Abstract
Sodium-glucose co-transporter 2 inhibitors (SGLT2i) are emerging as a new treatment strategy for heart failure with reduced ejection fraction (HFrEF) and—depending on the wistfully awaited results of two clinical trials (DELIVER and EMPEROR-Preserved)—may be the first drug class to improve cardiovascular outcomes in patients suffering from heart failure with preserved ejection fraction (HFpEF). Proposed mechanisms of action of this class of drugs are diverse and include metabolic and hemodynamic effects as well as effects on inflammation, neurohumoral activation, and intracellular ion homeostasis. In this review we focus on the growing body of evidence for SGLT2i-mediated effects on cardiac intracellular Na+ as an upstream mechanism. Therefore, we will first give a short overview of physiological cardiomyocyte Na+ handling and its deterioration in heart failure. On this basis we discuss the salutary effects of SGLT2i on Na+ homeostasis by influencing NHE1 activity, late INa as well as CaMKII activity. Finally, we highlight the potential relevance of these effects for systolic and diastolic dysfunction as well as arrhythmogenesis.
1. Introduction
SGLT2 inhibitors (SGLT2i) were designed as antidiabetic drugs lowering blood glucose levels by selective inhibition of the sodium-glucose cotransporter 2 (SGLT2) in the renal proximal tubule with consequent increase in glycosuria [1]. Although tight glycemic control, which can be achieved by the addition of SGLT2i to the antidiabetic treatment regimen, has the potential to positively affect major adverse cardiovascular events (MACE) [2,3], the risk of myocardial infarction or stroke was not significantly reduced by treatment with SGLT2i [4,5,6]. Moreover, a mediation analysis of EMPA-REG OUTCOME by Inzucchi et al. demonstrated that the reduction in HbA1c levels by empagliflozin contributed only slightly to the observed cardioprotective effects [7]. These data are corroborated by recent studies in patients with heart failure, in whom SGLT2i prevented hospitalization for heart failure and cardiovascular death in diabetic and nondiabetic patients alike [8,9]. Furthermore, a direct SGLT2-dependent effect on cardiomyocytes can be excluded due to the lack of SGLT2 expression in the heart [10,11,12]. Therefore, many (pre-) clinical trials were performed to gain further mechanistic insights. Consequently, a myriad of potential mechanisms was proposed including direct cardiac effects such as inhibition of cardiac Na+/H+ exchanger 1 (NHE1) [13,14,15], Ca2+/calmodulin-dependent protein kinase II (CaMKII) [10,16], and late Na+ current (late INa) [17]. Intriguingly, these proteins are centrally involved in cardiac Na+ homeostasis, which is fundamentally disrupted in heart failure [18,19,20], making it an interesting target for heart failure treatment.
In this narrative review we shortly summarize the current knowledge on Na+ homeostasis in heart failure and elaborate on potential pathways by which SGLT2i could improve cellular Na+ handling and thus prevent heart failure progression.
2. Cardiac Na+ Handling
2.1. Na+ Handling in the Healthy Heart
In cardiomyocytes Na+ homeostasis is finely tuned by an orchestrated activity of several transporters, ion channels and kinases reflecting its great importance for fundamental cellular processes as excitation-contraction coupling and mitochondrial metabolism.
During the cardiac action potential (AP), activation of voltage-gated Na+ channels (Nav1.5) mediates the upstroke (phase 0) of the cardiac AP and thus allows for the activation of voltage-gated L-type Ca2+ channels (LTCC), leading to Ca2+-induced Ca2+ release (CICR) from the sarcoplasmic reticulum (SR) and finally activation of the contractile apparatus. Since voltage-gated Na+ channels undergo rapid inactivation (few ms) the magnitude of Na+ influx mediated by peak Na+ current (peak INa) is small. However, beside peak INa, a small persistent INa component has been described that is generated by Na+ channels exhibiting a sustained bursting activity even at negative membrane potentials [21]. In addition, during the plateau phase of the cardiac action potential, Na+ current steady-state inactivation and activation can overlap resulting in the generation of a long-lasting INa window current. Both latter Na+ current components (together labelled as late INa) have a low amplitude, but their long duration leads to a significant amount of Na+ entry into the cell [20,22,23,24,25].
Furthermore, Na+ influx can be mediated by the cardiac isoform of the Na+/H+ exchanger (NHE1), which couples the extrusion of one H+ with the influx of one Na+ and is thus also centrally involved in the regulation of intracellular pH (pHi) [26]. In addition, Na+ homeostasis is tightly coupled with cellular Ca2+ homeostasis via the electrogenic sarcolemmal Na2+/Ca2+ exchanger (NCX), which exchanges 3 Na+ for 1 Ca2+ and constitutes—together with SR Ca2+ ATPase 2a (SERCA2a)—the main means of cytosolic Ca2+ removal during diastole [27,28]. Indeed, in the healthy heart, NCX is responsible for the extrusion of all Ca2+ that entered the cell via LTCC in order to maintain a steady-state condition [28,29]. Importantly, NCX can operate in a bidirectional fashion depending on ion gradients across the membrane as well as membrane potential. In “forward mode,” which is favored by increased [Ca2+]i and membrane potentials negative to the Nernst potential of NCX, Ca2+ is removed from the cell, as mentioned above, while Na+ enters the cell. The opposite is true for NCX “reverse mode,” which is promoted by an increase in [Na+]i and positive membrane potentials. Apart from these transporters, which are accountable for most of the Na+-entry (NCX > Nav > NHE) [28,30,31] further Na+ influx is mediated by Na+/HCO3− cotransporter, Na+/K+/2Cl− cotransporter, as well as Na+/Mg2+ exchanger [31,32]. Importantly, while SGLT2 is not expressed in the heart, SGLT1 expression was repeatedly confirmed in healthy hearts and is even upregulated in cardiac disease potentially contributing to Na+ influx [33,34].
The inward gradient for Na+ is maintained by the energy consuming sarcolemmal Na+/K+-ATPase transporting 3 Na+ out of the cell in exchange with 2 K+ and simultaneously building up the negative resting membrane potential, which is primarily dictated by the K+ conductivity of the cell membrane.
Besides its importance in cardiac excitation-contraction coupling, [Na]i is also involved in cellular energy supply as well as mitochondrial redox regulation, as recently reviewed by Bertero et al. [35]. In brief, mitochondrial Ca2+ concentration ([Ca2+]m), which is elevated in parallel to cytosolic Ca2+ levels, e.g., during β-adrenergic stimulation, activates Krebs cycle dehydrogenases to increase the production of reducing equivalents (NADH and NADPH) in order to meet the energetic demand and also to preserve the anti-oxidative capacity of the cell [36]. While mitochondrial Ca2+ uptake is mediated via the mitochondrial Ca2+ uniporter (MCU) [37], activity of the mitochondrial Na+/Ca2+ exchanger (NCLX) primarily causes Ca2+ extrusion linking [Na]i with [Ca2+]m [28,38]. Consequently, an increase in [Na]i results in a decrease in [Ca2+]m, which in turn hampers Krebs cycle activity [36].
Given the role of Na+ homeostasis for cardiac excitation-contraction coupling as well as cardiac metabolism it comes as no surprise that dysregulation of Na+ handling is centrally involved in the development and progression of heart failure.
2.2. Na+ Dysregulation in Heart Failure
Heart failure is characterized by cellular Na+ overload as a consequence of a dysbalance between Na+ in- and efflux. Indeed, bulk cytosolic [Na+]i from failing compared to non-failing myocytes is elevated by ~2 to 6 mM [18,24,31,39,40,41]. An increase in Na+ influx is predominantly caused by (1) an increase in late Na+ current (late INa), (2) an increase in NHE1 activity and (3) increased NCX forward mode due to cytosolic Ca2+ overload in failing cardiomyocytes [31] (Figure 1).
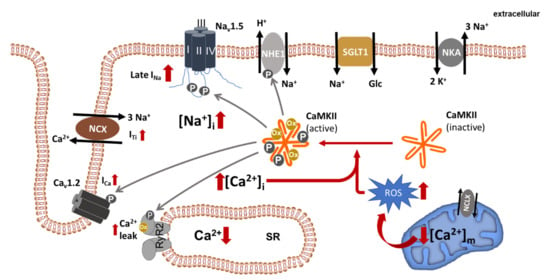
Figure 1.
Dysregulated ion homeostasis in failing cardiomyocytes. Increased late INa as well as increased forward mode NCX (constituting ITi), NHE1- and SGLT1 activity contribute to cellular Na+ overload ([Na+]i). Na+ overload, in turn, promotes cytosolic Ca2+ overload ([Ca2+]i) and mitochondrial Ca2+ depletion ([Ca2+]m) (with increased ROS generation), both of which are able to activate CaMKII, which is centrally involved in the regulation of multiple Na+ and Ca2+ channels/transporters. The solid grey lines indicate known CaMKII phosphorylation targets. NKA, Na+/K+ ATPase; RyR2, ryanodine receptor 2; SR, sarcoplasmic reticulum; NCX, Na+/Ca2+ exchanger; NCLX, mitochondrial Na+/Ca2+ exchanger; ROS, reactive oxygen species.
