
Understanding PITX2-Dependent Atrial Fibrillation Mechanisms through Computational Models


 and
and 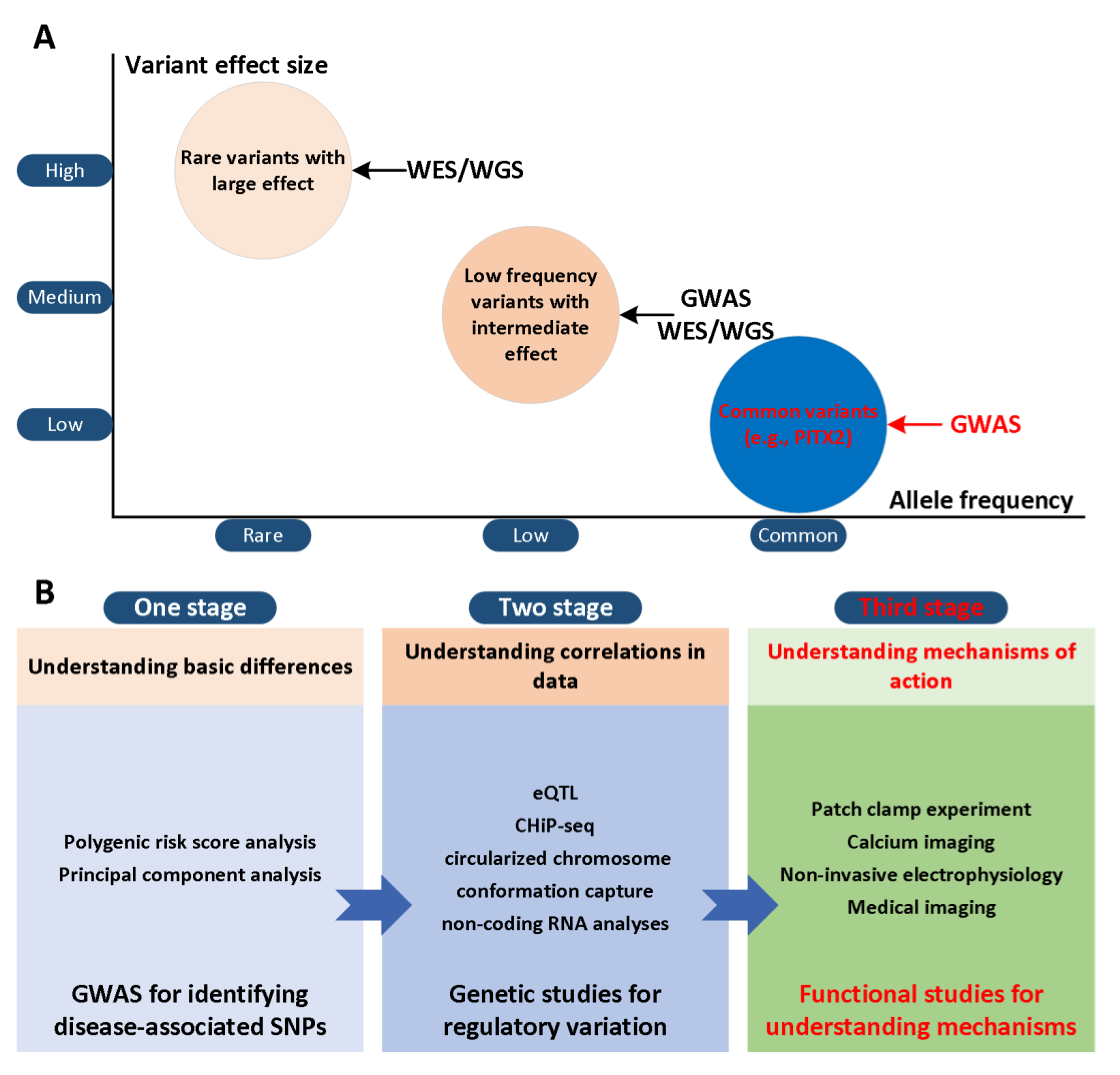{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Role of PITX2 in Cardiogenesis and Arrhythmogenesis
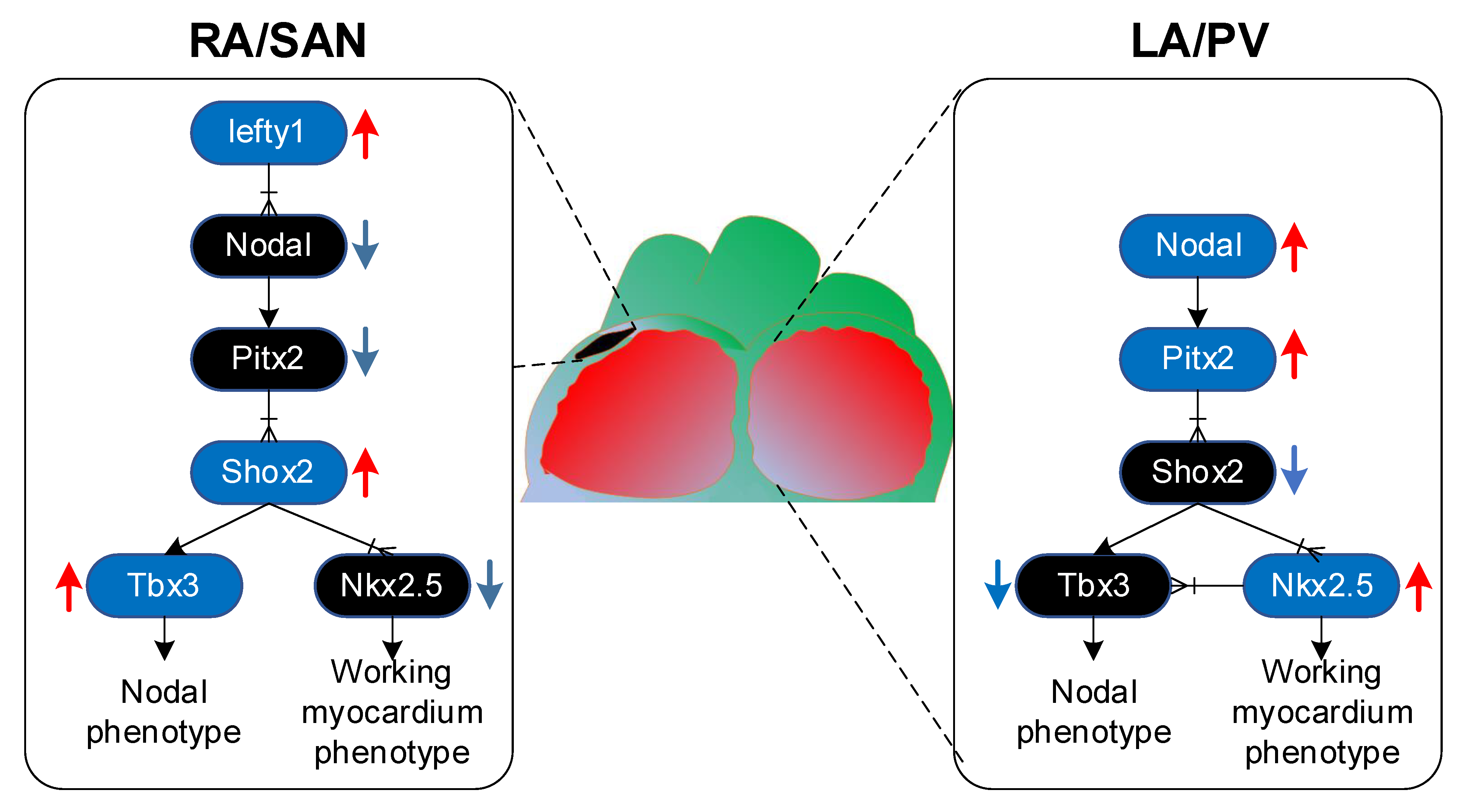
2.1. Pitx2 Promotes Left–Right Asymmetry
2.2. Gene Regulatory Mechanisms Driven by PITX2
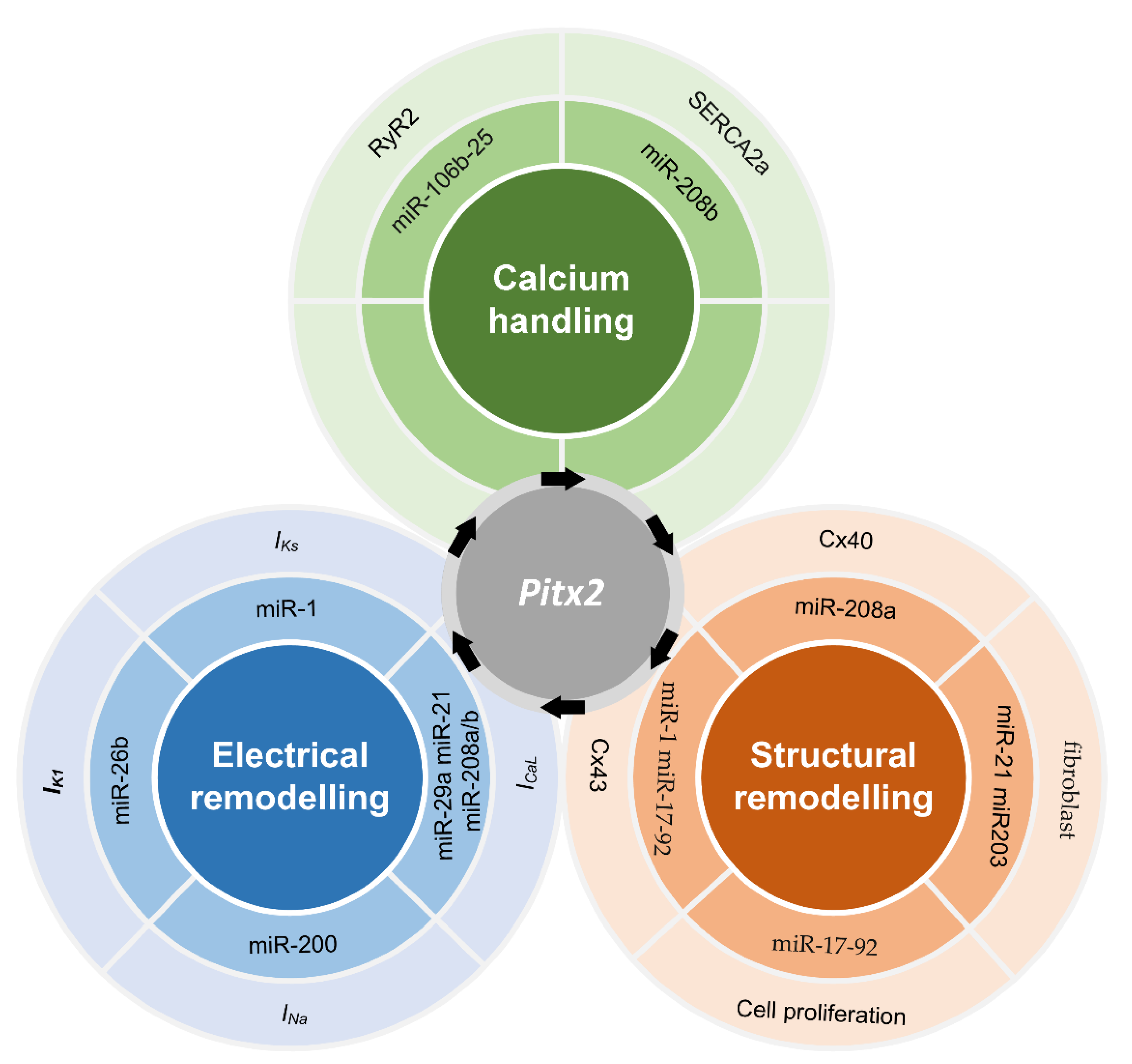
2.3. microRNAs Regulated by PITX2
2.4. Membrane Effector Genes Regulated by PITX2
2.5. Remodelling Linked Impaired PITX2 to AF
3. Recent Advances in Atrial Modelling
