
Partitioning Entropy with Action Mechanics: Predicting Chemical Reaction Rates and Gaseous Equilibria of Reactions of Hydrogen from Molecular Properties


Abstract
:1. Introduction
1.1. The Virial Theorem
1.2. Two Kinds of Virial Effects
- (i)
- The virial theorem has been adopted by physicists for systems of particles such as the hydrogen atom responsible for the solar spectrum and the gravitational structure of stars. The theorem defines the kinetic and potential energy of particles, for variations involving absorption or emission of quanta travelling at light speed. For verification of modelling in quantum theory, variations in kinetic energy must be set equal to the energy of the quanta, with their absorption resulting in increased potential energy equal to the decrease in kinetic energy. So, the change in potential energy is considered as equal to the sum of the decrease in the kinetic energy plus the quantum absorbed into the local field. Considering a single particle subject to quantum excitation [Σhv, J] in a central field, the variation in potential energy can be expressed as the sum of the quanta absorbed plus the decrease in the kinetic energy.The variation in potential energy of collapsing stars can be expressed by the same equation in reverse, leading to emission of radiation with an equivalent increase in kinetic energy and temperature of the stars. We have claimed elsewhere [8] that molecules in the Earth’s atmosphere are subject to this form of the virial theorem, yielding a lapse rate for change of temperature with altitude from balancing thermal and gravitational energies of 6.9 K per km from the land surface, which is consistent with observations, a gradient far from isothermal, nor the result of adiabatic expansion.
- (ii)
- By contrast, in systems of molecules moving as material points in a heat bath providing a multiplicity of central forces, as in an ideal gas, the relationship between kinetic and potential energy differs as a function of temperature and pressure. This is illustrated in the case of water where the molecules can exist frozen at low temperature, aggregated as clusters in liquid able to flow under gravity, and as separate molecules in the gas or vapor phase. All three forms exist on the Earth’s surface, in contrast with all other gaseous constituents of the atmosphere, only existing as vapor. Heat transmitted as quanta does reversible work on all three phases of water, melting or vaporising water while raising the temperature of the gas. For systems in a common heatbath, the absorption of quanta no longer results in decreased kinetic energy and temperature of orbiting particles, in contrast to matter bound by a dominant central force, such as electrons in atoms or matter by gravity. Clausius’ Equation (8) describes [6] the reversible effect of heat on the system. This equates the effect of heat dQ [J] on the variation in the quantity of sensible heat in the body dH plus the internal work or potential of the ergal dJ and the external work dW.dQ = dH + dJ + dW
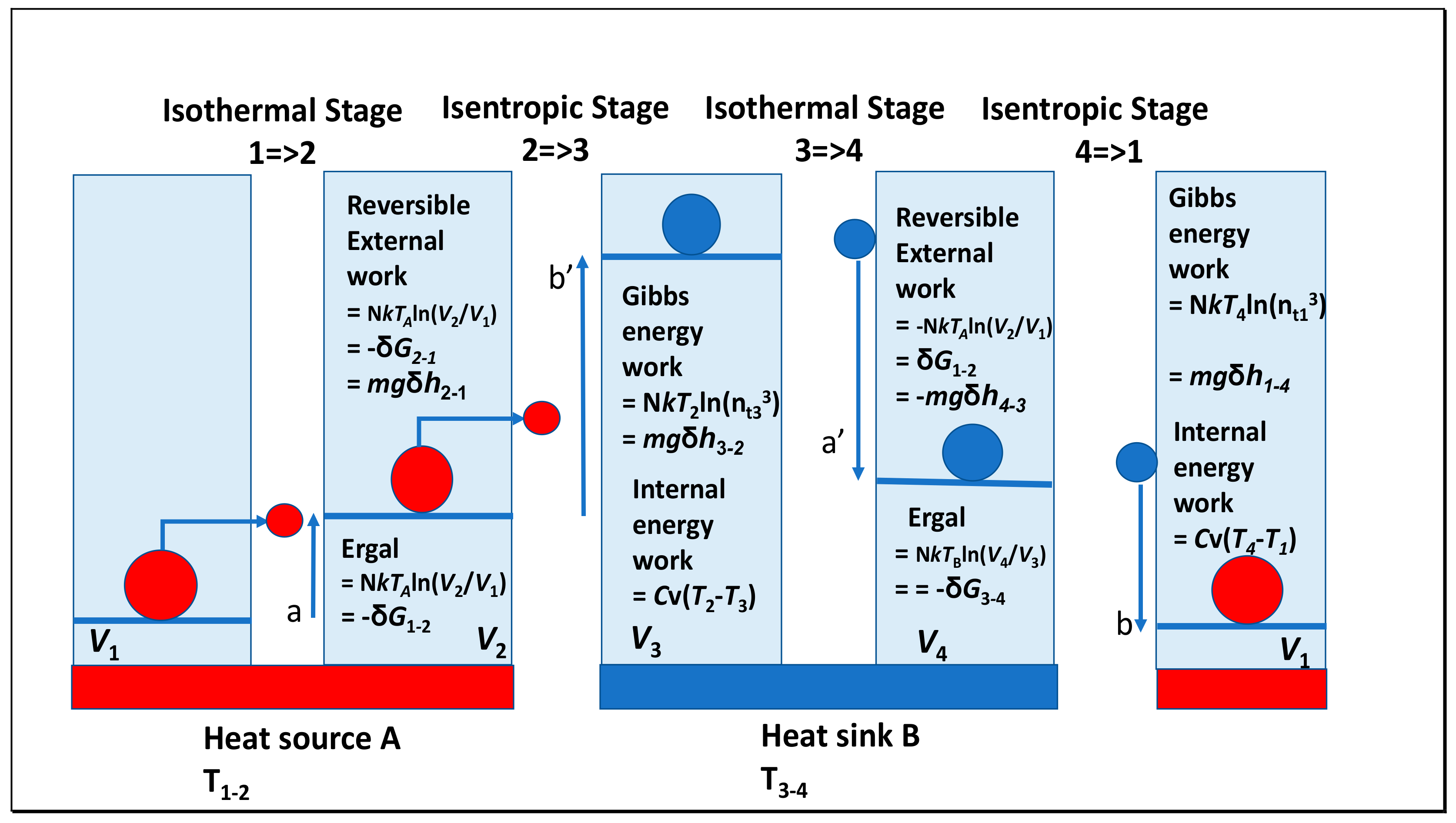
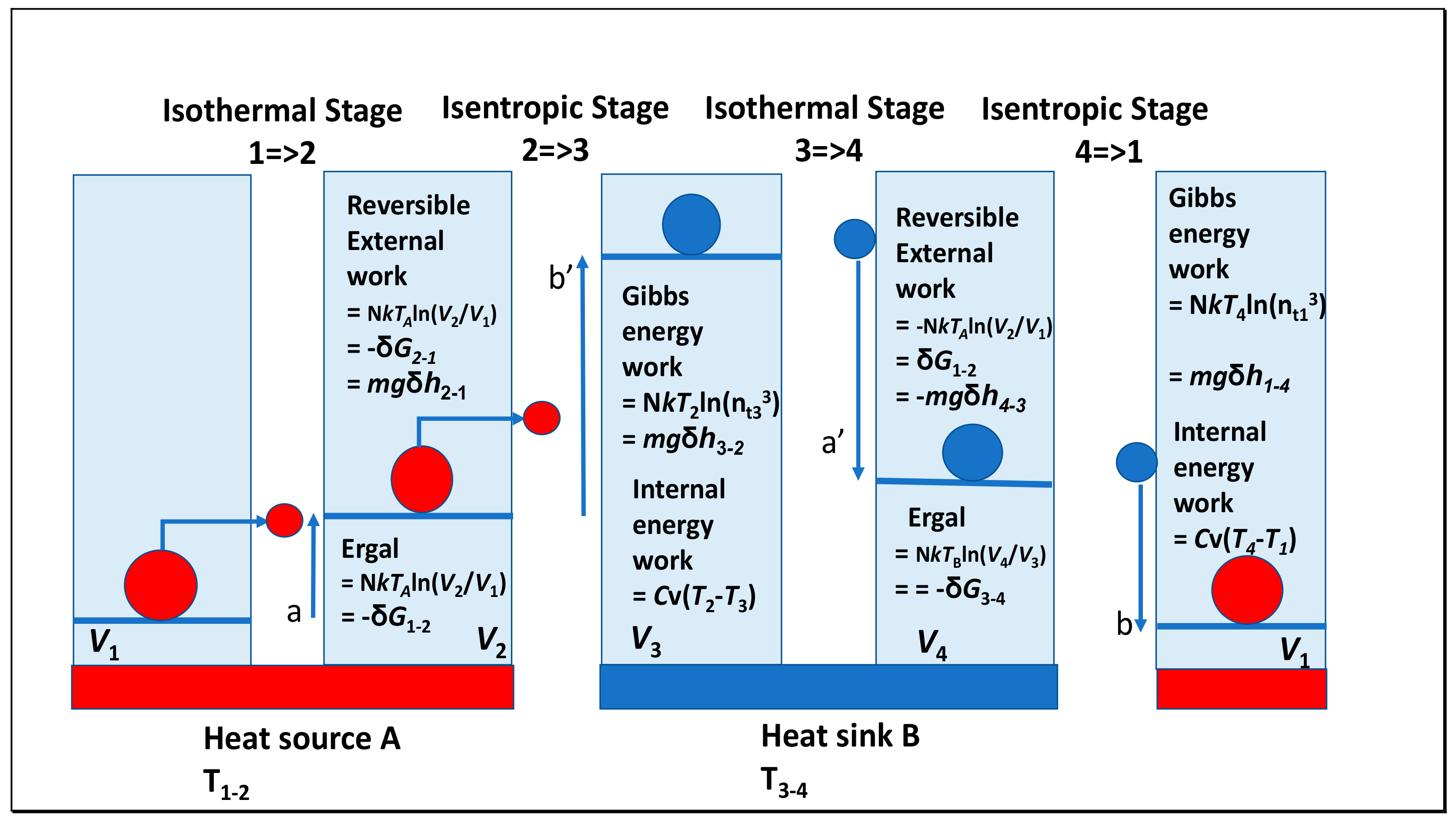
1.3. Some Lessons from Revising the Carnot Cycle
1.4. Clausius’ Entropy and the Ergal
2. Action Mechanics
A = E − ST
H = E + RT
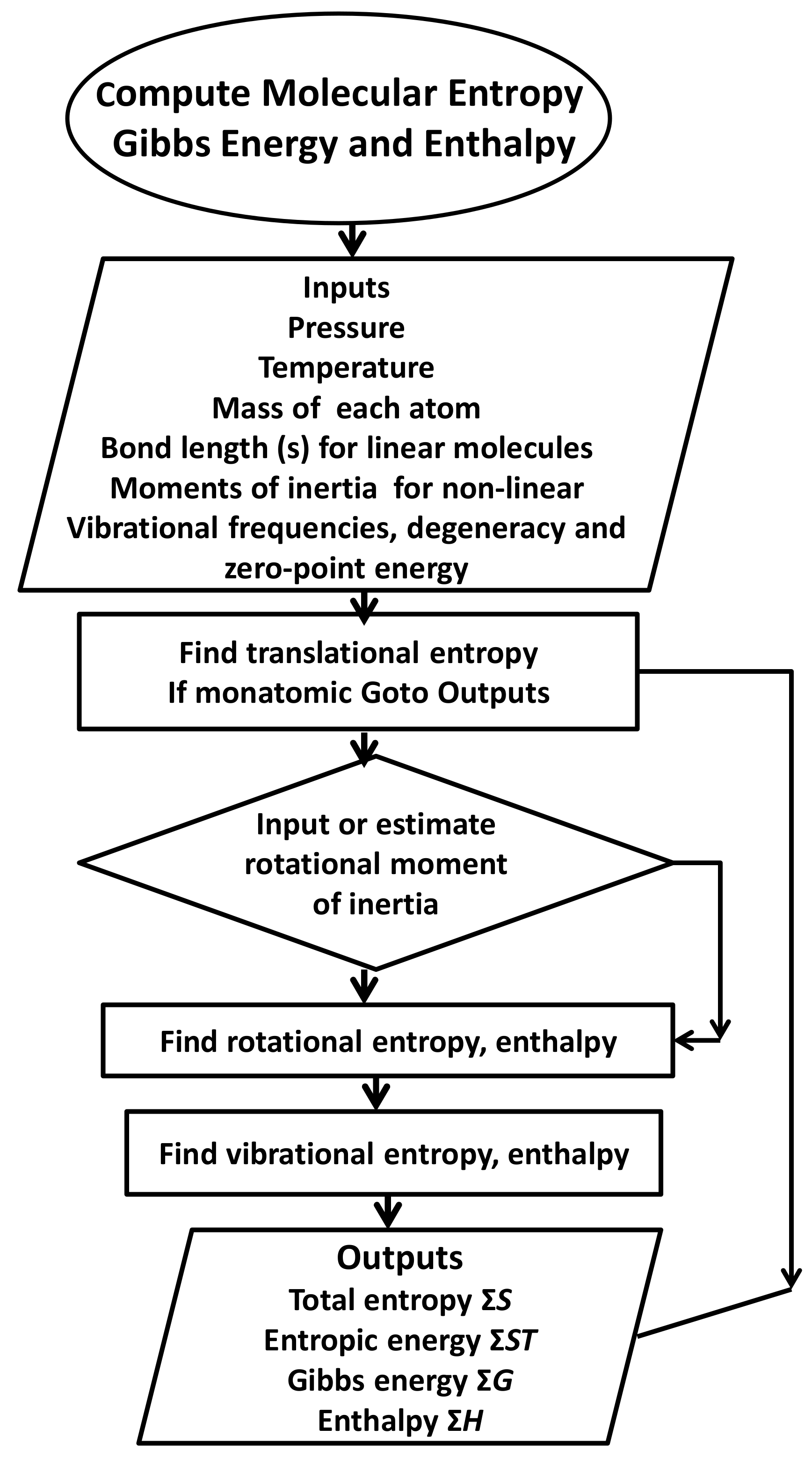
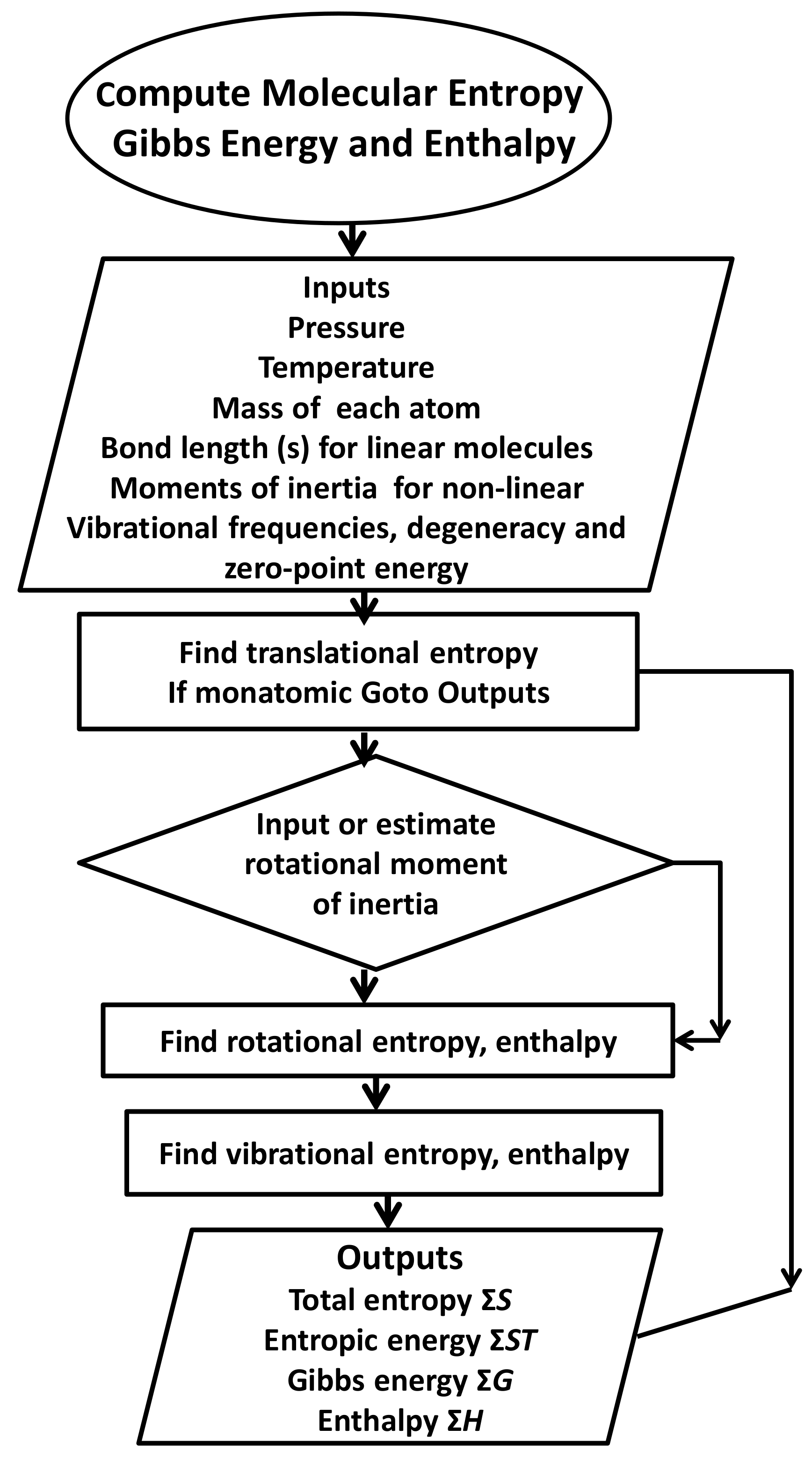
2.1. Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Relevant Equations |
|---|---|
| Entropic energy | ST = RTln[Qee7/2(3kTI/ħ)3(2kTI/ħ)2/(σzt)]; H2, O2, N2, CO2 ST = RTln[Qee7/2(@t/ħ)3(@r/ħ)2] |
| Translational action | @t = (3kTI)0.5/zt1/3; zt = 23∙(1.085)3 = 10.2297 |