2.2.1. Late INa
As stated above, the vast majority of voltage-gated Na+ channels inactivate rapidly leading to a marked membrane depolarization but only little Na+ influx. In contrast, there is a small proportion of channels, which remain activated or close and quickly reopen, mediating a sustained Na+ current termed late INa [20,22,23,24,25,42]. The molecular mechanisms for this peculiar gating behavior are not fully understood. Interestingly, in the recent years it has become evident that beside Nav1.5 also neuronal Nav isoforms (e.g., Nav 1.8) [43,44,45,46] are involved in the generation of late INa. Even though this current is small under physiological conditions (~0.5% of peak INa), it is upregulated in heart failure, where it consequently gains further impact on AP duration and is a major source of cytosolic Na+ overload [19,20,23,25,47]. Several mechanisms might account for the increase in late INa in the failing heart including hypoxia [48], mechanical [49] and oxidative stress [19,23,25]. In this context, Ca2+/Calmodulin-dependent kinase IIδ (CaMKIIδ), a serine/threonine kinase that is markedly upregulated in heart failure and involved in heart failure development and progression [50,51,52], occupies a central role. In the recent years, a plethora of evidence has accumulated for CaMKII-dependent stimulation of late INa in a variety of cardiac diseases [24,47,53]. For example, augmentation of late INa by reactive oxygen species (ROS) with consequent disturbances of Na+ and Ca2+ homeostasis requires oxidative activation of CaMKII [25,54]. In accordance, three CaMKII phosphorylation sites were detected at the Nav1.5 I-II linker loop (S516, S571 and T594) [55,56]. Interestingly, the reported shift of INa steady state inactivation (SSI) to more negative potentials and increase in late INa induced by CaMKII-dependent Nav1.5 phosphorylation [24] could only be reproduced by phosphorylation of S571 [56], while phosphorylation of S516 and T594 resulted in a leftward shift of SSI without increasing late INa [55]. To add further complexity, Na+ channel β-subunit composition might additionally be relevant for the regulation of late INa [57]. In conclusion, while the relevance of specific phosphorylation sites is still under debate, there is consensus that CaMKII-dependent Na+ channel phosphorylation is the major driver for late INa augmentation in heart failure. However, although inhibition of late INa e.g., by ranolazine has been shown to significantly decrease [Na+]i in human heart failure [19], this current alone cannot account for the observed Na+ overload in failing cardiomyocytes [24,58].
2.2.2. NHE1
Indeed, several studies demonstrated an upregulation of NHE1 activity in heart failure [59,60]. In a rabbit model of combined pressure and volume overload induced heart failure, Baartscheer and colleagues showed an increased Na+ influx, which could be blunted by inhibition of the increased NHE1 activity with the specific inhibitor cariporide [60]. Interestingly, cariporide could also partly rescue the disturbed Ca2+ handling in these myocytes [60]. In a follow-up experiment using the same rabbit model, this working group was also able to show that chronic cariporide treatment (for 3 months after operation) prevented heart failure development by alleviating structural and cellular remodeling [61]. It is still unclear, if the observed increase of NHE1 activity in heart failure is due to an increase in channel expression, posttranslational modification of the channel, or both. We have recently reported that NHE1 expression is markedly upregulated in ventricular tissue of patients with end-stage heart failure as compared to patients with left ventricular hypertrophy and normal systolic function [15]. Intriguingly, NHE1 expression has also been shown to be upregulated in the failing right ventricles of a rat model of pulmonary hypertension [62]. Furthermore, treatment of neonatal rat ventricular cardiomyocytes with aldosterone, a known driver of cardiac hypertrophy and heart failure, results in an increased NHE1 expression with consequent stimulation of hypertrophic signaling, which was ameliorated by cariporide treatment [63]. On the other hand, Yokoyama et al. did not observe any alteration in cardiomyocyte NHE1 expression in a small cohort of patients with chronic end-stage heart failure undergoing heart transplantation compared to non-used donor hearts, despite a markedly increased NHE activity in the diseased hearts [59] pointing to a relevance of post-translational modification. Importantly, NHE1 has been shown to be readily phosphorylated by several kinases—including CaMKII—resulting in a stimulation of channel function [64,65].
2.2.3. Na+/K+ ATPase
As stated above, Na+ overload can also result from a reduced capacity to extrude Na+ from the cytosol, a process, which is mainly mediated by the Na+/K+ ATPase. Several studies have shown a decrease in the expression level of the Na+/K+ ATPase in animal models of heart failure as well as human heart failure [66,67,68]. Furthermore there might be a shift in the composition of different isoforms of the α subunit (α1, α2, α3), which show different affinities to Na+, as well as the β subunits (β1 and β2, with β1 being the prevailing isoform in the heart) [39,67,68,69,70]. While these subunit shifts could result in different activity and affinity of the pump, the available data are inconsistent maybe due to species differences or differences in the etiology or stage of heart failure. The expression level and phosphorylation of phospholemman (PLM), the main regulator of Na+/K+ ATPase activity, by protein kinases A and C (PKA, PKC) adds a further layer of complexity, especially as data are again conflicting. In a rabbit model of volume-overload-induced heart failure PLM appeared to be hyperphosphorylated [69], while downregulation of inhibitor-1 (I-1) with consequent increase in protein phosphatase-1 (PP-1) activity resulted in PLM hypophosphorylation in human heart failure [71]. Intriguingly, several studies failed to observe a reduction in Na+/K+ ATPase activity in the failing heart [18,60] making the contribution of this enzyme to Na+ overload still a matter of debate.
2.2.4. NCX
In contrast to the Na+/K+ ATPase the relevance of NCX is better characterized. NCX has a special role in ion dysbalance in heart failure as its activity is tightly coupled to [Na+]i and [Ca2+]i, as described above (see Section 2.1). Importantly, a majority of studies have shown that NCX expression and activity are upregulated in heart failure [72,73,74] with potentially far-reaching consequences.
On the one hand, an increase in [Na+]i may induce NCX reverse mode in the early phase of the action potential and hamper Ca2+ extrusion via NCX forward mode later on potentially contributing to SR Ca2+ loading, which might in turn mitigate contractile dysfunction [75,76]. On the other hand slowing of diastolic Ca2+ removal from the cytosol contributes to diastolic dysfunction [77,78]. However, during the cardiac cycle NCX primarily operates in its forward mode, which in combination with a decreased SERCA activity and an increased SR Ca2+ leak unloads the SR resulting in decreased Ca2+ transient amplitude [72,79,80,81]. At the same time the NCX-mediated Na+-influx generates a depolarizing transient inward current (Iti) potentially causing cellular pro-arrhythmic events in form of delayed afterdepolarizations (DAD) [82,83]. Moreover, the increase in [Na+]i indirectly reduces the availability of reducing equivalents, which not only leads to a deficit in ATP but also contributes to oxidative stress (as outlined in Section 2.1). Consequently, a vicious cycle with ROS-induced CaMKII-activation and subsequent cellular Ca2+ and Na+ overload with its deleterious effects on cardiac function develops (Figure 1).
3. Effects of SGLT2i on Cardiac Na+ Homeostasis
There is overwhelming evidence that SGLT2i are cardioprotective in diabetic and non-diabetic patients. Consequently, a vast number of trials were conducted to decipher the underlying mechanisms. While the proposed mechanisms are multifaceted, a striking number of observations are directly or indirectly linked to effects on cellular Na+ handling, as set out below (Figure 2).
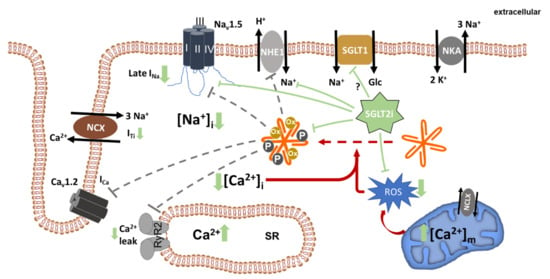
Figure 2.
Beneficial effects of SGLT2i on cellular ion homeostasis. SGLT2i directly or indirectly inhibit NHE1, late INa, and CaMKII, leading to a reduction in cellular Na+ and Ca2+ overload. As a consequence of reduced [Na+] and possibly also because of direct mitochondrial effects of SGLT2i, mitochondrial ROS generation is decreased. Dashed grey lines indicate potentially reduced interaction of CaMKII with its known targets. Solid green lines indicate inhibition mediated by SGLT2i. NKA, Na+/K+ ATPase; RyR2, ryanodine receptor 2; SR, sarcoplasmic reticulum; NCX, Na+/Ca2+ exchanger; NCLX, mitochondrial Na+/Ca2+ exchanger; ROS, reactive oxygen species.
3.1. Inhibition of NHE1
There are several studies showing that inhibition of NHE1 (usually by its specific inhibitor cariporide) prevents development or worsening of heart failure [60,61,84,85,86,87]. The first work assuming an effect of SGLT2i on NHE1 was by Baartscheer et al. who observed that treatment of isolated ventricular cardiomyocytes of healthy mice and rabbits with empagliflozin reduced [Na+]i as well as systolic and diastolic [Ca2+]i probably due to decreased NHE1 activity [13]. In addition [Ca2+]m, assessed by the use of a mitochondrially targeted fluorescence resonance energy transfer (FRET-)-based Ca2+ indicator (mitoCam), increased [13]. In a later work the same group could reproduce NHE1 inhibition by dapagliflozin and canagliflozin in healthy murine cardiomyocytes and suggested direct binding of SGLT2i to the Na+ binding site of NHE1 by applying molecular docking studies [14]. Furthermore, we could recently show inhibition of NHE1-dependent pH recovery also in isolated human atrial cardiomyocytes, suggesting a potential role for the observed cardioprotection in clinical trials [15]. Interestingly, in a HFpEF model using Dahl salt-sensitive rats fed with high salt diet, chronic treatment with dapagliflozin (6 weeks) attenuated Ca2+ and Na+ overload and increased Ca2+ transient amplitude [88]. Furthermore, upregulation of NHE1, SGLT1, CaMKII, Nav1.5, and NCX1 in the diseased hearts was found to be attenuated by chronic dapagliflozin treatment. Importantly, the authors could not detect acute effects of dapagliflozin treatment on [Na+]i or [Ca2+]i in isolated myocytes. Unfortunately, NHE1 function in cardiomyocytes was not assessed in this study. However, inhibition of NHE1 activity by dapaglifozin was demonstrated in human umbilical vein endothelial cells (HUVEC) [88]. Noteworthy, Chung et al. could not detect NHE1 inhibition or a decrease in [Na+]i by SGTL2i treatment neither in unpaced healthy human nor animal (mouse, rat and guinea pig) cardiomyocytes [89]. Interestingly, in patch clamp experiments they also assessed Na+/K+ ATPase current without differences between treatment groups. To address the conflicting data, Zuurbier et al. tested the influence of differences in the experimental setup (e.g., different pH-buffering systems, pacing of cells, extracellular pH, DMSO concentration) on the contradictory results and concluded that different conditions could not account for the discrepancies [90]. After transformation of the SBFI ratio-data of Chung et al. into [Na+]i they even suggest that there is also a detectable decrease in [Na+]i upon treatment with empagliflozin [90]. Overall, if NHE1 inhibition by SGLT2i can indeed convey a decrease in [Na+]i in healthy cardiomyocytes is debatable, especially when considering that NHE1 activity at normal intracellular pH is low and contributes only little to cellular Na+ loading [91]. In a mouse model of ischemia-reperfusion (IR), empagliflozin delayed the time to onset of contracture as well as IR injury similar to cariporide, again pointing to a NHE1-dependent effect [92]. However, if a potential NHE1 inhibition by SGLT2i might contribute to a decrease in Na+ influx in failing cardiomyocytes with disturbed pH regulation [93] has not been systematically investigated to date.
