
Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland



{kind=link}
{kind=link}
Abstract
:1. Introduction
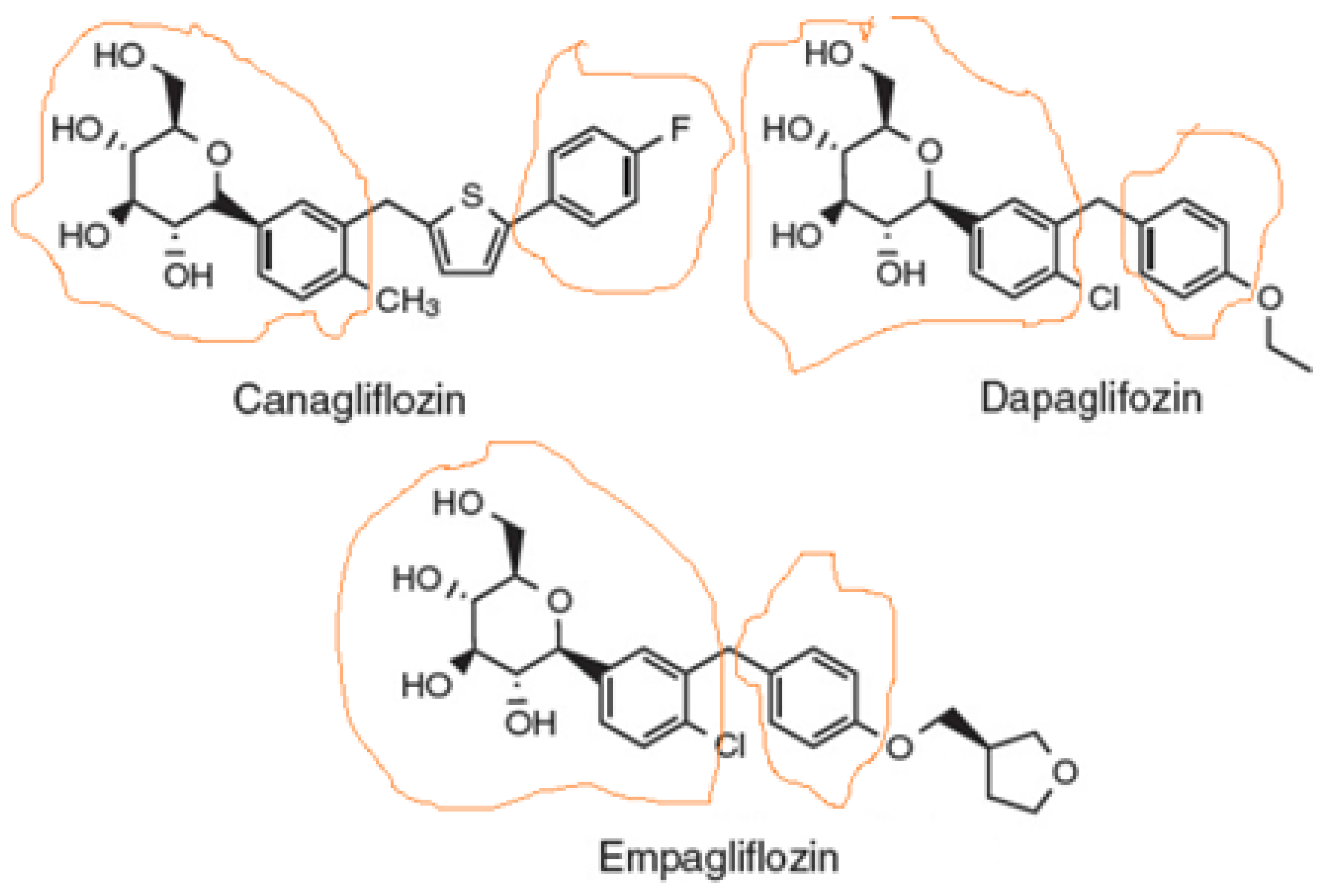
1.1. Chemistry & Structure-Activity Relationships of SGLT2 Inhibitors
1.2. Cardiovascular Benefits of SGLT2 Inhibitors
2. Potential Mechanisms Underlying the Cardiovascular Benefits of SGLT2 Inhibitors
Ketone Bodies and SGLT2 Inhibitor-Mediated Sympatholysis
3. Current Evidence for SGLT2 Inhibitor-Induced Sympatholysis
3.1. Evidence from Clinical Trials and Animal Models
3.2. Role of FFAR3
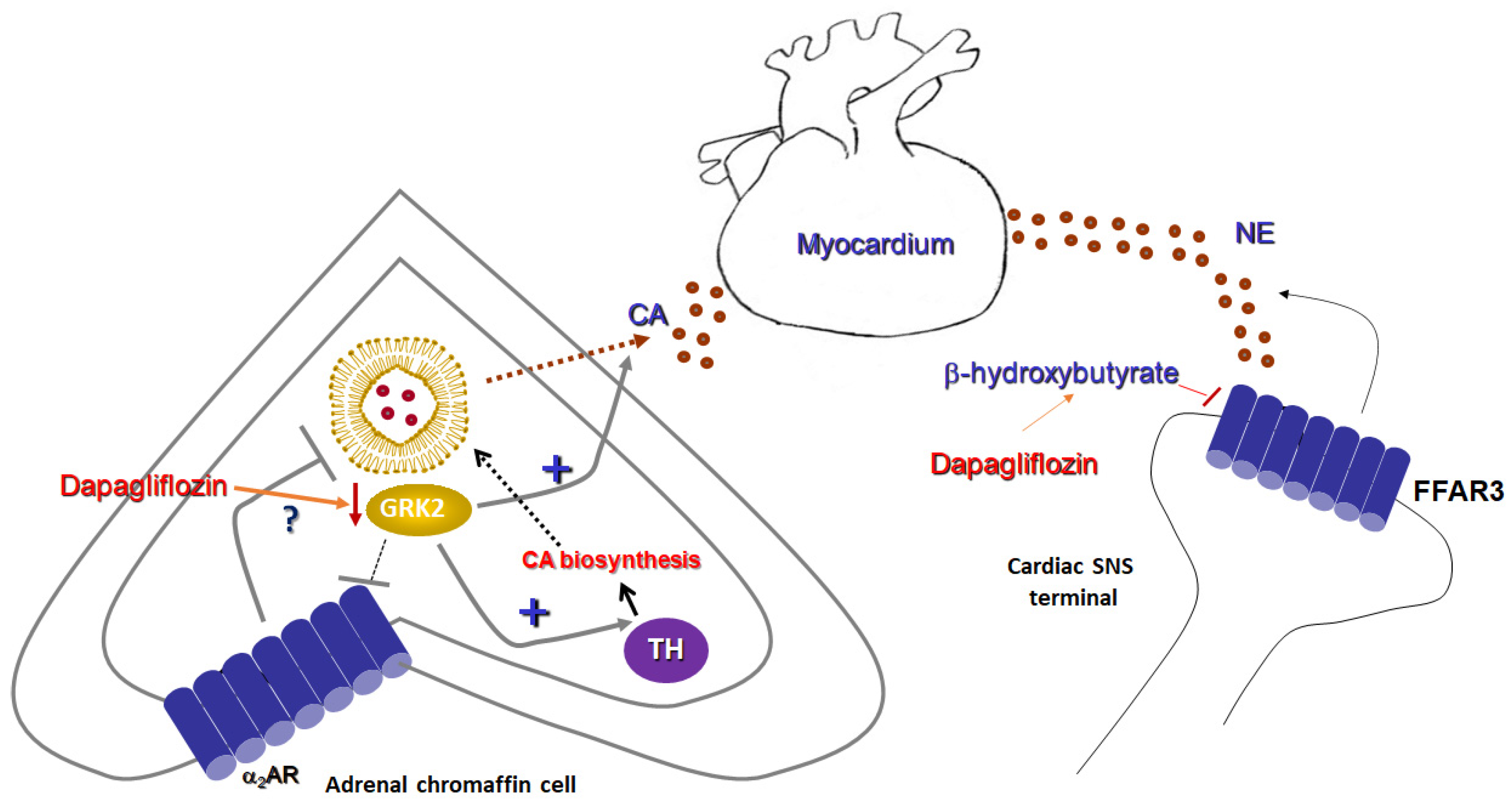
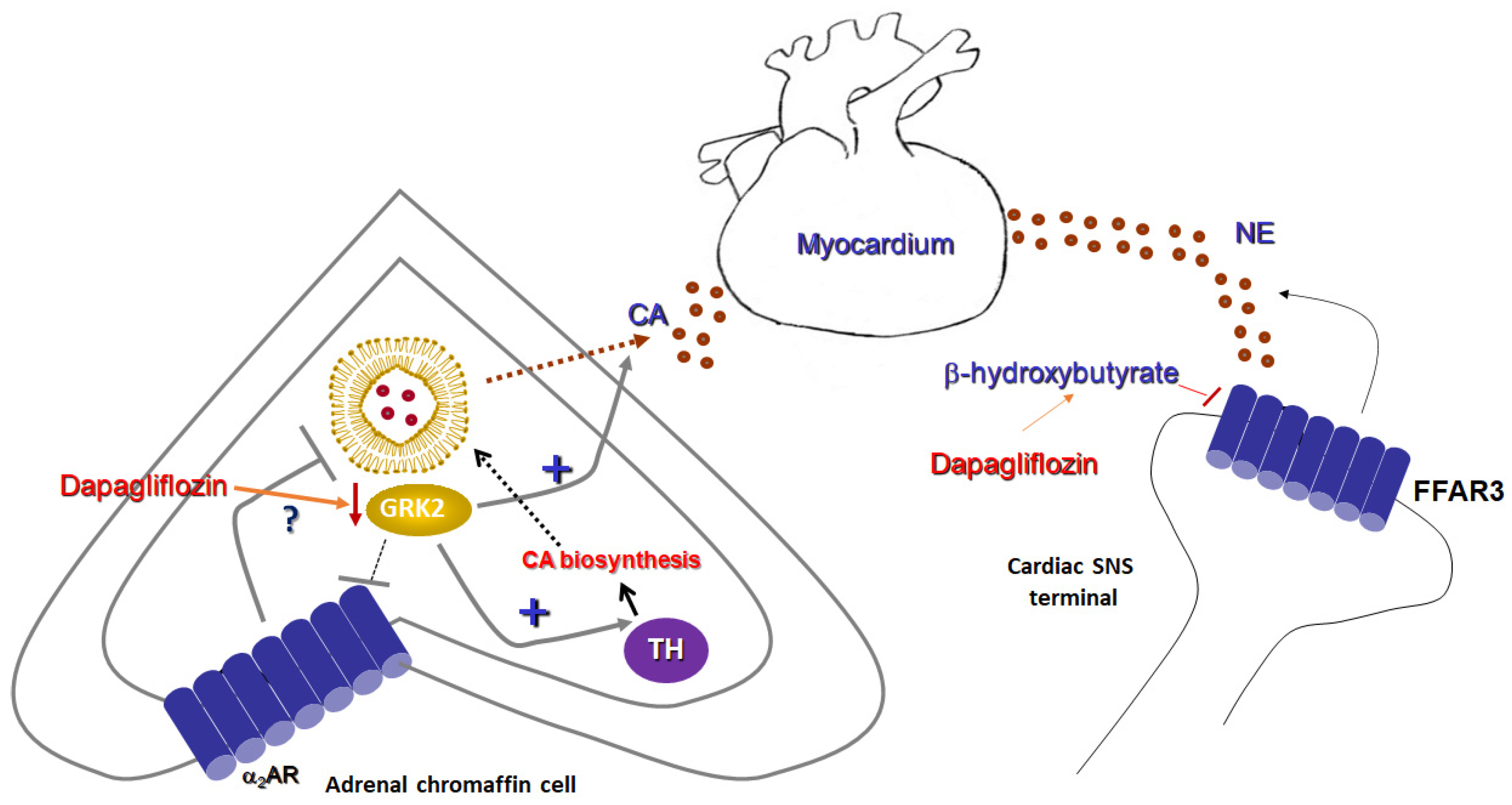
4. Research Hypothesis: Dapagliflozin Attenuates Adrenal Catecholamine Production
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scheen, A.J. Pharmacodynamics, efficacy and safety of sodium–glucose co-transporter type 2 (SGLT2) Inhibitors for the Treatment of Type 2 Diabetes Mellitus. Drugs 2015, 75, 33–59. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.S.; Siddiqi, T.J.; Memon, M.M.; Khan, M.S.; Rawasia, W.F.; Ayub, M.T.; Sreenivasan, J.; Golzar, Y. Sodium-glucose co-transporter 2 inhibitors and cardiovascular outcomes: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2018, 25, 495–502. [Google Scholar] [CrossRef]
- Bonora, B.M.; Avogaro, A.; Fadini, G.P. Extraglycemic Effects of SGLT2 Inhibitors: A Review of the Evidence. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 161–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.-R.; Zhang, J.; Greenberg, J.; Lee, T.; Liu, J. Discovery of non-glucoside SGLT2 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2472–2475. [Google Scholar] [CrossRef] [PubMed]
- Hummel, C.S.; Lu, C.; Liu, J.; Ghezzi, C.; Hirayama, B.A.; Loo, D.D.; Kepe, V.; Barrio, J.R.; Wright, E.M. Structural selectivity of human SGLT inhibitors. Am. J. Physiol. Cell Physiol. 2012, 302, C373–C382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.L.; Li, H.C.; Lau, K.M.; Chan, A.; Lau, C.B.S.; Shing, T.K.M. Concise and Stereodivergent Synthesis of Carbasugars Reveals Unexpected Structure-Activity Relationship (SAR) of SGLT2 Inhibition. Sci. Rep. 2017, 7, 5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.S.; Lee, S.H.; Kim, M.J.; Seo, H.J.; Lee, J.; Lee, S.H.; Jung, M.E.; Son, E.J.; Lee, M.; Kim, J.; et al. Synthesis and SAR of Thiazolylmethylphenyl Glucoside as Novel C-Aryl Glucoside SGLT2 Inhibitors. ACS Med. Chem. Lett. 2010, 2, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef]
