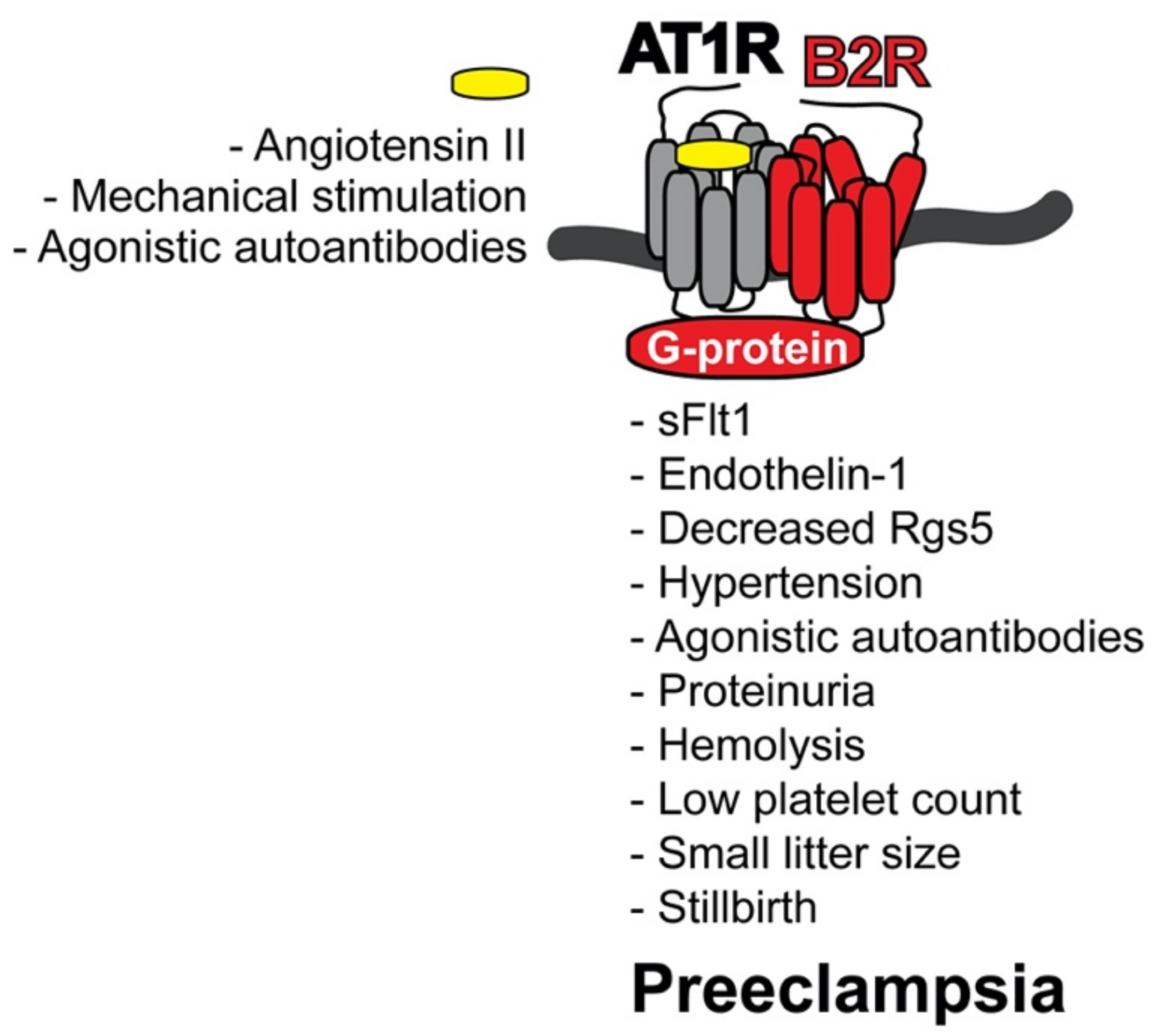
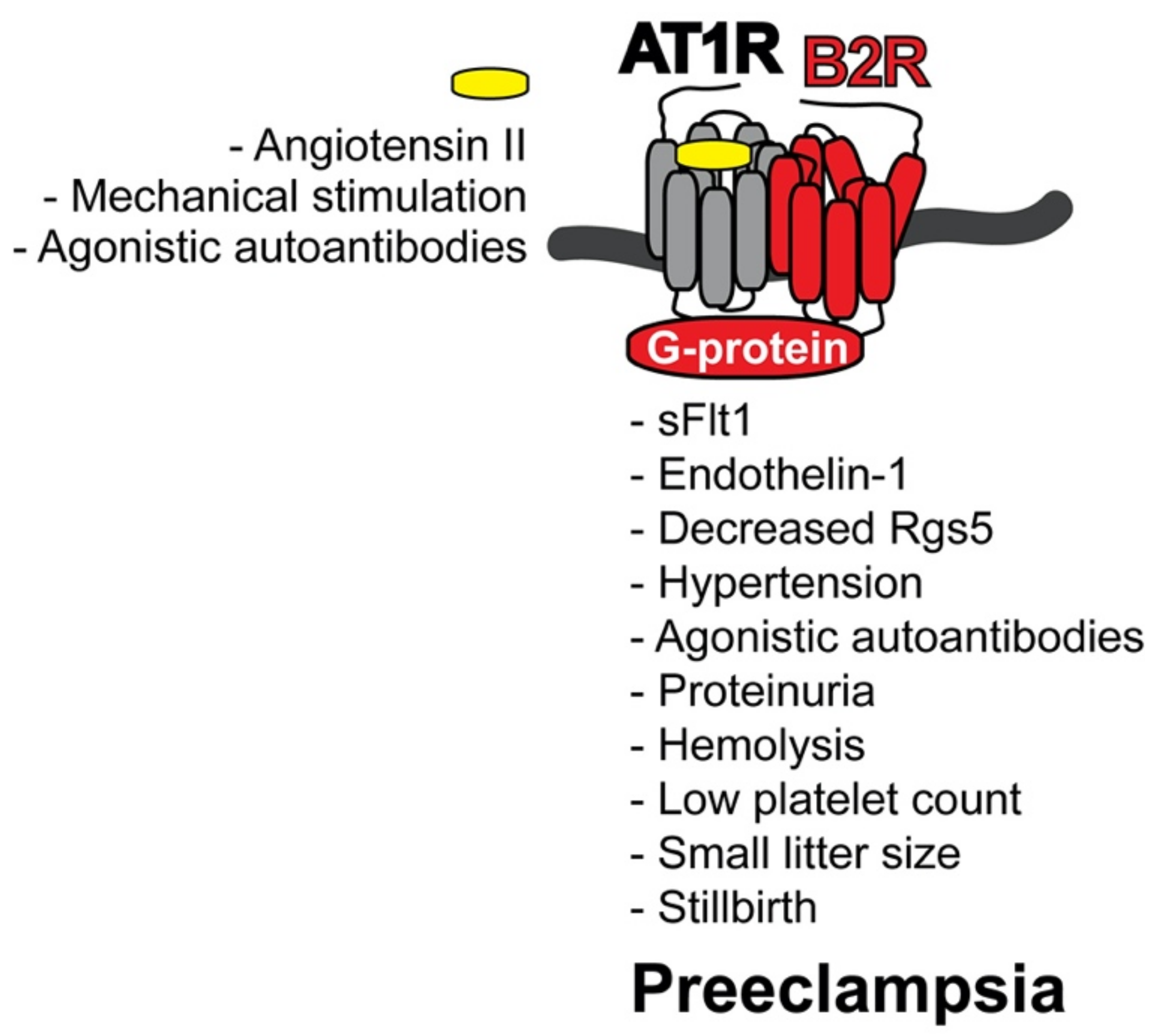
4.2. AT1R-B2R Heteromeric Protein Complexes Trigger Major Symptoms of Preeclampsia
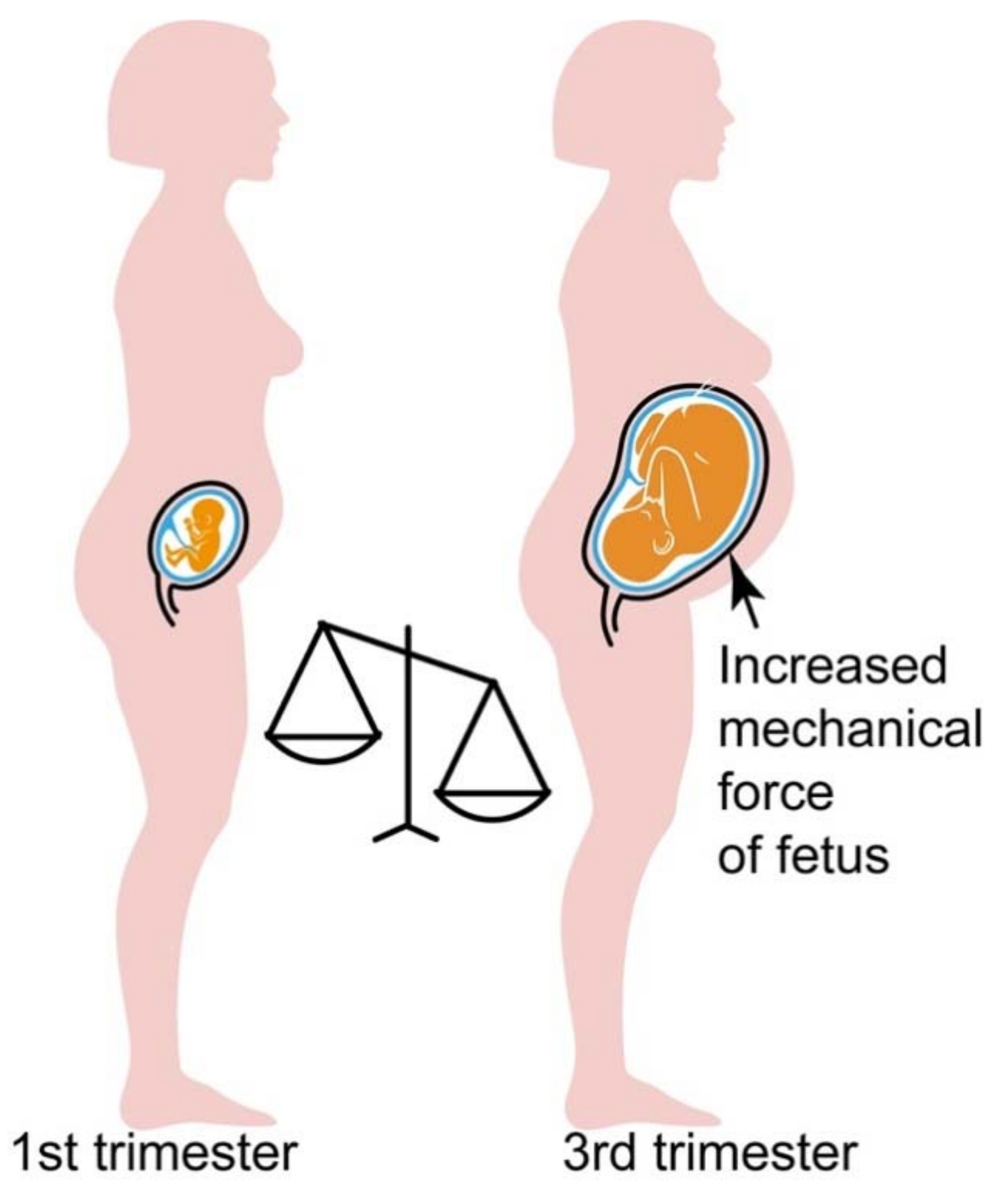
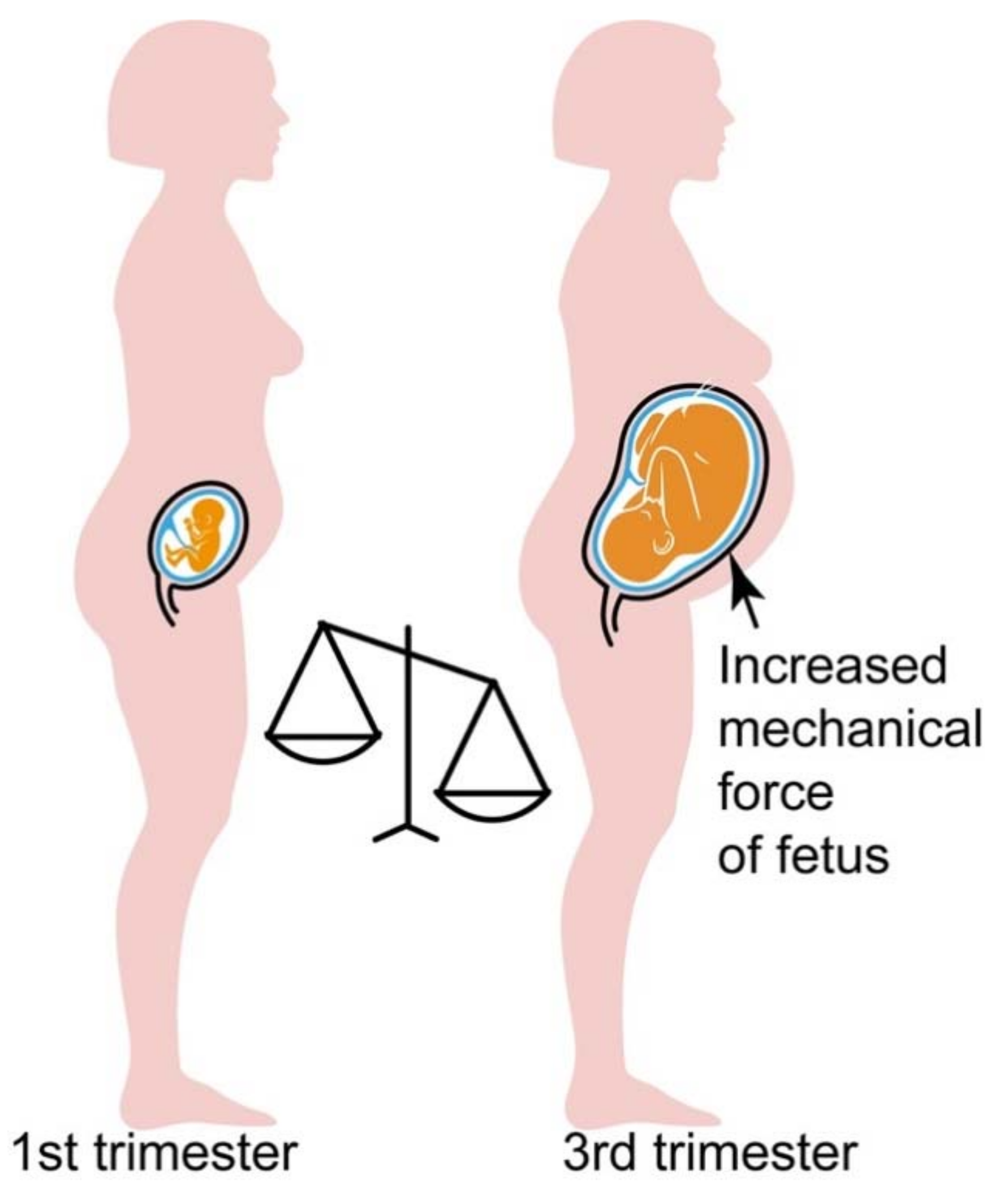
4.3. Hypersensitive AT1R-B2R Heteromers Are Activated by Mechanical Forces, Which Increase during Pregnancy
4.4. Inducers of AT1R-B2R Heteromerization in Preeclampsia
5. Treatment Approaches of Preeclampsia
5.1. Aspirin for Prevention of Preeclampsia
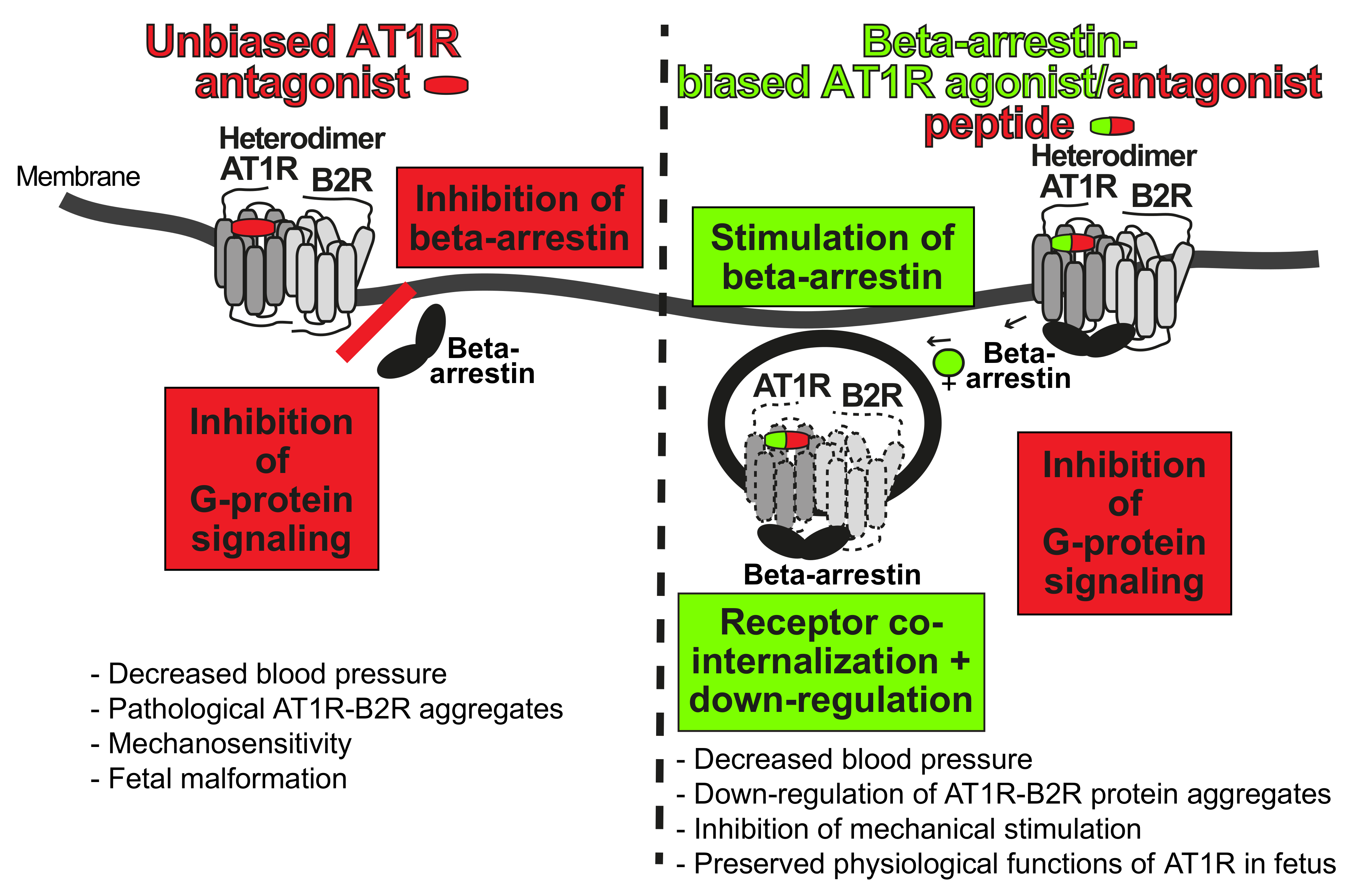
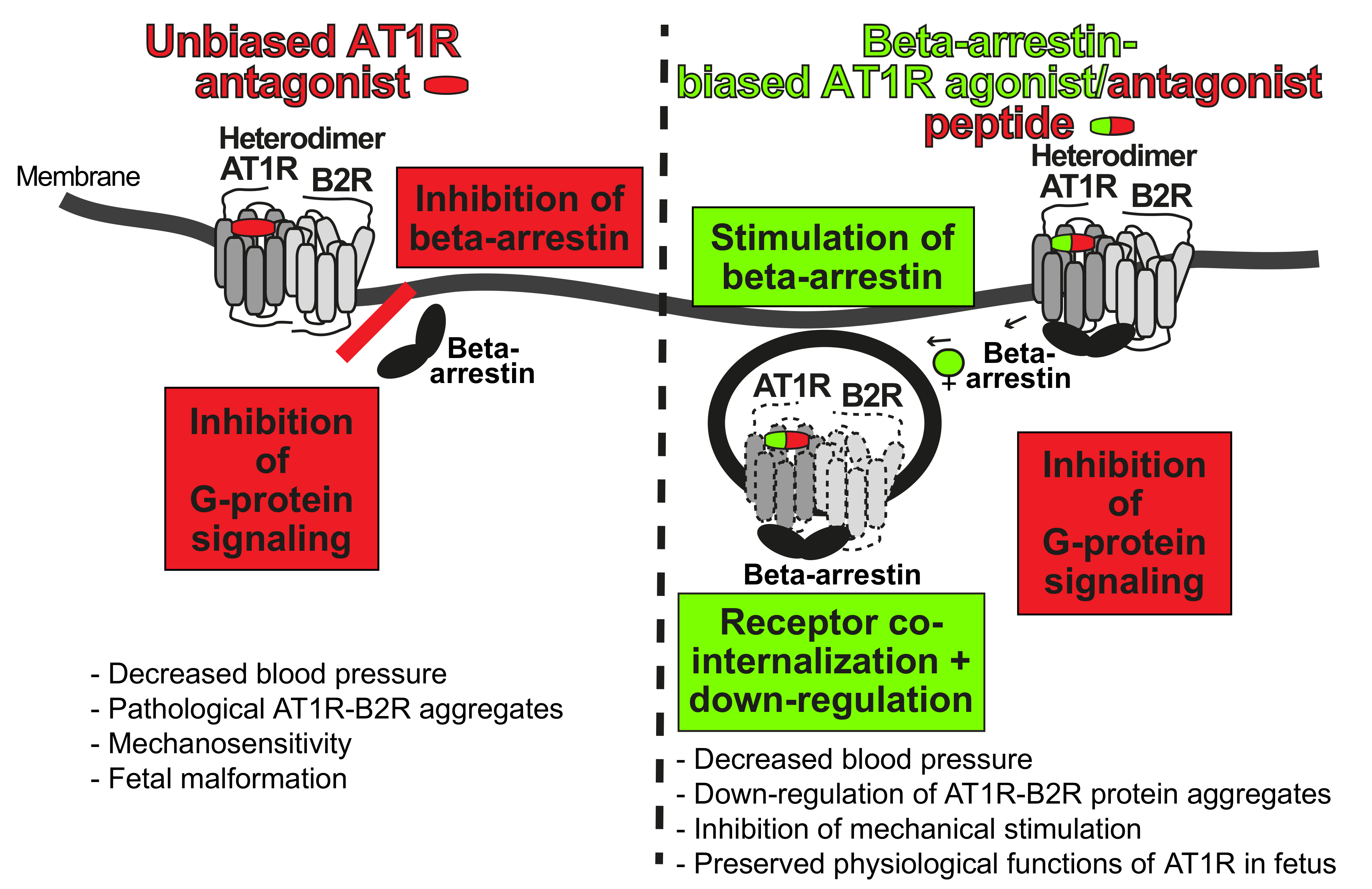
5.2. Targeting of Preeclampsia Symptoms and AT1R-B2R by Beta-Arrestin-Biased Agonism at the AT1 Receptor
5.3. Inhibition of Exaggerated AT1R-B2R-Stimulated Calcium Signaling of Preeclampsia
6. Long-Term Complications of Preeclampsia
6.1. Preeclampsia Increases the Risk of Cardiovascular and Renal Dysfunction Later in Life
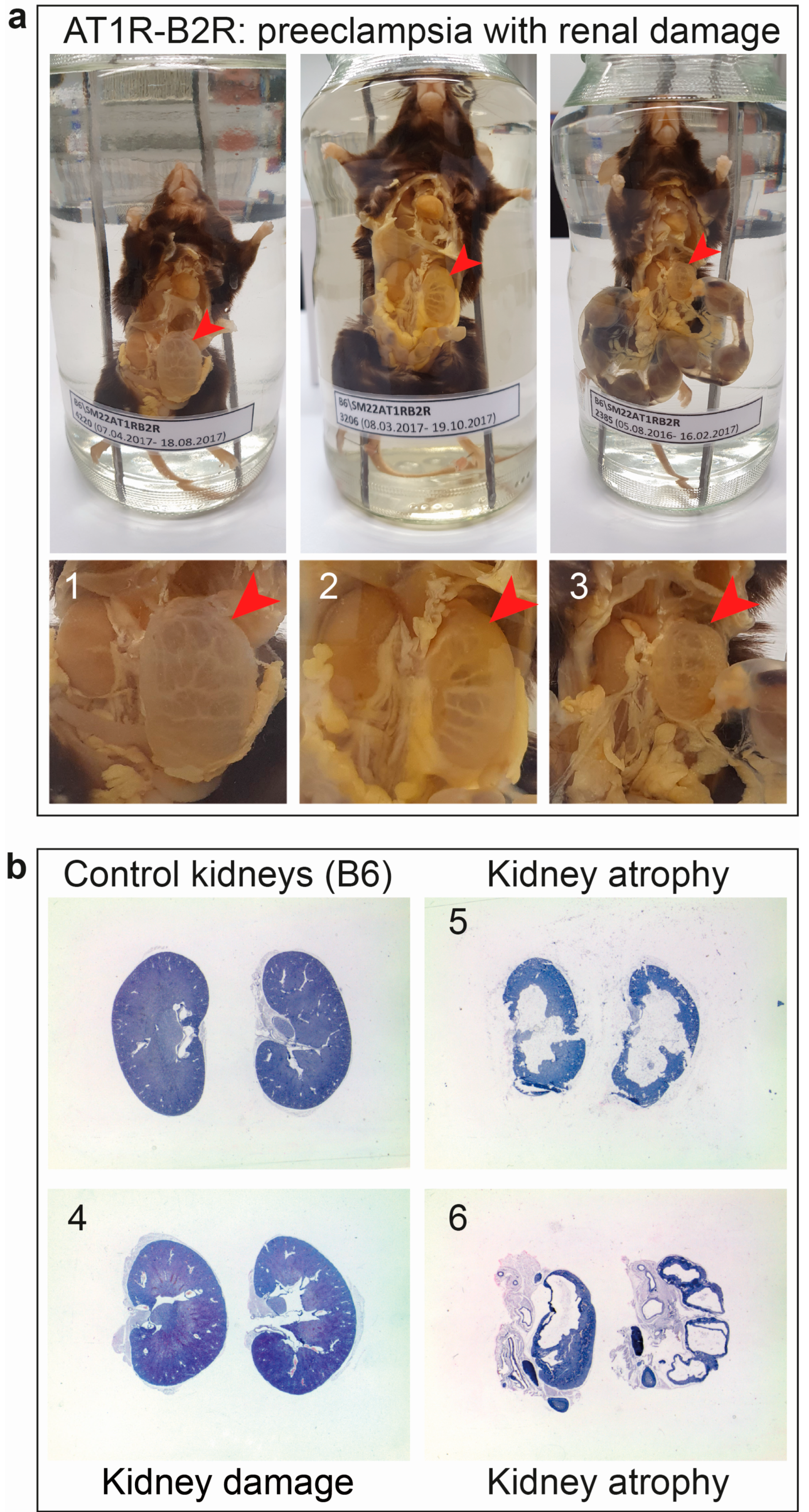