3.1. Ion Channel Modelling
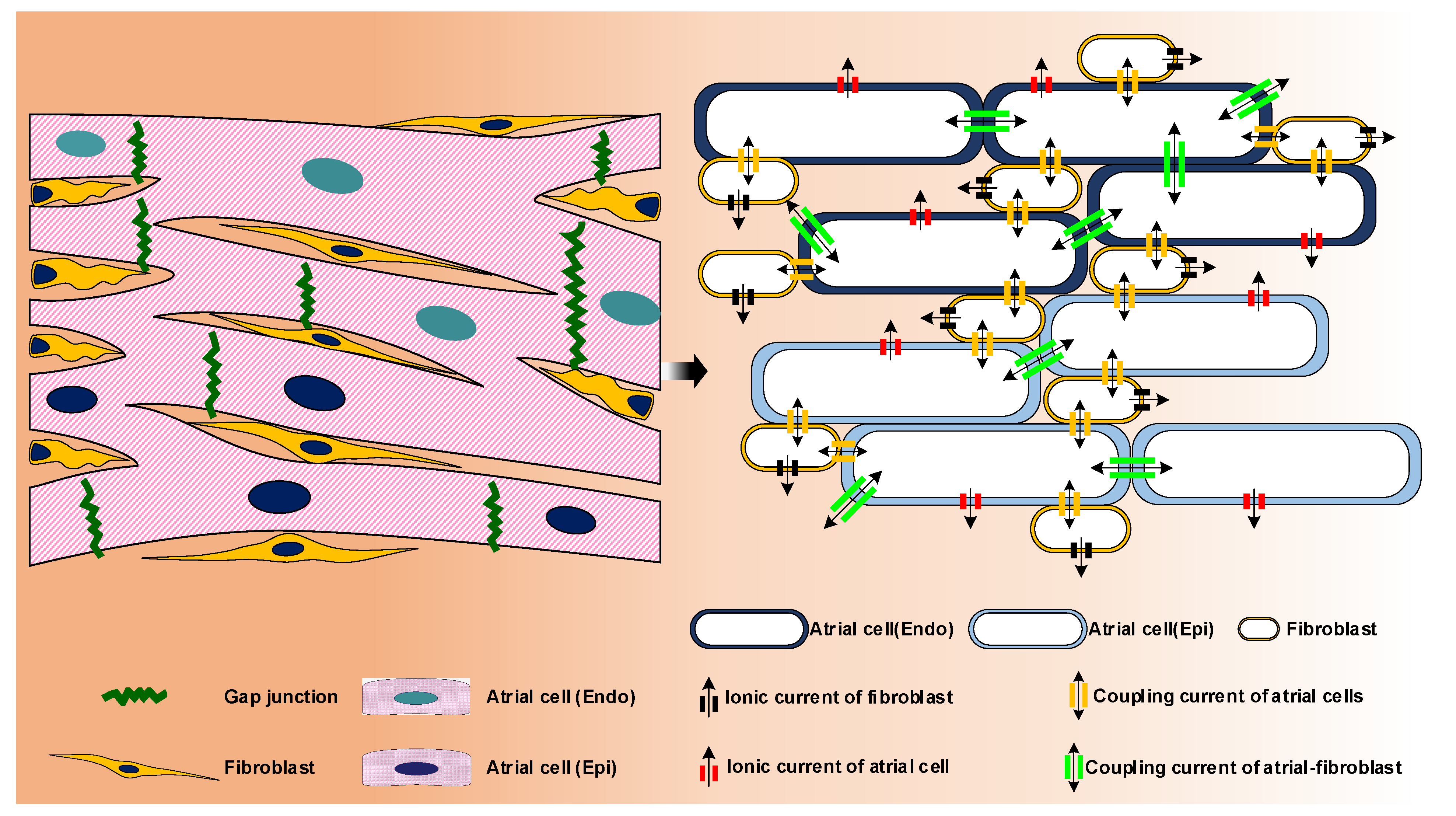
3.2. Computational Modelling of Atrial Cell
3.3. Geometric and Image-Based Atrial Modeling
4. Mechanistic Insights into PITX2-Dependent AF Using Computational Models
4.1. Calcium Handling Abnormalities and PITX2-Dependent AF
4.2. Electrical Remodelling and PITX2-Dependent AF
4.3. Electrical Heterogeneity and PITX2-Dependent AF
4.4. PITX2 Mutation and Familial AF
4.5. Clinical Relevance and Challenges
- (1)
- Each action potential model has different advantages and disadvantages, with numerous results being model specific.