- Lytvyn, Y.; Bjornstad, P.; Udell, J.; Lovshin, J.A.; Cherney, D.Z. Sodium Glucose Cotransporter-2 Inhibition in Heart Failure. Circulation 2017, 136, 1643–1658. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Zannad, F. Effects of Sodium-Glucose Cotransporter 2 Inhibitors for the Treatment of Patients With Heart Failure. JAMA Cardiol. 2017, 2, 1025–1029. [Google Scholar] [CrossRef]
- Zaccardi, F.; Webb, D.R.; Htike, Z.Z.; Youssef, D.; Khunti, K.; Davies, M.J. Efficacy and safety of sodium-glucose co-transporter-2 inhibitors in type 2 diabetes mellitus: Systematic review and network meta-analysis. Diabetes Obes. Metab. 2016, 18, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Ganguly, S.; Goh, S.-Y. Weight loss associated with sodium-glucose cotransporter-2 inhibition: A review of evidence and underlying mechanisms. Obes. Rev. 2018, 19, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Palmiero, G.; Cesaro, A.; Vetrano, E.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Moscarella, E.; Gragnano, F.; Salvatore, T.; Rinaldi, L.; et al. Impact of SGLT2 Inhibitors on Heart Failure: From Pathophysiology to Clinical Effects. Int. J. Mol. Sci. 2021, 22, 5863. [Google Scholar] [CrossRef] [PubMed]
- Jhund, P.S.; Ponikowski, P.; Docherty, K.F.; Gasparyan, S.B.; Böhm, M.; Chiang, C.E.; Desai, A.S.; Howlett, J.; Kitakaze, M.; Petrie, M.C.; et al. Dapagliflozin and Recurrent Heart Failure Hospitalizations in Heart Failure with Reduced Ejection Fraction: An Analysis of DAPA-HF. Circulation 2021, 143, 1962–1972. [Google Scholar] [CrossRef]
- Bertero, E.; Roma, L.P.; Ameri, P.; Maack, C. Cardiac effects of SGLT2 inhibitors: The sodium hypothesis. Cardiovasc. Res. 2018, 114, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitchett, D.; Inzucchi, S.E.; Cannon, C.P.; McGuire, D.K.; Scirica, B.M.; Johansen, O.E.; Sambevski, S.; Kaspers, S.; Pfarr, E.; George, J.T.; et al. Empagliflozin Reduced Mortality and Hospitalization for Heart Failure Across the Spectrum of Cardiovascular Risk in the EMPA-REG OUTCOME Trial. Circulation 2019, 139, 1384–1395. [Google Scholar] [CrossRef]
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (2002). Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs. diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002, 288, 2981–2997. [Google Scholar] [CrossRef]
