
Heuristic Approach to Understanding the Accumulation Process in Hydrothermal Pores


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
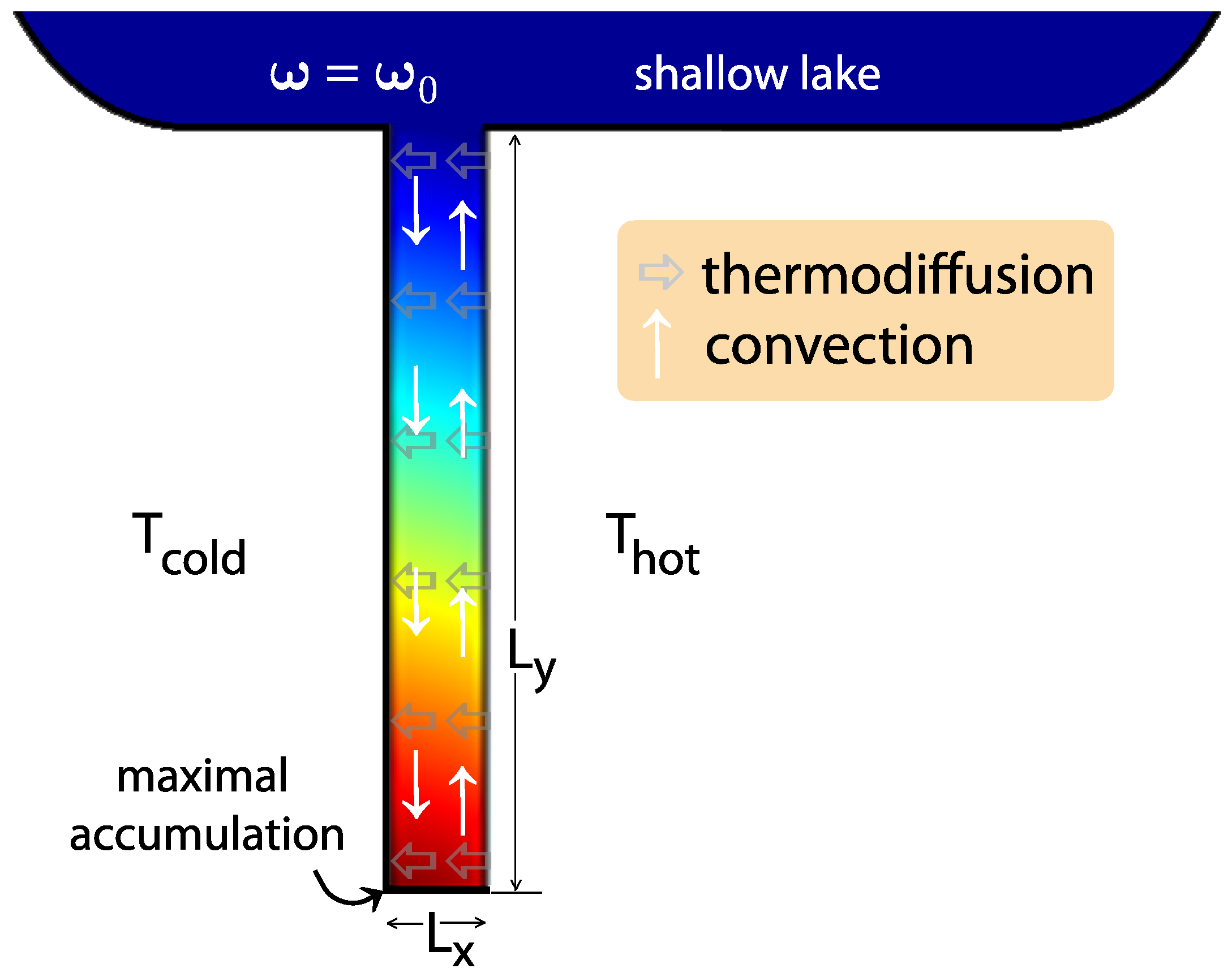
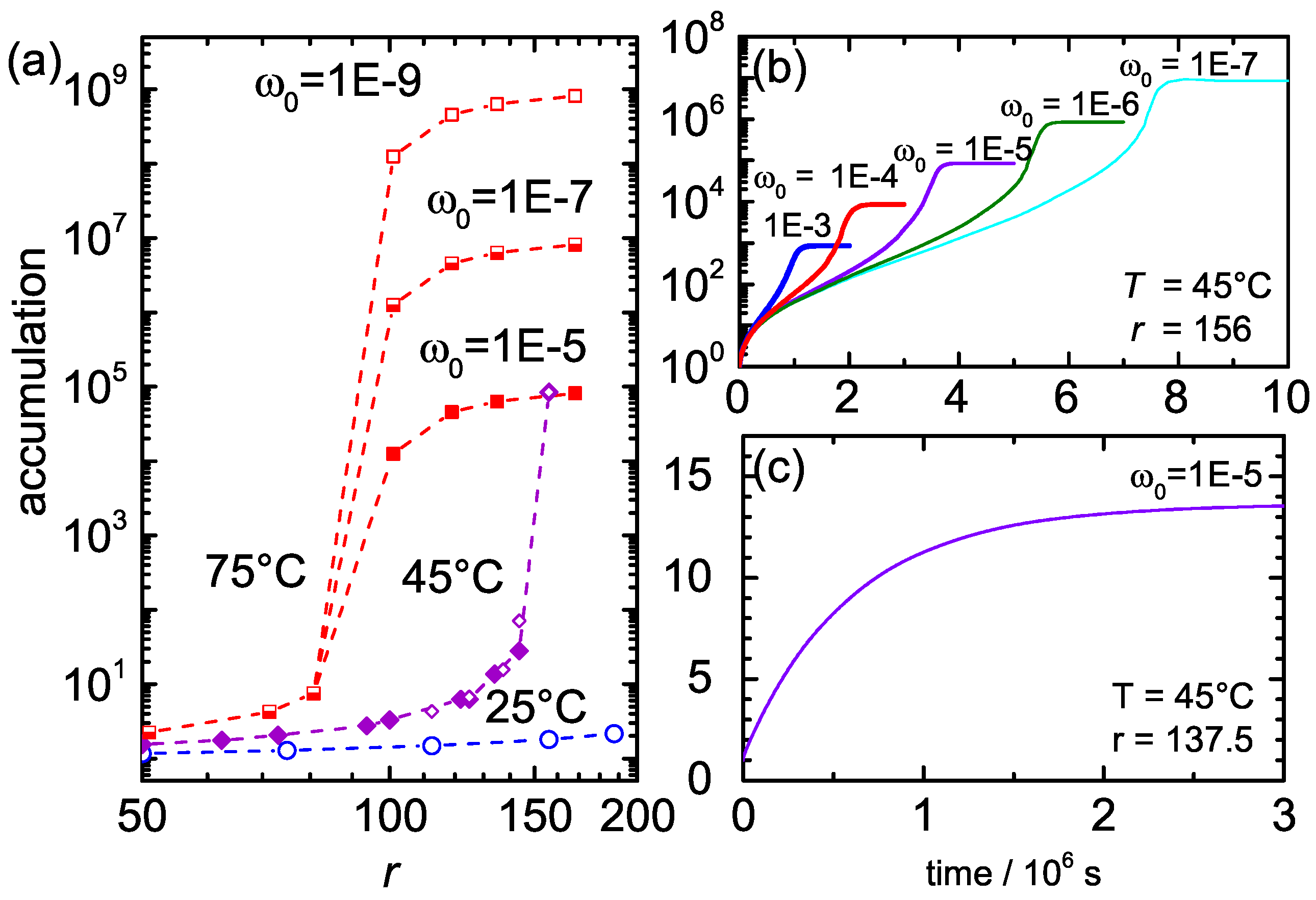
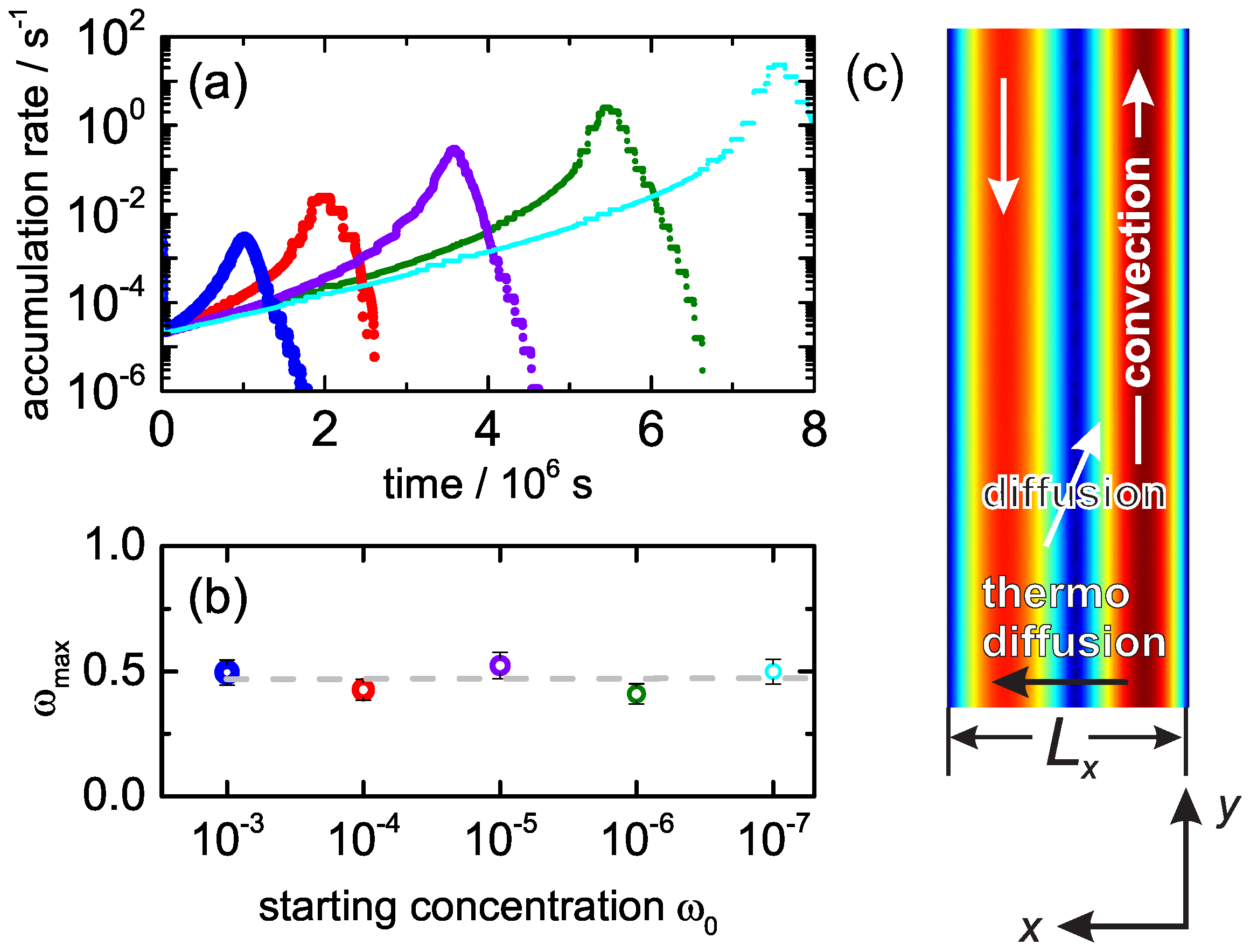
2. Accumulation in Hydrothermal Pores
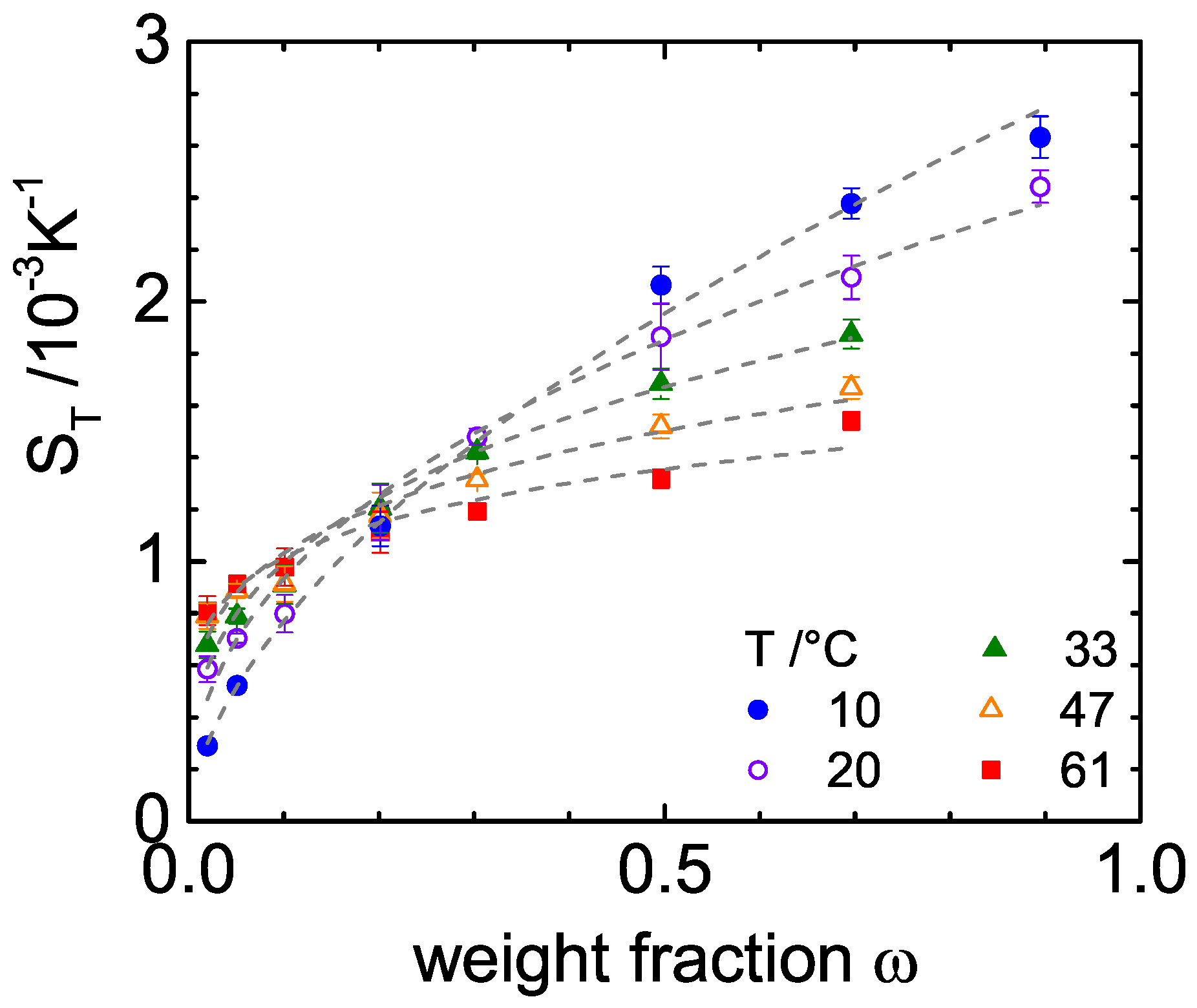
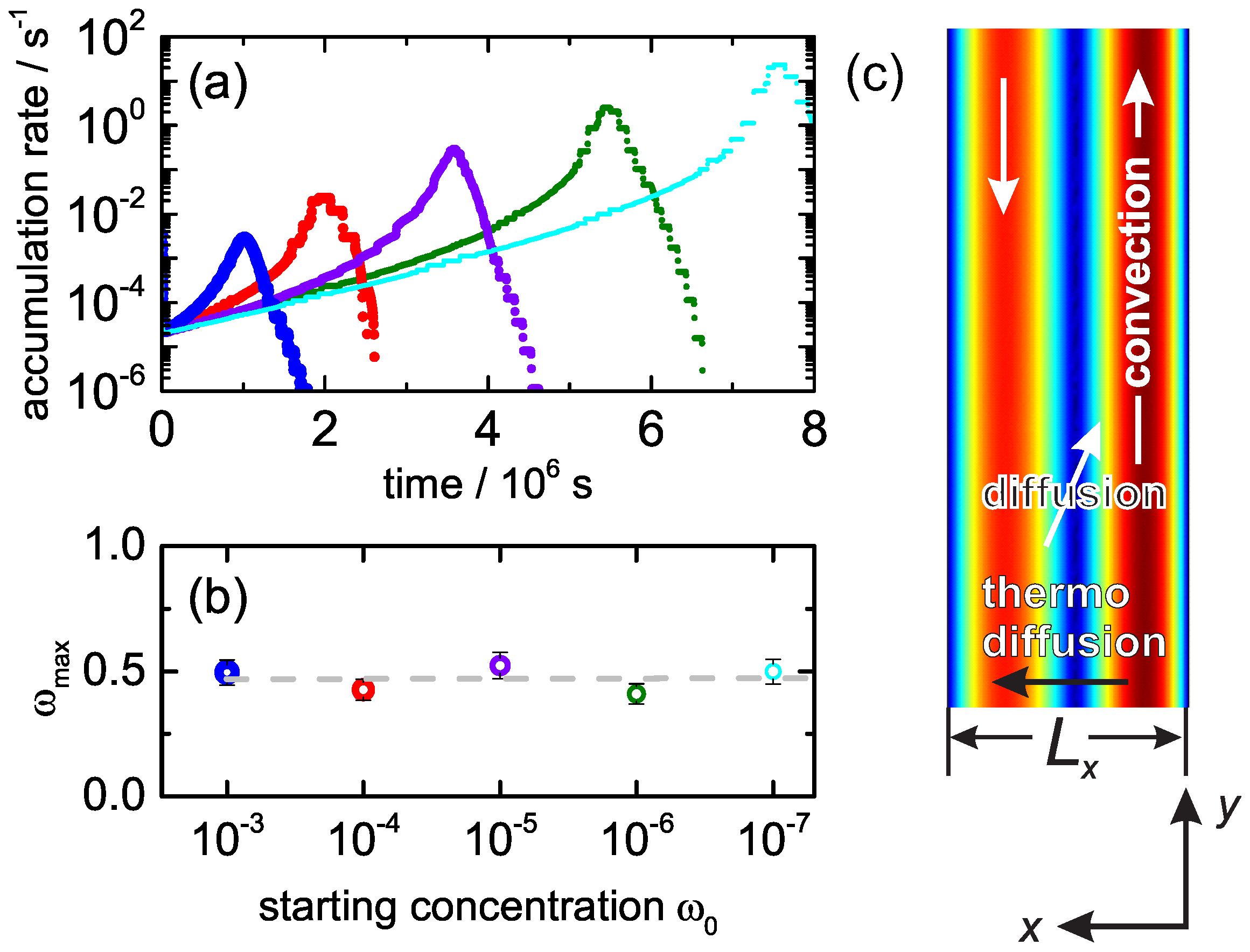
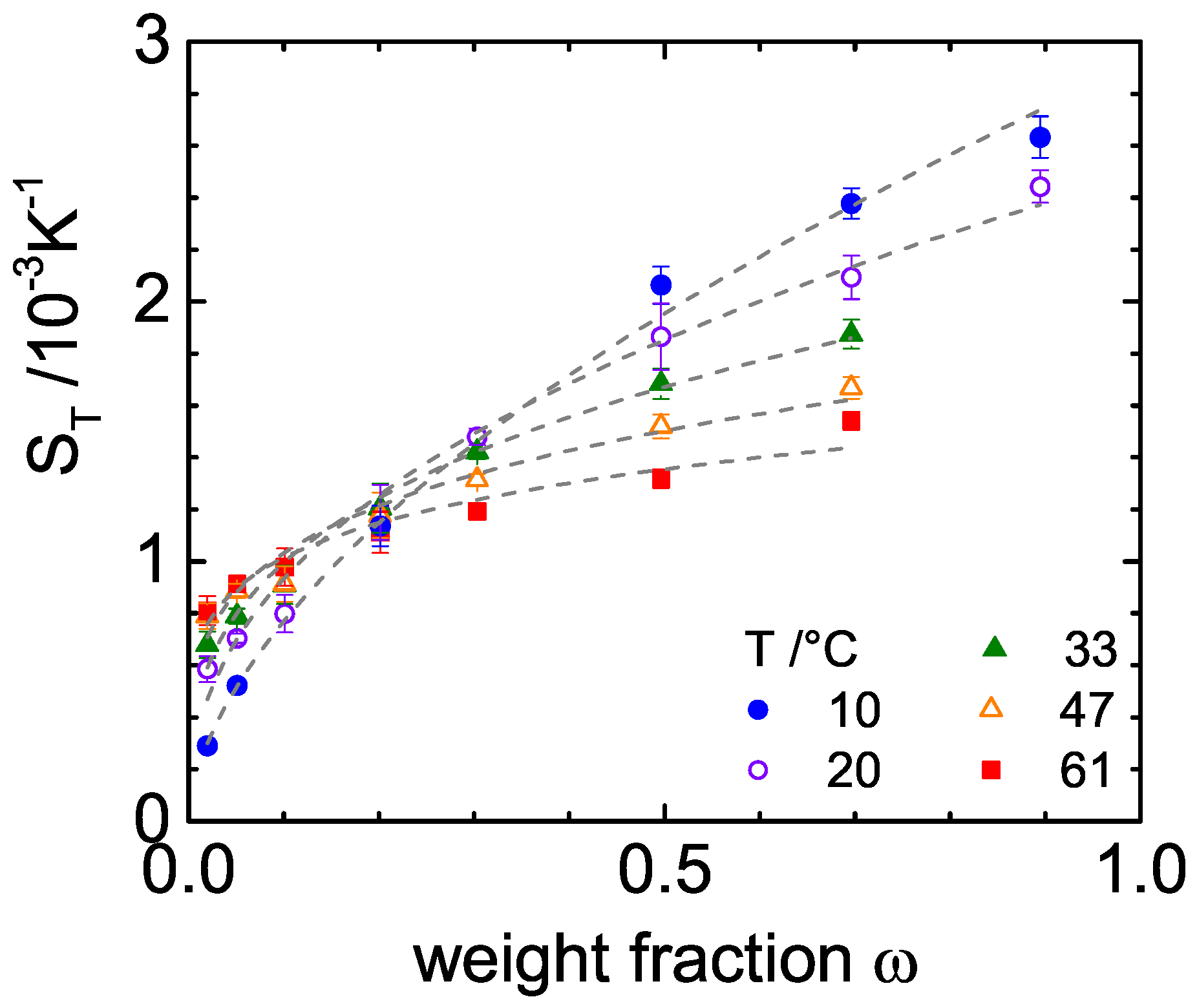
3. Experimental Results
4. Discussion
5. Conclusions
6. Materials and Methods
6.1. Sample Preparation and IR-TDFRS Measurements
6.2. Finite Element Calculations
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| IR-TDFRS | infrared thermal diffusion forced Rayleigh scattering |
| FA | formamide |
| RNA | ribonucleic acid |
| DNA | deoxyribonucleic acid |
References
- Baaske, P.; Weinert, F.M.; Duhr, S.; Lemke, K.H.; Russell, M.J.; Braun, D. Extreme Accumulation of Nucleotides in Simulated Hydrothermal Pore Systems. Proc. Natl. Acad. Sci. USA 2007, 104, 9346–9351. [Google Scholar] [CrossRef] [PubMed]
- Niether, D.; Afanasenkau, D.; Dhont, J.K.G.; Wiegand, S. Accumulation of Formamide in Hydrothermal Pores to Form Prebiotic Nucleobases. Proc. Natl. Acad. Sci. USA 2016, 113, 4272–4277. [Google Scholar] [CrossRef] [PubMed]