- (2)
- The etiology of AF is diverse, but currently available cardiomyocyte models only have limited options for tailoring models to specific clinical conditions.
- (3)
- Only a handful of labs worldwide have the available expertise, computing power and required collaboration between clinicians, scientists and engineers to apply mechanistic whole-atria models in the clinical setting.
- (4)
- The extent of personalization of whole-atria models, particularly with regard to electrophysiological properties, remains very limited.
- (5)
- Current patient-level models do not incorporate fundamental mechanistic patterns of AF pathophysiology.
- (6)
- Integration of mechanistic modeling with “big data” approaches might help to improve AF diagnosis and management.
5. Open Questions Regarding Research into PITX2-Dependent AF
- (1)
- A robust method for the identification of the precise spatial distribution of PITX2 throughout human atria is needed. Cardiomyocytes from different atrial locations may exhibit spatial heterogeneities in AP properties reported in humans. The impact of changes in spatial heterogeneities due to impaired PITX2 should be further investigated.
- (2)
- Well-designed experiments to assess the PITX2 dependence of changes in electrophysiological properties, such as ionic concentrations and additional ionic currents, could help to re-evaluate anti-arrhythmic drugs that have often been ineffective thus far. Based on these findings, the effectiveness of anti-arrhythmic drugs can be assessed using computational models.
- (3)
- There is a need for more accurate PV ectopy models. Future studies should focus on development of accurate models of PV electrophysiology, structure and fibrosis distribution, that can be used to investigate how patient-specific predisposition to PV ectopy, in conjunction with patient-specific substrate, result in the onset and maintenance of PITX2-dependent AF.
- (4)
- Adipose-like tissue was found in PITX2 conditional-knockout hearts and morphological analysis of patient left atrial appendage tissue biopsies revealed tissue heterogeneity with marked fatty deposits and fibrosis in some specimens, and high myocardium content in others. Changes in PITX2 -induced remodelling (including adipose tissue deposition, atrial stretch with mechano-electrical feedback, the fibrotic atrial substrate, atrial wall thickness heterogeneity and so on) should be incorporated into the 3D human atria and simulations increase the AF mechanisms due to impaired PITX2.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Namboodiri, N.; Stiles, M.K.; Young, G.D.; Sanders, P. Electrophysiological features of atrial flutter in cardiac sarcoidosis: A report of two cases. Indian Pacing Electrophysiol. J. 2012, 12, 284–289. [Google Scholar] [CrossRef]
- Calkins, H.; Hindricks, G.; Cappato, R.; Kim, Y.-H.; Saad, E.B.; Aguinaga, L.; Akar, J.G.; Badhwar, V.; Brugada, J.; Camm, J. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Europace 2018, 20, e1–e160. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Franco, D.; Aranega, A.; Daimi, H. Genetics and epigenetics of atrial fibrillation. Int. J. Mol. Sci. 2020, 21, 5717. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, L.A.; Alonso, M.E.; Badía-Careaga, C.; Rollán, I.; Arias, C.; Fernández-Miñán, A.; López-Jiménez, E.; Aránega, A.; Gómez-Skarmeta, J.L.; Franco, D.; et al. Long-range regulatory interactions at the 4q25 atrial fibrillation risk locus involve PITX2c and ENPEP. BMC Biol. 2015, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Chinchilla, A.; Aránega, A.E. Transgenic insights linking pitx2 and atrial arrhythmias. Front. Physiol. 2012, 3, 206. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Arnar, D.O.; Helgadottir, A.; Gretarsdottir, S.; Holm, H.; Sigurdsson, A.; Jonasdottir, A.; Baker, A.; Thorleifsson, G.; Kristjansson, K. Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 2007, 448, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Kornej, J.; Börschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of atrial fibrillation in the 21st century: Novel methods and new insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Chen, S.A.; Ernst, S.; Guzman, C.E.; Han, S.; Kalarus, Z.; Labadet, C.; Lin, Y.J.; Lo, L.W.; Nogami, A. 2019 APHRS expert consensus statement on three-dimensional mapping systems for tachycardia developed in collaboration with HRS, EHRA, and LAHRS. J. Arrhythmia 2020, 36, 215. [Google Scholar] [CrossRef]
- Foo, F.S.; Stiles, M.K.; Clare, G.C.; Lever, N.; Hooks, D.; Heaven, D.; Boddington, D.; Zealand, H.R.N. Recent trends in cardiac electrophysiology and catheter ablation in New Zealand. Intern. Med. J. 2020, 50, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.V.; Kalyanasundaram, A.; Li, N.; Scott, S.S.; Artiga, E.J.; Subr, M.M.; Zhao, J.; Hansen, B.J.; Hummel, J.D.; Fedorov, V.V. Comprehensive evaluation of electrophysiological and 3D structural features of human atrial myocardium with insights on atrial fibrillation maintenance mechanisms. J. Mol. Cell. Cardiol. 2021, 151, 56–71. [Google Scholar] [CrossRef]
- Li, N.; Kalyanasundaram, A.; Hansen, B.J.; Artiga, E.J.; Sharma, R.; Abudulwahed, S.H.; Helfrich, K.M.; Rozenberg, G.; Wu, P.J.; Zakharkin, S.; et al. Impaired neuronal sodium channels cause intranodal conduction failure and reentrant arrhythmias in human sinoatrial node. Nat. Commun. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Aslanidi, O.V.; Colman, M.A.; Varela, M.; Zhao, J.; Smaill, B.H.; Hancox, J.C.; Boyett, M.R.; Zhang, H. Heterogeneous and anisotropic integrative model of pulmonary veins: Computational study of arrhythmogenic substrate for atrial fibrillation. Interface Focus 2013, 3, 20120069. [Google Scholar] [CrossRef] [PubMed]