- Takasu, T.; Takakura, S. Effect of ipragliflozin, an SGLT2 inhibitor, on cardiac histopathological changes in a non-diabetic rat model of cardiomyopathy. Life Sci. 2019, 230, 19–27. [Google Scholar] [CrossRef]
- Min, S.H.; Oh, T.J.; Baek, S.I.; Lee, D.H.; Kim, K.M.; Moon, J.H.; Choi, S.H.; Park, K.S.; Jang, H.C.; Lim, S. Degree of keto-naemia and its association with insulin resistance after dapagliflozin treatment in type 2 diabetes. Diabetes Metab. 2018, 44, 73–76. [Google Scholar] [CrossRef]
- Okamoto, A.; Yokokawa, H.; Sanada, H.; Naito, T. Changes in Levels of Biomarkers Associated with Adipocyte Function and Insulin and Glucagon Kinetics During Treatment with Dapagliflozin Among Obese Type 2 Diabetes Mellitus Patients. Drugs R D 2016, 16, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Polidori, D.; Iijima, H.; Goda, M.; Maruyama, N.; Inagaki, N.; Crawford, P.A. Intra- and inter-subject variability for increases in serum ketone bodies in patients with type 2 diabetes treated with the sodium glucose co-transporter 2 inhibitor canagliflozin. Diabetes Obes. Metab. 2018, 20, 1321–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Novikov, A.; Vallon, V. Ketosis and diabetic ketoacidosis in response to SGLT2 inhibitors: Basic mechanisms and therapeutic perspectives. Diabetes Metab. Res. Rev. 2017, 33, e2886. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy Production in Diabetes: Novel Translational Insights into the Heart Failure Benefits of SGLT2 Inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Voros, G.; Ector, J.; Garweg, C.; Droogne, W.; Van Cleemput, J.; Peersman, N.; Vermeersch, P.; Janssens, S. Increased Cardiac Uptake of Ketone Bodies and Free Fatty Acids in Human Heart Failure and Hypertrophic Left Ventricular Remodeling. Circ. Hear. Fail. 2018, 11, e004953. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; Harada, E.; Nakagawa, H.; Morikawa, Y.; Shono, M.; Kugimiya, F.; Yoshimura, M.; Yasue, H. The diabetic heart utilizes ketone bodies as an energy source. Metabolism 2017, 77, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hu, X.; Jiang, H. HDAC inhibition: A novel therapeutic approach for attenuating heart failure by suppressing cardiac remodeling. Int. J. Cardiol. 2016, 214, 41–42. [Google Scholar] [CrossRef]
