
Role of Aberrantly Activated Lysophosphatidic Acid Receptor 1 Signaling Mediated Inflammation in Renal Aging

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vivo Experiments
2.2. Cell Culture and In Vitro Experiments
2.3. Renal Function
2.4. Histological and Microscopic Analyses
2.5. Immunohistochemistry
2.6. Immunofluorescence Analysis
2.7. Western Blot Analysis
2.8. Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
2.9. Small Interfering RNA (siRNA) Transfection in HRPTEpiC
2.10. Statistical Analysis
3. Results
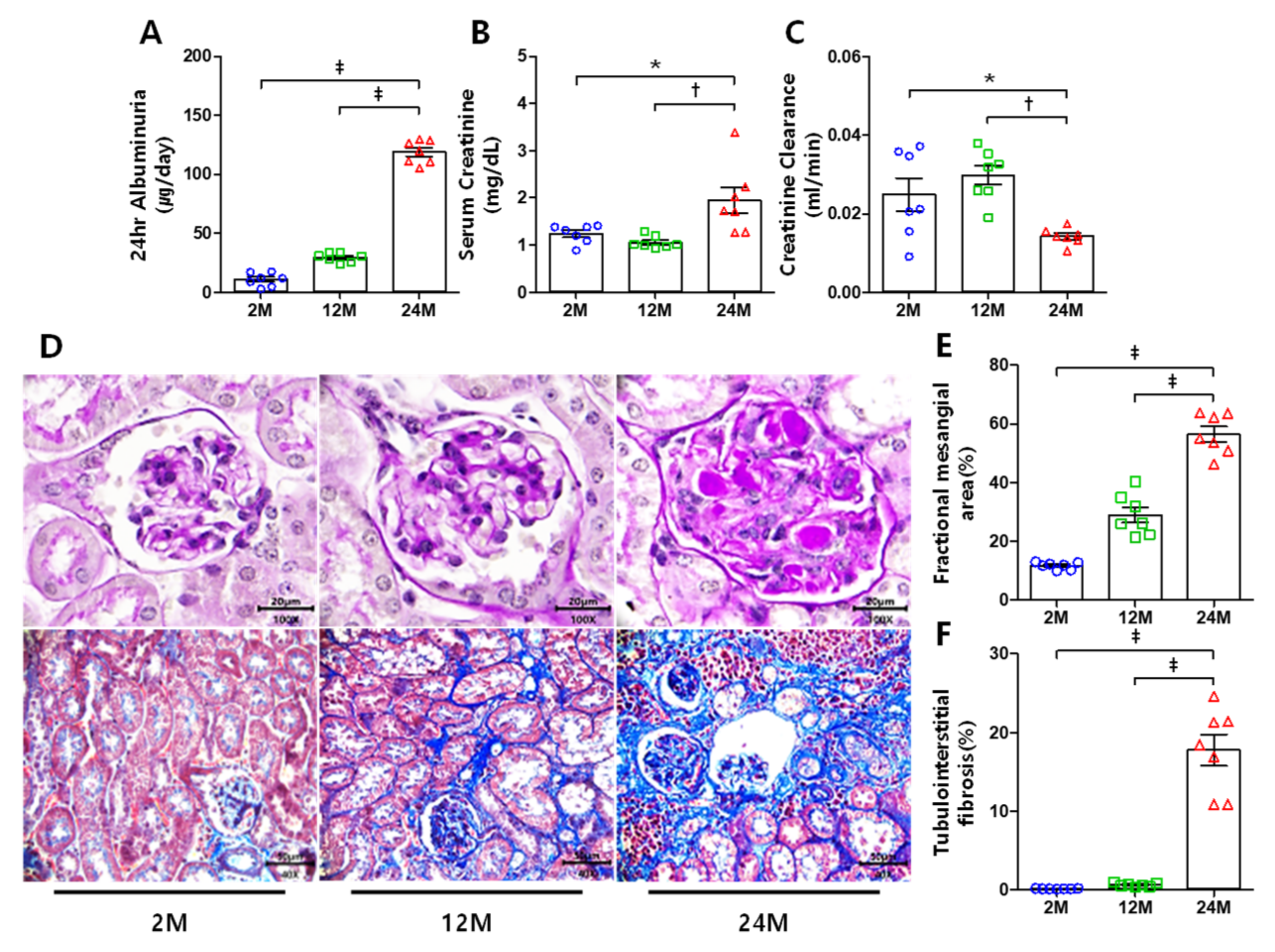
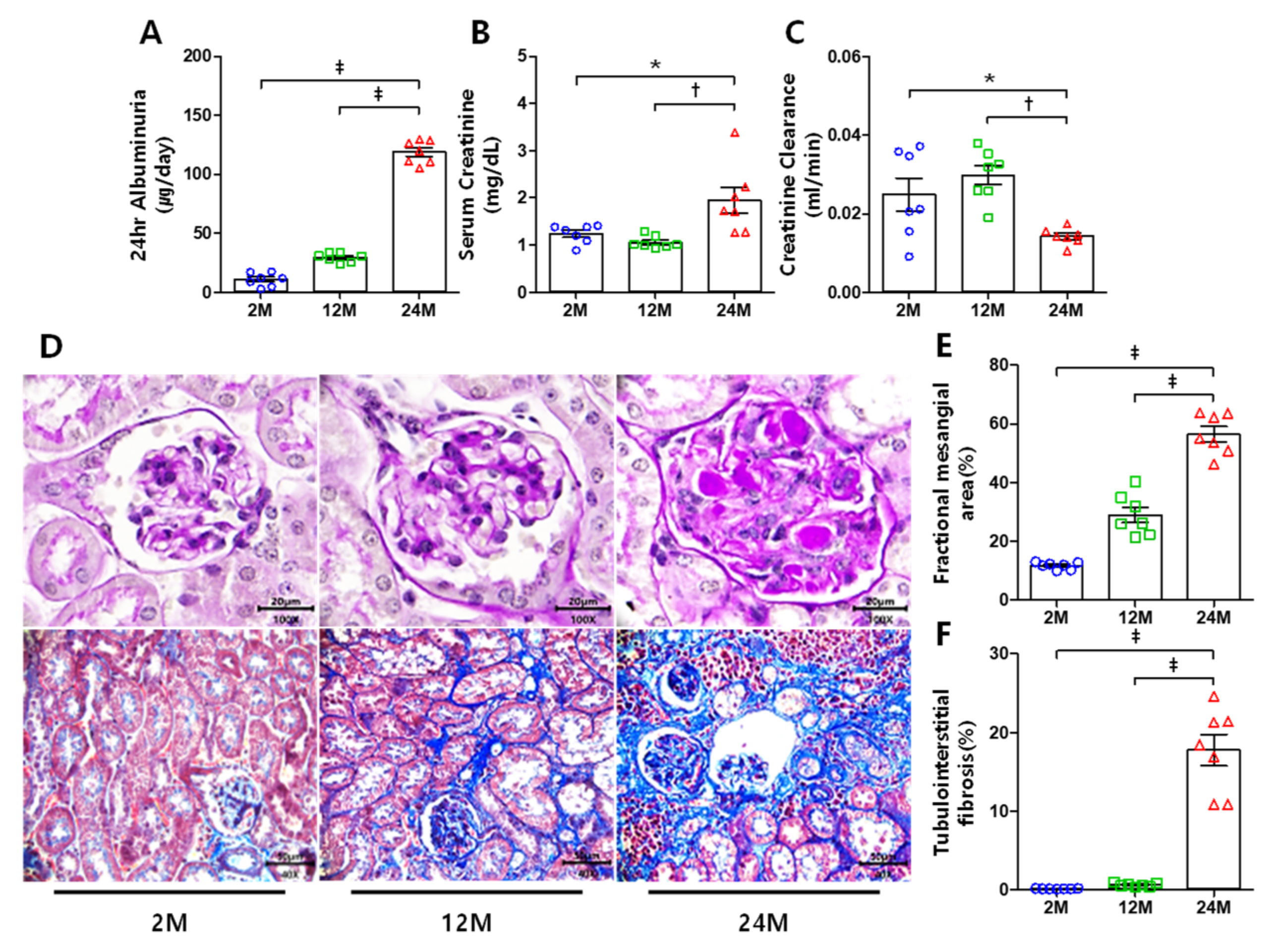
3.1. Renal Function and Histological Changes
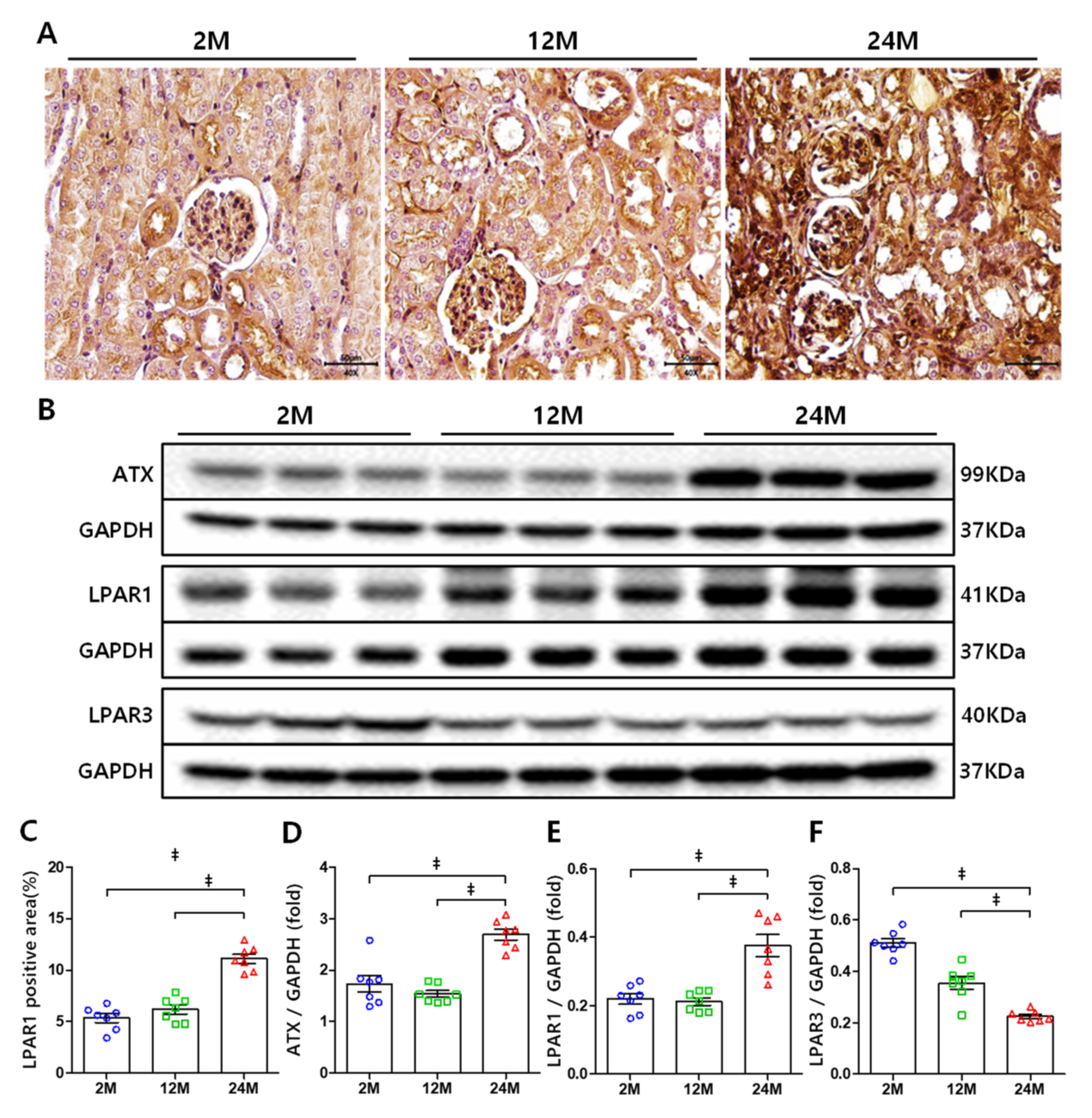
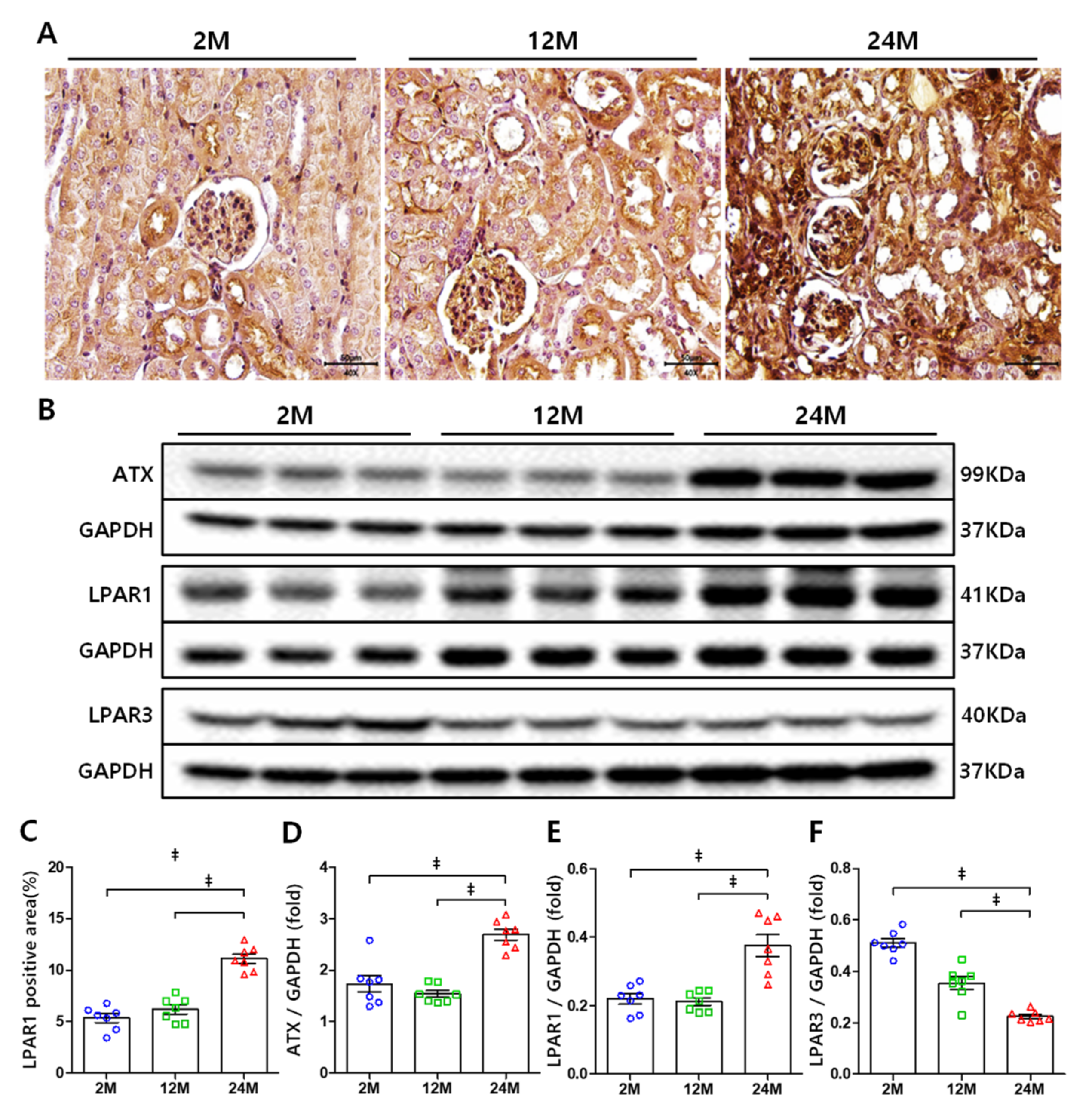
3.2. ATX and LPAR1 Expression Increases in Aged Mice Kidney, but LPAR3 Expression Decreases
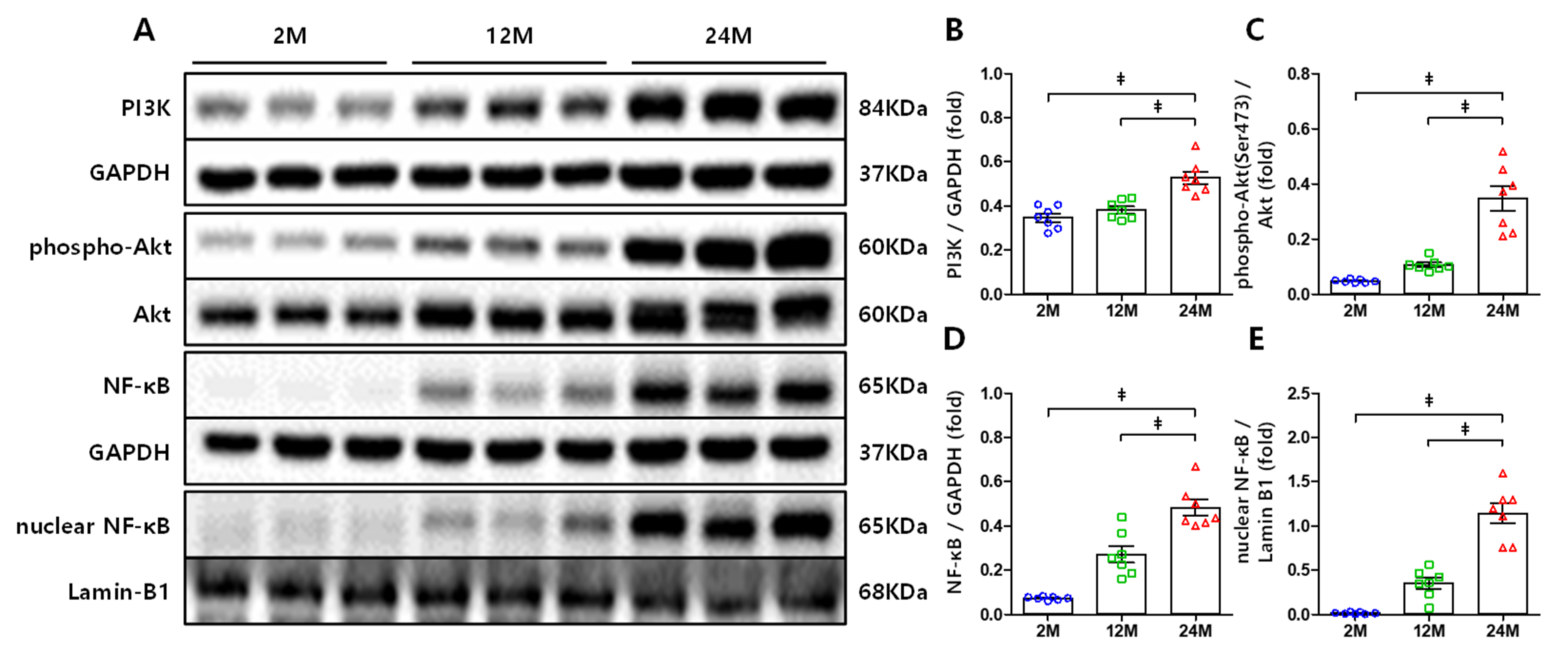
3.3. PI3K, Akt, and NF-κB Expression Increased in the Kidneys of Aged Mice
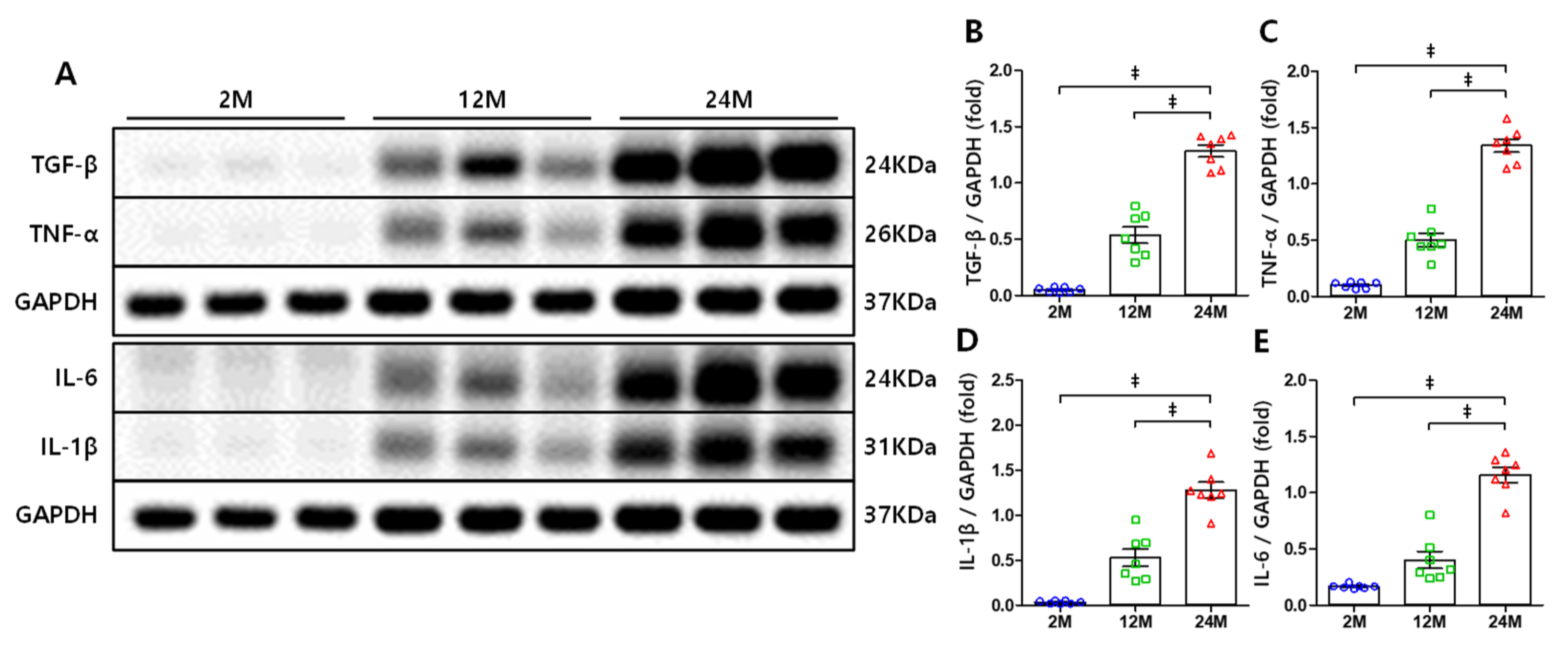
3.4. NF-κB-Mediated Increase in the Expression Levels of TNF-α, IL-1β, and IL-6