- Aslanidi, O.V.; Colman, M.A.; Stott, J.; Dobrzynski, H.; Boyett, M.R.; Holden, A.V.; Zhang, H. 3D virtual human atria: A computational platform for studying clinical atrial fibrillation. Prog. Biophys. Mol. Biol. 2011, 107, 156–168. [Google Scholar] [CrossRef]
- Alday, E.A.P.; Colman, M.A.; Langley, P.; Butters, T.D.; Higham, J.; Workman, A.J.; Hancox, J.C.; Zhang, H. A new algorithm to diagnose Atrial ectopic origin from multi lead ECG systems-insights from 3D virtual human Atria and Torso. PLoS Comput. Biol. 2015, 11, e1004026. [Google Scholar] [CrossRef]
- Zhu, Y.; Bai, J.; Lo, A.; Lu, Y.; Zhao, J. Mechanisms underlying pro-arrhythmic abnormalities arising from Pitx2-induced electrical remodelling: An in silico intersubject variability study. Ann. Transl. Med. 2021, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhu, Y.; Lo, A.; Gao, M.; Lu, Y.; Zhao, J.; Zhang, H. In Silico Assessment of Class I Antiarrhythmic Drug Effects on Pitx2-Induced Atrial Fibrillation: Insights from Populations of Electrophysiological Models of Human Atrial Cells and Tissues. Int. J. Mol. Sci. 2021, 22, 1265. [Google Scholar] [CrossRef]
- Bai, J.; Zhu, Y.; Lo, A.; Lu, Y.; Zhao, J. In Silico Assessment of Genetic Variation in PITX2 Reveals the Molecular Mechanisms of Calcium-Mediated Cellular Triggered Activity in Atrial Fibrillation. Annu. Int. Conf. IEEE Eng. Med. Biol. 2020, 2020, 2353–2356. [Google Scholar]
- Lo, A.C.Y.; Bai, J.; Gladding, P.A.; Fedorov, V.V.; Zhao, J. Afterdepolarizations and abnormal calcium handling in atrial myocytes with modulated SERCA uptake: A sensitivity analysis of calcium handling channels. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2020, 378, 20190557. [Google Scholar] [CrossRef]
- Bai, J.; Lu, Y.; Lo, A.; Zhao, J.; Zhang, H. PITX2 upregulation increases the risk of chronic atrial fibrillation in a dose-dependent manner by modulating I(Ks) and I(CaL) -insights from human atrial modelling. Ann. Transl. Med. 2020, 8, 191. [Google Scholar] [CrossRef]
- Bai, J.; Lu, Y.; Zhang, H. In silico study of the effects of anti-arrhythmic drug treatment on sinoatrial node function for patients with atrial fibrillation. Sci. Rep. 2020, 10, 305. [Google Scholar] [CrossRef]
- Bai, J.; Lo, A.; Gladding, P.A.; Stiles, M.K.; Fedorov, V.V.; Zhao, J. In silico investigation of the mechanisms underlying atrial fibrillation due to impaired Pitx2. PLoS Comput. Biol. 2020, 16, e1007678. [Google Scholar] [CrossRef]
- Bai, J.; Lu, Y.; Lo, A.; Zhao, J.; Zhang, H. Proarrhythmia in the p.Met207Val PITX2c-Linked Familial Atrial Fibrillation-Insights From Modeling. Front. Physiol. 2019, 10, 1314. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Wang, K.; Li, Q.; Bai, J.; Zhang, H. Influence of the distribution of fibrosis within an area of myocardial infarction on wave propagation in ventricular tissue. Sci. Rep. 2019, 9, 14151. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Wang, K.; Liu, Y.; Li, Y.; Liang, C.; Luo, G.; Dong, S.; Yuan, Y.; Zhang, H. Computational Cardiac Modeling Reveals Mechanisms of Ventricular Arrhythmogenesis in Long QT Syndrome Type 8: CACNA1C R858H Mutation Linked to Ventricular Fibrillation. Front. Physiol. 2017, 8, 771. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Yin, R.; Wang, K.; Zhang, H. Mechanisms Underlying the Emergence of Post-acidosis Arrhythmia at the Tissue Level: A Theoretical Study. Front. Physiol. 2017, 8, 195. [Google Scholar] [CrossRef]
- Bai, J.; Wang, K.; Li, Q.; Yuan, Y.; Zhang, H. Pro-arrhythmogenic effects of CACNA1C G1911R mutation in human ventricular tachycardia: Insights from cardiac multi-scale models. Sci. Rep. 2016, 6, 31262. [Google Scholar] [CrossRef]
- Xiong, Z.; Nash, M.P.; Cheng, E.; Fedorov, V.V.; Stiles, M.K.; Zhao, J. ECG signal classification for the detection of cardiac arrhythmias using a convolutional recurrent neural network. Physiol. Meas. 2018, 39, 094006. [Google Scholar] [CrossRef]
- Hernandez-Torres, F.; Rodríguez-Outeiriño, L.; Franco, D.; Aranega, A.E. Pitx2 in Embryonic and Adult Myogenesis. Front. Cell Dev. Biol. 2017, 5, 46. [Google Scholar] [CrossRef]
- Franco, D.; Sedmera, D.; Lozano-Velasco, E. Multiple Roles of Pitx2 in Cardiac Development and Disease. J. Cardiovasc. Dev. Dis. 2017, 4, 16. [Google Scholar] [CrossRef]
- Franco, D.; Garcia-Padilla, C.; Dominguez, J.N.; Lozano-Velasco, E.; Aranega, A. Cardiac Development: A Glimpse on Its Translational Contributions. Hearts 2021, 2, 87–118. [Google Scholar] [CrossRef]
- Clauss, S.; Kääb, S. Is Pitx2 growing up? Circ. Cardiovasc. Genet. 2011, 4, 105–107. [Google Scholar] [CrossRef]
- Vallejo, D.; Hernández-Torres, F.; Lozano-Velasco, E.; Rodriguez-Outeiriño, L.; Carvajal, A.; Creus, C.; Franco, D.; Aránega, A.E. PITX2 enhances the regenerative potential of dystrophic skeletal muscle stem cells. Stem. Cell Rep. 2018, 10, 1398–1411. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Jongbloed, M.R.; Gittenberger-de Groot, A.C. Pitx2: A challenging teenager. Circ. Res. 2008, 102, 749–751. [Google Scholar] [CrossRef]
- Tessari, A.; Pietrobon, M.; Notte, A.; Cifelli, G.; Gage, P.J.; Schneider, M.D.; Lembo, G.; Campione, M. Myocardial Pitx2 differentially regulates the left atrial identity and ventricular asymmetric remodeling programs. Circ. Res. 2008, 102, 813–822. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Smits, G.J.; Kispert, A.; Moorman, A.F. Development of the pacemaker tissues of the heart. Circ. Res. 2010, 106, 240–254. [Google Scholar] [CrossRef]
- Franco, D.; Christoffels, V.M.; Campione, M. Homeobox transcription factor Pitx2: The rise of an asymmetry gene in cardiogenesis and arrhythmogenesis. Trends Cardiovasc. Med. 2014, 24, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, W.; Lu, M.F.; Brown, N.A.; Martin, J.F. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development 2001, 128, 2039–2048. [Google Scholar] [CrossRef] [PubMed]