- Lkhagva, B.; Lin, Y.K.; Kao, Y.H.; Chazo, T.F.; Chung, C.C.; Chen, S.A.; Chen, Y.J. Novel Histone Deacetylase Inhibitor Modulates Cardiac Peroxisome Proliferator-Activated Receptors and Inflammatory Cytokines in Heart Failure. Pharmacology 2015, 96, 184–191. [Google Scholar] [CrossRef]
- Sano, M.; Chen, S.; Imazeki, H.; Ochiai, H.; Seino, Y. Changes in heart rate in patients with type 2 diabetes mellitus after treatment with luseogliflozin: Sub-analysis of placebo-controlled, double-blind clinical trials. J. Diabetes Investig. 2018, 9, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in Heart Failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R. beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000, 101, 558–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, X.; Simonavicius, N.; Tian, H.; Ling, L. Medium-chain Fatty Acids as Ligands for Orphan G Protein-coupled Receptor GPR84. J. Biol. Chem. 2006, 281, 34457–34464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G Protein-coupled Receptors GPR41 and GPR43 Are Activated by Propionate and Other Short Chain Carboxylic Acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [Green Version]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, L.A.; Smith, N.J.; Jenkins, L.; Brown, A.J.; Milligan, G. Conserved Polar Residues in Transmembrane Domains V, VI, and VII of Free Fatty Acid Receptor 2 and Free Fatty Acid Receptor 3 Are Required for the Binding and Function of Short Chain Fatty Acids. J. Biol. Chem. 2008, 283, 32913–32924. [Google Scholar] [CrossRef] [Green Version]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.-Y.; Lannoy, V.; Decobecq, M.-E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional Characterization of Human Receptors for Short Chain Fatty Acids and Their Role in Polymorphonuclear Cell Activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- Capote, L.A.; Perez, R.M.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Al-Lahham, S.H.; Roelofsen, H.; Priebe, M.; Weening, D.; Dijkstra, M.; Hoek, A.; Rezaee, F.; Venema, K.; Vonk, R.J. Regulation of adipokine production in human adipose tissue by propionic acid. Eur. J. Clin. Investig. 2010, 40, 401–407. [Google Scholar] [CrossRef]
- Xiong, Y.; Miyamoto, N.; Shibata, K.; Valasek, M.A.; Motoike, T.; Kedzierski, R.M.; Yanagisawa, M. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc. Natl. Acad. Sci. USA 2004, 101, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511. [Google Scholar] [CrossRef]