- Harada, K. Formation of Amino-Acids by Thermal Decomposition of Formamide—Oligomerization of Hydrogen Cyanide. Nature 1967, 214, 479–480. [Google Scholar] [CrossRef]
- Miyakawa, S.; Cleaves, H.J.; Miller, S.L. The Cold Origin of Life: A. Implications Based on the Hydrolytic Stabilities of Hydrogen Cyanide and Formamide. Orig. Life Evol. Biosph. 2002, 32, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Saladino, R.; Crestini, C.; Pino, S.; Costanzo, G.; di Mauro, E. Formamide and the Origin of Life. Phys. Life Rev. 2012, 9, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Origin of First Cells at Terrestrial, Anoxic Geothermal Fields. Proc. Natl. Acad. Sci. USA 2012, 109, E821–E830. [Google Scholar] [CrossRef] [PubMed]
- Ferus, M.; Nesvorny, D.; Sponer, J.; Kubelik, P.; Michalcikova, R.; Shestivska, V.; Sponer, J.E.; Civis, S. High-Energy Chemistry of Formamide: A Unified Mechanism of Nucleobase Formation. Proc. Natl. Acad. Sci. USA 2015, 112, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Pino, S.; Sponer, J.E.; Costanzo, G.; Saladino, R.; di Mauro, E. From Formamide to RNA, the Path Is Tenuous but Continuous. Life 2015, 5, 372–384. [Google Scholar] [CrossRef] [PubMed]
