
Dissipation-Driven Selection under Finite Diffusion: Hints from Equilibrium and Separation of Time Scales


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
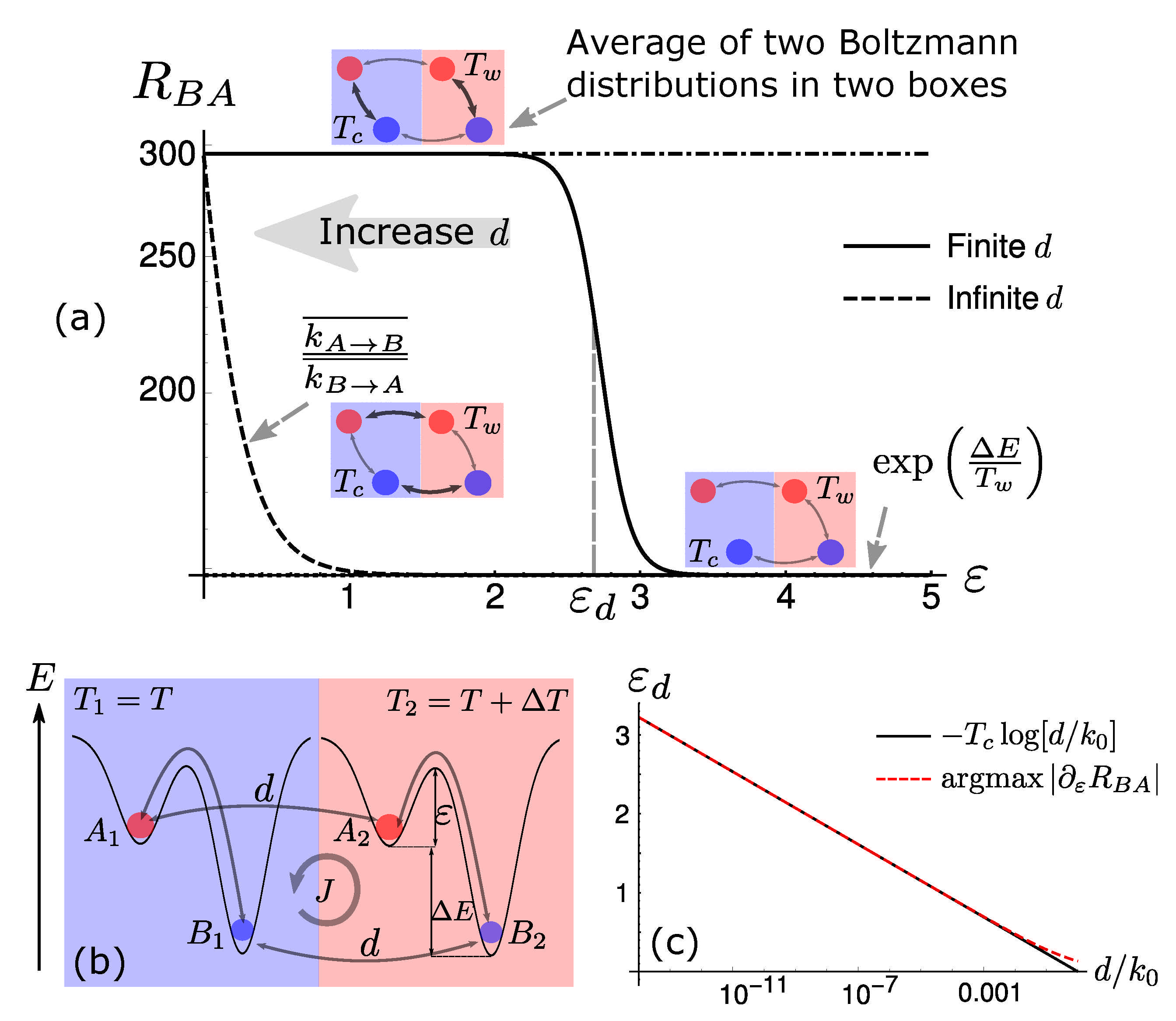
2. Phase-Transition for Selection in Two-State Systems
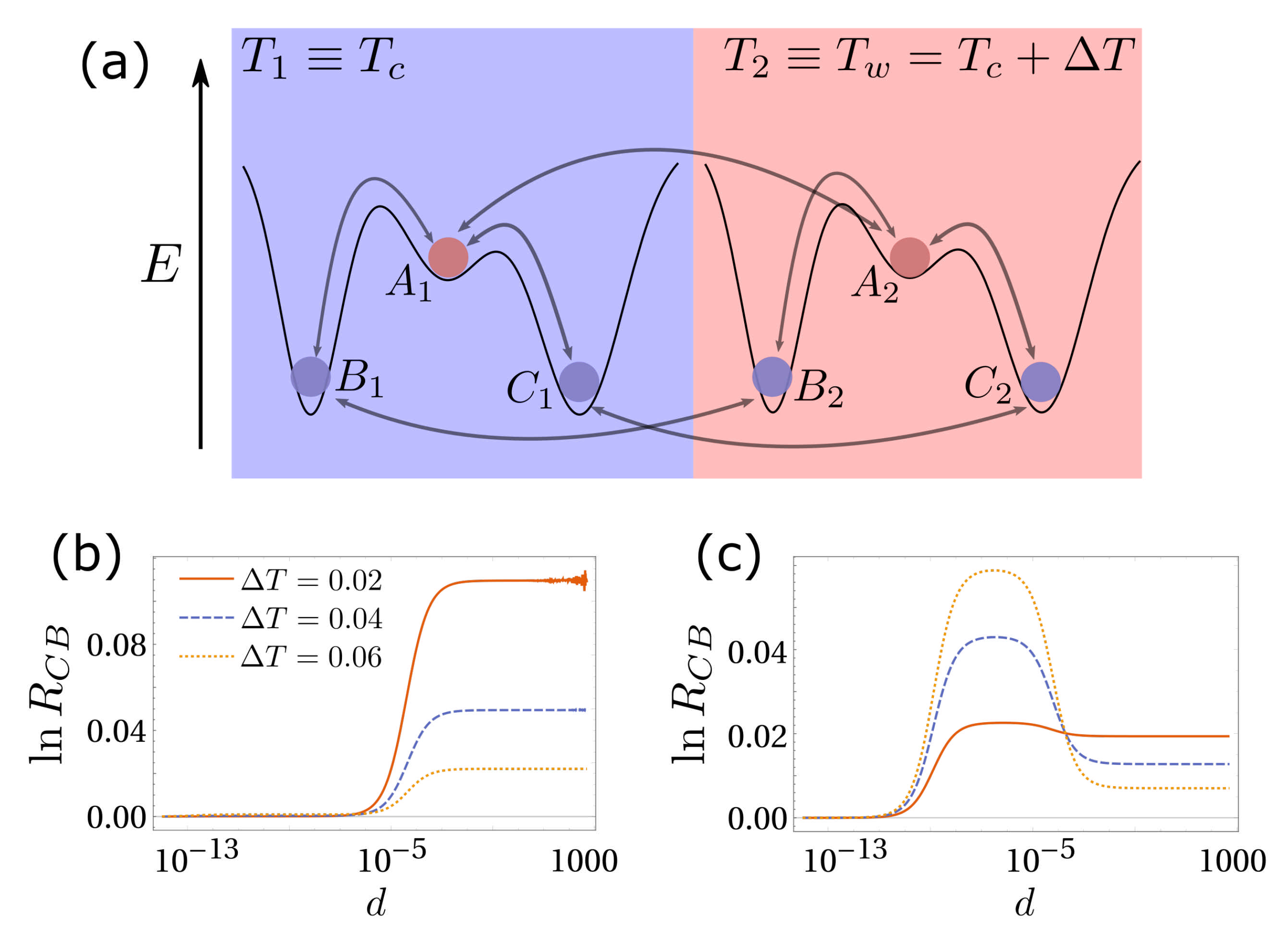
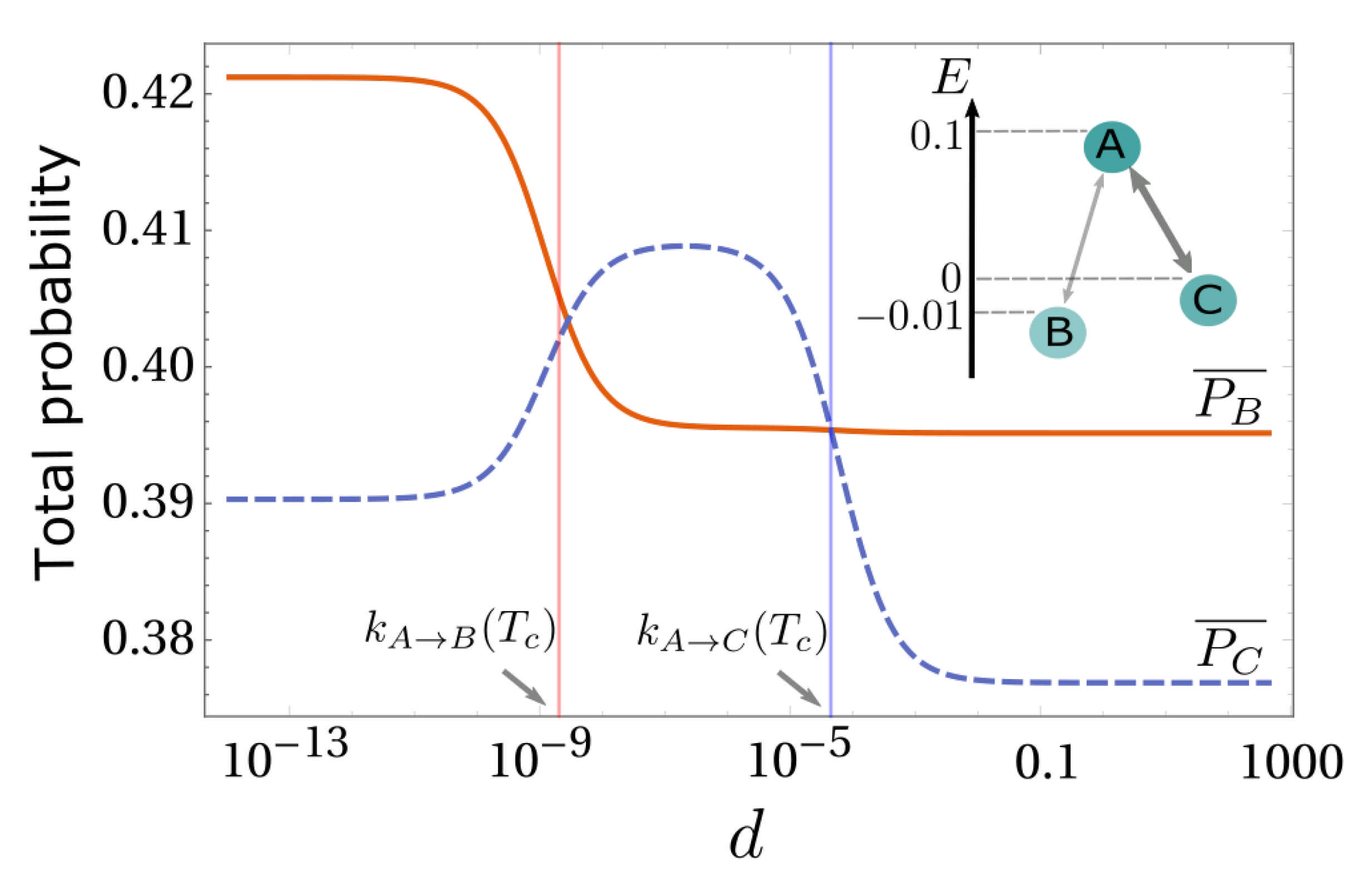
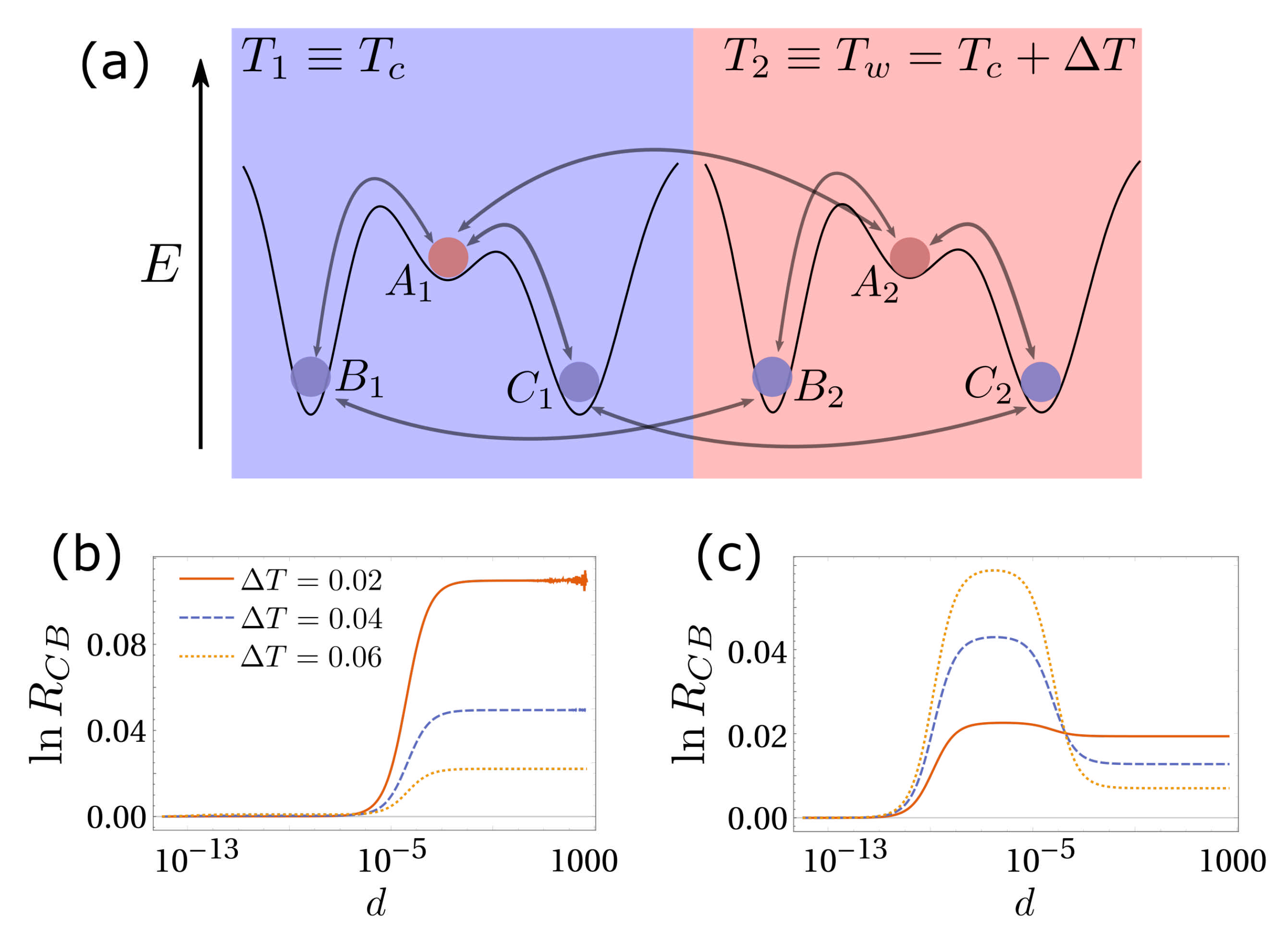
3. Simplest Case for Selection: A Three-State System
4. Time-Scale Separation and Equilibrium Hints
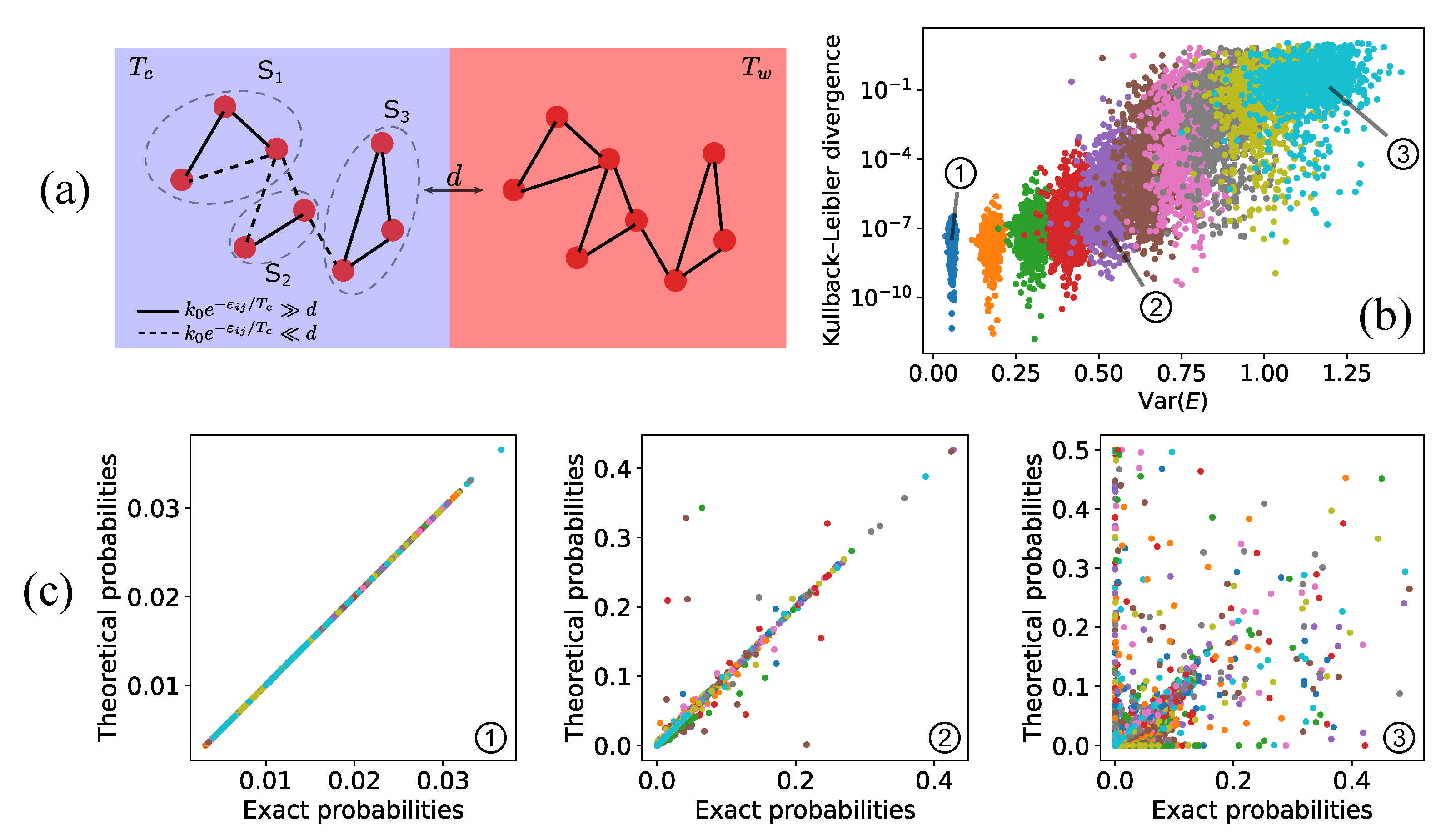
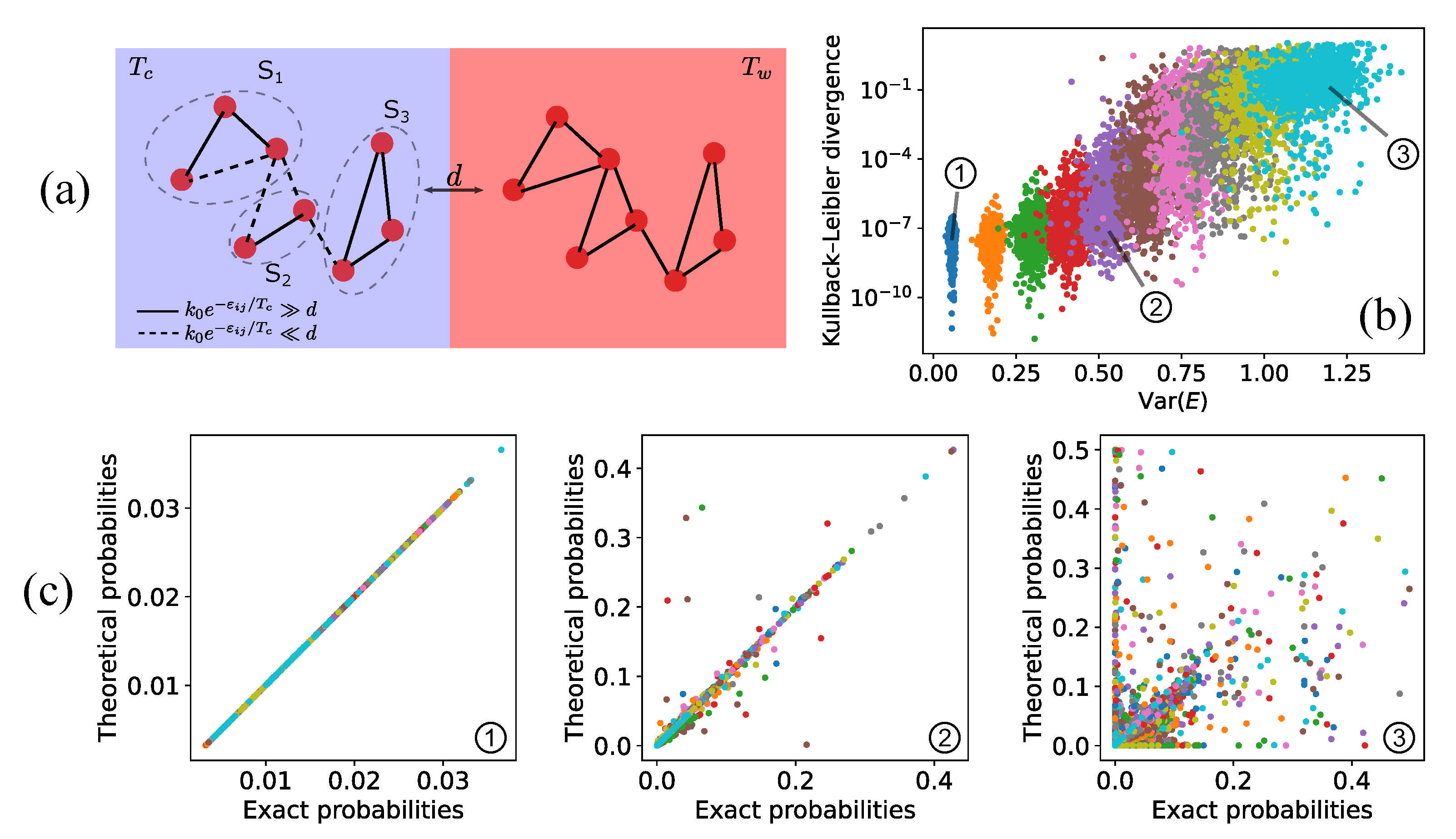
4.1. Fast-Dissipation Chemical Sub-Networks in Two-Box Models
4.2. Fast-Dissipation Ensemble Distribution in Two-Box Models
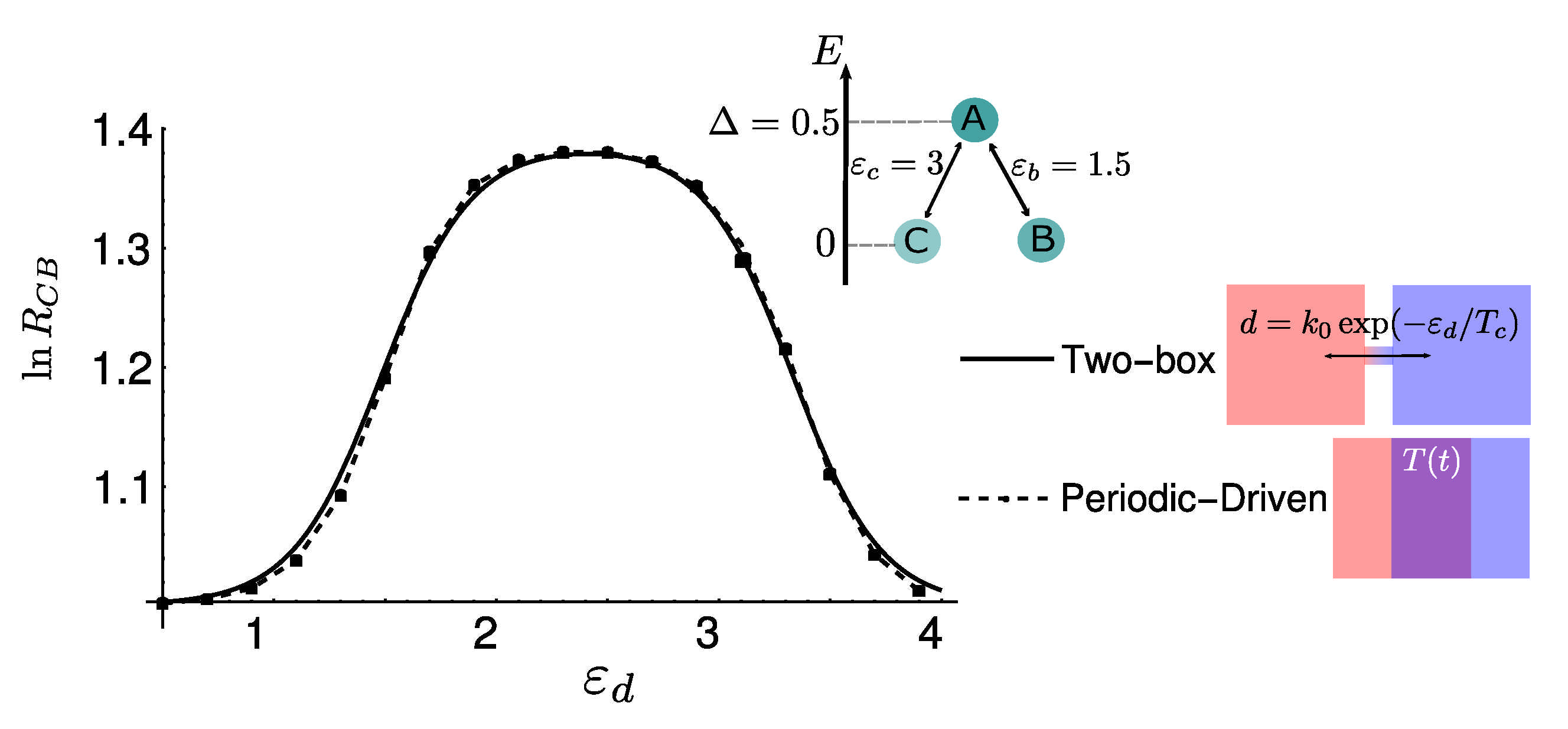
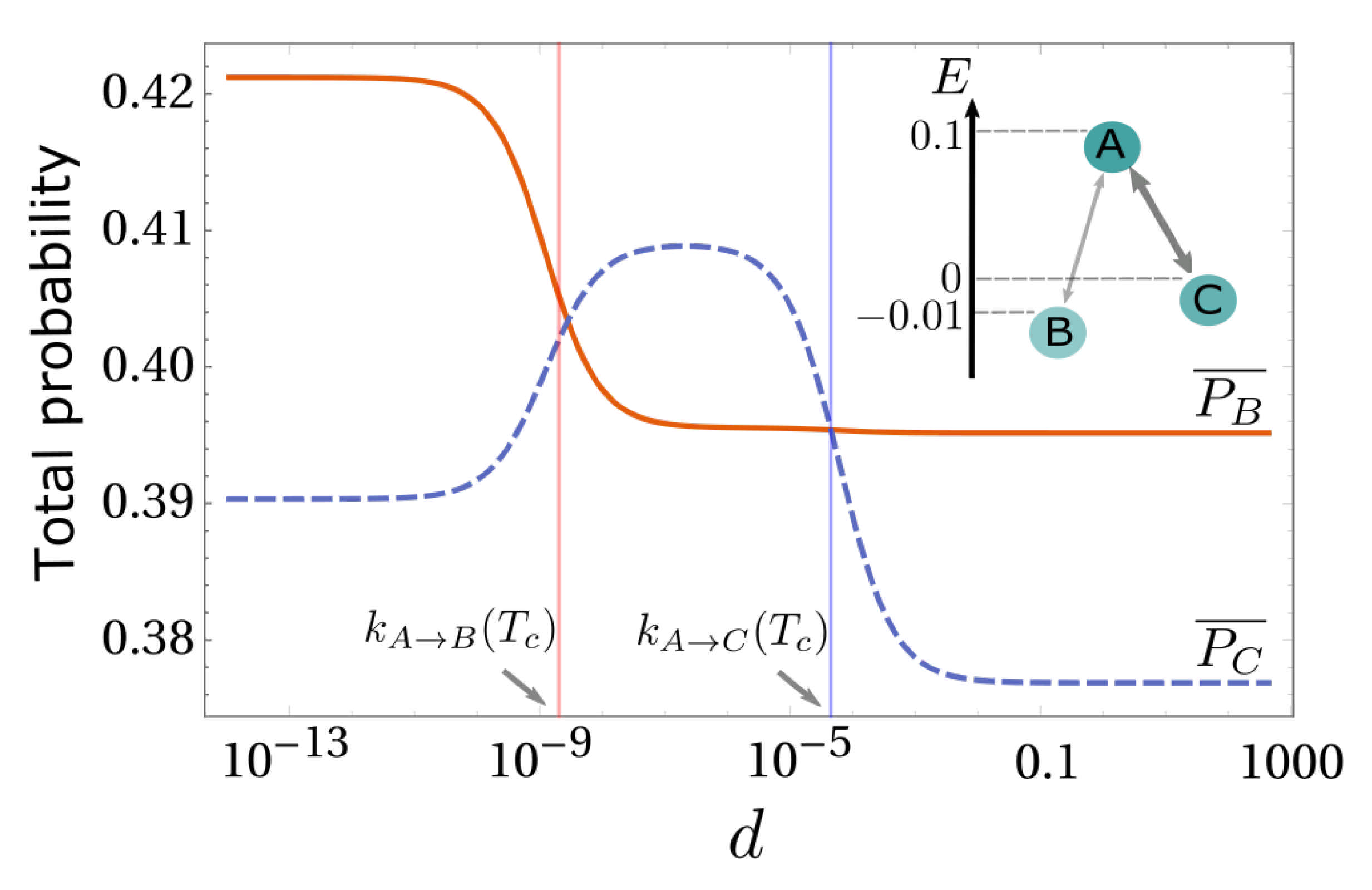
4.3. Numerical Results and Energy Landscapes
4.4. Fast-Dissipation Chemical Sub-Networks for Continuous Systems
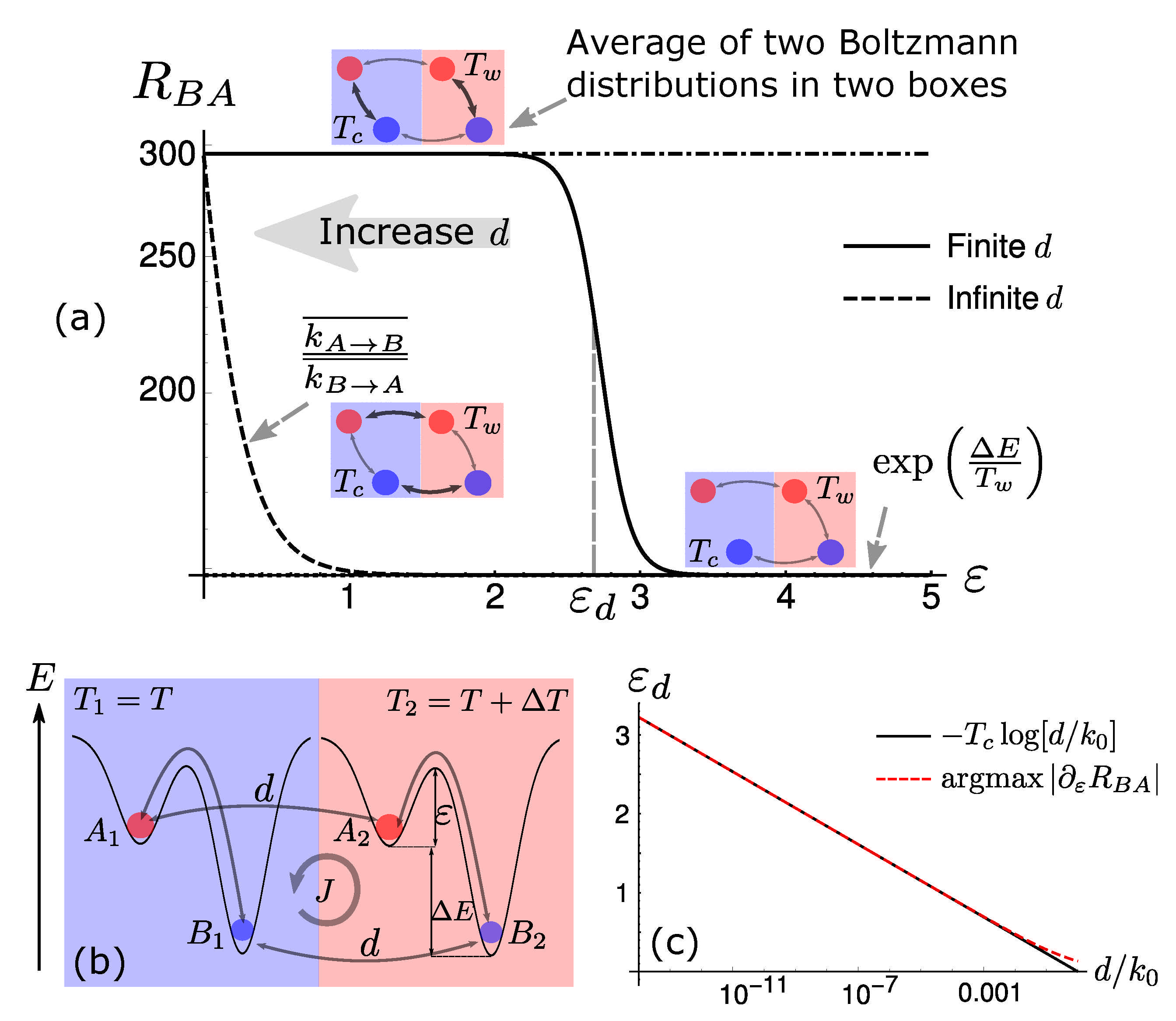
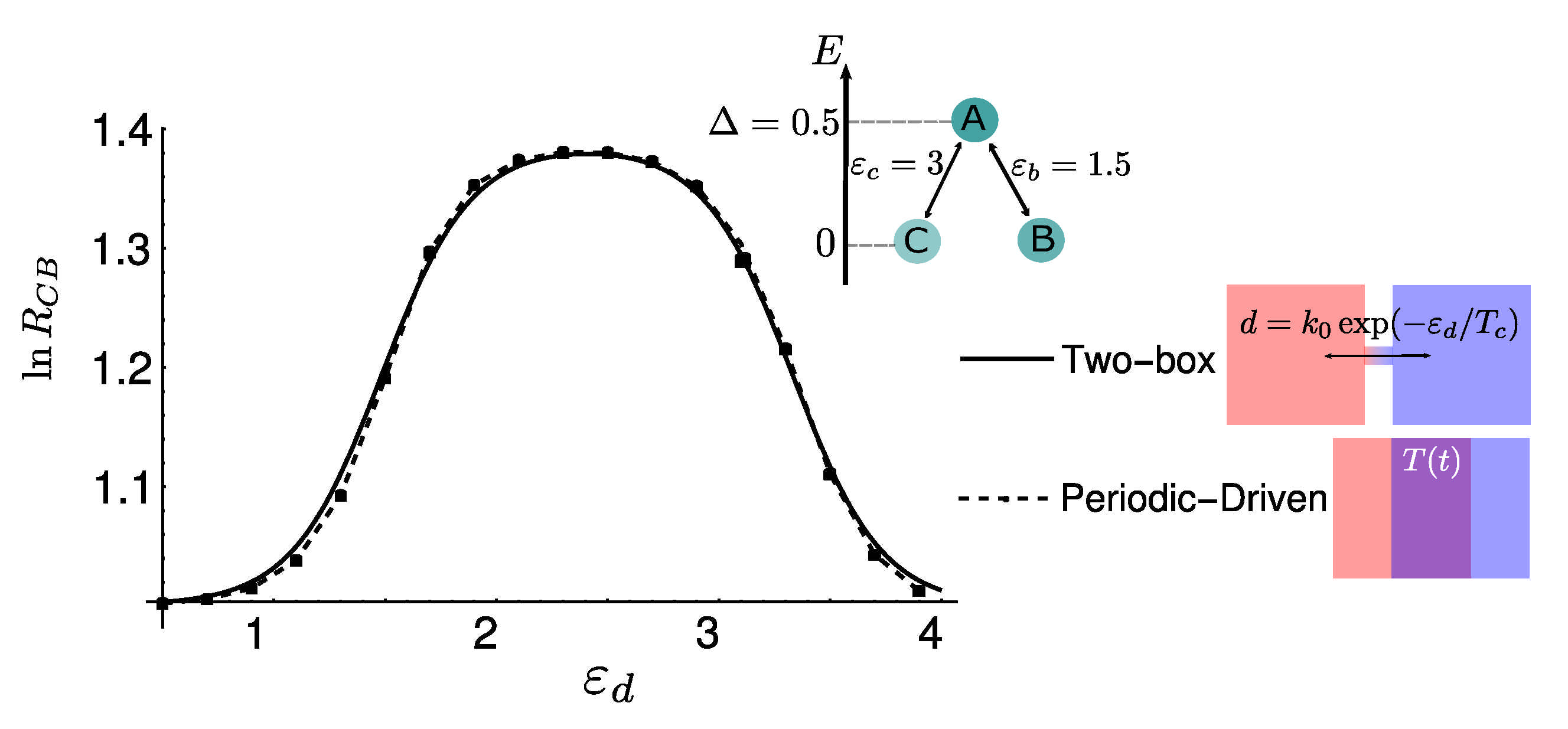
5. Diffusion-Controlled Switch of Selection