| Rotational action | @r = (2kTI)0.5/σ |
| Qe | Electron multiplicity partitioning translational entropy |
| σ,zt | Rotational and translational symmetry constants |
| Vibrational action | Σk[x/(ex − 1) − ln(1 − e−x)] x = hν/kT |
| Vibrational heat capacity | Σ{kTx2/[2(coshx − 1)] x = hν/kT |
| Glossary | Action [@, J.sec]—a scalar or non-directional measure with dimensions [MRVδθ], a finite spatial segment of momentum; action is relativistically invariant. Angular momentum [J.sec]—a conservative vector with dimensions [MRV]. Enthalpy [H, J]—consists of internal energy as sensible heat and heat of atmospheric expansion RT. Entropy [S, J K−1]—a pure statistic for cumulative sensible and latent heat content per Kelvin or to indicate information. Ergal [J]—heat required for internal work of dissociating bound molecules and for raising their quantum state required by the temperature. Gibbs energy [G, J]—the potential to store thermal energy as work, decreasing in magnitude as this is achieved. Radial inertia [MR]—denotes the radial path of matter, the product of momentum by time, a component of action. |
3. Activated Transition States
3.1. Reaction Kinetics
- (i)
- Absorption of energy quanta or photons, which activate the electrons in the absorbing molecules (N) to excited states (N*), present in concentration ratios given by the Boltzmann distribution, where ε is the difference in energy between the ground and excited levels.N*/N0 = e−ε/kT
- (ii)
- The larger the quantum of activation energy required for activation (ε = hν), the lower the concentration of the activated state N*. In general, the rate of the chemical reaction resulting from activation is dictated by the concentration or frequency of occurrence of the activated transition state, a state of lower free energy and higher entropic energy. According to Planck [14], any such activation as the result of the absorption of quanta increases the entropy, decreasing the Gibbs energy.
- (iii)
- The absorption of photons in photosynthesis [1] raising electrons in the two photosystems of green plants (PSII and PSI) to higher action states of greater mobility is one important example of this process. The absorption of resonant infrared photons exciting vibrations in gaseous molecules such as those shown in Table 2, emitted from the earth’s surface by polyatomic gas molecules such as H2O, CO2, CH4, or N2O, is another.
- (iv)
- As indicated by Equation (17), raising the temperature so that the ratio ћω/kT declines provides another means of increasing the proportion of molecules in the activated state needed to allow a chemical transition to take place. This requirement explains why life only exists within an optimal temperature range. If conditions are too cold, below freezing, biological molecules will be too immobile for life. If too hot, the lifetime of biological bondings will be too short for stable organisms to occur.
- (v)
- The most common means employed by living organisms to mobilise chemical species is by providing catalytic agents or coupling mechanisms, lowering the activation energy for reaction, or by providing a forceful mechanism to achieve it; thus, this raises the frequency or likelihood of activated transition state species and increasing the reaction rate as discussed below. This lowers the size of the quantum of energy hν needed to reach an activated state allowing transition.
- (vi)
- This process of reducing the magnitude of the activation energy (actually reducing the negative Gibbs energy barrier) is known as catalysis, and the biological catalysts involved are colloidal proteins called enzymes.