- De Groot, S.; Mazur, P. Non-Equilibrium Thermodynamics; Dover: New York, NY, USA, 1984. [Google Scholar]
- Morozov, K.I.; Köhler, W. Thermophoresis of Polymers: Nondraining vs. Draining Coil. Langmuir 2014, 30, 6571–6576. [Google Scholar] [CrossRef] [PubMed]
- Würger, A. Thermal Non-Equilibrium Transport in Colloids. Rep. Prog. Phys. 2010, 73, 126601. [Google Scholar] [CrossRef]
- Dhont, J.K.G.; Briels, W.J. Single-Particle Thermal Diffusion of Charged Colloids: Double-Layer Theory in a Temperature Gradient. Eur. Phys. J. E 2008, 25, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Köhler, W.; Morozov, K.I. The Soret Effect in Liquid Mixtures—A Review. J. Non-Equilb. Thermodyn. 2016, 41, 151–197. [Google Scholar] [CrossRef]
- Yang, M.C.; Ripoll, M. Driving Forces and Polymer Hydrodynamics in the Soret Effect. J. Phys. Condens. Matter 2012, 24, 195101. [Google Scholar] [CrossRef] [PubMed]
- Galliero, G.; Volz, S. Thermodiffusion in Model Nanofluids by Molecular Dynamics Simulations. J. Chem. Phys. 2008, 128, 064505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Müller-Plathe, F. The Soret Effect in Dilute Polymer Solutions: Influence of Chain Length, Chain Stiffness, and Solvent Quality. J. Chem. Phys. 2006, 125, 124903. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, B.; Nieto-Draghi, C.; Avalos, J.B. The Role of Molecular Interactions in the Change of Sign of the Soret Coefficient. Europhys. Lett. 2004, 67, 976–982. [Google Scholar] [CrossRef]
- Römer, F.; Wang, Z.; Wiegand, S.; Bresme, F. Alkali Halide Solutions under Thermal Gradients: Soret Coefficients and Heat Transfer Mechanisms. J. Phys. Chem. B 2013, 117, 8209–8222. [Google Scholar] [CrossRef] [PubMed]
- Naumann, P.; Datta, S.; Sottmann, T.; Arlt, B.; Frielinghaus, H.; Wiegand, S. Isothermal Behavior of the Soret Effect in Nonionic Microemulsions: Size Variation by Using Different n-Alkanes. J . Phys. Chem. B 2014, 118, 3451–3460. [Google Scholar] [CrossRef] [PubMed]
- Parola, A.; Piazza, R. A Microscopic Approach to Thermophoresis in Colloidal Suspensions. J. Phys. Condens. Matter 2005, 17, S3639–S3643. [Google Scholar] [CrossRef]
- Ning, H.; Dhont, J.K.G.; Wiegand, S. Thermal-Diffusive Behavior of a Dilute Solution of Charged Colloids. Langmuir 2008, 24, 2426–2432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kriegs, H.; Buitenhuis, J.; Dhont, J.K.G.; Wiegand, S. Thermophoresis of Charged Colloidal Rods. Soft Matter 2013, 9, 8697–8704. [Google Scholar] [CrossRef]