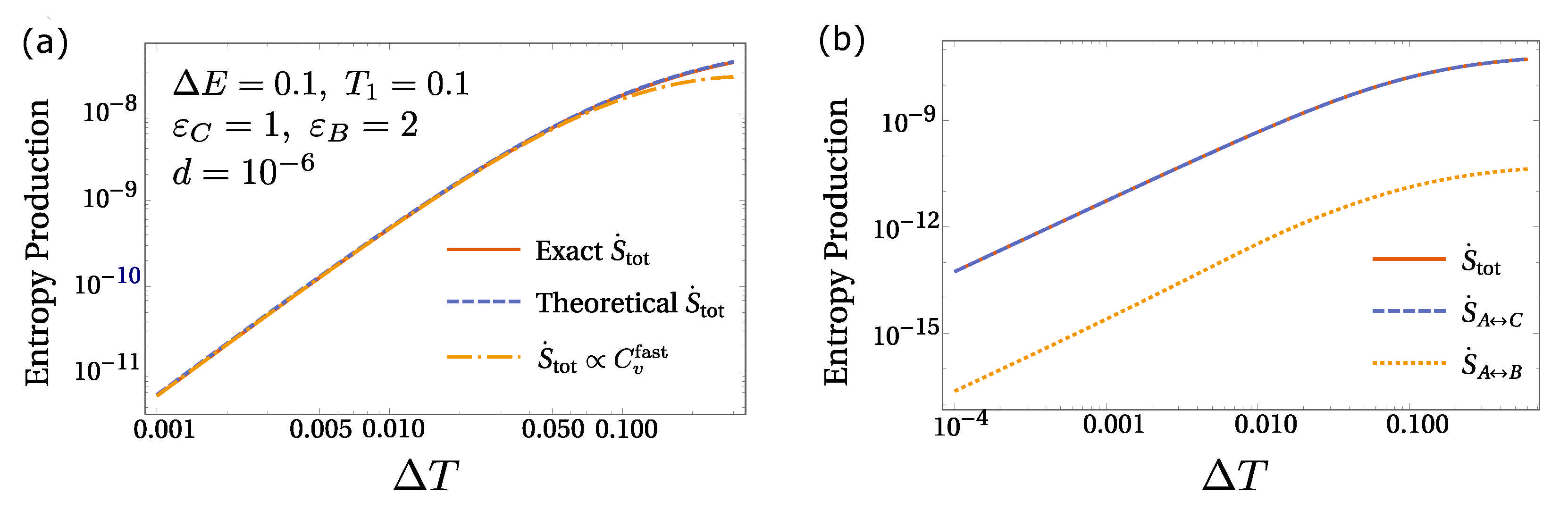
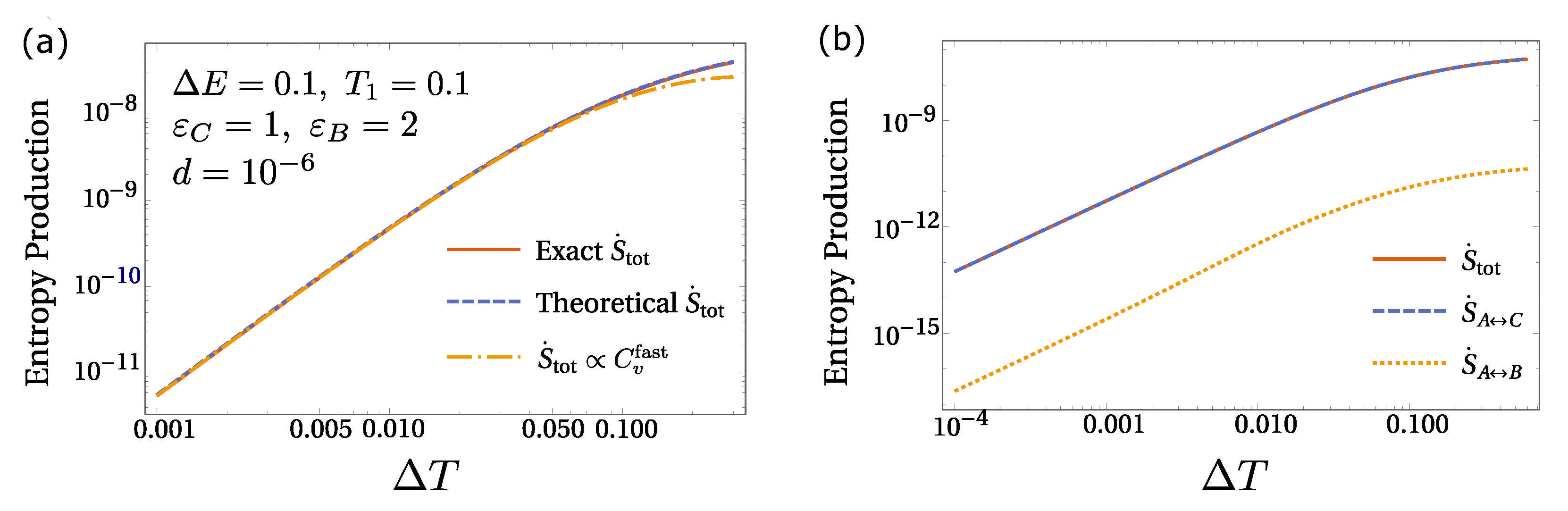
6. Equilibrium Hints for Entropy Production
7. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. The Maximum Possible Ratio of Two-State System
References
- Gardiner, C. Stochastic Methods; Springer: Berlin, Germany, 2009; Volume 4. [Google Scholar]
- Rao, R.; Esposito, M. Nonequilibrium thermodynamics of chemical reaction networks: Wisdom from stochastic thermodynamics. Phys. Rev. X 2016, 6, 041064. [Google Scholar] [CrossRef] [Green Version]
- Pascal, R.; Pross, A.; Sutherland, J.D. Towards an evolutionary theory of the origin of life based on kinetics and thermodynamics. Open Biol. 2013, 3, 130156. [Google Scholar] [CrossRef] [Green Version]
- Assenza, S.; Sassi, A.S.; Kellner, R.; Schuler, B.; De Los Rios, P.; Barducci, A. Efficient conversion of chemical energy into mechanical work by Hsp70 chaperones. elife 2019, 8, e48491. [Google Scholar] [CrossRef]