- Albert, P.R.; Robillard, L. G protein specificity: Traffic direction required. Cell. Signal. 2002, 14, 407–418. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. GRK2 Inhibition in Heart Failure: Something Old, Something New. Curr. Pharm. Des. 2012, 18, 186–191. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Rosmaninho-Salgado, J.; Araújo, I.M.; Álvaro, A.R.; Duarte, E.P.; Cavadas, C. Intracellular signaling mechanisms mediating catecholamine release upon activation of NPY Y1receptors in mouse chromaffin cells. J. Neurochem. 2007, 103, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Jun, D.J.; Hur, E.M.; Lee, S.K.; Suh, B.-S.; Kim, K.T. Activity-dependent potentiation of large dense-core vesicle release modulated by mitogen-activated protein kinase/extracellularly regulated kinase signaling. Endocrinology 2006, 147, 1349–1356. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Kuwahara, K. Sodium-Glucose Cotransporter-2 inhibitors are potential therapeutic agents for treatment of non-diabetic heart failure patients. J. Cardiol. 2020, 76, 123–131. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef]
- Jafferjee, M.; Valero, T.R.; Marrero, C.; McCrink, K.A.; Brill, A.; Lymperopoulos, A. GRK2 Up-Regulation Creates a Positive Feedback Loop for Catecholamine Production in Chromaffin Cells. Mol. Endocrinol. 2016, 30, 372–381. [Google Scholar] [CrossRef] [Green Version]
- Lymperopoulos, A.; Brill, A.; McCrink, K.A. GPCRs of adrenal chromaffin cells & catecholamines: The plot thickens. Int. J. Biochem. Cell Biol. 2016, 77, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Gao, E.; Ebert, S.N.; Dorn, G.W., II; Koch, W.J. Reduction of sympathetic activity via adren-al-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 2010, 285, 16378–16386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Soltys, S.; Koch, W.J. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol. Ther. 2008, 16, 302–307. [Google Scholar] [CrossRef]