- Ammirabile, G.; Tessari, A.; Pignataro, V.; Szumska, D.; Sutera Sardo, F.; Benes, J., Jr.; Balistreri, M.; Bhattacharya, S.; Sedmera, D.; Campione, M. Pitx2 confers left morphological, molecular, and functional identity to the sinus venosus myocardium. Cardiovasc. Res. 2012, 93, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Mommersteeg, M.T.; Brown, N.A.; Prall, O.W.; de Gier-de Vries, C.; Harvey, R.P.; Moorman, A.F.; Christoffels, V.M. Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ. Res. 2007, 101, 902–909. [Google Scholar] [CrossRef]
- Wang, J.; Klysik, E.; Sood, S.; Johnson, R.L.; Wehrens, X.H.; Martin, J.F. Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl. Acad. Sci. USA 2010, 107, 9753–9758. [Google Scholar] [CrossRef]
- Herraiz-Martínez, A.; Tarifa, C.; Lozano-Velasco, E.; Jiménez-Sábado, V.; Casabella, S.; Hernández-Torres, F.; Daimi, H.; Vázquez Ruiz de Castroviejo, E.; Delpón, E.; Caballero, R. Novel PITX2 Homeodomain-Contained Mutations from ATRIAL Fibrillation Patients Deteriorate Calcium Homeostasis. Hearts 2021, 2, 251–269. [Google Scholar] [CrossRef]
- Kirchhof, P.; Kahr, P.C.; Kaese, S.; Piccini, I.; Vokshi, I.; Scheld, H.H.; Rotering, H.; Fortmueller, L.; Laakmann, S.; Verheule, S.; et al. PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ. Cardiovasc. Genet. 2011, 4, 123–133. [Google Scholar] [CrossRef]
- Liu, C.; Liu, W.; Palie, J.; Lu, M.F.; Brown, N.A.; Martin, J.F. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development 2002, 129, 5081–5091. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Velasco, E.; Hernández-Torres, F.; Daimi, H.; Serra, S.A.; Herraiz, A.; Hove-Madsen, L.; Aránega, A.; Franco, D. Pitx2 impairs calcium handling in a dose-dependent manner by modulating Wnt signalling. Cardiovasc. Res. 2016, 109, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Velasco, E.; Wangensteen, R.; Quesada, A.; Garcia-Padilla, C.; Osorio, J.A.; Ruiz-Torres, M.D.; Aranega, A.; Franco, D. Hyperthyroidism, but not hypertension, impairs PITX2 expression leading to Wnt-microRNA-ion channel remodeling. PLoS ONE 2017, 12, e0188473. [Google Scholar] [CrossRef]
- Tian, J.; Popal, M.S.; Huang, R.; Zhang, M.; Zhao, X.; Zhang, M.; Song, X. Caveolin as a Novel Potential Therapeutic Target in Cardiac and Vascular Diseases: A Mini Review. Aging Dis. 2020, 11, 378–389. [Google Scholar] [CrossRef]
- Falk, N.; Joachimsthaler, A.; Kessler, K.; Lux, U.T.; Noegel, A.A.; Kremers, J.; Brandstätter, J.H.; Gießl, A.; Falk, N.; Joachimsthaler, A.; et al. Lack of a Retinal Phenotype in a Syne-2/Nesprin-2 Knockout Mouse Model. Cells 2019, 8, 1238. [Google Scholar] [CrossRef]
- Franco, D.; Lozano-Velasco, E.; Aranega, A. Gene regulatory networks in atrial fibrillation. World J. Med. Genet. 2016, 6, 1–16. [Google Scholar] [CrossRef]
- Franco, D.; Aranega, A.; Dominguez, J.N. Non-coding RNAs and atrial fibrillation. In Non-Coding RNAs in Cardiovascular Diseases; Springer: Berlin/Heidelberg, Germany, 2020; pp. 311–325. [Google Scholar]
- Petkova, M.; Atkinson, A.J.; Yanni, J.; Stuart, L.; Aminu, A.J.; Ivanova, A.D.; Pustovit, K.B.; Geragthy, C.; Feather, A.; Li, N.; et al. Identification of Key Small Non-Coding MicroRNAs Controlling Pacemaker Mechanisms in the Human Sinus Node. J. Am. Heart Assoc. 2020, 9, e016590. [Google Scholar] [CrossRef]
- García-Padilla, C.; Aránega, A.; Franco, D. The role of long non-coding RNAs in cardiac development and disease. AIMS Genet. 2018, 5, 124–140. [Google Scholar] [CrossRef]
- Torrado, M.; Franco, D.; Lozano-Velasco, E.; Hernández-Torres, F.; Calviño, R.; Aldama, G.; Centeno, A.; Castro-Beiras, A.; Mikhailov, A. A microRNA-transcription factor blueprint for early atrial arrhythmogenic remodeling. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Wang, J.; Bai, Y.; Li, N.; Ye, W.; Zhang, M.; Greene, S.B.; Tao, Y.; Chen, Y.; Wehrens, X.H.; Martin, J.F. Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc. Natl. Acad. Sci. USA 2014, 111, 9181–9186. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, N.W.E.; Kawasaki, M.; Berger, W.R.; Neefs, J.; Meulendijks, E.; Tijsen, A.J.; de Groot, J.R. MicroRNAs in Atrial Fibrillation: From Expression Signatures to Functional Implications. Cardiovasc. Drugs 2017, 31, 345–365. [Google Scholar] [CrossRef] [PubMed]
- Syeda, F.; Holmes, A.P.; Yu, T.Y.; Tull, S.; Kuhlmann, S.M.; Pavlovic, D.; Betney, D.; Riley, G.; Kucera, J.P.; Jousset, F.; et al. PITX2 Modulates Atrial Membrane Potential and the Antiarrhythmic Effects of Sodium-Channel Blockers. J. Am. Coll Cardiol. 2016, 68, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, A.; Daimi, H.; Lozano-Velasco, E.; Dominguez, J.N.; Caballero, R.; Delpón, E.; Tamargo, J.; Cinca, J.; Hove-Madsen, L.; Aranega, A.E.; et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ. Cardiovasc. Genet. 2011, 4, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Reyat, J.S.; Chua, W.; Cardoso, V.R.; Witten, A.; Kastner, P.M.; Kabir, S.N.; Sinner, M.F.; Wesselink, R.; Holmes, A.P.; Pavlovic, D.; et al. Reduced left atrial cardiomyocyte PITX2 and elevated circulating BMP10 predict atrial fibrillation after ablation. JCI Insight 2020, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; Matamoros, M.; Barana, A.; Amorós, I.; Gómez, R.; Núñez, M.; Sacristán, S.; Pinto, Á.; Fernández-Avilés, F.; Tamargo, J.; et al. Pitx2c increases in atrial myocytes from chronic atrial fibrillation patients enhancing IKs and decreasing ICa,L. Cardiovasc. Res. 2016, 109, 431–441. [Google Scholar] [CrossRef]