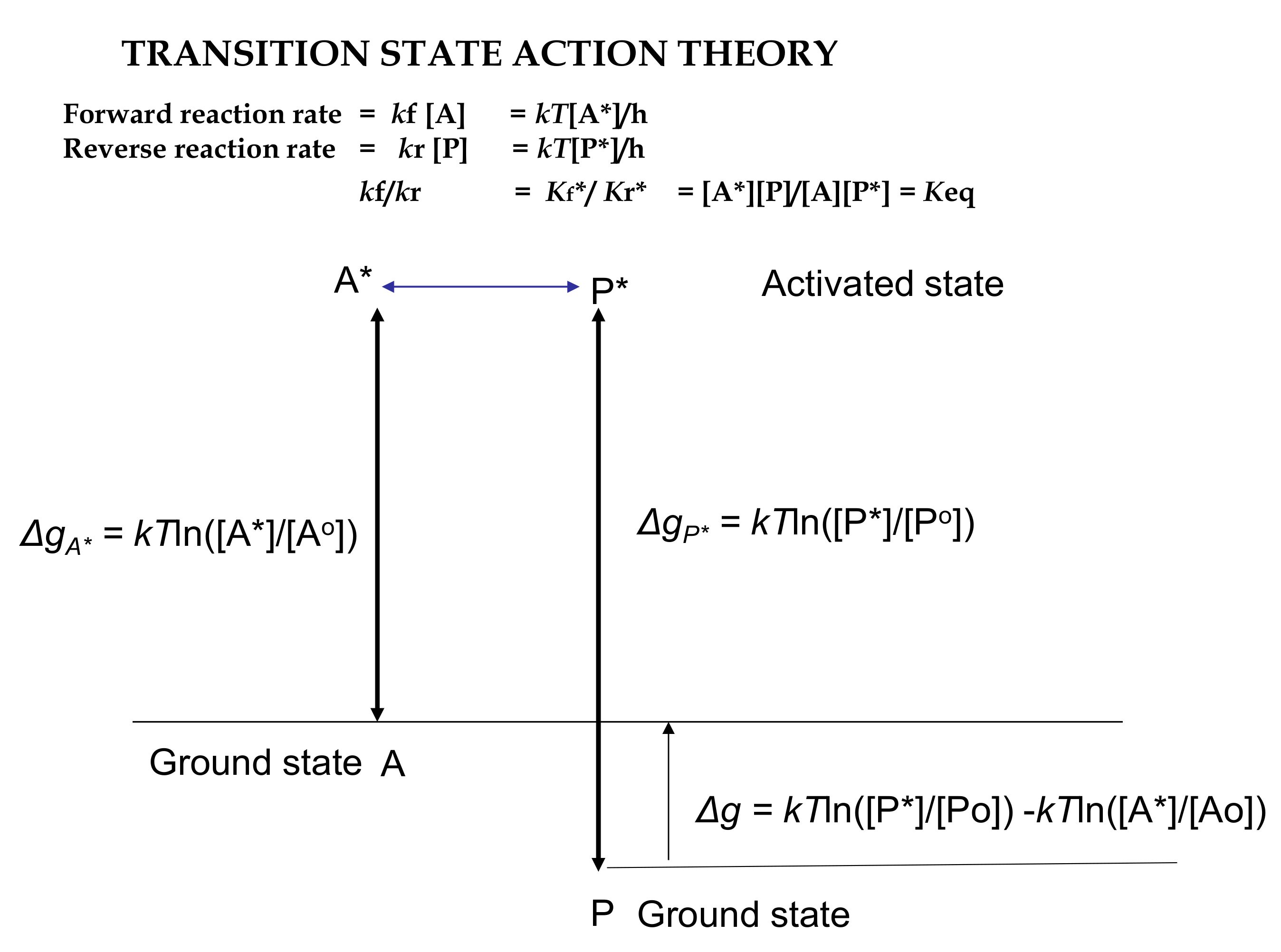
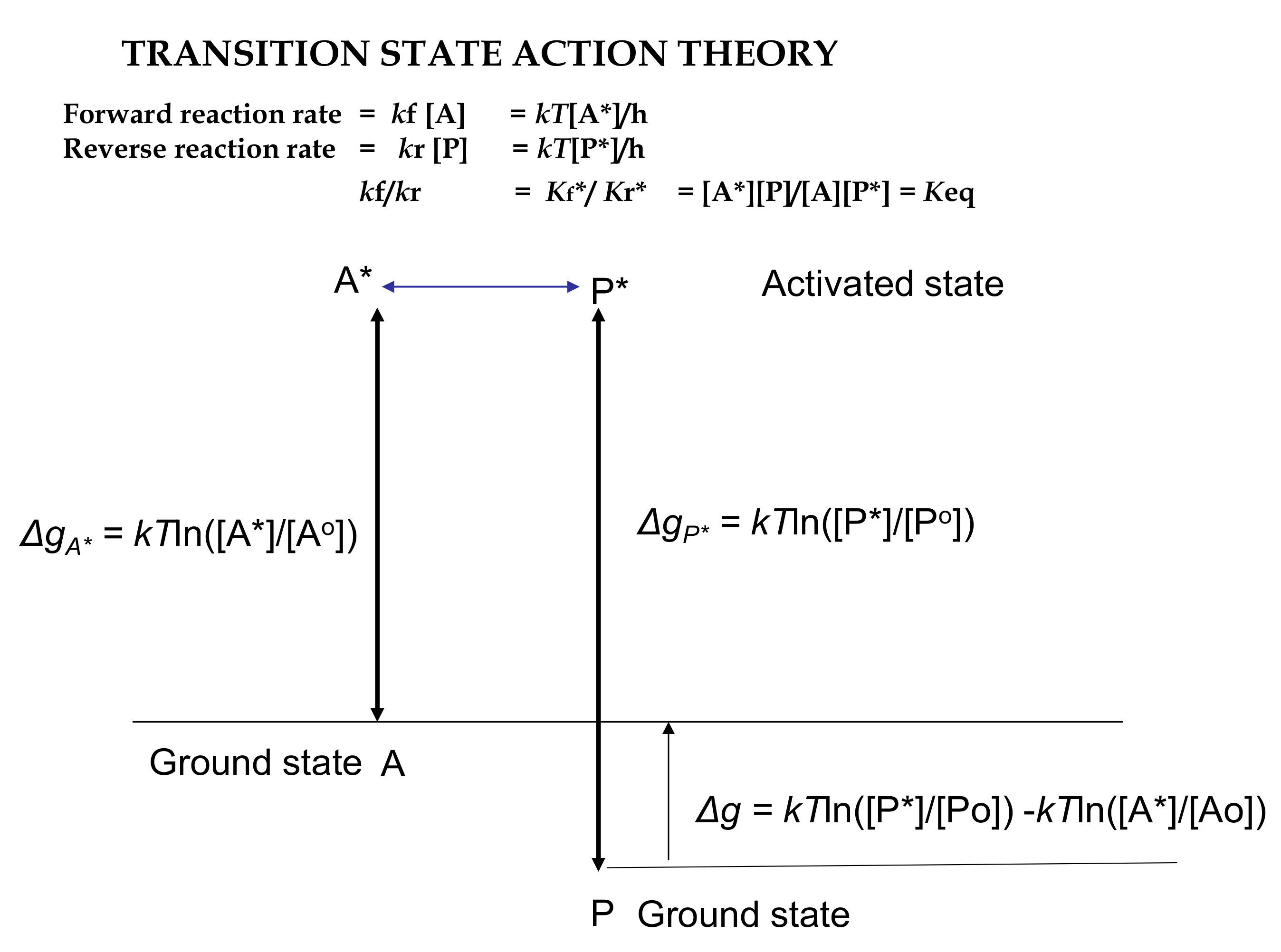
3.2. Action Revision of Eyring’s Absolute Transition State Theory
= Ea/RT2
= RTln[(*@t/o@t)3]
= −RTln[(o@t/*@t)3]
= −RTln[(N*)/(No)]
3.3. Radial Action
3.4. Chemical Potential and Work
= kTln([P*]/[A*]) = kTln1/K = −kTlnK
= kTln([P*][A]/[P][A*]) + kTln([P]/[A])
= −kTlnK + kTlnq
= RTln(q/K)
= −RTln([A*]/[P*]), just as under standard conditions.
3.5. Comparing Eyring’s Theory and Action Mechanics Transition States
- (i)
- In action mechanics, the activated transition state A* more realistically cannot be considered as actually in a standard state. The concentration is considered to be set as a Boltzmann exponential by the magnitude of the bonding energy of the reactant molecules.
- (ii)
- While Eyring’s theory usually analyses reaction rate in terms of Gibbs energy differences between the ground state reactant and the activated transition state molecules, in action mechanics, there can be no difference in chemical potential between the two states. This follows because the increased enthalpy and potential energy and vibrational amplitude of molecules in the activated transition state are corresponding to the decreased chemical or Gibbs energy in each activated molecule, according to the quanta required to be absorbed; this decrease in potential is paid for by the potential of the ground-state molecules. This is essential, given the Boltzmann equilibrium between ground and activated states for the ensemble of molecules.
- (iii)
- Proponents of Eyring’s transition state theory often fail to consider the reverse reaction as having a significant role in determining reaction rates. This may be valid in reactions proceeding far from equilibrium. However, action mechanics implies that in reactions closer to equilibrium, the chemical potentials for both reaction directions must be considered, given that the net reaction rate depends on the ratio of the transitions states A* and P*. Furthermore, the relative rates of forward and reverse reactions must always comply with the overall thermodynamic properties of the reaction, as shown above.