3.2. Inhibition of Late INa
Since late INa is a major contributor to Na+ overload in heart failure and inhibition of this current has proven to be cardioprotective in many models of cardiac disease [19,94,95], the idea of SGLT2i-mediated inhibition of this current seems appealing. Indeed, a recent study using mice with TAC (transverse aortic constriction)-induced heart failure as well as HEK293T cells transfected with Nav 1.5 channels harboring LQT3 mutations (causing increased late INa) showed that empagliflozin was able to significantly reduce late INa without affecting peak INa under the chosen experimental conditions (holding potential −120 mV, pacing frequency 1 Hz) [17]. This desirable preference for late INa over peak INa is shared by class Ib anti-arrhythmic drugs as well as ranolazine, but might—as for these drugs—become less pronounced under more physiological membrane potentials and at higher heart rates [96,97]. In a next step they observed that the incidence of spontaneous calcium transients evoked by treatment of healthy murine cardiomyocytes with the alkaloid veratridine, a specific stimulator of late INa, was rapidly and reversibly reduced. Applying a homology-modeling approach based on the structure of human Nav1.4 Philippaert and colleagues developed a transmembrane model of human Nav1.5. Molecular docking simulations and introduction of targeted mutations revealed the amino acids F1760 and W1345 in the fDIII-DIV site as putative interaction partners with SGLT2i [17]. Intriguingly, F1760 was also reported to be elementary in the binding of local anesthetics (e.g., lidocaine) [97,98] as well as ranolazine [99]. However, apart from a direct interaction between SGLT2i and Nav1.5 channels, there is also the possibility of an indirect inhibition of late INa by interference with current regulation e.g., by CaMKII, a possibility, which was not investigated so far.
3.3. Inhibition of CaMKII
CaMKII is a serine/threonine kinase, which is markedly upregulated in the failing heart and contributes fundamentally to the development and progression of heart failure due to detrimental effects on cardiac excitation-contraction as well as excitation-transcription coupling [16,50,51,52,81,100]. Apart from the above mentioned augmentation of late INa by direct channel phosphorylation (see Section 2.2.1), CaMKII is able to phosphorylate ryanodine receptors (RyR2) at Serine 2814 thus increasing channel open probability with consequent spontaneous diastolic Ca2+ release events (i.e., Ca2+ sparks) that in turn may prompt further diastolic SR Ca2+ release of adjacent RyR2 clusters giving rise to diastolic Ca2+ waves [100]. Ca2+ sparks as well as waves result in an elevated diastolic [Ca2+]i and activate NCX forward mode resulting in a depolarizing Na+ influx (Iti) that triggers delayed afterdepolarizations (DAD) [83] and contributes to cellular Na+ overload. Intriguingly, CaMKII is also known to stimulate NHE1 activity [64]. Thus, CaMKII inhibition could also be accountable for the observed inhibition of NHE1 activity upon SGLT2i treatment.
Importantly, we could recently show that acute exposure of isolated healthy and failing (5 weeks after TAC) murine cardiomyocytes to empagliflozin at a clinically relevant dose (1 µmol/L) for 24 h reduced CaMKII activity as assessed by HDAC4 pulldown assays [10]. This was accompanied by a decreased CaMKII-dependent RyR2 phosphorylation at S2814 not only in murine but also in human failing cardiomyocytes. In line with this, empagliflozin treatment reduced Ca2+ spark frequency and increased SR Ca2+ load as well as Ca2+ transient amplitude, as a cellular surrogate for improved contractile function. Furthermore, measurement of SBFI ratios revealed a reduction in bulk [Na+]i in healthy murine ventricular myocytes after 24 h of treatment. Interestingly, while the effects on CaMKII activity as well as Ca2+ transient amplitude were only observable after incubation with empagliflozin for 24 h, we could detect a decrease in subsarcolemmal [Na+] as assessed by application of the Nernst equation to the reversal potential of NCX currents, which were measured by whole cell patch clamp, as early as 30 min after start of treatment. Consistently, Pabel et al. reported no change in systolic Ca2+ transient amplitude, Ca2+ transient decay kinetics, or diastolic Ca2+ concentration in human failing cardiomyocytes upon acute empagliflozin exposure (15 min) [101]. The effects of prolonged SGLT2i treatment on Ca2+ handling were reproduced in a rat model of metabolic syndrome, where two weeks of dapagliflozin treatment also improved Ca2+ transient amplitude, SR Ca2+ load and increased left ventricular developed pressure [102]. Surprisingly, peak INa was reported to be significantly reduced in this study, while bulk [Na+]i was not influenced by dapagliflozin [102]. In addition, the authors ascribed shortening of the prolonged QTc time and action potential duration to an increase in repolarizing K+ currents as well as the decrease in peak INa. However, if a reduction of late INa might be involved, remained elusive. Overall, the beneficial effects were attributed to an improved mitochondrial function with decreased ROS production. Unfortunately, the final link between oxidative stress and the observed effects, which might e.g., be a decrease in CaMKII oxidation, was not investigated [102].
In summary, to date it is unclear, how SGLT2i interact with CaMKII activity. However, given the described time course of SGLT2i effects on CaMKII activity, Ca2+- and Na+ -handling it is possible that reduced CaMKII activity might be downstream of the direct SGLT2i target. Potential upstream targets could be late INa, NHE1 and/or reduced oxidative stress.
3.4. Effect on Other Na+ Transporters (SGLT1 and SMIT1)
In contrast to SGLT2, which is not present in the heart, two other members of the SLC5A gene family, namely SGLT1 (SLC5A1) and the sodium-myoinositol cotransporter-1 (SMIT1, SLC5A3) are expressed in the heart and have been shown to contribute to Na+ influx in cardiac disease [33,34,103,104]. In a mouse model of MI (LAD ligation) induced heart failure, SGLT1 expression was significantly upregulated and pretreatment with the specific inhibitor KGA-2727 attenuated left ventricular systolic dysfunction and fibrosis [105]. Importantly, except for sotagliflozin, which was designed as a dual SGLT1/SGLT2 inhibitor, empagliflozin (2680-fold), dapagliflozin (1242-fold), and canagliflozin (155-fold) exhibit an increased selectivity for SGLT2 over SGLT1 making a considerable contribution of SGLT1 inhibition in vivo unlikely [106].
On the other hand, myocardial overexpression of SMIT1 was shown to mediate hyperglycemia-induced NOX2 activation with consequent ROS production [11]. If this is also relevant in heart failure without concomitant hyperglycemia, is still unknown. Noteworthy, IC50 of empagliflozin for SMIT1 is 8.3 µmol/L, which is beyond the average plasma concentration of the drug [107].
Consequently, although conceivable, a relevant contribution of SGLT1 and SMIT1 to SGLT2i-mediated decrease in [Na+]i seems unlikely, but might warrant further investigation.
4. Potential Role of Reduced [Na+]i for Cardioprotection by SGLT2i
Keeping in mind that SGLT2i-mediated cardioprotection is primarily based on reduction in heart failure events (e.g., hospitalization for heart failure) rather than on vascular endpoints (myocardial infarction, stroke) we, in the following, focus on direct effects on cardiac function.
In the light of the central role of Na+ dysregulation for heart failure and the growing number of studies reporting effects of SGLT2i on Na+ handling, it is tempting to speculate that restoration of Na+ homeostasis might be pivotal. However, which of the observed beneficial effects on cardiac function and structure can indeed be explained by improved Na+ handling?
4.1. SGLT2i and Oxidative Stress
Oxidative stress is a hallmark of cardiac disease including heart failure [108,109] and centrally involved in pathways promoting hypertrophy, fibrosis, cell death, as well as directly or indirectly (e.g., via oxidation of different kinases) involved in perturbation of cellular ion homeostasis [25,54,110]. Cytosolic Na+ overload results in an extrusion of Ca2+ from the mitochondria mediated by the NCLX. This mitochondrial Ca2+ deficit impairs Ca2+-induced stimulation of Krebs cycle dehydrogenases, which ultimately causes a shortage in the reducing equivalents NADH and NADPH [36]. Consequently, anti-oxidative enzymes such as glutathione peroxidase cannot be adequately regenerated. Importantly, as discussed above, oxidative stress can in turn cause cellular Na+ accumulation (e.g., by stimulation of late INa) leading to a vicious cycle [25]. Thus, inhibiting Na+-influx by SGLT2i might mitigate oxidative stress as a central player in cardiac pathogenesis.