- Sehnem, A.L.; Neto, A.M.F.; Aquino, R.; Campos, A.F.C.; Tourinho, F.A.; Depeyrot, J. Temperature Dependence of the Soret Coefficient of Ionic Colloids. Phys. Rev. E 2015, 92, 042311. [Google Scholar] [CrossRef] [PubMed]
- Syshchyk, O.; Afanasenkau, D.; Wang, Z.; Kriegs, H.; Buitenhuis, J.; Wiegand, S. Influence of Temperature and Charge Effects on Thermophoresis of Polystyrene Beads. Eur. Phys. J. E 2016, 39. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, R.; Wolf, B.A.; Kita, R. Thermal Diffusion of Dextran in Aqueous Solutions in the Absence and the Presence of Urea. Biomacromolecules 2006, 7, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kriegs, H.; Wiegand, S. Thermal Diffusion of Nucleotides. J. Phys. Chem. B 2012, 116, 7463–7469. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, Y.; Wiegand, S.; Kita, R. Temperature Dependence of Soret Coefficient in Aqueous and Nonaqueous Solutions of Pullulan. Biomacromolecules 2010, 11, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Iacopini, S.; Rusconi, R.; Piazza, R. The “Macromolecular Tourist”: Universal Temperature Dependence of Thermal Diffusion in Aqueous Colloidal Suspensions. Eur. Phys. J. E 2006, 19, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Shinyashiki, N.; Yagihara, S.; Wiegand, S.; Kita, R. Ludwig-Soret