- McCrink, K.A.; Brill, A.; Lymperopoulos, A. Adrenal G protein-coupled receptor kinase-2 in regulation of sympathetic nervous system activity in heart failure. World J. Cardiol. 2015, 7, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Rengo, G.; Lymperopoulos, A.; Zincarelli, C.; Femminella, G.; Liccardo, D.; Pagano, G.; de Lucia, C.; Cannavo, A.; Gargiulo, P.; Ferrara, N.; et al. Blockade of β-adrenoceptors restores the GRK2-mediated adrenal α2-adrenoceptor-catecholamine production axis in heart failure. Br. J. Pharmacol. 2012, 166, 2430–2440. [Google Scholar] [CrossRef] [Green Version]
- Karkoulias, G.; McCrink, K.A.; Maning, J.; Pollard, C.M.; Desimine, V.L.; Patsouras, N.; Psallidopoulos, M.; Taraviras, S.; Lymperopoulos, A.; Flordellis, C. Sustained GRK2-dependent CREB activation is essential for α2-adrenergic receptor-induced PC12 neuronal differentiation. Cell. Signal. 2020, 66, 109446. [Google Scholar] [CrossRef]
- Rangasamy, S.B.; Dasarathi, S.; Nutakki, A.; Mukherjee, S.; Nellivalasa, R.; Pahan, K. Stimulation of Dopamine Production by Sodium Benzoate, a Metabolite of Cinnamon and a Food Additive. J. Alzheimers Dis. Rep. 2021, 5, 295–310. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Bathgate, A. Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: The known and the unknown. Pharmacogenomics 2012, 13, 323–341. [Google Scholar] [CrossRef]
- Herat, L.; Magno, A.; Rudnicka, C.; Hricova, J.; Carnagarin, R.; Ward, N.; Arcambal, A.; Kiuchi, M.G.; Head, G.; Schlaich, M.P.; et al. SGLT2 Inhibitor–Induced Sympathoinhibition: A Novel Mechanism for Cardiorenal Protection. JACC Basic Transl. Sci. 2020, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Conde-Sieira, M.; Alvarez, R.; López-Patiño, M.A.; Míguez, J.M.; Flik, G.; Soengas, J.L. ACTH-stimulated cortisol release from head kidney of rainbow trout is modulated by glucose concentration. J. Exp. Biol. 2013, 216, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, E.C.; Liu, P.Y.; Wu, S.N. Effectiveness in the inhibition of dapagliflozin and canagliflozin on M-type K+ current and α-methylglucoside-induced current in pituitary tumor (GH3) and pheochromocytoma PC12 cells. Eur. J. Pharmacol. 2020, 879, 173141. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Pollard, C.M.; Noa, D.P.; E Ferraino, K.; Cora, N.; Pereyra, J.M.; Ghandour, J.; Valiente, R.; Maning, J.; Brill, A.R.; et al. Dapagliflozin Exerts Adrenal Sympatholysis via G protein-Coupled Receptor Kinase-2 & Tyrosine Hydroxylase Downregulation. J. Endocr. Soc. 2021, 5, A655. [Google Scholar] [CrossRef]