First reports of anti-oxidative effects of SGLT2i stem from diabetic mouse and rat models, where treatment with ipragliflozin reduced plasma and liver levels of biomarkers of inflammation (IL-6, TNFα, CRP) and oxidative stress (thiobarbituric acid reactive substances and protein carbonyl) [111,112].
Since then, several studies also reported anti-oxidative properties of SGLT2i in the heart. Intriguingly, in human ventricular cardiomyocytes isolated from HFpEF patients acute treatment with empagliflozin (60 min) significantly reduced markers of oxidative stress (H2O2, GSH, lipid peroxidation, and 3-nitrotyrosine) as well as inflammatory markers (ICAM-1, VCAM-1, TNFα, and IL-6) [113]. Furthermore, in a diabetic mouse model 8 weeks of empagliflozin treatment resulted in a decrease in oxidative stress by activation of Nrf2/ARE signaling and downregulation of NADPH-oxidase 4 (NOX4) [114]. Of note, in non-diabetic rats with left ventricular dysfunction after MI, treatment with empagliflozin for 2 weeks after surgery again decreased myocardial oxidative stress but also increased pyruvate dehydrogenase (PDH) activity and ATP levels, improved left ventricular systolic function, reduced myocardial fibrosis and hypertrophy [115]. In another interesting study by Durak et al. using metabolic syndrome rats dapagliflozin not only reduced oxidative stress by improving mitochondrial membrane potential as well as mitochondrial fusion-fission and increased ATP/ADP ratio, but also shortened QTc time, reduced peak INa, and improved cellular Ca2+ handling (e.g., increased Ca2+ transients, increased SR Ca2+ load) [102]. Although none of these studies provided a direct link between the reduction of oxidative stress and a decrease in [Na+]i, it is conceivable that restoration of Na+ homeostasis contributes to these observations (Figure 3). However, further studies on the interplay between SGLT2i, [Na+]i, mitochondrial function and generation of ROS in heart failure are warranted.
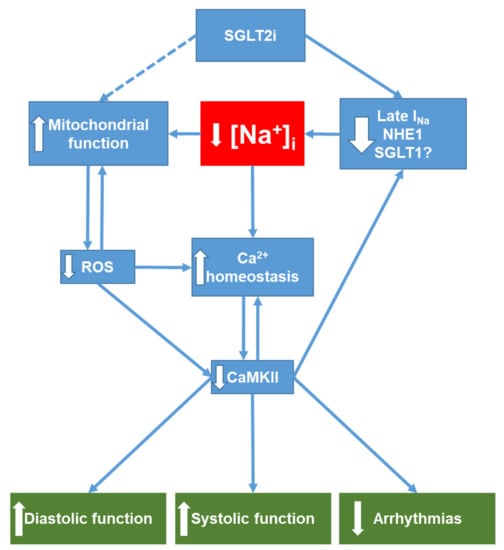
Figure 3.
Overview of potential SGLT2i targets relevant for ion homeostasis in the heart. SGLT2i directly reduces (via inhibition of Late INa, NHE1 and potentially SGLT1) intracellular Na+ accumulation in the failing heart. Decreased [Na+]i in turn improves mitochondrial function (see Section 4.1) and thus may contribute to the decrease in mitochondrial ROS formation observed with SGLT2i treatment. Reduction of oxidative stress attenuates oxidative CaMKII activation, which, together with decreased [Na+]i, improves cellular Ca2+ handling and leads to improved diastolic and systolic function. Moreover, both restoration of Ca2+ homeostasis and CaMKII inhibition are antiarrhythmic. Downward-pointing arrows denote reduction or inhibition of a signaling pathway. Upward-pointing arrows denote stimulation or enhancement of a pathway. NHE1, Na+/H+ exchanger 1; SGLT1, sodium-glucose co-transporter 1; ROS, reactive oxygen species; CaMKII, Ca2+/calmodulin-dependent protein kinase II; [Na+]i, cytosolic Na+ concentration.
4.2. SGLT2i and Contractile Function
When dealing with heart failure, it is important to differentiate between HFrEF and HFpEF. While patients with HFpEF display a normal systolic function but impaired diastolic function, HFrEF is defined by a decrease in cardiac systolic contractility (in addition to diastolic dysfunction) and the severity of contractile dysfunction is positively correlated with the occurrence of major adverse cardiac events including hospitalizations for heart failure as well as cardiovascular death [116,117]. Consequently, improving systolic contractile function (without increasing cardiac energy demand) in these patients, e.g., through ameliorated Na+ homeostasis, might be a key to improving cardiovascular outcomes [118,119,120].
There are several studies reporting an increase in LVEF upon SGLT2i treatment. In a small randomized, double-blind, and placebo-controlled clinical trial (n = 84) Santos-Gallego et al. reported an improvement in LVEF (6.0 ± 4.2 vs. −0.1 ± 3.9; p < 0.001) as well as a decrease in LV dimensions in non-diabetic HFrEF patients within 6 months of treatment with dapagliflozin [121]. A similar increase in LVEF was observed in a retrospective analysis of type II diabetic patients with HFrEF 24 months after initiation of SGLT2i treatment [122]. In a rat model of myocardial infarction (MI)-induced heart failure empagliflozin (start of administration 4 weeks after MI and continued for further 4 weeks) slightly improved contractile function and prevented renal insufficiency. Apart from normalization of increased SGLT2 expression these effects were primarily ascribed to inhibition of renal NHE3 [123]. Unfortunately, it remains unclear if the amelioration of cardiac dysfunction in this study of cardiorenal syndrome is due to improved renal function or due to direct cardiac effects [123]. Interestingly, in a non-diabetic mouse model of TAC-induced heart failure, empagliflozin treatment prevented worsening of LVEF assessed by echocardiography in vivo as well as ex vivo by an isolated perfused working heart system (excluding a role of extrinsic factors such as hemodynamics or nephroprotection) [124].
Basically, as mentioned above, improved contractility could, at least in part, be explained by the restoration of cardiac Na+-homeostasis with consequent beneficial effects on Ca2+ handling (Figure 3). However, only a few studies observing an improved contractile function in vivo also systematically investigated the underlying pathways. Proposed mechanisms include amelioration of adverse remodeling by activation of cardiac GTP enzyme cyclohydrolase 1 (cGCH1) with consequent activation of eNOS and nNOS resulting in an increase of NO levels and a decrease in O2− and nitrotyrosine levels [125]. In another study, dapagliflozin was shown to improve cardiac remodeling and contractile function by inhibition of the mitogen-activated protein kinases (MAPK) JNK and p38 [126]. Interestingly, as MAPK are readily activated by ROS [127], oxidative stress might again play a central role.
4.3. SGLT2i and Diastolic Function
Even broader pre-clinical evidence than for improved systolic function exists for SGLT2i mediated attenuation of diastolic dysfunction. For instance, Tanaka et al. showed that in a cohort of 53 type II diabetic patients with heart failure (HFpEF, HFmrEF, and HFrEF) administration of dapagliflozin improved left ventricular longitudinal function assessed by global longitudinal strain (via speckle-tracking strain analysis) and left ventricular diastolic function (E/e’) within 6 months [128]. Similar beneficial effects on diastolic function in patients with type II diabetes mellitus were reproduced in smaller trials using empagliflozin or canagliflozin [129,130]. In another interesting randomized, double-blind, and placebo-controlled study Rau et al. demonstrated that empagliflozin improved diastolic function (E/e´) as early as one day after initiation in diabetic patients, while non-invasively assessed hemodynamic parameters (stroke volume index, cardiac index, vascular resistance index or pulse rate) were unchanged [131]. While the results from two large clinical trials investigating the effects of empagliflozin (EMPEROR-Preserved [132]) or dapagliflozin (DELIVER [133]) in reducing cardiovascular death and heart failure events in non-diabetic patients with HFpEF are awaited for late 2021 and early 2022, respectively, there is pre-clinical evidence for an SGLT2i-mediated attenuation of diastolic dysfunction irrespective of a diabetic condition. Connelly and colleagues used unilateral nephrectomy or implantation of DOCA (deoxycorticosterone acetate) pellets in combination with high-salt diet to induce hypertension with a consequent HFpEF phenotype [134]. In this model empagliflozin treatment for 5 weeks improved diastolic function and reduced myocardial hypertrophy without affecting fibrosis [134]. Recently, in a porcine model of MI-induced HFrEF, treatment with empagliflozin for 2 months was shown to improve diastolic function as assessed by transthoracic echocardiography, cardiac magnetic resonance imaging as well as invasive hemodynamics [135]. This was associated with reduced myocardial fibrosis, reduced oxidative stress as well as improved eNOS-NO-cGMP-PKG signaling with consequent increase in titin phosphorylation, which contributes—when hypophosphorylated—to cardiomyocyte stiffness and diastolic dysfunction [135,136]. In line with this, recent studies in rodent as well as human HFrEF and HFpEF demonstrated improved diastolic function upon empagliflozin treatment due to reduced passive myofilament stiffness, which was also explained by enhanced titin phosphorylation [101,113]. Again improved NO-sGC-cGMP-PKG signaling was observed, which was, at least in part, due to attenuation of oxidative pathways [101,113]. However, the reasons for decreased oxidative stress remain elusive as the observed downregulation of inflammatory pathways could also be secondary [113]. Of note, reduced [Na+]i (e.g., due to decreased late INa) as well as inhibition of CaMKII can contribute to both reduced oxidative stress and inhibition of inflammatory pathways (e.g., by inhibition of the NLRP3 inflammasome) [17] and might thus be centrally involved in SGLT2i-mediated amelioration of diastolic dysfunction (Figure 3).