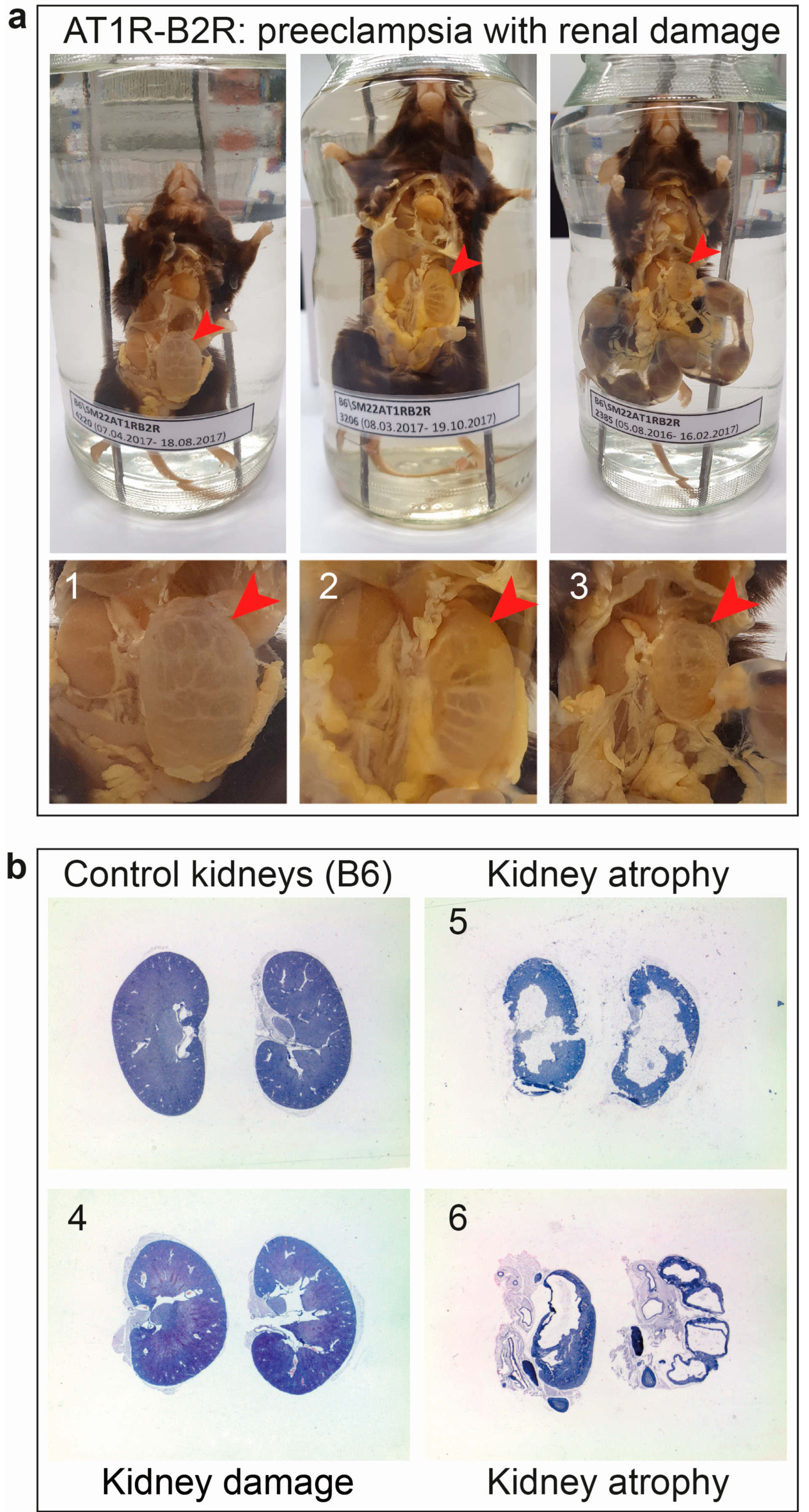
6.2. AT1R-B2R Aggregation Increases the Risk of Renal Dysfunction as a Long-Term Complication of Preeclampsia
6.3. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists. Gestational hypertension and preeclampsia: ACOG practice bulletin, number 222. Obstet. Gynecol. 2020, 135, e237–e260. [Google Scholar] [CrossRef]
- Bibbins-Domingo, K.; Grossman, D.C.; Curry, S.J.; Barry, M.J.; Davidson, K.W.; Doubeni, C.A.; Epling, J.W.; Kemper, A.R.; Krist, A.H.; Kurth, A.E.; et al. Screening for preeclampsia: US Preventive Services Task Force recommendation statement. JAMA 2017, 317, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.C.; Cluver, C.A.; Kingdom, J.; Tong, S. Pre-eclampsia. Lancet 2021, 398, 341–354. [Google Scholar] [CrossRef]
- Spong, C.Y.; Mercer, B.M.; D’Alton, M.; Kilpatrick, S.; Blackwell, S.; Saade, G. Timing of indicated late-preterm and early-term birth. Obstet. Gynecol. 