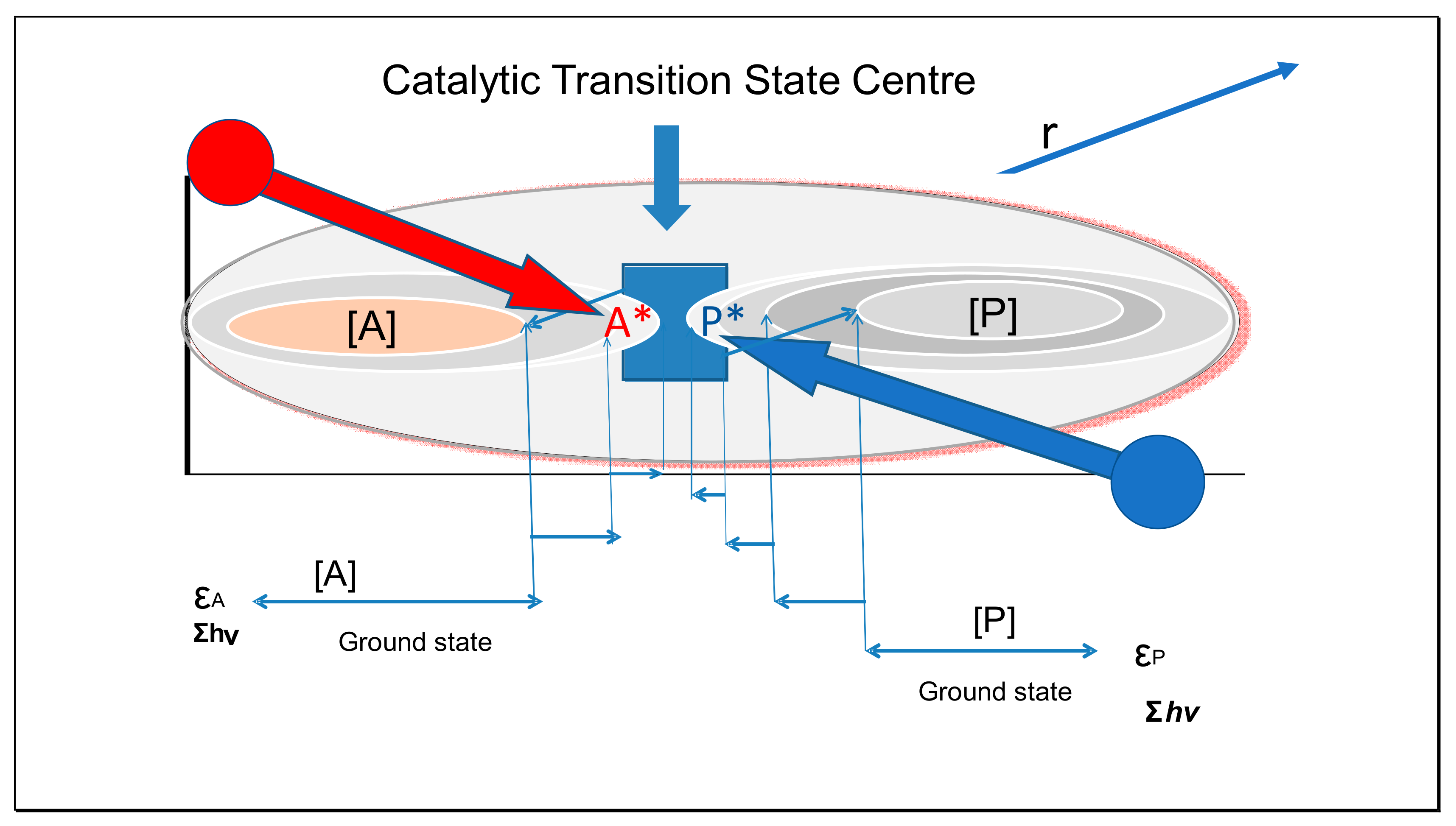
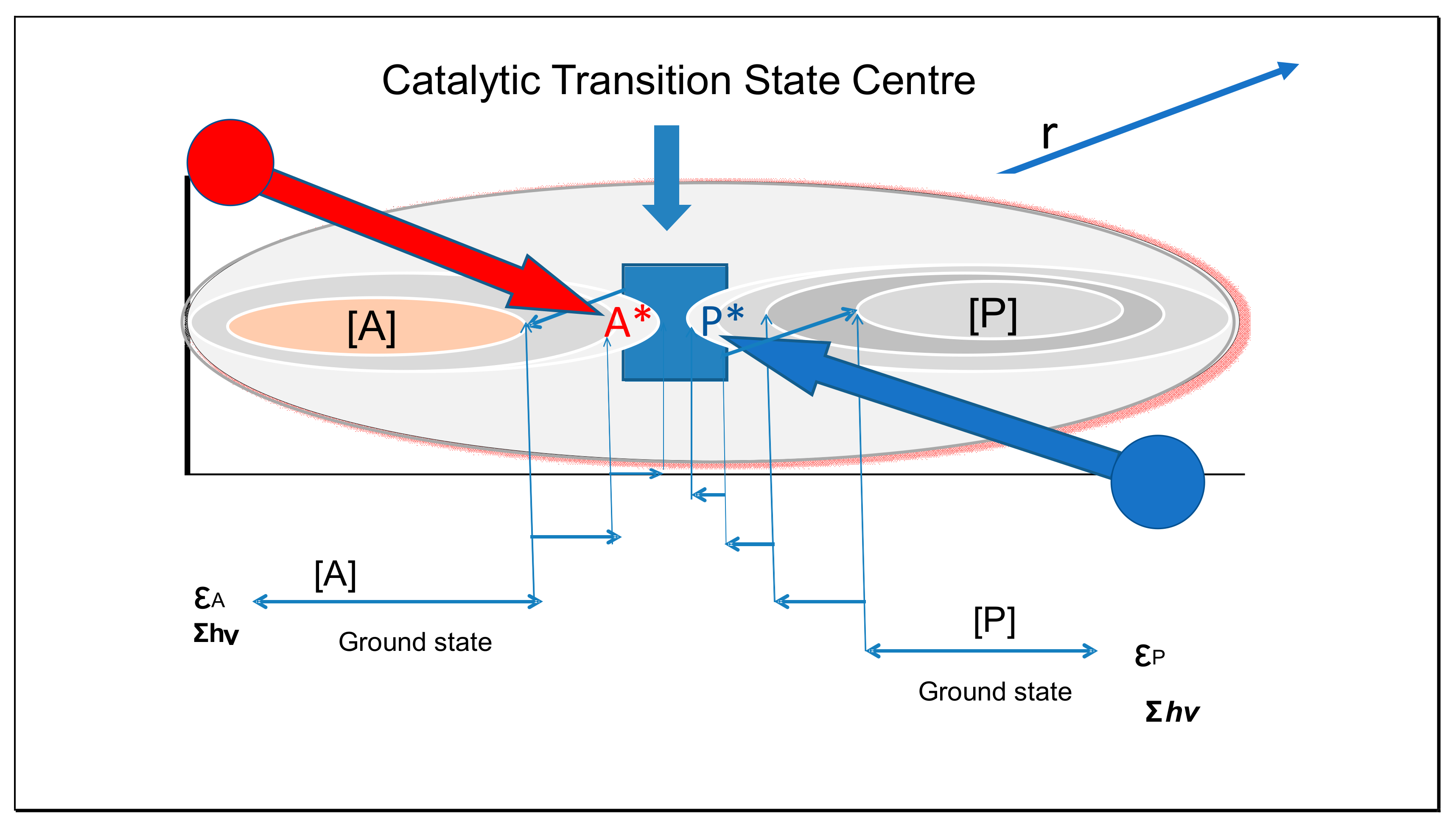
4. Catalytic Action
4.1. Are Colloidal Catalysts Also Inertial Anvils?
4.2. Effect of Binding Energy on Catalysis
4.3. Effect of Transition State Complementarity
5. Steady States and Equilibrium
Statistical and Action Mechanics
6. Illustrative Case Studies
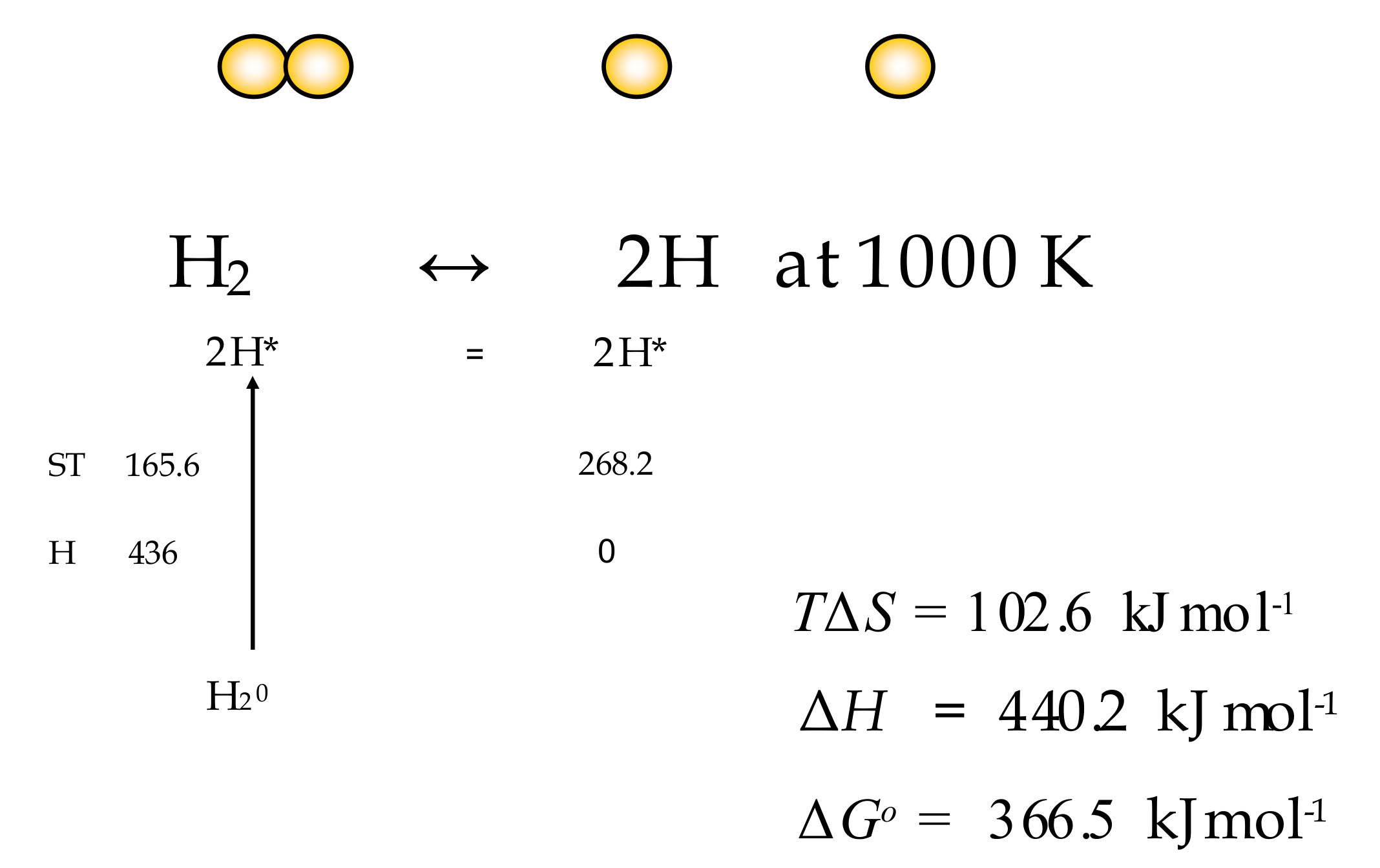
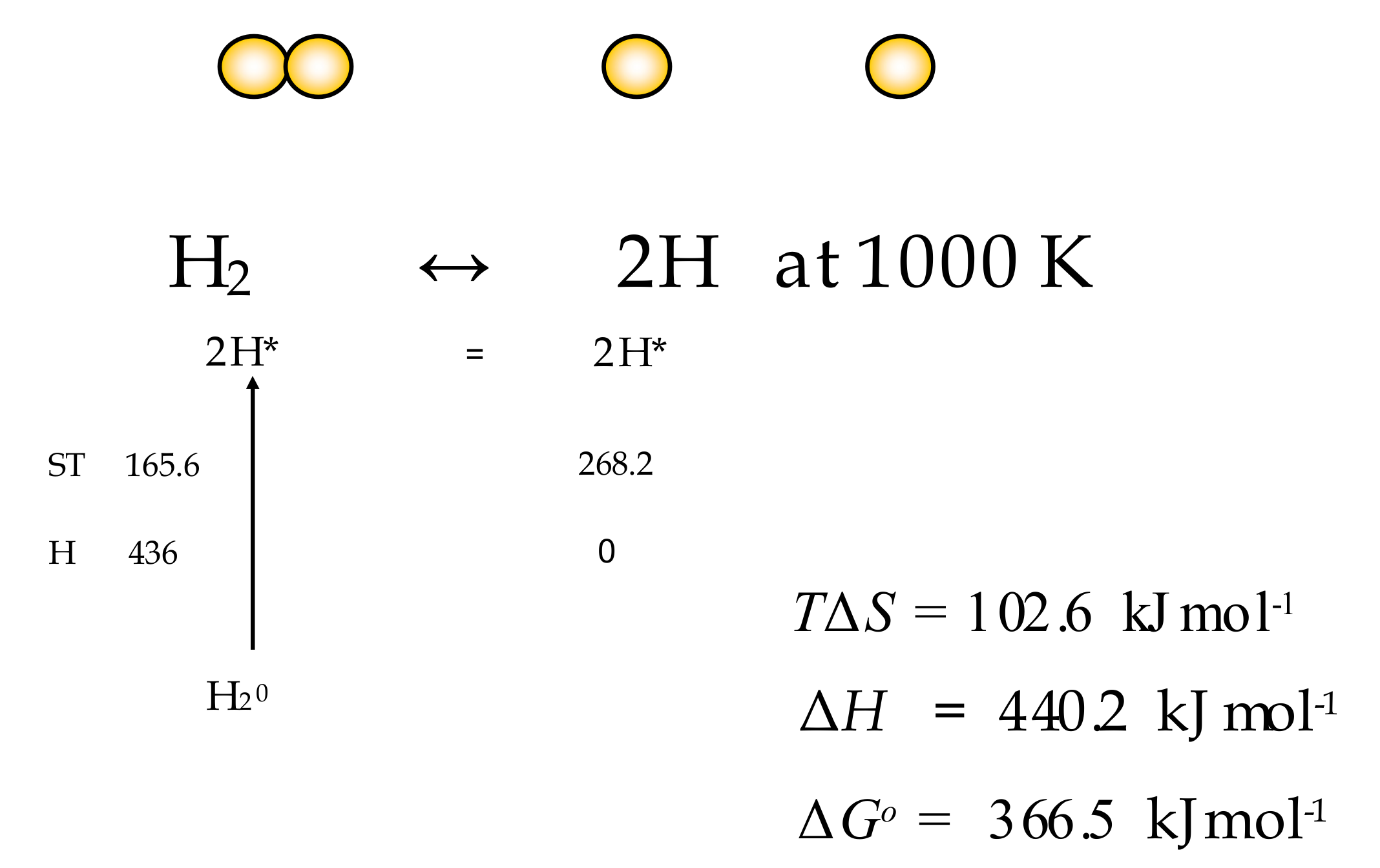
6.1. Dissociation of H2 to 2H
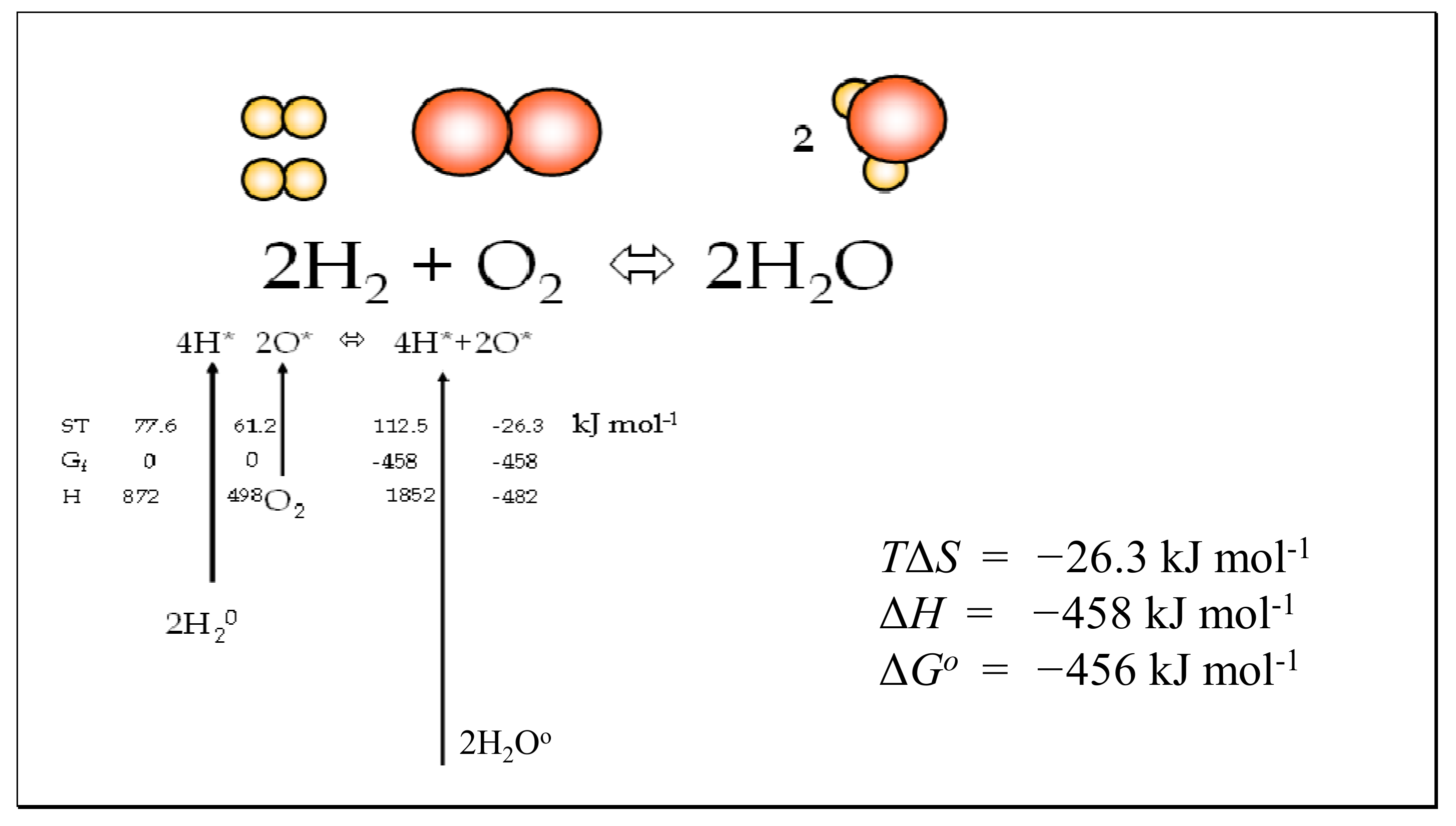
6.2. Association and Dissociation of Water
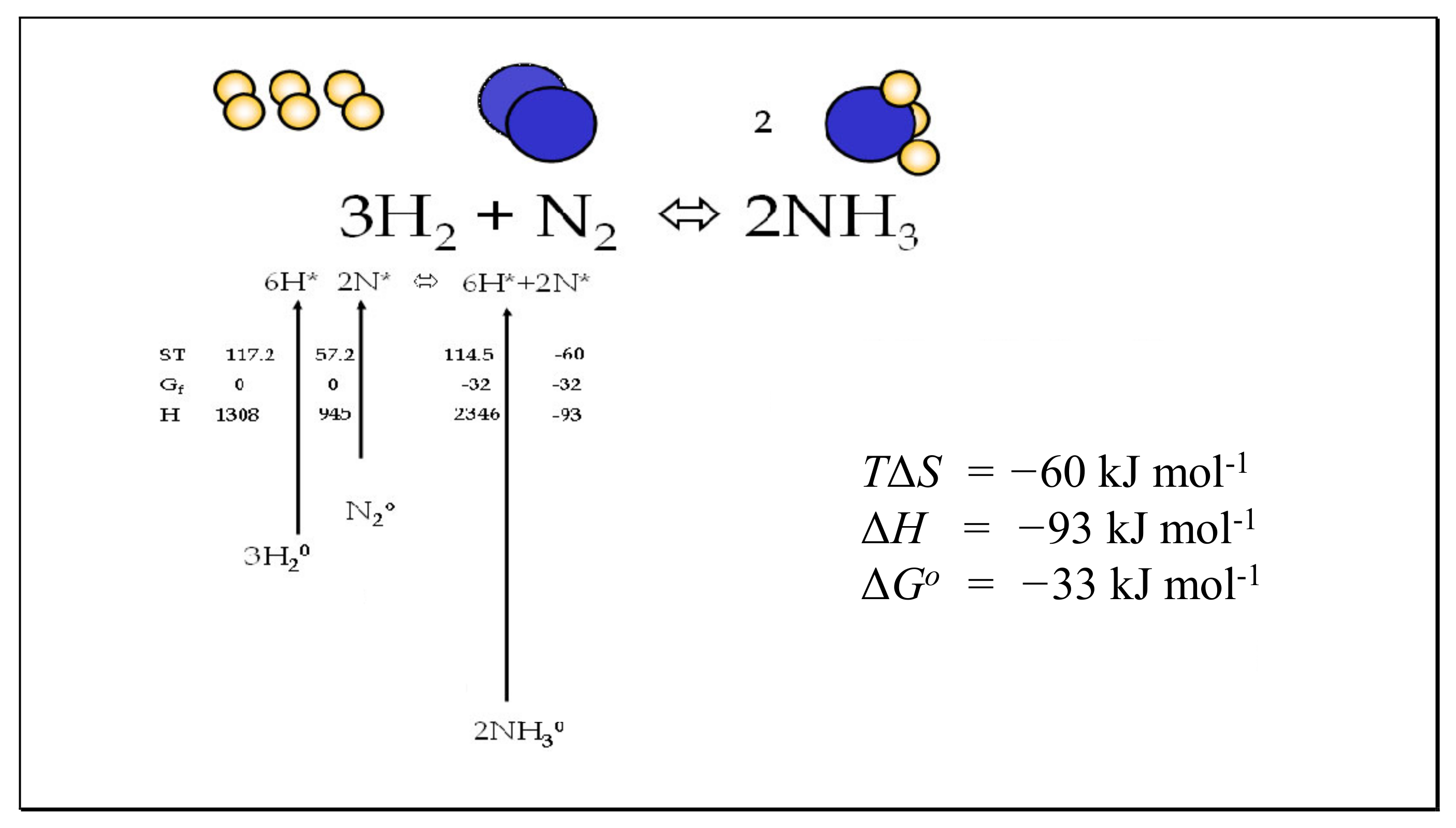
6.3. The Haber–Bosch Reaction
7. Conclusions
- (i)
- We have demonstrated the ease with which chemical potentials such as Gibbs energy can be estimated in action space, using simple algorithms combining structural details of molecules and environmental conditions of temperature and pressure. Values for vibrational, rotational, and translational action allow corresponding entropies and entropic energies, especially Gibbs energy, to be calculated from the prevailing temperature. These easily computed estimates of maximum entropy at varying temperatures allow the feasibility of gas phase reactions forming hydrogen from water or ammonia to be examined. The algorithms provide a good starting point for research into sources of renewable energy but have value in general, including in education.