4.4. SGLT2i and Arrhythmias
Heart failure patients are at an increased risk for atrial as well as ventricular arrhythmias. While there may be pronounced heterogeneities regarding the pathomechanisms of different types of arrhythmias, they all have one thing in common: arrhythmias occur when arrhythmic substrates (structural and/or electrical abnormalities, e.g., fibrosis) and triggers (early (EADs) and delayed afterdepolarizations (DADs)) coincide [137]. Typically, EADs result from prolongation of the APD allowing for the reactivation of LTCC. APD prolongation in turn is caused either by a decrease in repolarizing currents (i.e., outward K+ currents) and/or an increase in depolarizing currents (e.g., late INa) [23,40,110,138]. On the other hand, DADs result from spontaneous diastolic Ca2+ release from the SR via RyR2, which activates a transient inward current that is mainly mediated by NCX. Spontaneous SR Ca2+ leak is either due to RyR2 dysfunction or an increased SR Ca2+ load. The latter one usually results from Na+ overload with consequent cellular Ca2+ overload [139,140]. Importantly, oxidative stress as well as increased CaMKII activity (partly due to oxidative activation) play a critical role in the underlying electrophysiological alterations [25,54,81,141]. Against this background it is tempting to speculate that SGLT2i, which were shown to reduce [Na+]i as well as oxidative stress and inhibit CaMKII, also possess anti-arrhythmic effects (Figure 3). Apart from its potential effects on arrhythmic triggers SGLT2i have repeatedly been shown to also positively affect the arrhythmic substrate, especially by reduction of fibrosis, which can be explained by anti-inflammatory and anti-fibrotic effects that might also be mediated by restoration of intracellular Ca2+ and Na+ homeostasis [17,142]. In this regard it is important to mention that CaMKII activity in the diseased heart also regulates inflammation e.g., by stimulation of NFκB with consequent expression of complement factor B within cardiomyocytes [143,144,145].
Interestingly, in an explorative analysis of the DECLARE-TIMI 58 trial the incidence of atrial fibrillation (AF)/atrial flutter (AFL) in the dapagliflozin group was reduced by ~19% in this high-risk collective of type II diabetic patients independent of a history of heart failure, cardiovascular disease, or pre-existing AF [146]. In addition, in a huge cohort study with almost 160,000 diabetic patients the adjusted hazard ratio for new onset of arrhythmia (including supraventricular and ventricular arrhythmias) was 0.83 (CI 0.751–0.916) in the SGLT2i group [147]. Finally, a recently published meta-analysis including 22 trials with in sum 52,115 patients suggests that SGLT2i reduce the risk of AF, embolic stroke, and ventricular tachycardia (VT) with consistent associations for patients with diabetes mellitus, heart failure, or chronic kidney disease [148]. Thus, although evidence from prospective trials investigating the effects of SGLT2i on arrhythmic events as a primary outcome is still lacking, one can assume that SGLT2i have anti-arrhythmic properties.
This notion is further supported by a pre-clinical study. In a rat model of streptozotocin-induced type II diabetes 8 weeks of empagliflozin treatment reduced AF inducibility, reduced atrial interstitial fibrosis, and improved mitochondrial function [149]. These alterations were accompanied by an increased expression of PGC-1a, Nrf1, and Tfam, which are involved in mitochondrial biogenesis, as well as an increase in the expression of Mfn-1, OPA-1, and Drp-1, which are central to the regulation of mitochondrial fission/fusion [149].
Intriguingly, empagliflozin also reduced the occurrence of spontaneous calcium transients induced by the late INa activator veratridine in isolated healthy murine cardiomyocytes [17].
Overall, although anti-arrhythmic effects of SGLT2i seem plausible from our current understanding of the pharmacodynamic features of this class of drugs, further clinical and preclinical studies are warranted to corroborate this assumption and to clarify the potential underlying mechanisms.
5. Conclusions
In this narrative review we focused on the effects of SGLT2i on cardiomyocyte Na+ handling and its potential implications for the observed cardioprotective effects in vivo.
Due to the great scientific interest in the identification of the cellular targets mediating the cardioprotective effects of SGLT2i a plethora of potential mechanisms of action was proposed in the recent years. As it is unlikely that one substance interacts with an array of pathway specific targets, it is tempting to speculate that SGLT2i positively influence maybe only a couple of targets that are far upstream in the pathological processes of cardiac disease in general and heart failure in particular. CaMKII overactivation, Na+- and Ca2+-overload, and oxidative stress are hallmarks of heart failure, tightly interrelated and common basis for features of cardiac disease such as contractile and diastolic dysfunction, cardiac hypertrophy, and fibrosis. Consequently, interference of SGLT2i with these pathways as one of maybe a few major mechanisms is an appealing thought. Indeed, as outlined in this review, a growing body of evidence has shown that SGLT2i directly or indirectly inhibit CaMKII, reduce oxidative stress, and restore [Na+]i (potentially due to inhibition of late INa, NHE1 and/or other Na+ -transporters (SGLT1, SMIT1)) and [Ca2+]i. However, in regard to these effects, it is still unclear what is chicken and what is egg. Does inhibition of CaMKII normalize ion homeostasis and decrease ROS levels or is it the other way around? To address this issue further research, e.g., using specific knock-out models, is required and will also aid to understand which of the observed beneficial effects of SGLT2i in vivo indeed rely on this common pathway.
Author Contributions
Conceptualization, M.T. and S.W.; writing—original draft preparation, M.T.; writing—review and editing, S.W., M.T. and J.R.; visualization, M.T. and S.W. All authors have read and agreed to the published version of the manuscript.
Funding
M.T. is funded by the Else Kröner-Fresenius-Stiftung (EKFS). J.R. is funded by the German Cardiac Society (DGK). S.W. is supported by the German Research Foundation (DFG) [WA 2539/4-1, 5-1, 7-1, and 8-1] and by the DFG SFB 1350 grant (Project Number 387509280, TPA6).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: Characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Rinaldi, L.; Lascar, N.; Marrone, A.; Pafundi, P.C.; Adinolfi, L.E.; Marfella, R. Role of Tight Glycemic Control during Acute Coronary Syndrome on CV Outcome in Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 3106056. [Google Scholar] [CrossRef]
- Marfella, R.; Sasso, F.C.; Cacciapuoti, F.; Portoghese, M.; Rizzo, M.R.; Siniscalchi, M.; Carbonara, O.; Ferraraccio, F.; Torella, M.; Petrella, A.; et al. Tight glycemic control may increase regenerative potential of myocardium during acute infarction. J. Clin. Endocrinol. Metab. 2012, 97, 933–942. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights From a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018, 41, 356–363. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.P.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef]
- Van Steenbergen, A.; Balteau, M.; Ginion, A.; Ferté, L.; Battault, S.; de Ravenstein, C.M.; Balligand, J.-L.; Daskalopoulos, E.-P.; Gilon, P.; Despa, F.; et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci. Rep. 2017, 7, 41166. [Google Scholar] [CrossRef]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2010, 1, 57–92. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wüst, R.C.I.; Fiolet, J.W.T.; Stienen, G.J.M.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef]
- Trum, M.; Riechel, J.; Lebek, S.; Pabel, S.; Sossalla, S.T.; Hirt, S.; Arzt, M.; Maier, L.S.; Wagner, S. Empagliflozin inhibits Na+ /H+ exchanger activity in human atrial cardiomyocytes. ESC Heart Fail. 2020, 7, 4429–4437. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.; Wagner, S.; Maier, L.S.; Mustroph, J. CaMKII and GLUT1 in heart failure and the role of gliflozins. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165729. [Google Scholar] [CrossRef] [PubMed]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. The Cardiac Late Sodium Channel Current is a Molecular Target for the Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef]
- Sossalla, S.; Wagner, S.; Rasenack, E.C.L.; Ruff, H.; Weber, S.L.; Schöndube, F.A.; Tirilomis, T.; Tenderich, G.; Hasenfuss, G.; Belardinelli, L.; et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts--role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 2008, 45, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, C.R.; Chu, W.W.; Pu, J.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.J.; Makielski, J.C. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Undrovinas, A.I.; Fleidervish, I.A.; Makielski, J.C. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ. Res. 1992, 71, 1231–1241. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 1998, 98, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Shryock, J.C.; Wagner, S.; Maier, L.S.; Belardinelli, L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 2006, 318, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Ruff, H.M.; Weber, S.L.; Bellmann, S.; Sowa, T.; Schulte, T.; Anderson, M.E.; Grandi, E.; Bers, D.M.; Backs, J.; et al. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ. Res. 2011, 108, 555–565. [Google Scholar] [CrossRef]