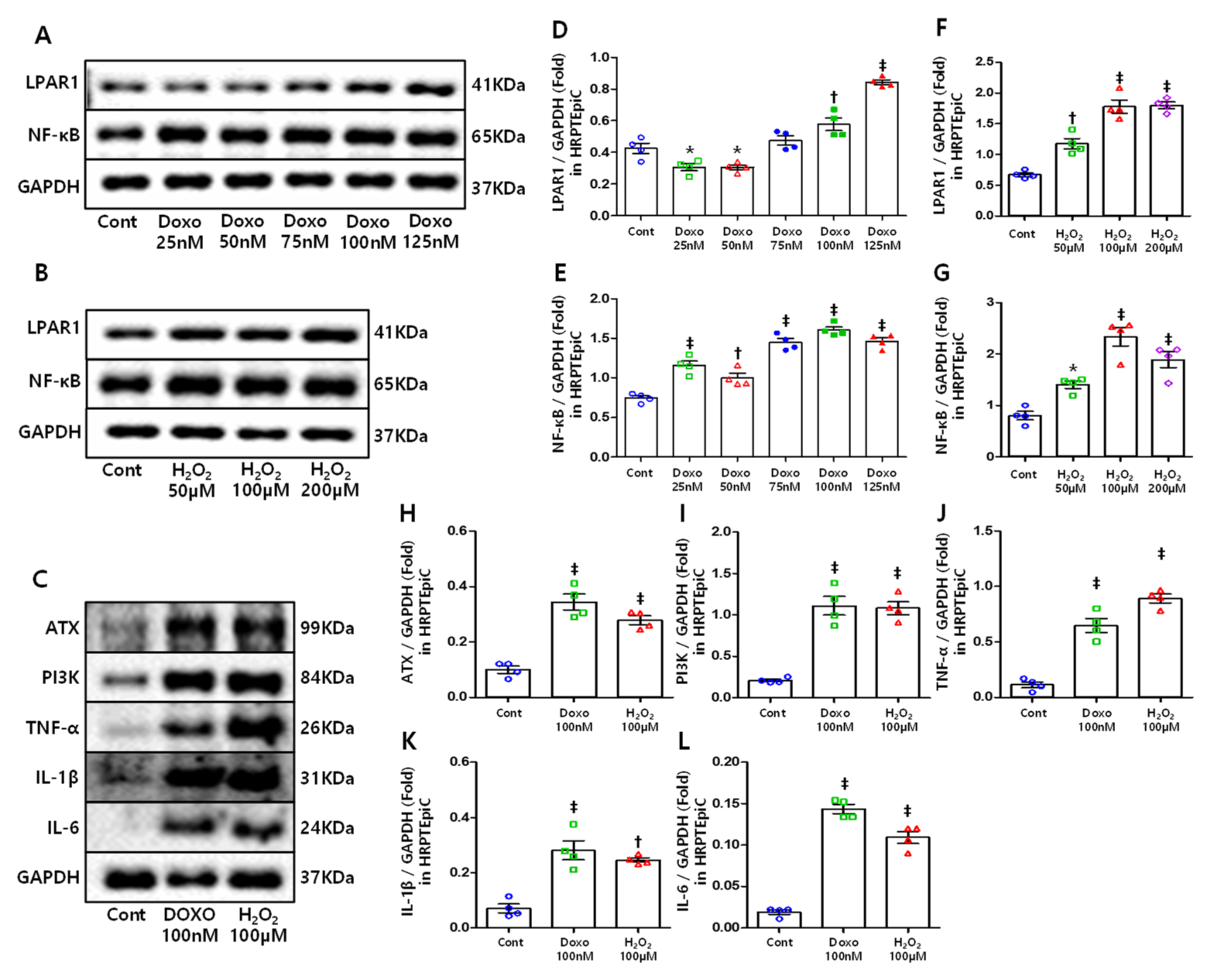
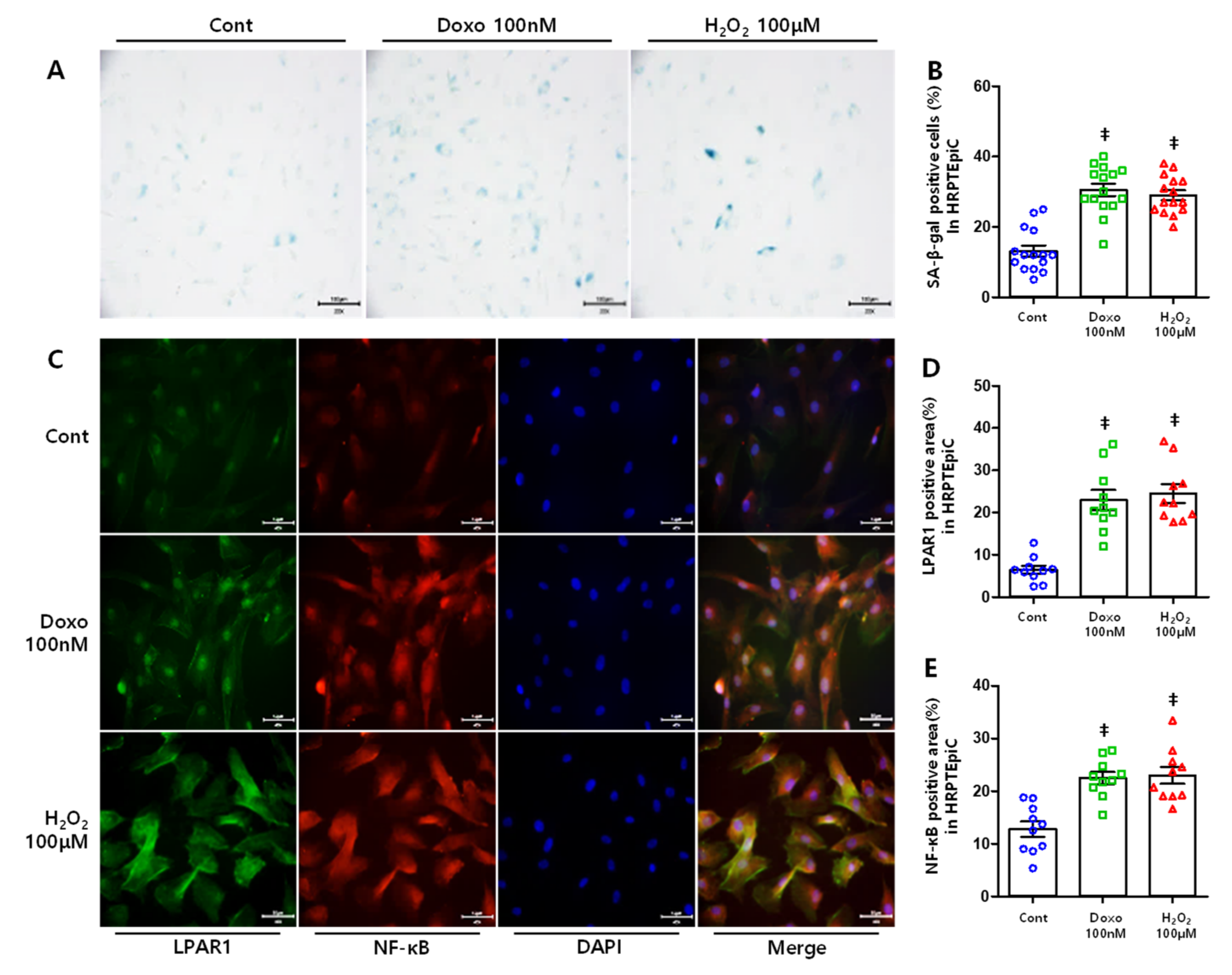
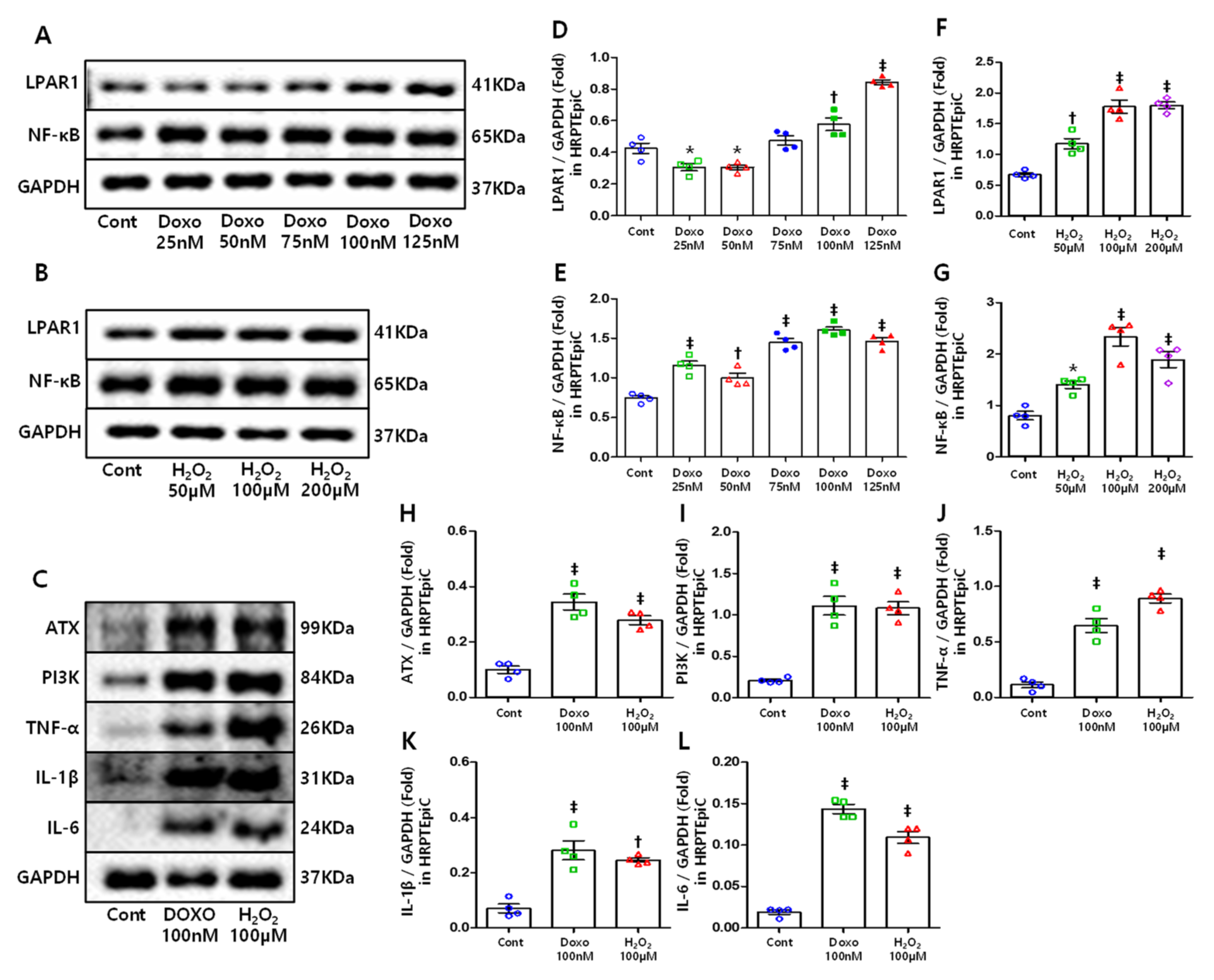
3.5. DOXO and H2O2 Treatment Induced Cellular Senescence Resulting in Increased LPAR1 and NF-κB Levels
3.6. ATX, PI3K, and Inflammatory Cytokines Increased in Senescent Cells
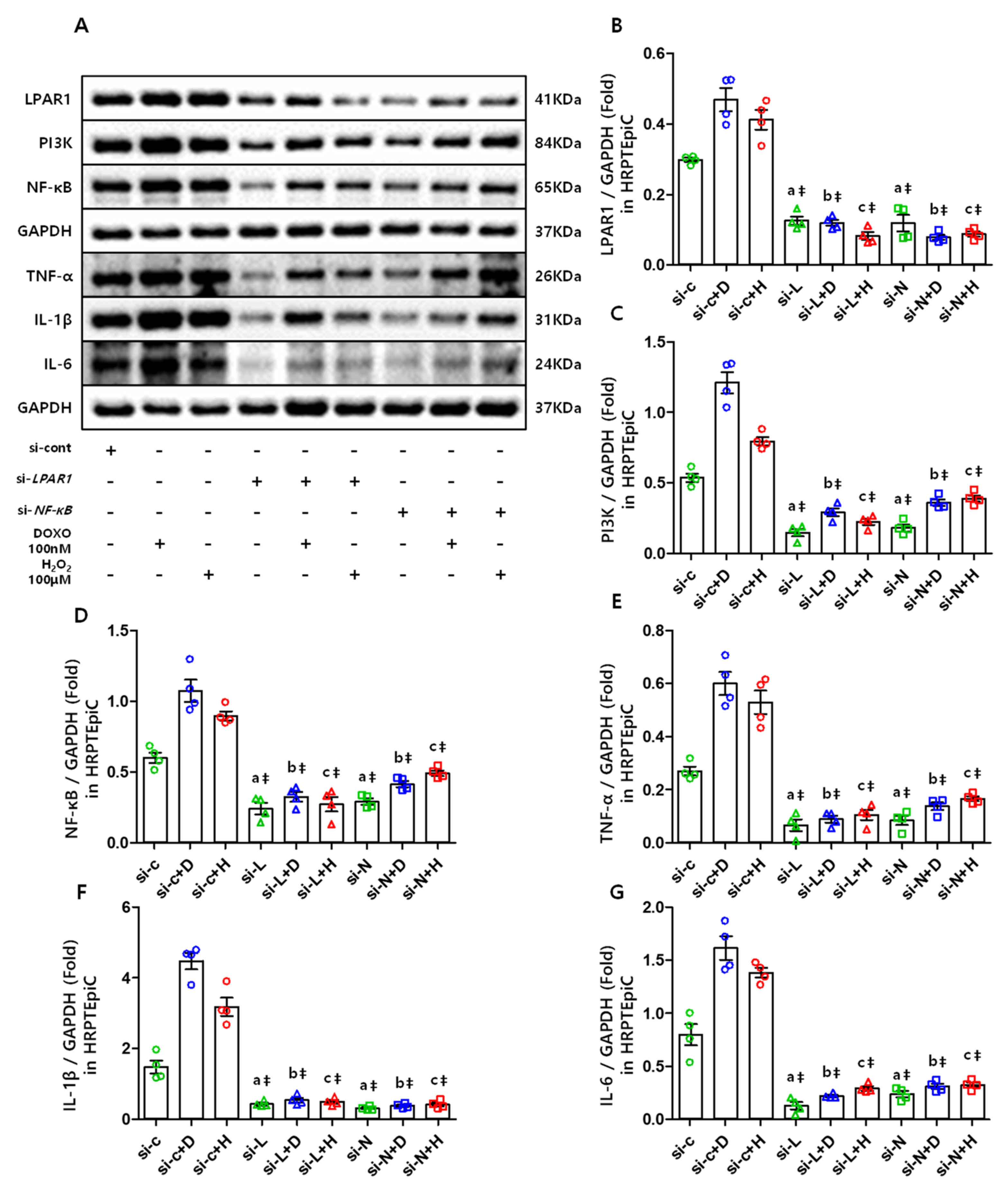
3.7. The LPAR1 Regulates NF-κB and Inflammatory Cytokines Expression Via PI3K, and NF-κB Is Also Essential in Maintaining the LPAR1 Level
4. Discussion
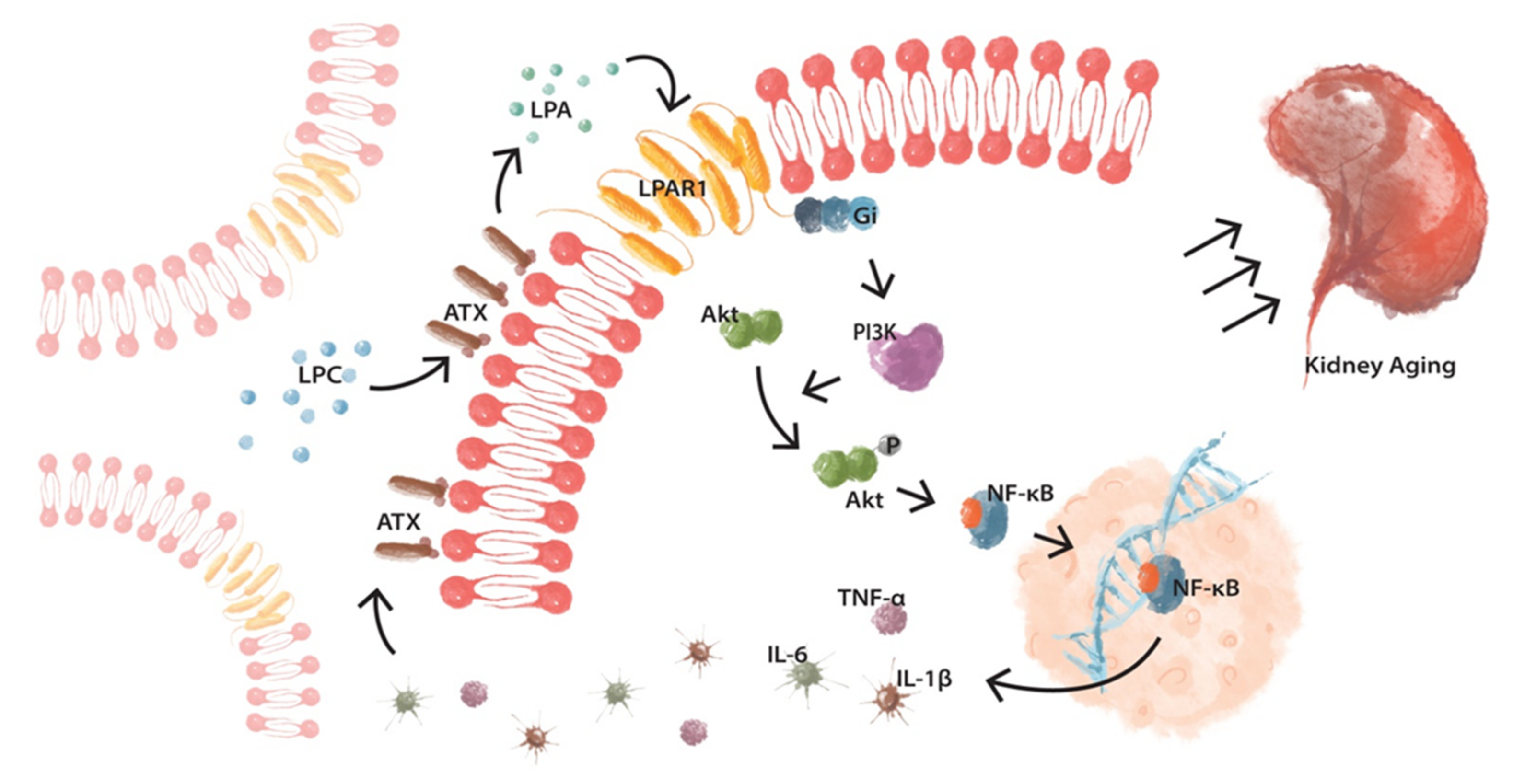
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Yoon, H.E.; Chung, B.H.; Choi, B.S.; Park, C.W.; Yang, C.W.; Kim, Y.S.; Hong, Y.A.; Kim, S.Y.; Chang, Y.K.; et al. Clinical outcomes and effects of treatment in older patients with idiopathic membranous nephropathy. Korean J. Intern. Med. 2019, 34, 1091–1099. [Google Scholar] [CrossRef]
- Medina-Liabres, K.R.P.; Kim, S. Continuous renal replacement therapy in elderly with acute kidney injury. Korean J. Intern. Med. 2020, 35, 740. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Bae, E.H.; Ma, S.K.; Kim, S.W. Association between initial vascular access and survival in hemodialysis according to age. Korean J. Intern. Med. 2019, 34, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.A.; Kim, J.H.; Kim, Y.K.; Chang, Y.K.; Park, C.W.; Kim, S.Y.; Kim, Y.S.; Kang, S.W.; Kim, N.H.; Kim, Y.L.; et al. Low parathyroid hormone level predicts infection-related mortality in incident dialysis patients: A prospective cohort study. Korean J. Intern. Med. 2020, 35, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Ping, H.; Zhang, X.; Xing, N. Prevalence of chronic kidney disease in China. Lancet 2012, 380, 216. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Rothenbacher, D. Prevalence of chronic kidney disease in population-based studies: Systematic review. BMC Public Health 2008, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Collaboration, G.B.D.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Helenius, M.; Hanninen, M.; Lehtinen, S.K.; Salminen, A. Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem J. 1996, 318 Pt 2, 603–608. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-kappaB in Aging and Disease. Aging Dis 2011, 2, 449–465. [Google Scholar]
- Adler, A.S.; Sinha, S.; Kawahara, T.L.; Zhang, J.Y.; Segal, E.; Chang, H.Y. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007, 21, 3244–3257. [Google Scholar] [CrossRef] [Green Version]
- Benesch, M.G.K.; MacIntyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of Age for Autotaxin and Lysophosphatidate Signaling: Clinical Applications for Preventing, Detecting and Targeting Tumor-Promoting Inflammation. Cancers 2018, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, D.; Oh, Y.S.; Jun, H.S. Lysophosphatidic Acid Signaling in Diabetic Nephropathy. Int J. Mol. Sci 2019, 20, 2850. [Google Scholar] [CrossRef] [Green Version]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [Green Version]
- Berkenkamp, B.; Susnik, N.; Baisantry, A.; Kuznetsova, I.; Jacobi, C.; Sorensen-Zender, I.; Broecker, V.; Haller, H.; Melk, A.; Schmitt, R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 2014, 9, e88071. [Google Scholar] [CrossRef] [Green Version]