- Goloubinoff, P.; Sassi, A.S.; Fauvet, B.; Barducci, A.; De Los Rios, P. Chaperones convert the energy from ATP into the nonequilibrium stabilization of native proteins. Nat. Chem. Biol. 2018, 14, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Zwicker, D.; Seyboldt, R.; Weber, C.A.; Hyman, A.A.; Jülicher, F. Growth and division of active droplets provides a model for protocells. Nat. Phys. 2017, 13, 408–413. [Google Scholar] [CrossRef]
- Horowitz, J.M.; England, J.L. Spontaneous fine-tuning to environment in many-species chemical reaction networks. Proc. Natl. Acad. Sci. USA 2017, 114, 7565–7570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, C.; Netočnỳ, K.; de Galway, W.O. Low temperature behavior of nonequilibrium multilevel systems. J. Phys. Math. Theor. 2013, 47, 035002. [Google Scholar] [CrossRef] [Green Version]
- Basu, U.; Maes, C. Nonequilibrium response and frenesy. J. Phys. Conf. Ser. 2015, 638, 012001. [Google Scholar] [CrossRef] [Green Version]
- Maes, C. Non-Dissipative Effects in Nonequilibrium Systems; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Busiello, D.M.; Liang, S.; Piazza, F.; De Los Rios, P. Dissipation-driven selection of states in non-equilibrium chemical networks. Commun. Chem. 2021, 4, 1–7. [Google Scholar] [CrossRef]
- Dass, A.V.; Georgelin, T.; Westall, F.; Foucher, F.; De Los Rios, P.; Busiello, D.M.; Liang, S.; Piazza, F. Equilibrium and nonequilibrium furanose selection in the ribose isomerisation network. Nat. Commun. 2021, 12, 2749. [Google Scholar] [CrossRef]
- Astumian, R.D. Kinetic asymmetry allows macromolecular catalysts to drive an information ratchet. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Schnakenberg, J. Network theory of microscopic and macroscopic behavior of master equation systems. Rev. Mod. Phys. 1976, 48, 571. [Google Scholar] [CrossRef]
- Hänggi, P.; Talkner, P.; Borkovec, M. Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 1990, 62, 251. [Google Scholar] [CrossRef]
- Kustova, E.; Giordano, D. Cross-coupling effects in chemically non-equilibrium viscous compressible flows. Chem. Phys. 2011, 379, 83–91. [Google Scholar] [CrossRef]