- (ii)
- The hypotheses advanced on catalysis and equilibrium are consistent with our previous research [4] on the Carnot cycle showing that the Gibbs energy (G, J) can be identified as an index of internal work comprising the field of energy referred to by Clausius as the ergal. We identify this field of statistically elevated mean values of quantum states physically sustaining the configuration of the material particles. This energy field sustaining its action complements the much smaller kinetic energy in an ideal system of unbound particles.
- (iii)
- Action mechanics also allows a useful revision of the Eyring transition state theory for the catalysis of chemical reactions. For uncatalysed reactions, it proposes a Boltzmann equilibrium between reactant molecules and their more or less activated species, with the reaction rate determined by the frequency of occurrence of the transition state, which is able to form products. This frequency depends uniquely on the strength of the bonding enthalpy and the temperature, determining the rate of reaction; the larger the barrier, the slower the rate. Uniquely, action mechanics proposes a reaction trajectory of quasi-equilibrium between substrates and species on binding sites on catalysts of similar Gibbs energy or chemical potential. We report the novel result that for simple reactions with unit steady-state activities of reactants and products, the ratio of the activated transition states must be equal to the equilibrium constant for the reaction. This result should be investigated with more complex reactions involving more than one reactant and product.
- (iv)
- Reactants bound on colloids are proposed to share their greater inertia and will therefore be subject to larger action impulses in collisions, either with free co-reactants or with co-reactants similarly bound on colloidal particles. The much lower density or frequency of catalytic colloids will ensure more sustained stresses in collisions, moving on straighter trajectories reflecting their greater inertial mass (mr), acting as microscopic anvils. Catalysis by enzymes remains mysterious; some enzymologists consider that many enzymes are “too big” for their binding functions alone [35], although the evolution of regulatory functions can also logically contribute to added size. However, if their inertial impetus in collisions is significant in forceful encounters with substrates, the size can be explained statistically in terms of enhancing the probability of increased stress for bound substrates in collisions. We suggest that the methods of action mechanics can also allow Gibbs energies to be calculated for such colloids, acting as forceful anvils, with respect to impulses affecting their vibrational, rotational and translational actions.
- (v)
- The ease of calculation of the number and frequency of virtual field quanta from translational and rotational action (Table 6), at least as mean values, was surprising. The mean value of the magnitude of virtual quanta shown in the action fields is effectively expressing the ergal. In principle, the values calculated indicate the field energy sustaining the action states of reactants and products in chemical reactions. It should be possible to obtain direct evidence of the frequency and the intensity of these quantum fields by using resonant detectors, since they lie in the range of infra-red, microwave, and radiowaves. There may be significant scope for controlling rates of reactions or even positions of equilibrium with sufficiently intense action fields such as laser beams.
- (vi)
- Recognising the magnitude of the field energy as the ergal may have profound importance for quantitative accounting of energy in many scenarios. This energy field is rarely considered or even hidden in areas as diverse as climate science, such as the potential or work energy stored in coherent cells of air such as anticyclones and cyclones, or in ‘calorie counting’ for human weight loss and nutrition. This fundamental principle of energy conservation was enunciated by Clausius almost two centuries ago in the equivalence of heat and work, but it is largely neglected. To what extent are the increasing obstructions to smooth air flow in modern cities and the ubiquitous wind farms of Europe and further afield causing local warming by reversible processes of internal work followed by turbulent release of radiant heat, particularly when masses of air activated in heat stored as vortical entropy [4] collide? The ability of the magnitude of changes in the ergal to release equivalent amounts of temperature-raising heat seems to have no role in the global circulation models on which the Intergovernmental Panel for Climate Change (IPCC) depends.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, I.R. Action in Ecosystems: Biothermodyamics for Sustainability; Research Studies Press/John Wiley: Baldock, UK, 2001; p. 251. [Google Scholar]
- Kennedy, I.R.; Geering, H.; Rose, M.T.; Crossan, A.N. A simple method to estimate entropy and free energy of atmospheric gases from their action. arXiv 2015, arXiv:1504.03866. [Google Scholar]
- Kennedy, I.R.; Geering, H.; Rose, M.; Crossan, A. A simple method to estimate entropy and free energy of atmospheric gases from their action. Entropy 2019, 21, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, I.R.; Hodzic, M. Action and entropy in heat engines: An action revision of the Carnot Cycle. Entropy 2021, 23, 860. [Google Scholar] [CrossRef] [PubMed]
- Einstein, A. Investigations of the Theory of the Brownian Movement; Dover Publications: Mineola, NY, USA, 1956. [Google Scholar]
- Clausius, R.J.E. On a mechanical theorem applicable to heat. Philos. Mag. 1870, 4, 122–127. [Google Scholar] [CrossRef]
- Clausius, R. The Mechanical Theory of Heat; MacMillan and Co.: London, UK, 1875. [Google Scholar]
- Kennedy, I.R. Computation of planetary atmospheres by action mechanics using temperature gradients consistent with the virial theorem. Int. J. Energy Environ. 2015, 9, 129–146. [Google Scholar]
- Boltzmann, L. Lectures in Gas Theory; Barth, J.A., Ed.; Dover Publications: New York, NY, USA, 1964; p. 342. [Google Scholar]
- Gray, J.M. Regnault’s determination of the specific heat of steam. Philos. Mag. 1882, 13, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Clausius, R. On the motive power of heat, and on the laws which can be deduced from it for the theory of heat (1850). In Reflections on the Motive Power of Fire; Mendoza, E., Ed.; Dover Publications: New York, NY, USA, 1960. [Google Scholar]