- Cingolani, H.E.; Ennis, I.L. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Despa, S.; Bers, D.M. Na+ transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Bridge, J.H.; Smolley, J.R.; Spitzer, K.W. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science 1990, 248, 376–378. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ and Na+ pump rate in rat and rabbit ventricular myocytes. J. Physiol. 2002, 539, 133–143. [Google Scholar] [CrossRef]
- Grandi, E.; Herren, A.W. CaMKII-dependent regulation of cardiac Na(+) homeostasis. Front. Pharmacol. 2014, 5, 41. [Google Scholar] [CrossRef]
- Bers, D. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc. Res. 2003, 57, 897–912. [Google Scholar] [CrossRef]
- Banerjee, S.K.; McGaffin, K.R.; Pastor-Soler, N.M.; Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 2009, 84, 111–118. [Google Scholar] [CrossRef]
- Lambert, R.; Srodulski, S.; Peng, X.; Margulies, K.B.; Despa, F.; Despa, S. Intracellular Na+ Concentration (Na+i) Is Elevated in Diabetic Hearts Due to Enhanced Na+-Glucose Cotransport. J. Am. Heart Assoc. 2015, 4, e002183. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Prates Roma, L.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Bay, J.; Kohlhaas, M.; Maack, C. Intracellular Na+ and cardiac metabolism. J. Mol. Cell. Cardiol. 2013, 61, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S. Intracellular Na in animal models of hypertrophy and heart failure: Contractile function and arrhythmogenesis. Cardiovasc. Res. 2003, 57, 887–896. [Google Scholar] [CrossRef]
- Shryock, J.C.; Song, Y.; Rajamani, S.; Antzelevitch, C.; Belardinelli, L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc. Res. 2013, 99, 600–611. [Google Scholar] [CrossRef]
- Pieske, B.; Maier, L.S.; Piacentino, V.; Weisser, J.; Hasenfuss, G.; Houser, S. Rate dependence of Na+i and contractility in nonfailing and failing human myocardium. Circulation 2002, 106, 447–453. [Google Scholar] [CrossRef]
- Horvath, B.; Bers, D.M. The late sodium current in heart failure: Pathophysiology and clinical relevance. ESC Heart Fail. 2014, 1, 26–40. [Google Scholar] [CrossRef]
- Yang, T.; Atack, T.C.; Stroud, D.M.; Zhang, W.; Hall, L.; Roden, D.M. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 2012, 111, 322–332. [Google Scholar] [CrossRef]
- Biet, M.; Barajas-Martínez, H.; Ton, A.-T.; Delabre, J.-F.; Morin, N.; Dumaine, R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na(+) channels. J. Mol. Cell. Cardiol. 2012, 53, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Bengel, P.; Ahmad, S.; Tirilomis, P.; Trum, M.; Dybkova, N.; Wagner, S.; Maier, L.S.; Hasenfuß, G.; Sossalla, S. Contribution of the neuronal sodium channel NaV1.8 to sodium- and calcium-dependent cellular proarrhythmia. J. Mol. Cell. Cardiol. 2020, 144, 35–46. [Google Scholar] [CrossRef]
- Pabel, S.; Ahmad, S.; Tirilomis, P.; Stehle, T.; Mustroph, J.; Knierim, M.; Dybkova, N.; Bengel, P.; Holzamer, A.; Hilker, M.; et al. Inhibition of NaV1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res. Cardiol. 2020, 115, 20. [Google Scholar] [CrossRef] [PubMed]
- Toischer, K.; Hartmann, N.; Wagner, S.; Fischer, T.H.; Herting, J.; Danner, B.C.; Sag, C.M.; Hund, T.J.; Mohler, P.J.; Belardinelli, L.; et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell. Cardiol. 2013, 61, 111–122. [Google Scholar] [CrossRef]
- Tang, Q.; Ma, J.; Zhang, P.; Wan, W.; Kong, L.; Wu, L. Persistent sodium current and Na+/H+ exchange contributes to the augmentation of the reverse Na+/Ca2+ exchange during hypoxia or acute ischemia in ventricular myocytes. Pflug. Arch. Eur. J. Physiol. 2012, 463, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Beyder, A.; Strege, P.R.; Reyes, S.; Bernard, C.E.; Terzic, A.; Makielski, J.; Ackerman, M.J.; Farrugia, G. Ranolazine decreases mechanosensitivity of the voltage-gated sodium ion channel Na(v)1.5: A novel mechanism of drug action. Circulation 2012, 125, 2698–2706. [Google Scholar] [CrossRef] [PubMed]
- Hoch, B.; Meyer, R.; Hetzer, R.; Krause, E.G.; Karczewski, P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 1999, 84, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Kirchhefer, U. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc. Res. 1999, 42, 254–261. [Google Scholar] [CrossRef]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA 2009, 106, 2342–2347. [Google Scholar] [CrossRef]
- Koval, O.M.; Snyder, J.S.; Wolf, R.M.; Pavlovicz, R.E.; Glynn, P.; Curran, J.; Leymaster, N.D.; Dun, W.; Wright, P.J.; Cardona, N.; et al. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 2012, 126, 2084–2094. [Google Scholar] [CrossRef]
- Wagner, S.; Dantz, C.; Flebbe, H.; Azizian, A.; Sag, C.M.; Engels, S.; Möllencamp, J.; Dybkova, N.; Islam, T.; Shah, A.M.; et al. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J. Mol. Cell. Cardiol. 2014, 75, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [PubMed]
- Hund, T.J.; Koval, O.M.; Li, J.; Wright, P.J.; Qian, L.; Snyder, J.S.; Gudmundsson, H.; Kline, C.F.; Davidson, N.P.; Cardona, N.; et al. A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Investig. 2010, 120, 3508–3519. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Kyle, J.W.; Undrovinas, A. Late Na+ current produced by human cardiac Na+ channel isoform Nav1.5 is modulated by its beta1 subunit. J. Physiol. Sci. JPS 2009, 59, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Puglisi, J.L.; Wagner, S.; Maier, L.S.; Severi, S.; Bers, D.M. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys. J. 2007, 93, 3835–3847. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, H.; Gunasegaram, S.; Harding, S.E.; Avkiran, M. Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J. Am. Coll. Cardiol. 2000, 36, 534–540. [Google Scholar] [CrossRef]
- Baartscheer, A. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc. Res. 2003, 57, 1015–1024. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; van Borren, M.M.G.J.; Belterman, C.N.W.; Coronel, R.; Opthof, T.; Fiolet, J.W.T. Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc. Res. 2005, 65, 83–92. [Google Scholar] [CrossRef]
- Chen, L.; Gan, X.T.; Haist, J.V.; Feng, Q.; Lu, X.; Chakrabarti, S.; Karmazyn, M. Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline-induced pulmonary vascular injury by the Na+-H+ exchange inhibitor cariporide. J. Pharmacol. Exp. Ther. 2001, 298, 469–476. [Google Scholar]
- Karmazyn, M.; Liu, Q.; Gan, X.T.; Brix, B.J.; Fliegel, L. Aldosterone increases NHE-1 expression and induces NHE-1-dependent hypertrophy in neonatal rat ventricular myocytes. Hypertension 2003, 42, 1171–1176. [Google Scholar] [CrossRef]
- Vila-Petroff, M.; Mundiña-Weilenmann, C.; Lezcano, N.; Snabaitis, A.K.; Huergo, M.A.; Valverde, C.A.; Avkiran, M.; Mattiazzi, A. Ca(2+)/calmodulin-dependent protein kinase II contributes to intracellular pH recovery from acidosis via Na(+)/H(+) exchanger activation. J. Mol. Cell. Cardiol. 2010, 49, 106–112. [Google Scholar] [CrossRef]
- Voelkl, J.; Pasham, V.; Ahmed, M.S.E.; Walker, B.; Szteyn, K.; Kuhl, D.; Metzler, B.; Alesutan, I.; Lang, F. Sgk1-dependent stimulation of cardiac Na+/H+ exchanger Nhe1 by dexamethasone. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2013, 32, 25–38. [Google Scholar] [CrossRef]
- Dixon, I.M.; Hata, T.; Dhalla, N.S. Sarcolemmal Na(+)-K(+)-ATPase activity in congestive heart failure due to myocardial infarction. Am. J. Physiol. 1992, 262, C664–C6671. [Google Scholar] [CrossRef]
- Schwinger, R.H.; Wang, J.; Frank, K.; Müller-Ehmsen, J.; Brixius, K.; McDonough, A.A.; Erdmann, E. Reduced sodium pump alpha1, alpha3, and beta1-isoform protein levels and Na+,K+-ATPase activity but unchanged Na+-Ca2+ exchanger protein levels in human heart failure. Circulation 1999, 99, 2105–2112. [Google Scholar] [CrossRef]
- Shamraj, O.I.; Grupp, I.L.; Grupp, G.; Melvin, D.; Gradoux, N.; Kremers, W.; Lingrel, J.B.; de Pover, A. Characterisation of Na/K-ATPase, its isoforms, and the inotropic response to ouabain in isolated failing human hearts. Cardiovasc. Res. 1993, 27, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, J.; Ai, X.; Moorman, J.R.; Pogwizd, S.M.; Bers, D.M. Expression and phosphorylation of the na-pump regulatory subunit phospholemman in heart failure. Circ. Res. 2005, 97, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.B.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef] [PubMed]
- El-Armouche, A.; Wittköpper, K.; Fuller, W.; Howie, J.; Shattock, M.J.; Pavlovic, D. Phospholemman-dependent regulation of the cardiac Na/K-ATPase activity is modulated by inhibitor-1 sensitive type-1 phosphatase. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 4467–4475. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Qi, M.; Yuan, W.; Samarel, A.M.; Bers, D.M. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999, 85, 1009–1019. [Google Scholar] [CrossRef]