- Docherty, M.H.; O’Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular Senescence in the Kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.Z.; Wang, X.; Yang, H.; Fogo, A.B.; Murphy, B.J.; Kaltenbach, R.; Cheng, P.; Zinker, B.; Harris, R.C. Lysophosphatidic Acid Receptor Antagonism Protects against Diabetic Nephropathy in a Type 2 Diabetic Model. J. Am. Soc. Nephrol. 2017, 28, 3300–3311. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Klein, J.; Gres, S.; Guigne, C.; Neau, E.; Valet, P.; Calise, D.; Chun, J.; Bascands, J.L.; Saulnier-Blache, J.S.; et al. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2007, 18, 3110–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehira, M.; Fujiwara, T.; Nakajima, S.; Okitsu, Y.; Onishi, Y.; Fukuhara, N.; Ichinohasama, R.; Okada, Y.; Harigae, H. An Lysophosphatidic Acid Receptors 1 and 3 Axis Governs Cellular Senescence of Mesenchymal Stromal Cells and Promotes Growth and Vascularization of Multiple Myeloma. Stem Cells 2017, 35, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.U.; Chou, C.H.; Chao, K.H.; Lee, H.; Lin, C.W.; Lu, H.F.; Yang, Y.S. Lysophosphatidic acid up-regulates expression of growth-regulated oncogene-alpha, interleukin-8, and monocyte chemoattractant protein-1 in human first-trimester trophoblasts: Possible roles in angiogenesis and immune regulation. Endocrinology 2010, 151, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Sapkota, A.; Gaire, B.P.; Choi, J.W. NLRP3 Inflammasome Activation Is Involved in LPA1-Mediated Brain Injury after Transient Focal Cerebral Ischemia. Int J. Mol. Sci 2020, 21, 8595. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.R.; Qi, Z.; Bottinger, E.P.; Breyer, M.D.; Sharma, K. Utility of endogenous creatinine clearance as a measure of renal function in mice. Kidney Int. 2004, 65, 1959–1967. [Google Scholar] [CrossRef] [Green Version]
- Brosius, F.C., 3rd; Alpers, C.E.; Bottinger, E.P.; Breyer, M.D.; Coffman, T.M.; Gurley, S.B.; Harris, R.C.; Kakoki, M.; Kretzler, M.; Leiter, E.H.; et al. Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 2503–2512. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wei, D.; Xiao, H. Methods of cellular senescence induction using oxidative stress. Methods Mol. Biol. 2013, 1048, 135–144. [Google Scholar] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Benesch, M.G.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef] [Green Version]
- Barbayianni, E.; Kaffe, E.; Aidinis, V.; Kokotos, G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 2015, 58, 76–96. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.G.; Ko, Y.M.; McMullen, T.P.; Brindley, D.N. Autotaxin in the crosshairs: Taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014, 588, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.M.; Chiang, J.C.; Lin, Y.C.; Lin, Y.N.; Chuang, P.Y.; Chang, Y.C.; Chen, C.C.; Wu, K.Y.; Hsieh, J.C.; Chen, S.K.; et al. Lysophosphatidic acid receptor LPA3 prevents oxidative stress and cellular senescence in Hutchinson-Gilford progeria syndrome. Aging Cell 2020, 19, e13064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Chan, K.T.; Blake, S.; Zhu, H.; Kang, J.; Trigos, A.S.; Madhamshettiwar, P.B.; Diesch, J.; Paavolainen, L.; Horvath, P.; Hannan, R.D.; et al. A functional genetic screen defines the AKT-induced senescence signaling network. Cell Death Differ. 2020, 27, 725–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Kim, E.N.; Lim, J.H.; Kim, H.D.; Ban, T.H.; Yang, C.W.; Park, C.W.; Choi, B.S. Role of Aberrantly Activated Lysophosphatidic Acid Receptor 1 Signaling Mediated Inflammation in Renal Aging. Cells 2021, 10, 2580. https://doi.org/10.3390/cells10102580
Jin Y, Kim EN, Lim JH, Kim HD, Ban TH, Yang CW, Park CW, Choi BS. Role of Aberrantly Activated Lysophosphatidic Acid Receptor 1 Signaling Mediated Inflammation in Renal Aging. Cells. 2021; 10(10):2580. https://doi.org/10.3390/cells10102580
Chicago/Turabian StyleJin, Yongjie, Eun Nim Kim, Ji Hee Lim, Hyung Duk Kim, Tae Hyun Ban, Chul Woo Yang, Cheol Whee Park, and Bum Soon Choi. 2021. "Role of Aberrantly Activated Lysophosphatidic Acid Receptor 1 Signaling Mediated Inflammation in Renal Aging" Cells 10, no. 10: 2580. https://doi.org/10.3390/cells10102580