- Gibbs, J.W. Elementary Principles in Statistical Mechanics; Charles Scribner’s Sons: New York, NY, USA, 1902. [Google Scholar]
- Brown, A. Statistical Physics; Edinburgh University Press: Edinburgh, UK, 1968. [Google Scholar]
- Planck, M. The Theory of Heat Radiation; Dover Publications: New York, NY, USA, 1959. [Google Scholar]
- Moore, W. Physical Chemistry; Longmans: London, UK, 1962. [Google Scholar]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Aylward, G.H.; Findlay, T.J.V. SI Chemical Data; John Wiley and Sons: Sydney, Australia, 1975. [Google Scholar]
- Leffler, J.E.; Grunwald, E. Rates and Equilibria of Organic Reactions; John Wiley and Sons: New York, NY, USA, 1963; p. 100. [Google Scholar]
- Eisenberg, D.; Crothers, D. Physical Chemistry with Applications to the Life Sciences; Benjamin/Cummings: Menlo Park, CA, USA, 1979. [Google Scholar]
- Fersht, A. Enzyme Structure and Mechanism; Freeman: Reading, UK, 1977. [Google Scholar]
- Kennedy, I.R. Acid Soil and Acid Rain; John Wiley and Sons Inc.: New York, NY, USA, 1992. [Google Scholar]
- Switala, J.; Loewen, P.C. Diversity of properties among catalases. Arch. Biochem. Biophys. 2002, 401, 145–154. [Google Scholar] [CrossRef]
- Jencks, W.P. Catalysis in Chemistry and Enzymology; Dover Publications: New York, NY, USA, 1987. [Google Scholar]
- Fersht, A. Structure and Mechanism in Protein Science; Freeman: New York, NY, USA, 1999. [Google Scholar]
- Pratt, G.L. Gas Kinetics; John Wiley & Sons Ltd.: London, UK, 1969. [Google Scholar]
- Pechukas, P.; McLafferty, F.J. On transition-state theory and the classical mechanics of collinear collisions. J. Chem. Phys. 1973, 58, 1622–1625. [Google Scholar] [CrossRef]
- Stone, S.R.; Copeland, L.; Kennedy, I.R. Glutamate dehydrogenase from lupin nodules: Purification and properties. Phytochemistry 1979, 18, 1273–1278. [Google Scholar] [CrossRef]
- Chen, J.; Kennedy, I.R. Purification and properties of lupin nodule glutamine synthetase. Phytochemistry 1985, 10, 2167–2172. [Google Scholar] [CrossRef]
- Langmuir, I.; Mackay, G.M.J. The dissociation of hydrogen into atoms. Part, I. Experimental. J. Am. Chem. Soc. 1914, 36, 1708–1722. [Google Scholar] [CrossRef] [Green Version]
- Langmuir, I. The dissociation of hydrogen into atoms. III. The mechanism of the reaction. J. Am. Chem. Soc. 1916, 38, 1145–1156. [Google Scholar] [CrossRef]
- Giauque, W.F. The entropy of hydrogen and the third law of thermodynamics the free energy and dissociation of hydrogen. J. Amer. Chem. Soc. 1930, 52, 4816–4831. [Google Scholar] [CrossRef]
- Johnston, H.L.; Walker, M.K. The dissociation of oxygen to 5000 K. The free energy of atomic oxygen. Trans. Electrochem. Soc. 1933, 55, 187–193. [Google Scholar] [CrossRef]
- Lovegrove, K.; Luzzi, A.; Soldiani, I.; Kreetz, H. Developing ammonia based thermochemical energy storage for dish power plants. Sol. Energy 2004, 76, 331–333. [Google Scholar] [CrossRef]
- Kennedy, I.R.; Ganguli, N.K. N2-fixing trees for profitable farm-forestry. Agric. For. 2016, 62, 39–46. [Google Scholar] [CrossRef]
- Benner, S.A. Enzyme kinetics and molecular evolution. Chem. Rev. 1989, 89, 789–806. [Google Scholar] [CrossRef]
- Lambert, F.L.; Leff, H.S. The correlation of standard entropy with enthalpy supplied from 0 to 298.15 K. J. Chem. Educ. 2009, 86, 94. [Google Scholar] [CrossRef]
- Mackay, D. Multimedia Environmental Models: The Fugacity Approach, 2nd ed.; Taylor and Francis: Boca Raton, FL, USA, 2001. [Google Scholar]
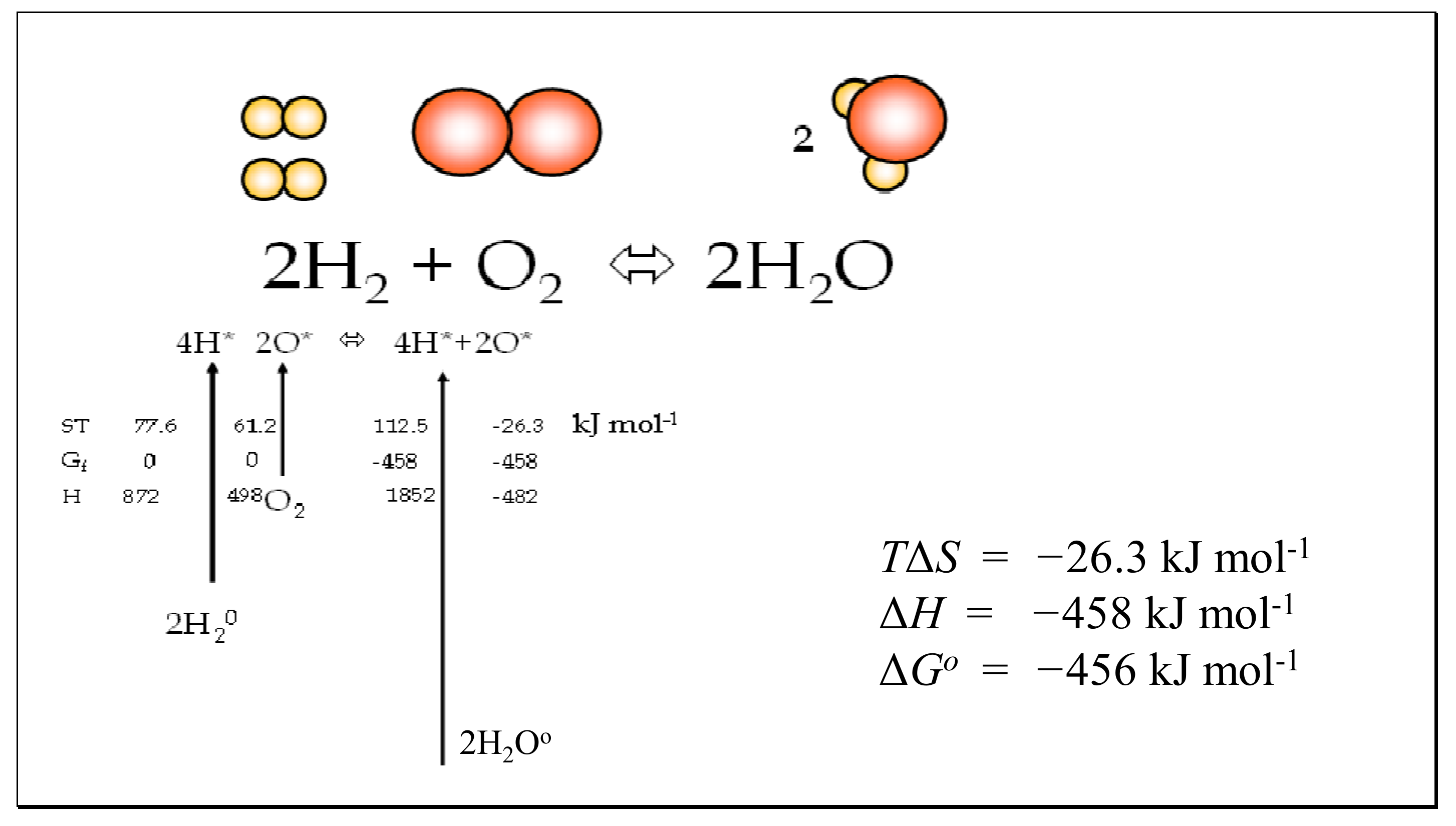
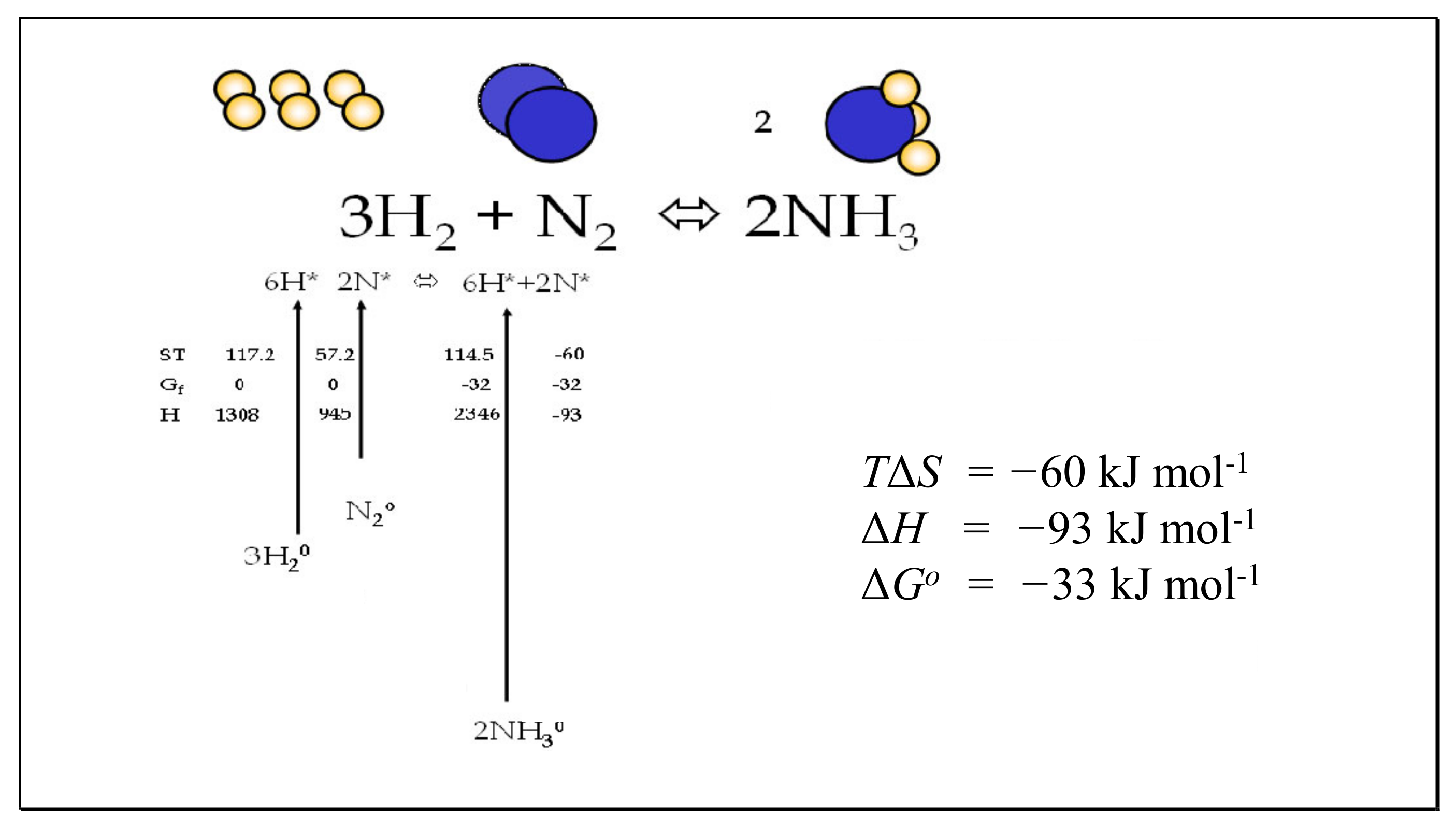
| Gas | Mass Daltons | Bond Length pm | Principal Moments of Inertia Ia, Ib, Ic ×1040 g cm2 | Vibration Frequencies cm−1 | Degeneracy | Multiplicity Symmetry Qe, Rotational Symmetry σ |
|---|---|---|---|---|---|---|
| H | 1.008 | - | - | - | - | |
| H2 | 2.016 | 74 | - | 4161 | - | 1, 2 |
| N2 | 28.014 | 110 | - | 2358 | - | 1, 2 |
| O2 | 16 + 16 = 32 | 121 | - | 1580 | 1 | 3, 2 |
| NH3 | 14 + 1 + 1 + 1 = 17 | 101 101 101 | 2.9638 2.9638 4.5176 | 3337 950 3447 1627 | 1 1 1 1 | 1, 3 |
| CO2 | 16 + 12 + 16 = 44 | 122 + 122 = 244 | Linear | 1388 667 2349 | 1 2 1 | 1, 2 |
| H2O | 16 + 1 + 1 = 18 | 74 | 1.024 | 3652 | 1 | 1, 2 |
| 74 | 1.920 | 1595 | 1 | |||
| 2.947 | 3756 | 1 | ||||
| N2O | 14 + 14 + 16 = 44 | NN 112 NO 119 | Linear | 2224 1285 589 | 1 1 2 | 1, 1 |
| CH4 | 12 + 1 + 1 + 1 + 1 = 16 | 108 108 108 108 | 5.27 5.27 5.27 | 2914 1526 3020 1306 | 1 2 3 3 | 1, 12 |
| Bond | ΔH at 298 K kJ/mol | ΔH Ergs Per Molecule ×1012 | Zero Point Vibrational Energy cm−1 | Characteristic Temperature Rotation Θrot K | Characteristic Temperature Vibration Θvib K |
|---|---|---|---|---|---|
| H-H = H2 | 436 | 7.19234576 | 2079.307 | 85.4 | 6210 |
| C-H = CH4 | 413 | 6.81293303 | |||
| N-H = NH3 | 391 | 6.45001650 | 7214.5 | ||
| O-H = H2O | 463 | 7.01302636 | |||
| C=O = CO2 | 745 | 11.2844953 | 0.561 | ||
| C-O- | 358 | 5.42259921 | |||
| C-N= | 305 | 4.61981217 | |||
| O-O- | 146 | 2.21145108 | |||
| O=O = O2 | 498 | 7.54316874 | 787.3797 | 2.07 | 2230 |
| O-N= | 201 | 3.04453196 | |||
| N=N- | 418 | 6.33141472 | |||
| N2 | 945 | 14.3138443 | 1175.778 | 2.86 | 3374 |
| N=O | 607 | 9.19418358 |
| 640 K N2 | N0/Nn = Vn/Vo | δɛ J | rn/ro = @tn/@t0 | ɛ ex Action kTln(ntn/nto)3 | 288 K N2 | N0/Nn = Vn/Vo | rn/ro = @tn/@t0 | ɛ ex Action kTln(ntn/nto)3 |
| e−δɛvib/kT | ×1020 | ×1020 | e−δɛvib | ×1020 | ×1020 | |||
| e−2hv/kT | 102,582 | 10.1954 | 46.812 | 10.1954 | 5hv/2 | 1.367e11 | 5151.40 | 10.1954 |
| e−hv/kT | 320.284 | 5.0977 | 6.842 | 5.0977 | 3hv/2 | 369605 | 77.771 | 5.0977 |
| eo | 1.000 | 0 | 1.000 | 0 | hv/2 | 0 | 1.000 | 0 |
| 640 K CO2 | No/Nn = Vn/Vo | δɛ ergs | rn/ro = @tn/@t0 | ɛ ex Action kTln(ntn/nto)3 | 288 K CO2 | No/Nn = Vn/Vo | rn/ro = @tn/@t0 | ɛ ex Action kTln(ntn/nto)3 |
| δɛvib | δɛvib | ×1013 | ×1013 | |||||
| e−2hv/kT | 20.065 | 2.6499 | 2.717 | 2.6499 | e−2hv/kT | 783.99 | 9.221 | 2.6499 |
| e−hv/kT | 4.479 | 1.3250 | 1.648 | 1.3250 | e−hv/kT | 28.000 | 3.037 | 1.3250 |
| eo | 1.000 | 0 | 1.000 | 0 | eo | 1.000 | 1.000 | 0 |
| Temp K | Entropy (oS) at 1 atm J/K | ST kJ/mole | Entropy (S) at 1 atm J/K | ST kJ/mole | −TΔS | −ΔH H2=>2H = 436 kJ | Gibbs Change ΔG |
|---|---|---|---|---|---|---|---|
| H | 2H | H2 | H2 | ||||
| 1000 | 134.088 | 268.176 | 165.562 | 165.562 | 102.614 | 440.157 | −366.543 |