2011, 118, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.; Kitt, J.; Leeson, P.; Aye, C.Y.L.; Lewandowski, A.J. Preeclampsia: Risk factors, diagnosis, management, and the cardiovascular impact on the offspring. J. Clin. Med. 2019, 8, 1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, L.M.; Cunningham, M.W.; Cornelius, D.C.; LaMarca, B. Preeclampsia: Long-term consequences for vascular health. Vasc. Health Risk Manag. 2015, 11, 403–415. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhao, Y.; Yu, A.; Zhao, B.; Gao, Y.; Niu, H. Diagnostic accuracy of the soluble Fms-like tyrosine kinase-1/placental growth factor ratio for preeclampsia: A meta-analysis based on 20 studies. Arch. Gynecol. Obstet. 2015, 292, 507–518. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; el Massiery, A.; Quitterer, U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med. 2001, 7, 1003–1009. [Google Scholar] [CrossRef]
- Quitterer, U.; Lother, H.; Abdalla, S. AT1 receptor heterodimers and angiotensin II responsiveness in preeclampsia. Semin. Nephrol. 2004, 24, 115–119. [Google Scholar] [CrossRef]
- Quitterer, U.; Fu, X.; Pohl, A.; Bayoumy, K.M.; Langer, A.; AbdAlla, S. Beta-arrestin1 prevents preeclampsia by downregulation of mechanosensitive AT1-B2 receptor heteromers. Cell 2019, 176, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Oliverio, M.I.; Mannon, P.J.; Best, C.F.; Maeda, N.; Smithies, O.; Coffman, T.M. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc. Natl. Acad. Sci. USA 1995, 92, 3521–3525. [Google Scholar] [CrossRef] [Green Version]
- Alfie, M.E.; Sigmon, D.H.; Pomposiello, S.I.; Carretero, O.A. Effect of high salt intake in mutant mice lacking bradykinin-B2 receptors. Hypertension 1997, 29, 483–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hypertension in Pregnancy: Diagnosis and Management. Available online: https://www.nice.org.uk/guidance/ng133 (accessed on 29 September 2021).