Effect of Aqueous Solutions of Ethylene Glycol Oligomers, Crown Ethers, and Glycerol: Temperature, Molecular Weight, and Hydrogen Bond Effect. J. Chem. Phys. 2015, 143, 124504. [Google Scholar] [CrossRef] [PubMed]
- Hill, A. The Possible Effects of the Aggregation of the Molecules of Haemoglobin on Its Dissociation Curves. J. Physiol. 1910, 40, 4–7. [Google Scholar]
- Goutelle, S.; Maurin, M.; Rougier, F.; Barbaut, X.; Bourguignon, L.; Ducher, M.; Maire, P. The Hill Equation: A Review of Its Capabilities in Pharmacological Modelling. Fundam. Clin. Pharmacol. 2008, 22, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Polyakov, P.; Wiegand, S. Systematic Study of the Thermal Diffusion in Associated Mixtures. J. Chem. Phys. 2008, 128, 034505. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.A.; Ferris, J.P.; Orgel, L.E. Studies in Prebiotic Synthesis: 2. Synthesis of Purine Precursors and Amino Acids from Aqueous Hydrogen Cyanide. J. Mol. Biol. 1967, 30, 223–253. [Google Scholar] [PubMed]
- Orgel, L.E. Polymerization on the Rocks: Theoretical Introduction. Orig. Life Evol. Biosph. 