| 2000 | 148.496 | 593.984 | 185.733 | 371.466 | 222.518 | 444.314 | −216.796 |
| 3000 | 156.924 | 941.544 | 197.533 | 592.599 | 348.445 | 448.471 | −99.526 |
| 4000 | 162.904 | 1303.232 | 205.904 | 823.616 | 479.616 | 481.628 | −2.012 |
| 5000 | 167.542 | 1675.420 | 212.348 | 1061.990 | 613.010 | 456.785 | +98.225 |
| 6000 | 171.332 | 2055.984 | 217.703 | 1306.221 | 749.763 | 460.942 | +230.821 |
| 7000 | 174.536 | 2443.504 | 222.189 | 1555.323 | 888.181 | 465.099 | +365.082 |
| 8000 | 177.312 | 2836.992 | 226.075 | 1808.600 | 1028.342 | 469.256 | +501.086 |
| 9000 | 179.760 | 3235.680 | 229.503 | 2065.520 | 1170.160 | 473.413 | +648.747 |
| Temp | Translational Ergal H J | Translational Virtual Quanta | Mean Energy H hω | Translational Ergal H2 | Translational Virtual Quanta | Mean Energy H2 hω | Rotational Ergal | Rotational Virtual Quanta | Mean Energy H2 hω |
|---|---|---|---|---|---|---|---|---|---|
| K | gt ×1019 | nt | ×1027 | gt ×1019 | nt | ×1021 | gr ×1020 | jr | ×1020 |
| 1000 | 1.8815 | 203.8 | 22.23 | 2.0250 | 288.3 | 0.702 | 4.8324 | 3.385 | 1.428 |
| 2000 | 4.2414 | 363.2 | 8.853 | 4.5285 | 513.7 | 0.882 | 9.4958 | 4.788 | 1.983 |
| 3000 | 6.7820 | 509.3 | 5.134 | 7.2127 | 720.2 | 1.001 | 11.7813 | 5.864 | 2.009 |
| 4000 | 9.4399 | 647.2 | 1.459 | 10.0141 | 915.3 | 1.094 | 17.2972 | 6.771 | 2.555 |
| 5000 | 12.1849 | 779.5 | 1.563 | 12.9028 | 1102.4 | 1.170 | 23.1626 | 7.570 | 3.060 |
| 6000 | 14.9996 | 907.4 | 1.653 | 15.8608 | 1283.3 | 1.236 | 29.3055 | 8.292 | 3.534 |
| 7000 | 17.8720 | 1031.8 | 1.732 | 18.8769 | 1459.2 | 1.293 | 35.6788 | 8.957 | 3.983 |
| 8000 | 20.7938 | 1153.2 | 1.803 | 21.9422 | 1630.9 | 1.345 | 42.2504 | 9.575 | 4.413 |
| 9000 | 23.7589 | 1272.2 | 1.868 | 25.0508 | 1799.1 | 1.392 | 48.9963 | 10.156 | 4.824 |
| K Temp | H2O, S, ST | O2, S, ST | H2, S, ST | TΔS 2H2 + O2 | −ΔH <> 2H2O | ΔG= ΔH − TΔS |
|---|---|---|---|---|---|---|
| J/K, kJ/mole | J/K, kJ/mole | J/K, kJ/mole | kJ/mole | 444 @ 0 | ||
| 1000 | 232.397, 232.397 | 243.403, 243.403 | 165.671, 165.671 | 109.951 | 463.112 | −353.161 |
| 2000 | 264.043, 528.086 | 267.976, 521.005 | 187.249, 374.498 | 213.829 | 474.273 | −260.444 |
| 3000 | 285.241, 855.724 | 283.116, 849.348 | 201.024, 603.071 | 344.042 | 485.208 | −141.166 |
| 4000 | 309.471, 1237.885 | 293.795, 1175.178 | 211.215, 844.858 | 389.124 | 496.715 | −107.591 |
| 5000 | 313.582, 1567.910 | 302.104, 1510.519 | 219.280, 1096.402 | 567.503 | 508.680 | +58.823 |
| 6000 | 323.942, 1943.651 | 308.904, 1853.422 | 225.943, 1335.658 | 637.436 | 520.770 | +116.666 |
| 7000 | 332.760, 2329.319 | 314.658, 2202.607 | 231.613, 1621.291 | 786.551 | 532.904 | +253.647 |
| 8000 | 340.431, 2723.448 | 319.646, 2557.165 | 236.545, 1892.359 | 894.987 | 545.178 | +349.809 |
| 9000 | 347.217, 3124.451 | 324.047, 2914.642 | 240.907, 2168.167 | 1002.074 | 557.541 | +444.533 |
| 10,000 | 353.299, 3533.00 | 327.985, 3279.850 | 244.818, 2448.176 | 1110.218 | 569.883 | +540.335 |
| Temperature | H2O kJ/mol | O2 | H2 | ΔH |
|---|---|---|---|---|
| 1000 | 37.141 | 34.598 | 29.398 | −19.112 |
| 2000 | 83.468 | 73.149 | 62.030 | −30.273 |
| 3000 | 130.953 | 111.084 | 96.015 | −41.208 |
| 4000 | 177.954 | 148.719 | 129.952 | −52.715 |
| 5000 | 224.562 | 186.366 | 163.719 | −64.680 |
| 6000 | 270.918 | 223.898 | 197.354 | −76.770 |
| 7000 | 317.144 | 261.396 | 230.898 | −88.904 |
| 8000 | 363.201 | 298.824 | 264.378 | −101.178 |
| 9000 | 409.213 | 336.339 | 297.814 | −113.541 |
| 10,000 | 455.171 | 373.793 | 331.216 | −125.883 |
| Temp. K | NH3, S, ST | N2, S, ST | H2, S, ST | TΔS | −ΔH 93 at 0 K | −ΔG |
|---|---|---|---|---|---|---|
| J/K, kJ/mole | J/K, kJ/mole | J/K, kJ/mole | kJ/mole | kJ/mole | kJ/mole | |
| 400 | 203.727, 81.491 | 200.189, 80.076 | 138.898, 55.559 | 83.771 | 97.924 | 14.153 |
| 500 | 212.331, 106.166 | 206.683, 103.342 | 145.391, 72.696 | 109.098 | 96.996 | −12.102 |
| 600 | 219.750, 131.850 | 211.989, 127.193 | 150.697, 90.418 | 134.747 | 95.300 | −39.447 |
| 700 | 226.328, 158.430 | 216.475, 151.533 | 155.183, 108.628 | 160.557 | 93.000 | −67.557 |
| 800 | 232.277, 185.822 | 220.361, 176.289 | 159.069, 127.255 | 186.410 | 90.094 | −93.316 |
| 900 | 237.732, 213.959 | 223.788, 201.409 | 162.496, 146.246 | 212.229 | 86.630 | −125.599 |
| 1000 | 242.789, 242.789 | 226.854, 226.854 | 165.562, 165.562 | 237.962 | 82.683 | −155.279 |
| NH3 | N2 | H2 | ΔH | |
|---|---|---|---|---|
| 400 | 14.998 | 11.640 | 11.640 | −4.924 |
| 500 | 19.842 | 14.550 | 14.550 | −3.996 |
| 600 | 25.040 | 17.460 | 17.460 | −2.300 |
| 700 | 30.555 | 20.370 | 20.370 | 0 |
| 800 | 36.373 | 23.280 | 23.280 | 2.906 |
| 900 | 42.470 | 26.190 | 26.190 | 6.370 |
| 1000 | 48.810 | 29.101 | 29.101 | 10.317 |
| Temp K | NH3, S, ST | N2, S, ST | H2, S, ST | TΔS + ΔCv |
|---|---|---|---|---|
| J/K, kJ/mole | J/K, kJ/mole | J/K, kJ/mole | kJ/mole | |
| 400 * | 207.964, 83.186 | 200.189, 80.076 | 138.898, 55.559 | −80.381 |
| 500 | 218.758, 109.379 | 206.683, 103.342 | 145.391, 72.696 | −102.672 |
| 600 | 228.224, 136.934 | 211.989, 127.193 | 150.697, 90.418 | −124.979 |
| 700 | 236.721, 165.705 | 216.475, 151.533 | 155.183, 108.628 | −146.007 |
| 800 | 244.485, 195.588 | 220.361, 176.289 | 159.069, 127.255 | −166.878 |
| 900 | 251.663, 226.497 | 223.788, 201.409 | 162.496, 146.246 | −196.153 |
| 1000 | 258.342, 258.342 | 226.854, 226.854 | 165.562, 165.562 | −206.856 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, I.R.; Hodzic, M. Partitioning Entropy with Action Mechanics: Predicting Chemical Reaction Rates and Gaseous Equilibria of Reactions of Hydrogen from Molecular Properties. Entropy 2021, 23, 1056. https://doi.org/10.3390/e23081056
Kennedy IR, Hodzic M. Partitioning Entropy with Action Mechanics: Predicting Chemical Reaction Rates and Gaseous Equilibria of Reactions of Hydrogen from Molecular Properties. Entropy. 2021; 23(8):1056. https://doi.org/10.3390/e23081056
Chicago/Turabian StyleKennedy, Ivan R., and Migdat Hodzic. 2021. "Partitioning Entropy with Action Mechanics: Predicting Chemical Reaction Rates and Gaseous Equilibria of Reactions of Hydrogen from Molecular Properties" Entropy 23, no. 8: 1056. https://doi.org/10.3390/e23081056
APA StyleKennedy, I. R., & Hodzic, M. (2021). Partitioning Entropy with Action Mechanics: Predicting Chemical Reaction Rates and Gaseous Equilibria of Reactions of Hydrogen from Molecular Properties. Entropy, 23(8), 1056. https://doi.org/10.3390/e23081056