- Nadadur, R.D.; Broman, M.T.; Boukens, B.; Mazurek, S.R.; Yang, X.; van den Boogaard, M.; Bekeny, J.; Gadek, M.; Ward, T.; Zhang, M.; et al. Pitx2 modulates a Tbx5-dependent gene regulatory network to maintain atrial rhythm. Sci. Transl. Med. 2016, 8, 354ra115. [Google Scholar] [CrossRef]
- Herraiz-Martínez, A.; Llach, A.; Tarifa, C.; Gandía, J.; Jiménez-Sabado, V.; Lozano-Velasco, E.; Serra, S.A.; Vallmitjana, A.; Vázquez Ruiz de Castroviejo, E.; Benítez, R.; et al. The 4q25 variant rs13143308T links risk of atrial fibrillation to defective calcium homoeostasis. Cardiovasc. Res. 2019, 115, 578–589. [Google Scholar] [CrossRef]
- Holmes, A.P.; Saxena, P.; Kabir, S.N.; O’Shea, C.; Kuhlmann, S.M.; Gupta, S.; Fobian, D.; Apicella, C.; O’Reilly, M.; Syeda, F.; et al. Atrial resting membrane potential confers sodium current sensitivity to propafenone, flecainide, and dronedarone. Heart Rhythm 2021, 18, 1212–1220. [Google Scholar] [CrossRef]
- Nattel, S.; Aguilar, M. Do Atrial Fibrillation-Promoting Gene Variants Act by Enhancing Atrial Remodeling? JACC Clin. Electrophysiol. 2020, 6, 1522–1524. [Google Scholar] [CrossRef]
- Wong, G.R.; Nalliah, C.J.; Lee, G.; Voskoboinik, A.; Prabhu, S.; Parameswaran, R.; Sugumar, H.; Anderson, R.D.; Ling, L.H.; McLellan, A.; et al. Genetic Susceptibility to Atrial Fibrillation Is Associated With Atrial Electrical Remodeling and Adverse Post-Ablation Outcome. JACC Clin. Electrophysiol. 2020, 6, 1509–1521. [Google Scholar] [CrossRef]
- Vilches, J.M.; Franco, D.; Aránega, A.E. Contribution of miRNAs to ion-channel remodelling in atrial fibrillation. World J. Hypertens. 2015, 5, 6–13. [Google Scholar] [CrossRef]
- Lozano-Velasco, E.; Vallejo, D.; Esteban, F.J.; Doherty, C.; Hernández-Torres, F.; Franco, D.; Aránega, A.E. A Pitx2-MicroRNA Pathway Modulates Cell Proliferation in Myoblasts and Skeletal-Muscle Satellite Cells and Promotes Their Commitment to a Myogenic Cell Fate. Mol. Cell. Biol. 2015, 35, 2892–2909. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, M.; Liu, H.F.; Wang, J.P. Effects of microRNA-21 targeting PITX2 on proliferation and apoptosis of pituitary tumor cells. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7547. [Google Scholar] [PubMed]
- Bai, J.; Wang, K.; Zhang, H. Potential Pathogenesis Discovery of Arrhythmia Based on Cardiac Electrophysiological Models: Research Progress. Prog. Biochem. Biophys 2016, 43, 128–140. [Google Scholar]
- Zhao, J.; Kharche, S.R.; Hansen, B.J.; Csepe, T.A.; Wang, Y.; Stiles, M.K.; Fedorov, V.V. Optimization of catheter ablation of atrial fibrillation: Insights gained from clinically-derived computer models. Int. J. Mol. Sci. 2015, 16, 10834–10854. [Google Scholar] [CrossRef] [PubMed]
- Niederer, S.A.; Lumens, J.; Trayanova, N.A. Computational models in cardiology. Nat. Rev. Cardiol. 2019, 16, 100–111. [Google Scholar] [CrossRef]
- Heijman, J.; Erfanian Abdoust, P.; Voigt, N.; Nattel, S.; Dobrev, D. Computational models of atrial cellular electrophysiology and calcium handling, and their role in atrial fibrillation. J. Physiol. 2016, 594, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Garratt, C.J.; Zhu, J.; Holden, A.V. Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc. Res. 2005, 66, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, K.; Li, Q.; Hancox, J.C.; Zhang, H. Reciprocal interaction between IK1 and If in biological pacemakers: A simulation study. PLoS Comput. Biol. 2021, 17, e1008177. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Zhang, H.; Grandi, E.; Narayan, S.M.; Giles, W.R. Transient outward K+ current can strongly modulate action potential duration and initiate alternans in the human atrium. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H527–H542. [Google Scholar] [CrossRef]
- Clerx, M.; Beattie, K.A.; Gavaghan, D.J.; Mirams, G.R. Four Ways to Fit an Ion Channel Model. Biophys. J. 2019, 117, 2420–2437. [Google Scholar] [CrossRef]
- Vagos, M.; van Herck, I.G.M.; Sundnes, J.; Arevalo, H.J.; Edwards, A.G.; Koivumäki, J.T. Computational Modeling of Electrophysiology and Pharmacotherapy of Atrial Fibrillation: Recent Advances and Future Challenges. Front. Physiol. 2018, 9, 1221. [Google Scholar] [CrossRef]
- Wilhelms, M.; Hettmann, H.; Maleckar, M.; Koivumäki, J.; Dössel, O.; Seemann, G. Benchmarking electrophysiological models of human atrial myocytes. Front. Physiol. 2013, 3, 487. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Onal, B.; Unudurthi, S.; Hund, T. Modeling CaMKII in cardiac physiology: From molecule to tissue. Front. Pharmacol. 2014, 5, 9. [Google Scholar] [CrossRef]
- McCabe, K.J.; Rangamani, P. Computational modeling approaches to cAMP/PKA signaling in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 154, 32–40. [Google Scholar] [CrossRef]
- Niederer, S.A.; Campbell, K.S.; Campbell, S.G. A short history of the development of mathematical models of cardiac mechanics. J. Mol. Cell. Cardiol. 2019, 127, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Courtemanche, M.; Ramirez, R.J.; Nattel, S. Ionic mechanisms underlying human atrial action potential properties: Insights from a mathematical model. Am. J. Physiol. 1998, 275, H301–H321. [Google Scholar] [CrossRef]
- Nygren, A.; Fiset, C.; Firek, L.; Clark, J.W.; Lindblad, D.S.; Clark, R.B.; Giles, W.R. Mathematical model of an adult human atrial cell: The role of K+ currents in repolarization. Circ. Res. 1998, 82, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Jacquemet, V. Steady-state solutions in mathematical models of atrial cell electrophysiology and their stability. Math. Biosci. 2007, 208, 241–269. [Google Scholar] [CrossRef] [PubMed]
- Maleckar, M.M.; Greenstein, J.L.; Giles, W.R.; Trayanova, N.A. K+ current changes account for the rate dependence of the action potential in the human atrial myocyte. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1398–H1410. [Google Scholar] [CrossRef] [PubMed]