1998, 28, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Hill, A.R.; Liu, R.H.; Orgel, L.E. Synthesis of Long Prebiotic Oligomers on Mineral Surfaces. Nature 1996, 381, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Franchi, M.; Gallori, E. A Surface-Mediated Origin of the RNA World: Biogenic Activities of Clay-Adsorbed RNA Molecules. Gene 2005, 346, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, S.; Ning, H.; Kriegs, H. Thermal Diffusion Forced Rayleigh Scattering Setup Optimized for Aqueous Mixtures. J. Phys. Chem. B 2007, 111, 14169–14174. [Google Scholar] [CrossRef] [PubMed]
- Egan, E.P.; Luff, B.B. Heat of Solution Heat Capacity and Density of Aqueous Formamide Solutions at 25 °C. J. Chem. Eng. Data 1966, 11, 194–196. [Google Scholar] [CrossRef]
- Akhtar, S.; Faruk, A.N.M.O.; Saleh, M.A. Viscosity of aqueous solutions of formamide, N-methylformamide and N,N-dimethylformamide. Phys. Chem. Liq. 2001, 39, 383–399. [Google Scholar] [CrossRef]
- Wohlfarth, C. Viscosity of the mixture (1) water; (2) formamide. In Supplement to IV/18; Lechner, M.D., Ed.; Landolt-Börnstein—Group IV Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 2009; pp. 709–711. [Google Scholar]
- Tobitani, A.; Tanaka, T. Predicting Thermal-Conductivity of Binary-Liquid Mixtures on Basis of Coordination-Number. Can. J. Chem. Eng. 1987, 65, 321–328. [Google Scholar] [CrossRef]