- Kolesnichenko, E.G.; Gorbachev, Y.E. Gas-dynamic equations for spatially inhomogeneous gas mixtures with internal degrees of freedom. I. General theory. Appl. Math. Model. 2010, 34, 3778–3790. [Google Scholar] [CrossRef]
- Busiello, D.M.; Gupta, D.; Maritan, A. Coarse-grained entropy production with multiple reservoirs: Unraveling the role of time scales and detailed balance in biology-inspired systems. Phys. Rev. Res. 2020, 2, 043257. [Google Scholar] [CrossRef]
- Klich, I.; Raz, O.; Hirschberg, O.; Vucelja, M. Mpemba index and anomalous relaxation. Phys. Rev. X 2019, 9, 021060. [Google Scholar] [CrossRef] [Green Version]
- Astumian, R.D. Adiabatic operation of a molecular machine. Proc. Natl. Acad. Sci. USA 2007, 104, 19715–19718. [Google Scholar] [CrossRef] [Green Version]
- Mandal, D.; Jarzynski, C. A proof by graphical construction of the no-pumping theorem of stochastic pumps. J. Stat. Mech. Theory Exp. 2011, 2011, P10006. [Google Scholar] [CrossRef] [Green Version]
- Busiello, D.M.; Hidalgo, J.; Maritan, A. Entropy production for coarse-grained dynamics. New J. Phys. 2019, 21, 073004. [Google Scholar] [CrossRef]
- Liang, S.; Busiello, D.M.; Rios, P.D.L. The intrinsic non-equilibrium nature of thermophoresis. arXiv 2021, arXiv:2102.03197. [Google Scholar]
- Piazza, R.; Parola, A. Thermophoresis in colloidal suspensions. J. Phys. Condens. Matter 2008, 20, 153102. [Google Scholar] [CrossRef]
- Rahman, M.; Saghir, M. Thermodiffusion or Soret effect: Historical review. Int. J. Heat Mass Transf. 2014, 73, 693–705. [Google Scholar] [CrossRef]
- Bo, S.; Celani, A. Multiple-scale stochastic processes: Decimation, averaging and beyond. Phys. Rep. 2017, 670, 1–59. [Google Scholar] [CrossRef] [Green Version]
- Raz, O.; Subaşı, Y.; Jarzynski, C. Mimicking nonequilibrium steady states with time-periodic driving. Phys. Rev. X 2016, 6, 021022. [Google Scholar] [CrossRef] [Green Version]