- Studer, R.; Reinecke, H.; Bilger, J.; Eschenhagen, T.; Böhm, M.; Hasenfuss, G.; Just, H.; Holtz, J.; Drexler, H. Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ. Res. 1994, 75, 443–453. [Google Scholar] [CrossRef]
- Wang, Z.; Nolan, B.; Kutschke, W.; Hill, J.A. Na+-Ca2+ exchanger remodeling in pressure overload cardiac hypertrophy. J. Biol. Chem. 2001, 276, 17706–17711. [Google Scholar] [CrossRef] [PubMed]
- Schillinger, W.; Teucher, N.; Christians, C.; Kohlhaas, M.; Sossalla, S.; van Nguyen, P.; Schmidt, A.G.; Schunck, O.; Nebendahl, K.; Maier, L.S.; et al. High intracellular Na+ preserves myocardial function at low heart rates in isolated myocardium from failing hearts. Eur. J. Heart Fail. 2006, 8, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Dipla, K.; Mattiello, J.A.; Margulies, K.B.; Jeevanandam, V.; Houser, S.R. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ. Res. 1999, 84, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Primessnig, U.; Schönleitner, P.; Höll, A.; Pfeiffer, S.; Bracic, T.; Rau, T.; Kapl, M.; Stojakovic, T.; Glasnov, T.; Leineweber, K.; et al. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2016, 18, 987–997. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Trafford, A.W.; Hutchings, D.C. The Control of Diastolic Calcium in the Heart: Basic Mechanisms and Functional Implications. Circ. Res. 2020, 126, 395–412. [Google Scholar] [CrossRef]
- Bers, D.M. Altered cardiac myocyte Ca regulation in heart failure. Physiology 2006, 21, 380–387. [Google Scholar] [CrossRef]
- Zhihao, L.; Jingyu, N.; Lan, L.; Michael, S.; Rui, G.; Xiyun, B.; Xiaozhi, L.; Guanwei, F. SERCA2a: A key protein in the Ca2+ cycle of the heart failure. Heart Fail. Rev. 2020, 25, 523–535. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef]
- Bers, D.M.; Pogwizd, S.M.; Schlotthauer, K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res. Cardiol. 2002, 97 (Suppl. 1), I36–I42. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Schlotthauer, K.; Li, L.; Yuan, W.; Bers, D.M. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 2001, 88, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Kilić, A.; Huang, C.X.; Rajapurohitam, V.; Madwed, J.B.; Karmazyn, M. Early and transient sodium-hydrogen exchanger isoform 1 inhibition attenuates subsequent cardiac hypertrophy and heart failure following coronary artery ligation. J. Pharmacol. Exp. Ther. 2014, 351, 492–499. [Google Scholar] [CrossRef]
- Aker, S.; Snabaitis, A.K.; Konietzka, I.; van de Sand, A.; Böngler, K.; Avkiran, M.; Heusch, G.; Schulz, R. Inhibition of the Na+/H+ exchanger attenuates the deterioration of ventricular function during pacing-induced heart failure in rabbits. Cardiovasc. Res. 2004, 63, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, S.; Hein, L.; Keller, U.; Klämbt, K.; Lohse, M.J. Inhibition of Na(+)-H(+) exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ. Res. 2002, 90, 814–819. [Google Scholar] [CrossRef]
- Kusumoto, K.; Haist, J.V.; Karmazyn, M. Na(+)/H(+) exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H738–H745. [Google Scholar] [CrossRef]
- Cappetta, D.; de Angelis, A.; Ciuffreda, L.P.; Coppini, R.; Cozzolino, A.; Miccichè, A.; Dell’Aversana, C.; D’Amario, D.; Cianflone, E.; Scavone, C.; et al. Amelioration of diastolic dysfunction by dapagliflozin in a non-diabetic model involves coronary endothelium. Pharmacol. Res. 2020, 157, 104781. [Google Scholar] [CrossRef]
- Chung, Y.J.; Park, K.C.; Tokar, S.; Eykyn, T.R.; Fuller, W.; Pavlovic, D.; Swietach, P.; Shattock, M.J. Off-target effects of SGLT2 blockers: Empagliflozin does not inhibit Na+/H+ exchanger-1 or lower Na+i in the heart. Cardiovasc. Res. 2020, cvaa323. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Coronel, R. SGLT2 inhibitor empagliflozin inhibits the cardiac Na+/H+ exchanger 1: Persistent inhibition under various experimental conditions. Cardiovasc. Res. 2021, cvab129. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Dhein, S.; Beuckelmann, D.J. Role of the cardiac Na(+)/H(+)exchanger in Ca(2+)(i)and Na(+)(i)handling during intracellular acidosis. Effect of cariporide (Hoe 642). Pharmacol. Res. 2002, 45, 35–41. [Google Scholar] [CrossRef]
- Uthman, L.; Nederlof, R.; Eerbeek, O.; Baartscheer, A.; Schumacher, C.; Buchholtz, N.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Delayed ischaemic contracture onset by empagliflozin associates with NHE1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc. Res. 2019, 115, 1533–1545. [Google Scholar] [CrossRef]
- Kemi, O.J.; Arbo, I.; Høydal, M.A.; Loennechen, J.P.; Wisløff, U.; Smith, G.L.; Ellingsen, Ø. Reduced pH and contractility in failing rat cardiomyocytes. Acta Physiol. 2006, 188, 185–193. [Google Scholar] [CrossRef]
- Sossalla, S.; Maurer, U.; Schotola, H.; Hartmann, N.; Didié, M.; Zimmermann, W.-H.; Jacobshagen, C.; Wagner, S.; Maier, L.S. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIδ(C) can be reversed by inhibition of late Na(+) current. Basic Res. Cardiol. 2011, 106, 263–272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Undrovinas, A.I.; Belardinelli, L.; Undrovinas, N.A.; Sabbah, H.N. Ranolazine improves abnormal repolarization and contraction in left ventricular myocytes of dogs with heart failure by inhibiting late sodium current. J. Cardiovasc. Electrophysiol. 2006, 17 (Suppl. 1), S169–S177. [Google Scholar] [CrossRef] [PubMed]
- Horváth, B.; Hézső, T.; Kiss, D.; Kistamás, K.; Magyar, J.; Nánási, P.P.; Bányász, T. Late Sodium Current Inhibitors as Potential Antiarrhythmic Agents. Front. Pharmacol. 2020, 11, 413. [Google Scholar] [CrossRef]
- Pless, S.A.; Galpin, J.D.; Frankel, A.; Ahern, C.A. Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat. Commun. 2011, 2, 351. [Google Scholar] [CrossRef] [PubMed]
- Ahern, C.A.; Eastwood, A.L.; Dougherty, D.A.; Horn, R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ. Res. 2008, 102, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Fredj, S.; Sampson, K.J.; Liu, H.; Kass, R.S. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: Evidence for site of action. Br. J. Pharmacol. 2006, 148, 16–24. [Google Scholar] [CrossRef]
- Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003, 92, 904–911. [Google Scholar] [CrossRef]
- Pabel, S.; Wagner, S.; Bollenberg, H.; Bengel, P.; Kovács, Á.; Schach, C.; Tirilomis, P.; Mustroph, J.; Renner, A.; Gummert, J.; et al. Empagliflozin directly improves diastolic function in human heart failure. Eur. J. Heart Fail. 2018, 20, 1690–1700. [Google Scholar] [CrossRef]
- Durak, A.; Olgar, Y.; Degirmenci, S.; Akkus, E.; Tuncay, E.; Turan, B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc. Diabetol. 2018, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, A.; Cantini, G.; Tani, A.; Coppini, R.; Zecchi-Orlandini, S.; Raimondi, L.; Luconi, M.; Mannucci, E. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: A new potential pharmacological target. Int. J. Cardiol. 2017, 243, 86–90. [Google Scholar] [CrossRef]
- Li, Z.; Agrawal, V.; Ramratnam, M.; Sharma, R.K.; D’Auria, S.; Sincoular, A.; Jakubiak, M.; Music, M.L.; Kutschke, W.J.; Huang, X.N.; et al. Cardiac sodium-dependent glucose cotransporter 1 is a novel mediator of ischaemia/reperfusion injury. Cardiovasc. Res. 2019, 115, 1646–1658. [Google Scholar] [CrossRef] [PubMed]
- Sawa, Y.; Saito, M.; Ishida, N.; Ibi, M.; Matsushita, N.; Morino, Y.; Taira, E.; Hirose, M. Pretreatment with KGA-2727, a selective SGLT1 inhibitor, is protective against myocardial infarction-induced ventricular remodeling and heart failure in mice. J. Pharmacol. Sci. 2020, 142, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes 2013, 62, 3324–3328. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2019, 6, 186. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.; Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ. Res. 2000, 86, 152–157. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.; Islam, M.M.T.; Lebek, S.; Baier, M.; Hegner, P.; Eaton, P.; Maier, L.S.; Wagner, S. Inhibition of cardiac potassium currents by oxidation-activated protein kinase A contributes to early afterdepolarizations in the heart. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1347–H1357. [Google Scholar] [CrossRef]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of sodium-glucose cotransporter 2 selective inhibitor ipragliflozin on hyperglycaemia, oxidative stress, inflammation and liver injury in streptozotocin-induced type 1 diabetic rats. J. Pharm. Pharmacol. 2014, 66, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.Á.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2021, 117, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Yurista, S.R.; Silljé, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.-M.; Pavez Giani, M.G.; Hillebrands, J.-L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Anavekar, N.; Skali, H.; McMurray, J.J.V.; Swedberg, K.; Yusuf, S.; Granger, C.B.; Michelson, E.L.; Wang, D.; Pocock, S.; et al. Influence of ejection fraction on cardiovascular outcomes in a broad spectrum of heart failure patients. Circulation 2005, 112, 3738–3744. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.S.; von Lueder, T.G.; Zannad, F.; Rossignol, P.; Duarte, K.; Chouihed, T.; Dickstein, K.; Atar, D.; Agewall, S.; Girerd, N. Relationship between left ventricular ejection fraction and mortality after myocardial infarction complicated by heart failure or left ventricular dysfunction. Int. J. Cardiol. 2018, 272, 260–266. [Google Scholar] [CrossRef]