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.P.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. International Society for the Study of Hypertension in Pregnancy (ISSHP). Hypertensive disorders of pregnancy: ISSHP classification, diagnosis, and management recommendations for international practice. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, E.; Medcalf, K.E.; Park, A.L.; Ray, J.G.; High Risk of Pre-eclampsia Identification Group. Clinical risk factors for pre-eclampsia determined in early pregnancy: Systematic review and meta-analysis of large cohort studies. BMJ 2016, 353, i1753. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, G.; Grewal, J.; Männistö, T.; Mendola, P.; Chen, Z.; Xie, Y.; Laughon, S.K. Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn. Dis. 2014, 24, 283–289. [Google Scholar] [PubMed]
- Levron, Y.; Dviri, M.; Segol, I.; Yerushalmi, G.M.; Hourvitz, A.; Orvieto, R.; Mazaki-Tovi, S.; Yinon, Y. The ‘immunologic theory’ of preeclampsia revisited: A lesson from donor oocyte gestations. Am. J. Obstet. Gynecol. 2014, 211, e1–e5. [Google Scholar] [CrossRef]
- Blazquez, A.; Garcia, D.; Rodriguez, A.; Vassena, R.; Figueras, F.; Vernaeve, V. Is oocyte donation a risk factor for preeclampsia? A systematic review and meta-analysis. J. Assist. Reprod. Genet. 2016, 33, 855–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaratinam, S.; Gallos, I.D.; Meah, N.; Usman, S.; Ismail, K.M.; Khan, K.S.; TIPPS (Tests in Prediction of Pre-eclampsia’s Severity) Review Group. How accurate are maternal symptoms in predicting impending complications in women with preeclampsia? A systematic review and meta-analysis. Acta Obstet Gynecol. Scand. 2011, 90, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Sakaguchi, K.; Shibasaki, T.; Takahashi, H.; Kawakami, Y.; Furuya, K.; Kikuchi, Y. Cerebral edema on MRI in severe preeclamptic women developing eclampsia. J. Perinat. Med. 2005, 33, 199–205. [Google Scholar] [CrossRef]
- Judy, A.E.; McCain, C.L.; Lawton, E.S.; Morton, C.H.; Main, E.K.; Druzin, M.L. Systolic hypertension, preeclampsia-related mortality, and stroke in California. Obstet. Gynecol. 2019, 133, 1151–1159. [Google Scholar] [CrossRef]
- Cunningham, F.G.; Fernandez, C.O.; Hernandez, C. Blindness associated with preeclampsia and eclampsia. Am. J. Obstet. Gynecol. 1995, 172, 1291–1298. [Google Scholar] [CrossRef]
- Arias, F.; Mancilla-Jimenez, R. Hepatic fibrinogen deposits in preeclampsia. Immunofluorescent evidence. N. Engl. J. Med. 1976, 295, 578–582. [Google Scholar] [CrossRef]
- Schneider, H. Leberpathologie im Rahmen des HELLP-Syndroms. Arch. Gynecol. Obstet. 1994, 255 (Suppl. 2), S245–S254. [Google Scholar] [CrossRef] [PubMed]
- Stanhewicz, A.E.; Nuckols, V.R.; Pierce, G.L. Maternal microvascular dysfunction during preeclamptic pregnancy. Clin. Sci. 2021, 135, 1083–1101. [Google Scholar] [CrossRef] [PubMed]
- Kattah, A. Preeclampsia and kidney disease: Deciphering cause and effect. Curr. Hypertens. Res. 2020, 22, 91. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J.; Ganiger, V.C. Acute kidney injury in pregnancy-specific disorders. Indian J. Nephrol. 2017, 27, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.E.; Makris, A.; Ogle, R.F.; Tooher, J.M.; Hennessy, A. Role of proteinuria in defining pre-eclampsia: Clinical outcomes for women and babies. Clin. Exp. Pharmacol. Physiol. 2010, 37, 466–470. [Google Scholar] [CrossRef]
- Lei, T.; Qiu, T.; Liao, W.; Li, K.; Lai, X.; Huang, H.; Yuan, R.; Chen, L. Proteinuria may be an indicator of adverse pregnancy outcomes in patients with preeclampsia: A retrospective study. Reprod. Biol. Endocrinol. 2021, 19, 71. [Google Scholar] [CrossRef]
- Sasamori, Y.; Tanaka, A.; Ayabe, T. Liver disease in pregnancy. Hepatol. Res. 2020, 50, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Huang, P.; Lyu, M.; Dong, J. Oxidative stress and preeclampsia-associated prothrombotic state. Antioxidants. 2020, 9, 1139. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Scully, M.; Provot, F.; Blasco, M.; Coppo, P.; Noris, M.; Paizis, K.; Kavanagh, D.; Pene, F.; Quezada, S.; et al. Management of thrombotic microangiopathy in pregnancy and postpartum: Report from an international working group. Blood 2020, 136, 2103–2117. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, U.; Heimdal, K. Pathogenesis of the syndrome of hemolysis, elevated liver enzymes, and low platelet count (HELLP): A review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 166, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.C.; Verma, R.P. Fetal Growth Restriction; StatPearls Publishing: Treasure Island, FL, USA, 14 August 2021. [Google Scholar]
- Mecacci, F.; Avagliano, L.; Lisi, F.; Clemenza, S.; Serena, C.; Vannuccini, S.; Rambaldi, M.P.; Simeone, S.; Ottanelli, S.; Petraglia, F. Fetal growth restriction: Does an integrated maternal hemodynamic-placental model fit better? Reprod. Sci. 2021, 28, 2422–2435. [Google Scholar] [CrossRef]
- Scott, G.; Gillon, T.E.; Pels, A.; von Dadelszen, P.; Magee, L.A. Guidelines-similarities and dissimilarities: A systematic review of international clinical practice guidelines for pregnancy hypertension. Am. J. Obstet. Gynecol. 2020, S0002-9378(20)30846-2. [Google Scholar] [CrossRef]
- Poon, L.C.; Shennan, A.; Hyett, J.A.; Kapur, A.; Hadar, E.; Divakar, H.; McAuliffe, F.; da Silva Costa, F.; von Dadelszen, P.; McIntyre, H.D.; et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: A pragmatic guide for first-trimester screening and prevention. Int. J. Gynaecol. Obstet. 2019, 145, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, R.G.; Ogunbodede, A.C. Hypertensive disorders of pregnancy. Emerg. Med. Clin. N. Am. 2019, 37, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Papageorghiou, A.T.; Yu, C.K.; Bindra, R.; Pandis, G.; Nicolaides, K.H.; Fetal Medicine Foundation Second Trimester Screening Group. Multicenter screening for pre-eclampsia and fetal growth restriction by transvaginal uterine artery Doppler at 23 weeks of gestation. Ultrasound Obstet. Gynecol. 2001, 18, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Nikuei, P.; Rajaei, M.; Roozbeh, N.; Mohseni, F.; Poordarvishi, F.; Azad, M.; Haidari, S. Diagnostic accuracy of sFlt1/PIGF ratio as a marker for preeclampsia. BMC Pregnancy Childbirth 2020, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Hurrell, A.; Beardmore-Gray, A.; Duhig, K.; Webster, L.; Chappell, L.C.; Shennan, A.H. Placental growth factor in suspected preterm pre-eclampsia: A review of the evidence and practicalities of implementation. BJOG 2020, 127, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Duhig, K.E.; Myers, J.; Seed, P.T.; Sparkes, J.; Lowe, J.; Hunter, R.M.; Shennan, A.H.; Chappell, L.C.; PARROT Trial Group. Placental growth factor testing to assess women with suspected pre-eclampsia: A multicentre, pragmatic, stepped-wedge cluster-randomized controlled trial. Lancet 2019, 393, 1807–1818. [Google Scholar] [CrossRef] [Green Version]
- Parchem, J.G.; Brock, C.O.; Chen, H.Y.; Kalluri, R.; Barton, J.R.; Sibai, B.M. Preeclampsia Triage by Rapid Assay Trial (PETRA) Investigators. Placental Growth Factor and the risk of adverse neonatal and maternal outcomes. Obstet. Gynecol. 2020, 135, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Barton, J.R.; Woelkers, D.A.; Newman, R.B.; Combs, C.A.; How, H.Y.; Boggess, K.A.; Martin, J.N.; Kupfer, K.; Sibai, B.M.; for the PETRA (Preeclampsia Triage by Rapid Assay) Trial. Placental growth factor predicts time to delivery in women with signs or symptoms of early preterm preeclampsia: A prospective multicenter study. Am. J. Obstet. Gynecol. 2020, 222, e1–e259. [Google Scholar] [CrossRef]
- Hurrell, A.; Duhig, K.; Vandermolen, B.; Shennan, A.H. Recent advances in the diagnosis and management of pre-eclampsia. Fac. Rev. 2020, 9, 10. [Google Scholar] [CrossRef]
- PlGF-Based Testing to Help Diagnose Suspected Pre-Eclampsia (Triage PlGF Test, Elecsys Immunoassay sFlt-1/PlGF Ratio, DELFIA Xpress PlGF 1-2-3 Test, and BRAHMS sFlt-1 Kryptor/BRAHMS PlGF Plus Kryptor PE Ratio). Available online: https://www.nice.org.uk/guidance/dg23 (accessed on 29 September 2021).