- Koivumäki, J.T.; Korhonen, T.; Tavi, P. Impact of sarcoplasmic reticulum calcium release on calcium dynamics and action potential morphology in human atrial myocytes: A computational study. PLoS Comput. Biol. 2011, 7, e1001067. [Google Scholar] [CrossRef] [PubMed]
- Koivumäki, J.T.; Seemann, G.; Maleckar, M.M.; Tavi, P. In silico screening of the key cellular remodeling targets in chronic atrial fibrillation. PLoS Comput. Biol. 2014, 10, e1003620. [Google Scholar] [CrossRef] [PubMed]
- Colman, M.A.; Aslanidi, O.V.; Kharche, S.; Boyett, M.R.; Garratt, C.; Hancox, J.C.; Zhang, H. Pro-arrhythmogenic effects of atrial fibrillation-induced electrical remodelling: Insights from the three-dimensional virtual human atria. J. Physiol. 2013, 591, 4249–4272. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Pandit, S.V.; Voigt, N.; Workman, A.J.; Dobrev, D.; Jalife, J.; Bers, D.M. Human atrial action potential and Ca2+ model: Sinus rhythm and chronic atrial fibrillation. Circ. Res. 2011, 109, 1055–1066. [Google Scholar] [CrossRef]
- Bai, J.; Gladding, P.A.; Stiles, M.K.; Fedorov, V.V.; Zhao, J. Ionic and cellular mechanisms underlying TBX5/PITX2 insufficiency-induced atrial fibrillation: Insights from mathematical models of human atrial cells. Sci. Rep. 2018, 8, 15642. [Google Scholar] [CrossRef]
- Voigt, N.; Heijman, J.; Trausch, A.; Mintert-Jancke, E.; Pott, L.; Ravens, U.; Dobrev, D. Impaired Na⁺-dependent regulation of acetylcholine-activated inward-rectifier K⁺ current modulates action potential rate dependence in patients with chronic atrial fibrillation. J. Mol. Cell. Cardiol. 2013, 61, 142–152. [Google Scholar] [CrossRef]
- Schmidt, C.; Wiedmann, F.; Voigt, N.; Zhou, X.B.; Heijman, J.; Lang, S.; Albert, V.; Kallenberger, S.; Ruhparwar, A.; Szabó, G.; et al. Upregulation of K(2P)3.1 K+ Current Causes Action Potential Shortening in Patients with Chronic Atrial Fibrillation. Circulation 2015, 132, 82–92. [Google Scholar] [CrossRef]
- Voigt, N.; Heijman, J.; Wang, Q.; Chiang, D.Y.; Li, N.; Karck, M.; Wehrens, X.H.T.; Nattel, S.; Dobrev, D. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 2014, 129, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, H.; van Sloun, B.; Schönleitner, P.; van Zandvoort, M.A.M.J.; Antoons, G.; Heijman, J. The Subcellular Distribution of Ryanodine Receptors and L-Type Ca2+ Channels Modulates Ca2+-Transient Properties and Spontaneous Ca2+-Release Events in Atrial Cardiomyocytes. Front. Physiol. 2018, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Colman, M.A. Arrhythmia mechanisms and spontaneous calcium release: Bi-directional coupling between re-entrant and focal excitation. PLoS Comput. Biol. 2019, 15, e1007260. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, A.; Li, N.; Gardner, M.L.; Artiga, E.J.; Hansen, B.J.; Webb, A.; Freitas, M.A.; Pietrzak, M.; Whitson, B.A.; Mokadam, N.A.; et al. Fibroblast-Specific Proteo-Transcriptomes Reveal Distinct Fibrotic Signatures of Human Sinoatrial Node in Non-Failing and Failing Hearts. Circulation 2021. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.H.; Bernus, O.; Cherry, E.M.; Dierckx, H.; Fenton, F.H.; Mirabella, L.; Panfilov, A.V.; Sachse, F.B.; Seemann, G.; Zhang, H. Models of cardiac tissue electrophysiology: Progress, challenges and open questions. Prog. Biophys. Mol. Biol. 2011, 104, 22–48. [Google Scholar] [CrossRef] [PubMed]
- Hansen, B.J.; Zhao, J.; Csepe, T.A.; Moore, B.T.; Li, N.; Jayne, L.A.; Kalyanasundaram, A.; Lim, P.; Bratasz, A.; Powell, K.A.; et al. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur. Heart J. 2015, 36, 2390–2401. [Google Scholar] [CrossRef] [PubMed]
- Aronis, K.N.; Ali, R.; Trayanova, N.A. The role of personalized atrial modeling in understanding atrial fibrillation mechanisms and improving treatment. Int. J. Cardiol. 2019, 287, 139–147. [Google Scholar] [CrossRef]
- Hansen, B.J.; Zhao, J.; Helfrich, K.M.; Li, N.; Iancau, A.; Zolotarev, A.M.; Zakharkin, S.O.; Kalyanasundaram, A.; Subr, M.; Dastagir, N.; et al. Unmasking Arrhythmogenic Hubs of Reentry Driving Persistent Atrial Fibrillation for Patient-Specific Treatment. J. Am. Heart Assoc. 2020, 9, e017789. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiong, Z.; Nalar, A.; Hansen, B.J.; Kharche, S.; Seemann, G.; Loewe, A.; Fedorov, V.V.; Zhao, J. A robust computational framework for estimating 3D Bi-Atrial chamber wall thickness. Comput. Biol. Med. 2019, 114, 103444. [Google Scholar] [CrossRef]
- Fu, X.; Liu, T.; Xiong, Z.; Smaill, B.H.; Stiles, M.K.; Zhao, J. Segmentation of histological images and fibrosis identification with a convolutional neural network. Comput. Biol. Med. 2018, 98, 147–158. [Google Scholar] [CrossRef]
- Hansen, B.J.; Zhao, J.; Fedorov, V.V. Fibrosis and Atrial Fibrillation: Computerized and Optical Mapping; A View into the Human Atria at Submillimeter Resolution. JACC Clin. Electrophysiol. 2017, 3, 531–546. [Google Scholar] [CrossRef]
- Zolotarev, A.M.; Hansen, B.J.; Ivanova, E.A.; Helfrich, K.M.; Li, N.; Janssen, P.M.L.; Mohler, P.J.; Mokadam, N.A.; Whitson, B.A.; Fedorov, M.V.; et al. Optical Mapping-Validated Machine Learning Improves Atrial Fibrillation Driver Detection by Multi-Electrode Mapping. Circ. Arrhythmia Electrophysiol. 2020, 13, e008249. [Google Scholar] [CrossRef] [PubMed]
- Csepe, T.A.; Zhao, J.; Sul, L.V.; Wang, Y.; Hansen, B.J.; Li, N.; Ignozzi, A.J.; Bratasz, A.; Powell, K.A.; Kilic, A.; et al. Novel application of 3D contrast-enhanced CMR to define fibrotic structure of the human sinoatrial node in vivo. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Fedorov, V.V.; Fu, X.; Cheng, E.; Macleod, R.; Zhao, J. Fully automatic left atrium segmentation from late gadolinium enhanced magnetic resonance imaging using a dual fully convolutional neural network. IEEE Trans. Med. Imaging 2018, 38, 515–524. [Google Scholar] [CrossRef]