- Swe, M.T.; Thongnak, L.-O.; Jaikumkao, K.; Pongchaidecha, A.; Chatsudthipong, V.; Lungkaphin, A. Dapagliflozin attenuates renal gluconeogenic enzyme expression in obese rats. J. Endocrinol. 2020, 245, 193–205. [Google Scholar] [CrossRef]
- Shi, L.; Zhu, D.; Wang, S.; Jiang, A.; Li, F. Dapagliflozin Attenuates Cardiac Remodeling in Mice Model of Cardiac Pressure Overload. Am. J. Hypertens. 2019, 32, 452–459. [Google Scholar] [CrossRef]
- Kim, J.H.; Ko, H.Y.; Wang, H.J.; Lee, H.; Yun, M.; Kang, E.S. Effect of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on gluconeogenesis in proximal renal tubules. Diabetes Obes. Metab. 2019, 22, 373–382. [Google Scholar] [CrossRef]
- Sun, X.; Cao, Z.; Ma, Y.; Shao, Y.; Zhang, J.; Yuan, G.; Guo, X. Resveratrol attenuates dapagliflozin-induced renal gluconeogenesis via activating the PI3K/Akt pathway and suppressing the FoxO1 pathway in type 2 diabetes. Food Funct. 2021, 12, 1207–1218. [Google Scholar] [CrossRef]
- Jia, Y.; He, J.; Wang, L.; Su, L.; Lei, L.; Huang, W.; Geng, X.; Zhang, S.; Meng, X.; Zhou, H.; et al. Dapagliflozin Aggravates Renal Injury via Promoting Gluconeogenesis in db/db Mice. Cell. Physiol. Biochem. 2018, 45, 1747–1758. [Google Scholar] [CrossRef]
- Yang, M.; Lin, Y.; Wang, Y.; Wang, Y. High-glucose induces cardiac myocytes apoptosis through Foxo1 /GRK2 signaling pathway. Biochem. Biophys. Res. Commun. 2019, 513, 154–158. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lymperopoulos, A.; Borges, J.I.; Cora, N.; Sizova, A. Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland. Int. J. Mol. Sci. 2021, 22, 7684. https://doi.org/10.3390/ijms22147684
Lymperopoulos A, Borges JI, Cora N, Sizova A. Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland. International Journal of Molecular Sciences. 2021; 22(14):7684. https://doi.org/10.3390/ijms22147684
Chicago/Turabian StyleLymperopoulos, Anastasios, Jordana I. Borges, Natalie Cora, and Anastasiya Sizova. 2021. "Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland" International Journal of Molecular Sciences 22, no. 14: 7684. https://doi.org/10.3390/ijms22147684
APA StyleLymperopoulos, A., Borges, J. I., Cora, N., & Sizova, A. (2021). Sympatholytic Mechanisms for the Beneficial Cardiovascular Effects of SGLT2 Inhibitors: A Research Hypothesis for Dapagliflozin’s Effects in the Adrenal Gland. International Journal of Molecular Sciences, 22(14), 7684. https://doi.org/10.3390/ijms22147684