- Cerdeira, A.S.; O’Sullivan, J.; Ohuma, E.O.; Harrington, D.; Szafranski, P.; Black, R.; Mackillop, L.; Impey, L.; Greenwood, C.; James, T.; et al. Randomized interventional study on prediction of preeclampsia/eclampsia in women with suspected preeclampsia: INSPIRE. Hypertension 2019, 74, 983–990. [Google Scholar] [CrossRef]
- Ormesher, L.; Johnstone, E.D.; Shawkat, E.; Dempsey, A.; Chmiel, C.; Ingram, E.; Higgins, L.E.; Myers, J.E. A clinical evaluation of placental growth factor in routine practice in high-risk women presenting with suspected pre-eclampsia and/or fetal growth restriction. Pregnancy Hypertens. 2018, 14, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Mather, A.R.; Dom, A.M.; Thorburg, L.L. Low-dose aspirin in pregnancy: Who? when? how much? and why? Curr. Opin. Obstet. Gynecol. 2021, 33, 65–71. [Google Scholar] [CrossRef]
- Wright, D.; Nicolaides, K.H. Aspirin delays the development of preeclampsia. Am. J. Obstet. Gynecol. 2019, 220, e1–e580. [Google Scholar] [CrossRef] [PubMed]
- ACOG Committee Opinion No. 743. Low-dose aspirin use during pregnancy. Obstet. Gynecol. 2018, 132, e44–e52. [Google Scholar] [CrossRef]
- Walsh, S.W.; Strauss, J.F. The road to low-dose aspirin therapy for the prevention of preeclampsia began with the placenta. Int. J. Mol. Sci. 2021, 22, 6985. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.P.; Alexander, B.T.; Llinas, M.T.; Bennett, W.A.; Khalil, R.A. Pathophysiology of hypertension during preeclampsia linking placental ischemia with endothelial dysfunction. Hypertension 2001, 38, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Khalil, R.A. Vascular mechanisms and molecular targets in hypertensive pregnancy and preeclampsia. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H661–H681. [Google Scholar] [CrossRef] [PubMed]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.N.; Oparil, S. Preeclampsia-pathophysiology and clinical presentations: JACC State-of-the-art review. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef]
- Hong, K.; Park, H.J.; Cha, D. Clinical implications of placenta-derived angiogenic/anti-angiogenic biomarkers in preeclampsia. Biomark. Med. 2021, 15, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.M.; Torrado, J.; Sosa, C.; Zocalo, Y.; Bia, D. Role of arterial impairment in preeclampsia: Should the paradigm shift? Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2011–H2030. [Google Scholar] [CrossRef]
- Melchiorre, K.; Giorgione, V.; Thilaganathan, B. The placenta and preeclampsia: Villain or victim? Am. J. Obstet. Gynecol 2021, S0002-9378(20)31198-4. [Google Scholar] [CrossRef]
- Mirabito Colafella, K.M.; Bovée, D.M.; Danser, A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp Eye Res. 2019, 186, 107680. [Google Scholar] [CrossRef]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quitterer, U.; AbdAlla, S. Improvements of symptoms of Alzheimer‘s disease by inhibition of the angiotensin system. Pharmacol. Res. 2020, 154, 104230. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.M. The role of RAS in the pathogenesis of preeclampsia. Curr. Hypertens. Rep. 2006, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Gant, N.F.; Daley, G.L.; Chand, S.; Whalley, P.J.; MacDonald, P.C. A study of angiotensin II pressor response throughout primigravid pregnancy. J. Clin. Investig. 1973, 52, 2682–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, H.; Oeney, T.; Hauck, U.; Distler, A.; Philipp, T. Increased intracellular free calcium and sensitivity to angiotensin II in platelets of preeclamptic women. Am. J. Hypertens. 1989, 2, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.D.; Zsengellér, Z.K.; Khankin, E.V.; Lo, A.S.; Rajakumar, A.; DuPont, J.J.; McCurley, A.; Moss, M.E.; Zhang, D.; Clark, C.D.; et al. Soluble fms-like tyrosine kinase 1 promotes angiotensin II sensitivity in preeclampsia. J. Clin. Investig. 2016, 126, 2561–2574. [Google Scholar] [CrossRef]
- Stanhewicz, A.E.; Jandu, S.; Santhanam, L.; Alexander, L.M. Increased angiotensin II sensitivity contributes to microvascular dysfunction in women who have had preeclampsia. Hypertension 2017, 70, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Irani, R.A.; Xia, Y. Renin angiotensin signaling in normal pregnancy and preeclampsia. Semin. Nephrol. 2011, 31, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jüpner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Investig. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Dechend, R.; Gratze, P.; Wallukat, G.; Shagdarsuren, E.; Plehm, R.; Bräsen, J.H.; Fiebeler, A.; Schneider, W.; Caluwaerts, S.; Vercruysse, L.; et al. Agonistic autoantibodies to the AT1 receptor in a transgenic rat model of preeclampsia. Hypertension 2005, 45, 742–746. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Kellems, R.E. Angiotensin receptor agonistic autoantibodies and hypertension: Preeclampsia and beyond. Circ. Res. 2013, 113, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.F.; Wang, J.; Jiao, X.Y.; Liu, H.R.; Zhao, R.R.; Zhi, J.M. Detection of serum autoantibodies against AT1A-receptor during the development of the four types of hypertensive rat models. Sheng Li Xue Bao 2006, 58, 90–94. [Google Scholar]
- McEachern, A.E.; Shelton, E.R.; Bhakta, S.; Obernolte, R.; Bach, C.; Zuppan, P.; Fujisaki, J.; Aldrich, R.W.; Jarnagin, K. Expression cloning of a rat B2 bradykinin receptor. Proc. Natl. Acad. Sci. USA 1991, 88, 7724–7728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkowski, J.A.; Ransom, R.W.; Seabrook, G.R.; Trumbauer, H.; Chen, H.; Hill, R.G.; Strader, C.D. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin action in smooth muscle and neurons. J. Biol. Chem. 1995, 270, 13706–13710. [Google Scholar] [CrossRef] [Green Version]
- Girolami, J.P.; Bouby, N.; Richer-Giudicelli, C.; Alhenc-Gelas, F. Kinins and kinin receptors in cardiovascular and renal diseases. Pharmaceuticals 2021, 14, 240. [Google Scholar] [CrossRef]
- Gainer, J.V.; Morrow, J.D.; Loveland, A.; King, D.J.; Brown, N.J. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N. Engl. J. Med. 1998, 339, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Guerby, P.; Tasta, O.; Swiader, A.; Pont, F.; Bujold, E.; Parant, O.; Vayssiere, C.; Salvayre, R.; Negre-Salvayre, A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox. Biol. 2021, 40, 101861. [Google Scholar] [CrossRef]
- Kawashima, S.; Yokoyama, M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 998–1005. [Google Scholar] [CrossRef]
- Perhal, A.; Wolf, S.; Jamous, Y.F.; Langer, A.; Abd Alla, J.; Quitterer, U. Increased reactive oxygen species generation contributes to the atherogenic activity of the B2 bradykinin receptor. Front. Med. 2019, 6, 32. [Google Scholar] [CrossRef]
- Ashworth, J.R.; Warren, A.Y.; Baker, P.N.; Johnson, I.R. Loss of endothelium-dependent relaxation in myometrial resistance arteries in pre-eclampsia. Br. J. Obstet. Gynaecol. 1997, 104, 1152–1158. [Google Scholar] [CrossRef]
- Svedas, E.; Nisell, H.; Vanwijk, M.J.; Nikas, Y.; Kublickiene, K.R. Endothelial dysfunction in uterine circulation in preeclampsia: Can estrogens improve it. Am. J. Obstet. Gynecol. 2002, 187, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, R.; Jalkanen, M.; Huotari, K. Urinary kallikrein excretion in normal and hypertensive pregnancy at term. Ann. Clin. Res. 1986, 18, 208–210. [Google Scholar] [PubMed]
