Molecular Conformational Manifolds between Gas-Liquid Interface and Multiphasic

Abstract
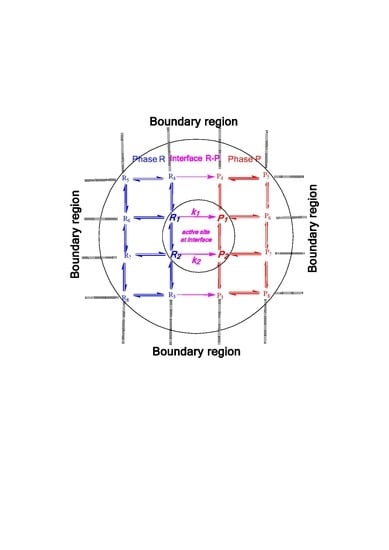
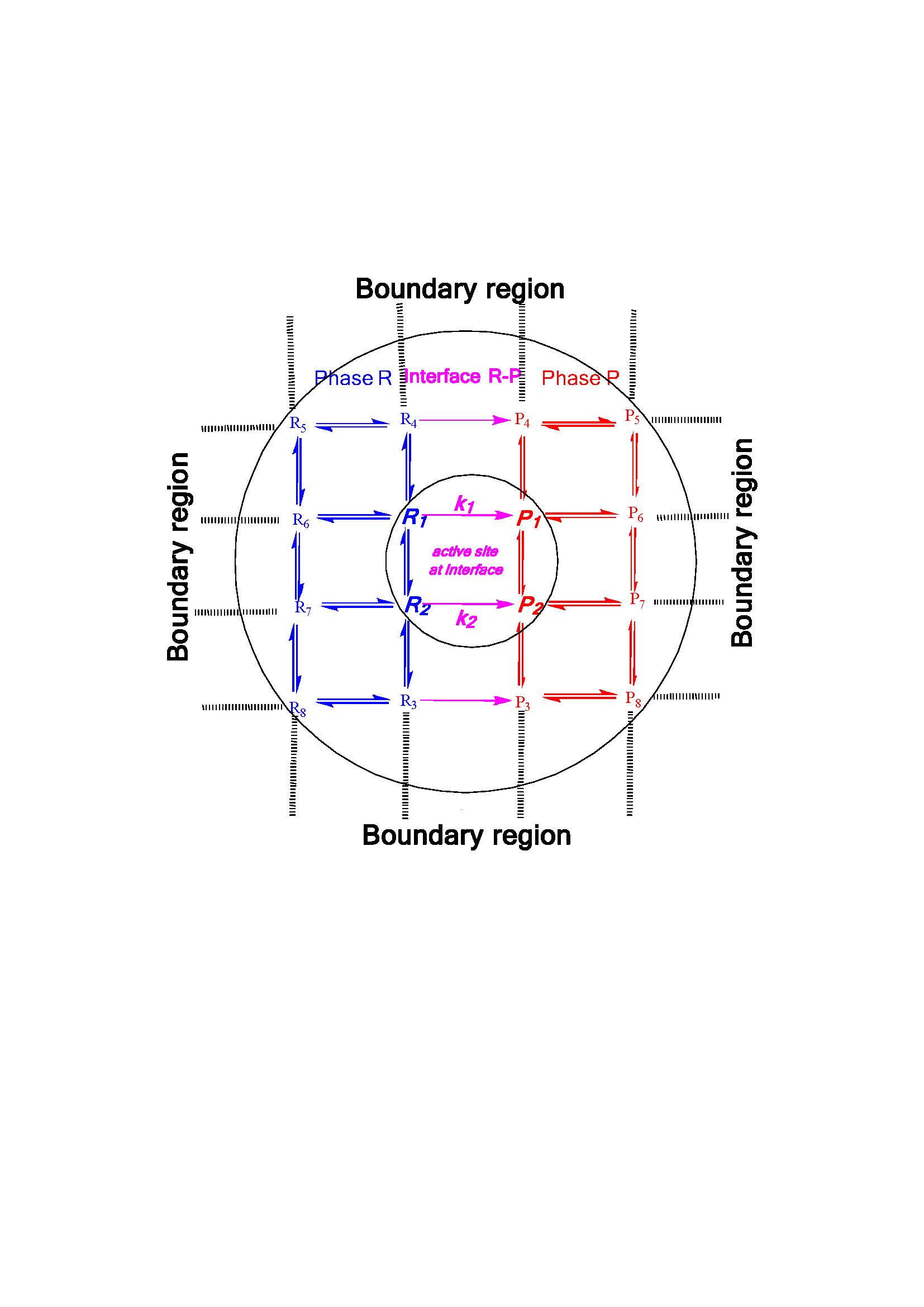
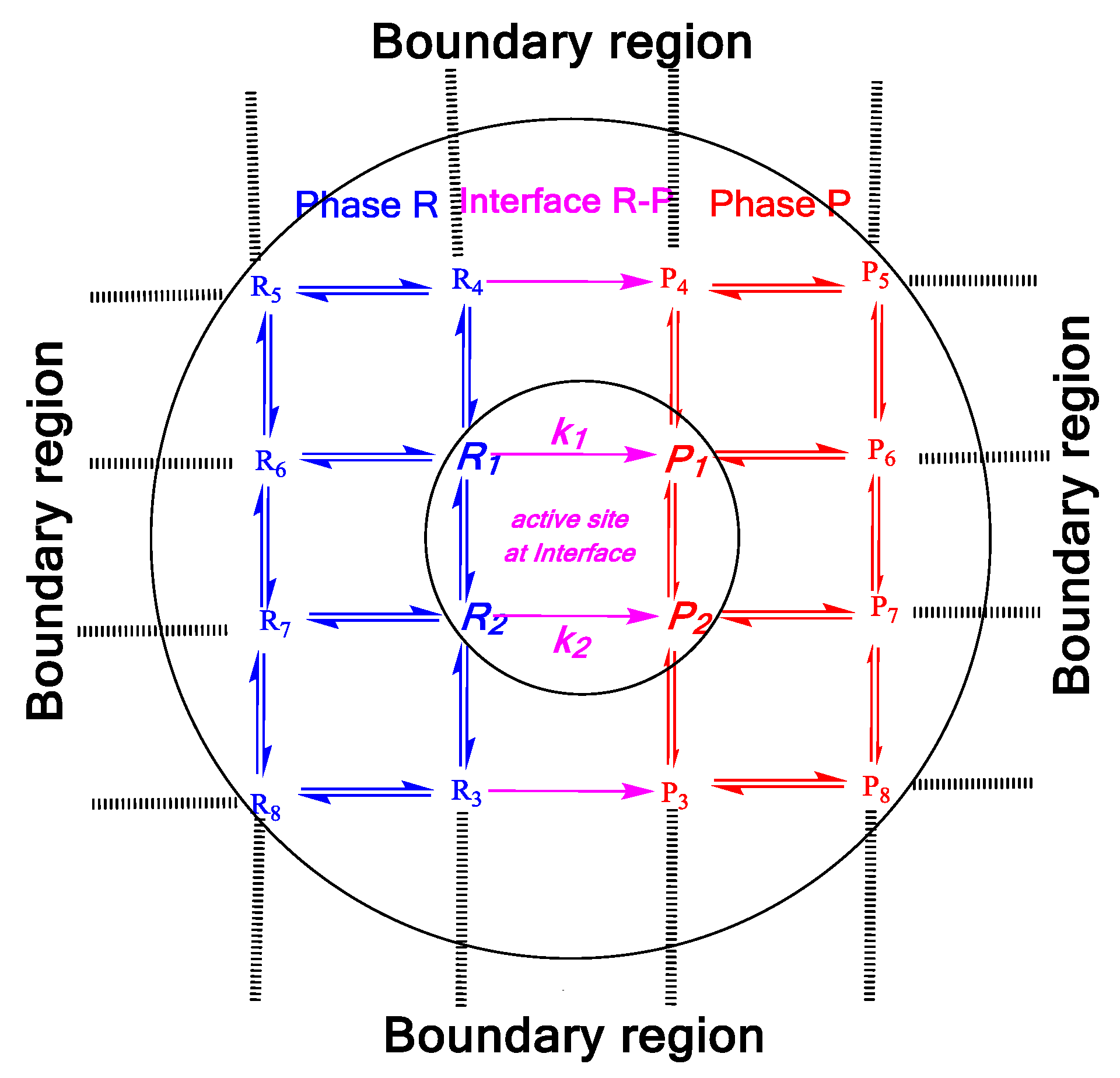
:
{kind=link}
{kind=link}
1. Introduction
2. Mass Evaporation Coefficient and Quantum/Statistical Mechanics Methods
3. Evaporation Rate of Fuel Droplets and Hybrid Kinetics Method
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gun’ko, V.M.; Nasiri, R.; Sazhin, S.S. Effects of the surroundings and conformerisation of n-dodecane molecules on evaporation/condensation processes. J. Chem. Phys. 2015, 142, 034502. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, R.; Gun’ko, V.M.; Sazhin, S.S. The effects of internal molecular dynamics on the evaporation/condensation of n-dodecane. Theor. Chem. Acc. 2015, 134, 83. [Google Scholar] [CrossRef]
- Nasiri, R. Revisiting kinetic boundary conditions at the surface of fuel droplet hydrocarbons: An atomistic computational fluid dynamics simulation. Sci. Rep. 2016, 6, 25572. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, R.; Luo, K.H. Specificity switching pathways in thermal and mass evaporation of multicomponent hydrocarbon droplets: A mesoscopic observation. Sci. Rep. 2017, 7, 5001. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, M.S. Fluids of dimerizing hard spheres, and fluid mixtures of hard spheres and dispheres. J. Chem. Phys. 1986, 85, 2929–2936. [Google Scholar] [CrossRef]
- Wertheim, M.S. Thermodynamic perturbation theory of polymerization. J. Chem. Phys. 1987, 87, 7323–7331. [Google Scholar] [CrossRef]
- Maghari, A.; Najafi, M. A novel approach for calculation of liquid—Vapor interfacial thickness. J. Stat. Mech. 2009, P05003. [Google Scholar] [CrossRef]
- Winget, P.; Dolney, D.M.; Giesen, D.J.; Cramer, C.J.; Truhlar, D.G. Minnesota Solvent Descriptor Database; University of Minnesota: Minneapolis, MN, USA, 2010. [Google Scholar]
- Yaws, C.L. Thermophysical Properties of Chemicals and Hydrocarbons; William Andrew Inc.: Norwich, NY, USA, 2008. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- NIST Chemistry WebBook. Saturation Properties for Dodecane—Temperature Increments. Available online: http://webbook.nist.gov/chemistry/ (accessed on 27 April 2016).
- Nasiri, R.; Field, M.J.; Zahedi, M.; Moosavi-Movahedi, A.A. Comparative DFT Study To Determine if α-Oxoaldehydes are Precursors for Pentosidine Formation. J. Phys. Chem. A 2012, 116, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, R.; Field, M.J.; Zahedi, M.; Moosavi-Movahedi, A.A. Cross-Linking Mechanisms of Arginine and Lysine with α,β-Dicarbonyl Compounds in Aqueous Solution. J. Phys. Chem. A 2011, 115, 13542–13555. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, R.; Zahedi, M.; Jamet, H.; Moosavi-Movahedi, A.A. Theoretical studies on models of lysine-arginine cross-links derived from α-oxoaldehydes: A new mechanism for glucosepane formation. J. Mol. Model. 2011, 18, 1645–1659. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasiri, R.; Luo, K.H. Molecular Conformational Manifolds between Gas-Liquid Interface and Multiphasic. Entropy 2017, 19, 695. https://doi.org/10.3390/e19120695
Nasiri R, Luo KH. Molecular Conformational Manifolds between Gas-Liquid Interface and Multiphasic. Entropy. 2017; 19(12):695. https://doi.org/10.3390/e19120695
Chicago/Turabian StyleNasiri, Rasoul, and Kai Hong Luo. 2017. "Molecular Conformational Manifolds between Gas-Liquid Interface and Multiphasic" Entropy 19, no. 12: 695. https://doi.org/10.3390/e19120695
APA StyleNasiri, R., & Luo, K. H. (2017). Molecular Conformational Manifolds between Gas-Liquid Interface and Multiphasic. Entropy, 19(12), 695. https://doi.org/10.3390/e19120695