- Busiello, D.M.; Jarzynski, C.; Raz, O. Similarities and differences between non-equilibrium steady states and time-periodic driving in diffusive systems. New J. Phys. 2018, 20, 093015. [Google Scholar] [CrossRef]
- Bai, S.; Ge, H.; Qian, H. Structure for energy cycle: A unique status of the second law of thermodynamics for living systems. Sci. China Life Sci. 2018, 61, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Lervik, A.; Kjelstrup, S.; Qian, H. Michaelis–Menten kinetics under non-isothermal conditions. Phys. Chem. Chem. Phys. 2015, 17, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ge, H. Nonequilibrium steady state of biochemical cycle kinetics under non-isothermal conditions. New J. Phys. 2018, 20, 043030. [Google Scholar] [CrossRef]
- England, J.L. Dissipative adaptation in driven self-assembly. Nat. Nanotechnol. 2015, 10, 919–923. [Google Scholar] [CrossRef]
- Schrödinger, E. What is Life? The Physical Aspect of the Living Cell and Mind; Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Prigogine, I.; Nicolis, G. Biological order, structure and instabilities. Q. Rev. Biophys. 1971, 4, 107–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, S.; De Los Rios, P.; Busiello, D.M. Dissipation-Driven Selection under Finite Diffusion: Hints from Equilibrium and Separation of Time Scales. Entropy 2021, 23, 1068. https://doi.org/10.3390/e23081068
Liang S, De Los Rios P, Busiello DM. Dissipation-Driven Selection under Finite Diffusion: Hints from Equilibrium and Separation of Time Scales. Entropy. 2021; 23(8):1068. https://doi.org/10.3390/e23081068
Chicago/Turabian StyleLiang, Shiling, Paolo De Los Rios, and Daniel Maria Busiello. 2021. "Dissipation-Driven Selection under Finite Diffusion: Hints from Equilibrium and Separation of Time Scales" Entropy 23, no. 8: 1068. https://doi.org/10.3390/e23081068
APA StyleLiang, S., De Los Rios, P., & Busiello, D. M. (2021). Dissipation-Driven Selection under Finite Diffusion: Hints from Equilibrium and Separation of Time Scales. Entropy, 23(8), 1068. https://doi.org/10.3390/e23081068