- Millar, J.G.; Campbell, S.K.; Albano, J.D.; Higgins, B.R.; Clark, A.D. Early prediction of pre-eclampsia by measurement of kallikrein and creatinine on a random urine sample. Br. J. Obstet. Gynaecol. 1996, 103, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Kyle, P.M.; Campbell, S.; Buckley, D.; Kissane, J.; de Swiet, M.; Albano, J.; Millar, J.G.; Redman, C.W. A comparison of the inactive urinary kallikrein:creatinine ratio and the angiotensin sensitivity test for the prediction of pre-eclampsia. Br. J. Obstet. Gynaecol. 1996, 103, 981–987. [Google Scholar] [CrossRef]
- Corthorn, J.; Germain, A.A.; Chacon, C.; Rey, S.; Soto, G.X.; Figueroa, C.D.; Müller-Esterl, W.; Duarte, I.; Valdes, G. Expression of kallikrein, bradykinin b2 receptor, and endothelial nitric oxide synthase in placenta in normal gestation, preeclampsia, and placenta accreta. Endocrine 2006, 29, 491–499. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 2000, 407, 94–98. [Google Scholar] [CrossRef]
- Quitterer, U.; AbdAlla, S. Discovery of pathologic GPCR aggregation. Front. Med. 2019, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 2004, 6, 499–506. [Google Scholar] [CrossRef]
- Berk, B.C.; Aronow, M.S.; Brock, T.A.; Cragoe, E.; Gimbrone, M.A.; Alexander, R.W. Angiotensin II-stimulated Na+/H+ exchange in cultured vascular smooth muscle cells. Evidence for protein kinase C-dependent and independent pathways. J. Biol. Chem. 1987, 262, 5057–5064. [Google Scholar] [CrossRef]
- Bulenger, S.; Marullo, S.; Bouvier, M. Emerging role of homo- and heterodimerization in G-protein-coupled receptor biosynthesis and maturation. Trends Pharmacol. Sci. 2005, 26, 131–137. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; Abdel-tawab, A.M.; Quitterer, U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 2001, 276, 39721–39726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.M.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Abadir, P.M.; Periasamy, A.; Carey, R.M.; Siragy, H.M. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension 2006, 48, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfie, M.E.; Yang, X.P.; Hess, F.; Carretero, O.A. Salt-sensitive hypertension in bradykinin B2 receptor knockout mice. Biochem. Biophys. Res. Commun. 1996, 224, 625–630. [Google Scholar] [CrossRef]
- AbdAlla, S.; Abdel-Baset, A.; Lother, H.; el Massiery, A.; Quitterer, U. Mesangial AT1/B2 receptor heterodimers contribute to angiotensin II hyperresponsiveness in experimental hypertension. J. Mol. Neurosci. 2005, 26, 185–192. [Google Scholar] [CrossRef]
- Saleh, L.; Verdonk, K.; Visser, W.; van den Meiracker, A.H.; Danser, A.H. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther. Adv. Cardiovasc. Dis. 2016, 10, 282–293. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Somlyo, A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003, 83, 1325–1358. [Google Scholar] [CrossRef] [Green Version]
- Wirth, A.; Benyo, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Orsy, P.; Horvath, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef]
- Zhao, M.; Li, L.; Yang, X.; Cui, J.; Li, H. FN1, FOS, and ITGA5 induce preeclampsia: Abnormal expression and methylation. Hypertens. Pregnancy 2017, 36, 302–309. [Google Scholar] [CrossRef]
- Murphy, S.R.; Cockrell, K. Regulation of soluble fms-like tyrosine kinase-1 production in response to placental ischemia/hypoxia: Role of angiotensin II. Physiol. Rep. 2015, 3, e12310. [Google Scholar] [CrossRef]
- Zhou, C.C.; Ahmad, S.; Mi, T.; Xia, L.; Abbasi, S.; Hewett, P.W.; Sun, C.; Ahmed, A.; Kellems, R.E.; Xia, Y. Angiotensin II induces soluble fms-Like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circ. Res. 2007, 100, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600–4614. [Google Scholar] [CrossRef] [PubMed]
- Holobotovskyy, V.; Chong, Y.S.; Burchell, J.; He, B.; Phillips, M.; Leader, L.; Murphy, T.V.; Sandow, S.L.; McKitrick, D.J.; Charles, A.K.; et al. Regulator of G protein signaling 5 is a determinant of gestational hypertension and preeclampsia. Sci. Transl. Med. 2015, 7, 290ra88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oats, J.N.; Broughton Pipkin, F.; Symonds, E.M.; Craven, D.J. A prospective study of plasma angiotensin-converting enzyme in normotensive primigravidae and their infants. Br. J. Obstet. Gynaecol. 1981, 88, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bdolah, Y.; Lam, C.; Rajakumar, A.; Shivalingappa, V.; Mutter, W.; Sachs, B.P.; Lim, K.H.; Bdolah-Abram, T.; Epstein, F.H.; Karumanchi, S.A. Twin pregnancy and the risk of preeclampsia: Bigger placenta or relative ischemia. Am. J. Obstet. Gynecol. 2008, 198, 428.e1–428.e6. [Google Scholar] [CrossRef]
- Anton, E.L.; Fernandes, D.; Assreuy, J.; da Silva-Santos, J.E. Bradykinin increases BP in endotoxemic rat: Functional and biochemical evidence of angiotensin II AT1/bradykinin B2 receptor heterodimerization. Br. J. Pharmacol. 2019, 176, 2608–2626. [Google Scholar] [CrossRef]
- Borzychowski, A.M.; Sargent, I.L.; Redman, C.W. Inflammation and pre-eclampsia. Semin. Fetal Neonatal Med. 2006, 11, 309–316. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Cottrell, J.; Amaral, L.M.; LaMarca, B. Inflammatory mediators: A causal link to hypertension during preeclampsia. Br. J. Pharmacol. 2019, 176, 1914–1921. [Google Scholar] [CrossRef]
- Wilson, P.C.; Lee, M.H.; Appleton, K.M.; El-Shewy, H.M.; Morinelli, T.A.; Peterson, Y.K.; Luttrell, L.M.; Jaffa, A.A. The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]-AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J. Biol. Chem. 2013, 288, 18872–18884. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; El-Shewy, H.M.; Luttrell, D.K.; Luttrell, L.M. Role of beta-arrestin-mediated desensitization and signaling in the control of angiotensin AT1a receptor-stimulated transcription. J. Biol. Chem. 2008, 283, 2088–2097. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.T.; Krueger, K.M.; Kendall, H.E.; Daaka, Y.; Fredericks, Z.L.; Pitcher, J.A.; Lefkowitz, R.J. Clathrin-mediated endocytosis of the beta-adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-arrestin1. J. Biol. Chem. 1997, 272, 31051–31057. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Harrison-Bernard, L.M.; Fuller, A.J.; Vanderpool, V.; Saifudeen, Z.; El-Dahr, S.S. The Bradykinin B2 receptor gene is a target of angiotensin II type 1 receptor signaling. J. Am. Soc. Nephrol. 2007, 18, 1140–1149. [Google Scholar] [CrossRef] [Green Version]
- Magnussen, E.B.; Vatten, L.J.; Lund-Nilsen, T.I.; Salvesen, K.A.; Davey Smith, G.; Romundstad, P.R. Prepregnancy cardiovascular risk factors as predictors of pre-eclampsia: Population based cohort study. BMJ 2007, 335, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaufils, M.; Uzan, S.; Donsimoni, R.; Colau, J.C. Prevention of pre-eclampsia by early antiplatelet therapy. Lancet 1985, 1, 840–842. [Google Scholar] [CrossRef]