- Belardinelli, L.; Shryock, J.C.; Fraser, H. Inhibition of the late sodium current as a potential cardioprotective principle: Effects of the late sodium current inhibitor ranolazine. Heart 2006, 92 (Suppl. 4), iv6–iv14. [Google Scholar] [CrossRef]
- Chandler, M.P.; Stanley, W.C.; Morita, H.; Suzuki, G.; Roth, B.A.; Blackburn, B.; Wolff, A.; Sabbah, H.N. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ. Res. 2002, 91, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Chandler, M.P.; Mishima, T.; Suzuki, G.; Chaudhry, P.; Nass, O.; Biesiadecki, B.J.; Blackburn, B.; Wolff, A.; Stanley, W.C. Ranolazine, a partial fatty acid oxidation (pFOX) inhibitor, improves left ventricular function in dogs with chronic heart failure. J. Card. Fail. 2002, 8, 416–422. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Vargas-Delgado, A.P.; Requena-Ibanez, J.A.; Garcia-Ropero, A.; Mancini, D.; Pinney, S.; Macaluso, F.; Sartori, S.; Roque, M.; Sabatel-Perez, F.; et al. Randomized Trial of Empagliflozin in Nondiabetic Patients with Heart Failure and Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2021, 77, 243–255. [Google Scholar] [CrossRef]
- Hwang, I.-C.; Cho, G.-Y.; Yoon, Y.E.; Park, J.J.; Park, J.-B.; Lee, S.-P.; Kim, H.-K.; Kim, Y.-J.; Sohn, D.-W. Different effects of SGLT2 inhibitors according to the presence and types of heart failure in type 2 diabetic patients. Cardiovasc. Diabetol. 2020, 19, 69. [Google Scholar] [CrossRef]
- Borges-Júnior, F.A.; Silva Dos Santos, D.; Benetti, A.; Polidoro, J.Z.; Wisnivesky, A.C.T.; Crajoinas, R.O.; Antônio, E.L.; Jensen, L.; Caramelli, B.; Malnic, G.; et al. Empagliflozin Inhibits Proximal Tubule NHE3 Activity, Preserves GFR, and Restores Euvolemia in Nondiabetic Rats with Induced Heart Failure. J. Am. Soc. Nephrol. JASN 2021, 32, 1616–1629. [Google Scholar] [CrossRef]
- Byrne, N.J.; Parajuli, N.; Levasseur, J.L.; Boisvenue, J.; Beker, D.L.; Masson, G.; Fedak, P.W.M.; Verma, S.; Dyck, J.R.B. Empagliflozin Prevents Worsening of Cardiac Function in an Experimental Model of Pressure Overload-Induced Heart Failure. JACC Basic Transl. Sci. 2017, 2, 347–354. [Google Scholar] [CrossRef]
- Del Carmen Asensio Lopez, M.; Lax, A.; Hernandez Vicente, A.; Saura Guillen, E.; Hernandez-Martinez, A.; Del Fernandez Palacio, M.J.; Bayes-Genis, A.; Pascual Figal, D.A. Author Correction: Empagliflozin improves post-infarction cardiac remodeling through GTP enzyme cyclohydrolase 1 and irrespective of diabetes status. Sci. Rep. 2020, 10, 17266. [Google Scholar] [CrossRef]
- Shi, L.; Zhu, D.; Wang, S.; Jiang, A.; Li, F. Dapagliflozin Attenuates Cardiac Remodeling in Mice Model of Cardiac Pressure Overload. Am. J. Hypertens. 2019, 32, 452–459. [Google Scholar] [CrossRef]
- Cao, J.; Qin, G.; Shi, R.; Bai, F.; Yang, G.; Zhang, M.; Lv, J. Overproduction of reactive oxygen species and activation of MAPKs are involved in apoptosis induced by PM2.5 in rat cardiac H9c2 cells. J. Appl. Toxicol. JAT 2016, 36, 609–617. [Google Scholar] [CrossRef]
- Tanaka, H.; Soga, F.; Tatsumi, K.; Mochizuki, Y.; Sano, H.; Toki, H.; Matsumoto, K.; Shite, J.; Takaoka, H.; Doi, T.; et al. Positive effect of dapagliflozin on left ventricular longitudinal function for type 2 diabetic mellitus patients with chronic heart failure. Cardiovasc. Diabetol. 2020, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Garg, A.; Yan, A.T.; Gupta, A.K.; Al-Omran, M.; Sabongui, A.; Teoh, H.; Mazer, C.D.; Connelly, K.A. Effect of Empagliflozin on Left Ventricular Mass and Diastolic Function in Individuals with Diabetes: An Important Clue to the EMPA-REG OUTCOME Trial? Diabetes Care 2016, 39, e212–e213. [Google Scholar] [CrossRef] [PubMed]
- Matsutani, D.; Sakamoto, M.; Kayama, Y.; Takeda, N.; Horiuchi, R.; Utsunomiya, K. Effect of canagliflozin on left ventricular diastolic function in patients with type 2 diabetes. Cardiovasc. Diabetol. 2018, 17, 73. [Google Scholar] [CrossRef]
- Rau, M.; Thiele, K.; Hartmann, N.-U.K.; Schuh, A.; Altiok, E.; Möllmann, J.; Keszei, A.P.; Böhm, M.; Marx, N.; Lehrke, M. Empagliflozin does not change cardiac index nor systemic vascular resistance but rapidly improves left ventricular filling pressure in patients with type 2 diabetes: A randomized controlled study. Cardiovasc. Diabetol. 2021, 20, 6. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Shahzeb Khan, M.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.P.; Choi, D.-J.; Chopra, V.; et al. Baseline characteristics of patients with heart failure with preserved ejection fraction in the EMPEROR-Preserved trial. Eur. J. Heart Fail. 2020, 22, 2383–2392. [Google Scholar] [CrossRef]
- Solomon, S.D.; de Boer, R.A.; DeMets, D.; Hernandez, A.F.; Inzucchi, S.E.; Kosiborod, M.N.; Lam, C.S.P.; Martinez, F.; Shah, S.J.; Lindholm, D.; et al. Dapagliflozin in Heart Failure with Preserved and Mildly Reduced Ejection Fraction: Rationale and Design of the DELIVER Trial. Eur. J. Heart Fail. 2021, 23, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.A.; Zhang, Y.; Visram, A.; Advani, A.; Batchu, S.N.; Desjardins, J.-F.; Thai, K.; Gilbert, R.E. Empagliflozin Improves Diastolic Function in a Nondiabetic Rodent Model of Heart Failure with Preserved Ejection Fraction. JACC Basic Transl. Sci. 2019, 4, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; San Antonio, R.; Garcia-Ropero, A.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Vargas-Delgado, A.P.; Flores-Umanzor, E.J.; Sanz, J.; et al. Empagliflozin Ameliorates Diastolic Dysfunction and Left Ventricular Fibrosis/Stiffness in Nondiabetic Heart Failure: A Multimodality Study. JACC Cardiovasc. Imaging 2021, 14, 393–407. [Google Scholar] [CrossRef]
- Hamdani, N.; Bishu, K.G.; von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Christ, T.; Fabritz, L.; Goette, A.; Hammwöhner, M.; Heijman, J.; Kockskämper, J.; Linz, D.; Odening, K.E.; Schweizer, P.A.; et al. German Cardiac Society Working Group on Cellular Electrophysiology state-of-the-art paper: Impact of molecular mechanisms on clinical arrhythmia management. Clin. Res. Cardiol. Off. J. Ger. Card. Soc. 2019, 108, 577–599. [Google Scholar] [CrossRef]
- Workman, A.J.; Marshall, G.E.; Rankin, A.C.; Smith, G.L.; Dempster, J. Transient outward K+ current reduction prolongs action potentials and promotes afterdepolarisations: A dynamic-clamp study in human and rabbit cardiac atrial myocytes. J. Physiol. 2012, 590, 4289–4305. [Google Scholar] [CrossRef] [PubMed]
- Wit, A.L. Afterdepolarizations and triggered activity as a mechanism for clinical arrhythmias. Pacing Clin. Electrophysiol. PACE 2018, in press. [Google Scholar] [CrossRef]
- Sedej, S.; Heinzel, F.R.; Walther, S.; Dybkova, N.; Wakula, P.; Groborz, J.; Gronau, P.; Maier, L.S.; Vos, M.A.; Lai, F.A.; et al. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc. Res. 2010, 87, 50–59. [Google Scholar] [CrossRef]
- Viatchenko-Karpinski, S.; Kornyeyev, D.; El-Bizri, N.; Budas, G.; Fan, P.; Jiang, Z.; Yang, J.; Anderson, M.E.; Shryock, J.C.; Chang, C.-P.; et al. Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 2014, 76, 247–256. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Singh, M.V.; Kapoun, A.; Higgins, L.; Kutschke, W.; Thurman, J.M.; Zhang, R.; Singh, M.; Yang, J.; Guan, X.; Lowe, J.S.; et al. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J. Clin. Investig. 2009, 119, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.V.; Anderson, M.E. Is CaMKII a link between inflammation and hypertrophy in heart? J. Mol. Med. 2011, 89, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Rusciano, M.R.; Sommariva, E.; Douin-Echinard, V.; Ciccarelli, M.; Poggio, P.; Maione, A.S. CaMKII Activity in the Inflammatory Response of Cardiac Diseases. Int. J. Mol. Sci. 2019, 20, 4374. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Bonaca, M.P.; Furtado, R.H.M.; Mosenzon, O.; Kuder, J.F.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; McGuire, D.K.; Wilding, J.P.H.; et al. Effect of Dapagliflozin on Atrial Fibrillation in Patients with Type 2 Diabetes Mellitus: Insights From the DECLARE-TIMI 58 Trial. Circulation 2020, 141, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Huang, J.-Y.; Siao, W.-Z.; Jong, G.-P. The association between SGLT2 inhibitors and new-onset arrhythmias: A nationwide population-based longitudinal cohort study. Cardiovasc. Diabetol. 2020, 19, 73. [Google Scholar] [CrossRef]
- Li, H.-L.; Lip, G.-Y.H.; Feng, Q.; Fei, Y.; Tse, Y.-K.; Wu, M.-Z.; Ren, Q.-W.; Tse, H.-F.; Cheung, B.-M.Y.; Yiu, K.-H. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).