- Xiong, Z.; Xia, Q.; Hu, Z.; Huang, N.; Bian, C.; Zheng, Y.; Vesal, S.; Ravikumar, N.; Maier, A.; Yang, X.; et al. A global benchmark of algorithms for segmenting the left atrium from late gadolinium-enhanced cardiac magnetic resonance imaging. Med. Image Anal. 2021, 67, 101832. [Google Scholar] [CrossRef]
- Jamart, K.; Xiong, Z.; Talou, G.D.M.; Stiles, M.K.; Zhao, J. Mini Review: Deep Learning for Atrial Segmentation From Late Gadolinium-Enhanced MRIs. Front. Cardiovasc. Med. 2020, 7, 86. [Google Scholar] [CrossRef]
- Hansen, B.J.; Zhao, J.; Li, N.; Zolotarev, A.; Zakharkin, S.; Wang, Y.; Atwal, J.; Kalyanasundaram, A.; Abudulwahed, S.H.; Helfrich, K.M.; et al. Human Atrial Fibrillation Drivers Resolved With Integrated Functional and Structural Imaging to Benefit Clinical Mapping. JACC Clin. Electrophysiol. 2018, 4, 1501–1515. [Google Scholar] [CrossRef]
- Oakes, R.S.; Badger, T.J.; Kholmovski, E.G.; Akoum, N.; Burgon, N.S.; Fish, E.N.; Blauer, J.J.; Rao, S.N.; DiBella, E.V.; Segerson, N.M.; et al. Detection and quantification of left atrial structural remodeling with delayed-enhancement magnetic resonance imaging in patients with atrial fibrillation. Circulation 2009, 119, 1758–1767. [Google Scholar] [CrossRef] [PubMed]
- Khurram, I.M.; Beinart, R.; Zipunnikov, V.; Dewire, J.; Yarmohammadi, H.; Sasaki, T.; Spragg, D.D.; Marine, J.E.; Berger, R.D.; Halperin, H.R.; et al. Magnetic resonance image intensity ratio, a normalized measure to enable interpatient comparability of left atrial fibrosis. Heart Rhythm 2014, 11, 85–92. [Google Scholar] [CrossRef]
- Fastl, T.E.; Tobon-Gomez, C.; Crozier, A.; Whitaker, J.; Rajani, R.; McCarthy, K.P.; Sanchez-Quintana, D.; Ho, S.Y.; O’Neill, M.D.; Plank, G.; et al. Personalized computational modeling of left atrial geometry and transmural myofiber architecture. Med. Image Anal. 2018, 47, 180–190. [Google Scholar] [CrossRef]
- Krueger, M.W.; Schmidt, V.; Tobón, C.; Weber, F.M.; Lorenz, C.; Keller, D.U.J.; Barschdorf, H.; Burdumy, M.; Neher, P.; Plank, G.; et al. Modeling Atrial Fiber Orientation in Patient-Specific Geometries: A Semi-Automatic Rule-Based Approach; Springer: Berlin/Heidelberg, Germany, 2011; pp. 223–232. [Google Scholar]
- Roy, A.; Varela, M.; Aslanidi, O. Image-Based Computational Evaluation of the Effects of Atrial Wall Thickness and Fibrosis on Re-entrant Drivers for Atrial Fibrillation. Front. Physiol. 2018, 9, 1352. [Google Scholar] [CrossRef]
- Seemann, G.; Höper, C.; Sachse, F.B.; Dössel, O.; Holden, A.V.; Zhang, H. Heterogeneous three-dimensional anatomical and electrophysiological model of human atria. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2006, 364, 1465–1481. [Google Scholar] [CrossRef]
- Zhao, J.; Hansen, B.J.; Wang, Y.; Csepe, T.A.; Sul, L.V.; Tang, A.; Yuan, Y.; Li, N.; Bratasz, A.; Powell, K.A. Three-dimensional integrated functional, structural, and computational mapping to define the structural “fingerprints” of heart-specific atrial fibrillation drivers in human heart ex vivo. J. Am. Heart Assoc. 2017, 6, e005922. [Google Scholar] [CrossRef]
- Hansen, B.J.; Li, N.; Helfrich, K.M.; Abudulwahed, S.H.; Artiga, E.J.; Joseph, M.E.; Mohler, P.J.; Hummel, J.D.; Fedorov, V.V. First In Vivo Use of High-Resolution Near-Infrared Optical Mapping to Assess Atrial Activation During Sinus Rhythm and Atrial Fibrillation in a Large Animal Model. Circ. Arrhythmia Electrophysiol. 2018, 11, e006870. [Google Scholar] [CrossRef] [PubMed]
- McLellan, A.J.; Ling, L.-H.; Azzopardi, S.; Lee, G.A.; Lee, G.; Kumar, S.; Wong, M.C.; Walters, T.E.; Lee, J.M.; Looi, K.-L. A minimal or maximal ablation strategy to achieve pulmonary vein isolation for paroxysmal atrial fibrillation: A prospective multi-centre randomized controlled trial (the Minimax study). Eur. Heart J. 2015, 36, 1812–1821. [Google Scholar] [CrossRef]
- Mechakra, A.; Footz, T.; Walter, M.; Aránega, A.; Hernández-Torres, F.; Morel, E.; Millat, G.; Yang, Y.Q.; Chahine, M.; Chevalier, P.; et al. A Novel PITX2c Gain-of-Function Mutation, p.Met207Val, in Patients with Familial Atrial Fibrillation. Am. J. Cardiol. 2019, 123, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Parvez, B.; Vaglio, J.; Rowan, S.; Muhammad, R.; Kucera, G.; Stubblefield, T.; Carter, S.; Roden, D.; Darbar, D. Symptomatic response to antiarrhythmic drug therapy is modulated by a common single nucleotide polymorphism in atrial fibrillation. J. Am. Coll Cardiol. 2012, 60, 539–545. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, J.; Lu, Y.; Zhu, Y.; Wang, H.; Yin, D.; Zhang, H.; Franco, D.; Zhao, J. Understanding PITX2-Dependent Atrial Fibrillation Mechanisms through Computational Models. Int. J. Mol. Sci. 2021, 22, 7681. https://doi.org/10.3390/ijms22147681
Bai J, Lu Y, Zhu Y, Wang H, Yin D, Zhang H, Franco D, Zhao J. Understanding PITX2-Dependent Atrial Fibrillation Mechanisms through Computational Models. International Journal of Molecular Sciences. 2021; 22(14):7681. https://doi.org/10.3390/ijms22147681
Chicago/Turabian StyleBai, Jieyun, Yaosheng Lu, Yijie Zhu, Huijin Wang, Dechun Yin, Henggui Zhang, Diego Franco, and Jichao Zhao. 2021. "Understanding PITX2-Dependent Atrial Fibrillation Mechanisms through Computational Models" International Journal of Molecular Sciences 22, no. 14: 7681. https://doi.org/10.3390/ijms22147681
APA StyleBai, J., Lu, Y., Zhu, Y., Wang, H., Yin, D., Zhang, H., Franco, D., & Zhao, J. (2021). Understanding PITX2-Dependent Atrial Fibrillation Mechanisms through Computational Models. International Journal of Molecular Sciences, 22(14), 7681. https://doi.org/10.3390/ijms22147681