- Atallah, A.; Lecarpentier, E.; Goffinet, F.; Doret-Dion, M.; Gaucherand, P.; Tsatsaris, V. Aspirin for prevention of preeclampsia. Drugs 2017, 77, 1819–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Doorn, R.; Mukhtarova, N.; Flyke, I.P.; Lasarev, M.; Kim, K.; Hennekens, C.H.; Hoppe, K.K. Dose of aspirin to prevent preterm preeclampsia in women with moderate or high-risk factors: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0247782. [Google Scholar] [CrossRef]
- Ding, Y.A.; MacIntyre, D.E.; Kenyon, C.J.; Semple, P.F. Potentiation of adrenaline-induced platelet aggregation by angiotensin II. Thromb. Haemost. 1985, 54, 717–720. [Google Scholar] [CrossRef]
- Swartz, S.L.; Moore, T.J. Effect of angiotensin II on collagen-induced platelet activation in normotensive subjects. Thromb. Haemost. 1990, 63, 87–90. [Google Scholar] [CrossRef]
- Kalinowski, L.; Matys, T.; Chabielska, E.; Buczko, W.; Malinski, T. Angiotensin II AT1 receptor antagonists inhibit platelet adhesion and aggregation by nitric oxide release. Hypertension 2002, 40, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Quan, A. Fetopathy associated with exposure to angiotensin converting enzyme inhibitors and angiotensin receptor antagonists. Early Hum. Dev. 2006, 82, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Shimada, C.; Akaishi, R.; Cho, K.; Morikawa, M.; Kaneshi, Y.; Yamda, T.; Minakami, H. Outomes of 83 fetuses exposed to angiotensin receptor blockers during the second or third trimesters: A literature review. Hypertens. Res. 2015, 38, 308–313. [Google Scholar] [CrossRef]
- Thomas, W.G.; Qian, H.; Chang, C.S.; Karnik, S. Agonist-induced phosphorylation of the angiotensin II (AT(1A)) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. J. Biol. Chem. 2000, 275, 2893–2900. [Google Scholar] [CrossRef] [Green Version]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 522, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Luttrell, L.M. Minireview: More than just a hammer: Ligand “bias” and pharmaceutical discovery. Mol. Endocrinol. 2014, 28, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Kumagai, H.; Motozawa, Y.; Suzuki, J.; Komuro, I. Biased agonism of the angiotensin II type I receptor. Int. Heart J. 2015, 56, 485–488. [Google Scholar] [CrossRef] [Green Version]
- Takezako, T.; Unal, H.; Karnik, S.S.; Node, K. Current topics in angiotensin II type 1 receptor research: Focus on inverse agonism, receptor dimerization and biased agonism. Pharmacol. Res. 2017, 123, 40–50. [Google Scholar] [CrossRef]
- Carvalho-Galvao, A.; Ogunlade, B.; Xu, J.; Silva-Alves, C.R.A.; Mendes-Junior, L.G.; Guimaraes, D.D.; Cruz, J.C.; Queiroz, T.M.; Balarini, C.M.; Braga, V.A.; et al. Central administration of TRV027 improves baroreflex sensitivity and vascular reactivity in spontaneously hypertensive rats. Clin. Sci. 2018, 132, 1513–1527. [Google Scholar] [CrossRef]
- Zanaty, M.; Seara, F.A.C.; Nakagawa, P.; Deng, G.; Mathieu, N.M.; Balapattabi, K.; Karnik, S.S.; Grobe, J.L.; Sigmund, C.D. Beta-arrestin-biased agonist targeting the brain AT1R (angiotensin II type 1 receptor) increases aversion to saline and lowers blood pressure in deoxycorticosterone acetate-salt hypertension. Hypertension 2021, 77, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Cotter, G.; Davison, B.A.; Butler, J.; Collins, S.P.; Ezekowitz, J.A.; Felker, G.M.; Filippatos, G.; Levy, P.D.; Metra, M.; Ponikowski, P.; et al. Relationship between baseline systolic blood pressure and long-term outcomes in acute heart failure patients treated with TRV027: An exploratory subgroup analysis of BLAST-AHF. Clin. Res. Cardiol. 2018, 107, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Liu, Q.; Liu, L.; Yang, W.W.; Zeng, Y. The effect of calcium channel blockers on prevention of preeclampsia in pregnant women with chronic hypertension. Clin, Exp. Obstet. Gynecol. 2015, 42, 79–81. [Google Scholar]
- Wu, H.; Wang, Y.; Wang, G.; Qiu, Z.; Hu, X.; Zhang, H.; Yan, X.; Ke, F.; Zou, A.; Wang, W.; et al. A bivalent antihypertensive vaccine targeting L-type calcium channels and angiotensin AT1 receptors. Br. J. Pharmacol. 2020, 177, 402–419. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Jara, Z.P.; Harford, T.; Saha, P.P.; Pardhi, T.R.; Desnoyer, R.; Karnik, S.S. Novel allosteric ligands of the angiotensin receptor AT1R as autoantibody blockers. Proc. Natl. Acad. Sci. USA 2021, 118, e2019126118. [Google Scholar] [CrossRef]
- Craici, I.; Wagner, S.; Garovic, V.D. Preeclampsia and future cardiovascular risk: Formal risk factor or failed stress test? Ther. Adv. Cardiovasc. Dis. 2008, 2, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paauw, N.D.; Luijken, K.; Franx, A.; Verhaar, M.C.; Lely, A.T. Long-term renal and cardiovascular risk after preeclampsia: Towards screening and prevention. Clin. Sci. 2016, 130, 239–246. [Google Scholar] [CrossRef]
- Hauspurg, A.; Ying, W.; Hubel, C.A.; Michos, E.D.; Ouyang, P. Adverse pregnancy outcomes and future maternal cardiovascular disease. Clin. Cardiol. 2018, 41, 239–246. [Google Scholar] [CrossRef]
- Frost, A.L.; Suriano, K.; Aye, C.Y.L.; Leeson, P.; Lewandowski, A.J. The immediate and long-term impact of preeclampsia on offspring vascular and cardiac physiology in the preterm infant. Front. Pediatr. 2021, 9, 625726. [Google Scholar] [CrossRef]
- Karatza, A.A.; Dimitriou, G. Preeclampsia emerging as a novel risk factor for cardiovascular disease in the offspring. Curr. Pediatr. Rev. 2020, 16, 194–199. [Google Scholar] [CrossRef]
- Barrett, P.M.; McCarthy, F.P.; Kublickiene, K.; Cormican, S.; Judge, C.; Evans, M.; Kublickas, M.; Perry, I.J.; Stenvinkel, P.; Kashan, A.S. Adverse pregnancy outcomes and long-term maternal kidney disease: A systematic review and meta-analysis. JAMA Netw. Open 2020, 3, e1920964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khashan, A.S.; Evans, M.; Kublickas, M.; McCarthy, F.P.; Kenny, L.C.; Stenvinkel, P.; Fitzgerald, T.; Kublickiene, K. Preeclampsia and risk of end stage kidney disease: A Swedish nationwide cohort study. PLoS Med. 2019, 16, e1002875. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, J.H.; Basit, S.; Wohlfahrt, J.; Damholt, M.B.; Boyd, H.A. Pre-eclampsia and risk of later kidney disease: Nationwide cohort study. BMJ 2019, 365, l1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Chen, S.H.; Ho, C.H.; Liang, F.W.; Chu, C.C.; Wang, H.Y.; Lu, Y.H. End-stage renal disease after hypertensive disorders in pregnancy. Am. J. Obstet. Gynecol. 2014, 210, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Vikse, B.E.; Irgens, L.M.; Leivestad, T.; Skjaerven, R.; Iversen, B.M. Preeclampsia and the risk of end-stage renal disease. N. Engl. J. Med. 2008, 359, 800–809. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quitterer, U.; AbdAlla, S. Pathological AT1R-B2R Protein Aggregation and Preeclampsia. Cells 2021, 10, 2609. https://doi.org/10.3390/cells10102609
Quitterer U, AbdAlla S. Pathological AT1R-B2R Protein Aggregation and Preeclampsia. Cells. 2021; 10(10):2609. https://doi.org/10.3390/cells10102609
Chicago/Turabian StyleQuitterer, Ursula, and Said AbdAlla. 2021. "Pathological AT1R-B2R Protein Aggregation and Preeclampsia" Cells 10, no. 10: 2609. https://doi.org/10.3390/cells10102609
APA StyleQuitterer, U., & AbdAlla, S. (2021). Pathological AT1R-B2R Protein Aggregation and Preeclampsia. Cells, 10(10), 2609. https://doi.org/10.3390/cells10102609