- Checoni, R.F.; Volpe, P.L.O. Measurements of the Molar Heat Capacities and Excess Molar Heat Capacities for Water plus Organic Solvents Mixtures at 288.15 K to 303.15 K and Atmospheric Pressure. J. Solut. Chem. 2010, 39, 259–276. [Google Scholar] [CrossRef]
- Ganiev, Y.A.; Rastorguev, Y.L. Thermal Conductivity of Organic Liquids. J. Eng. Phys. 1968, 15, 519–525. [Google Scholar]
- Young, H.D. University Physics; Addison-Wesley: Reading, MA, USA, 1992. [Google Scholar]
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niether, D.; Wiegand, S. Heuristic Approach to Understanding the Accumulation Process in Hydrothermal Pores. Entropy 2017, 19, 33. https://doi.org/10.3390/e19010033
Niether D, Wiegand S. Heuristic Approach to Understanding the Accumulation Process in Hydrothermal Pores. Entropy. 2017; 19(1):33. https://doi.org/10.3390/e19010033
Chicago/Turabian StyleNiether, Doreen, and Simone Wiegand. 2017. "Heuristic Approach to Understanding the Accumulation Process in Hydrothermal Pores" Entropy 19, no. 1: 33. https://doi.org/10.3390/e19010033
APA StyleNiether, D., & Wiegand, S. (2017). Heuristic Approach to Understanding the Accumulation Process in Hydrothermal Pores. Entropy, 19(1), 33. https://doi.org/10.3390/e19010033