Is Encephalopathy a Mechanism to Renew Sulfate in Autism? †


Abstract
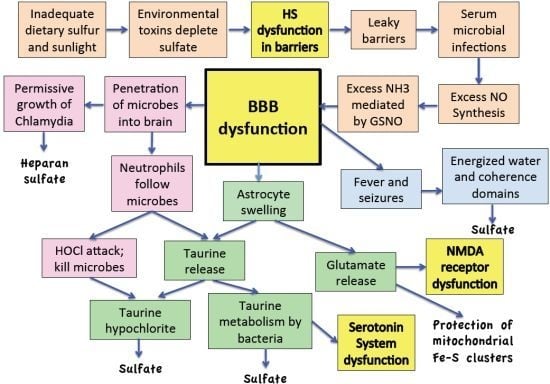
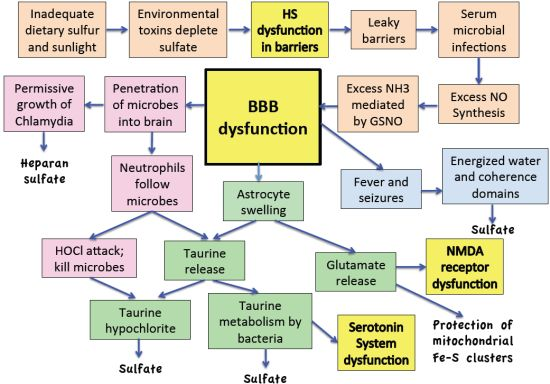
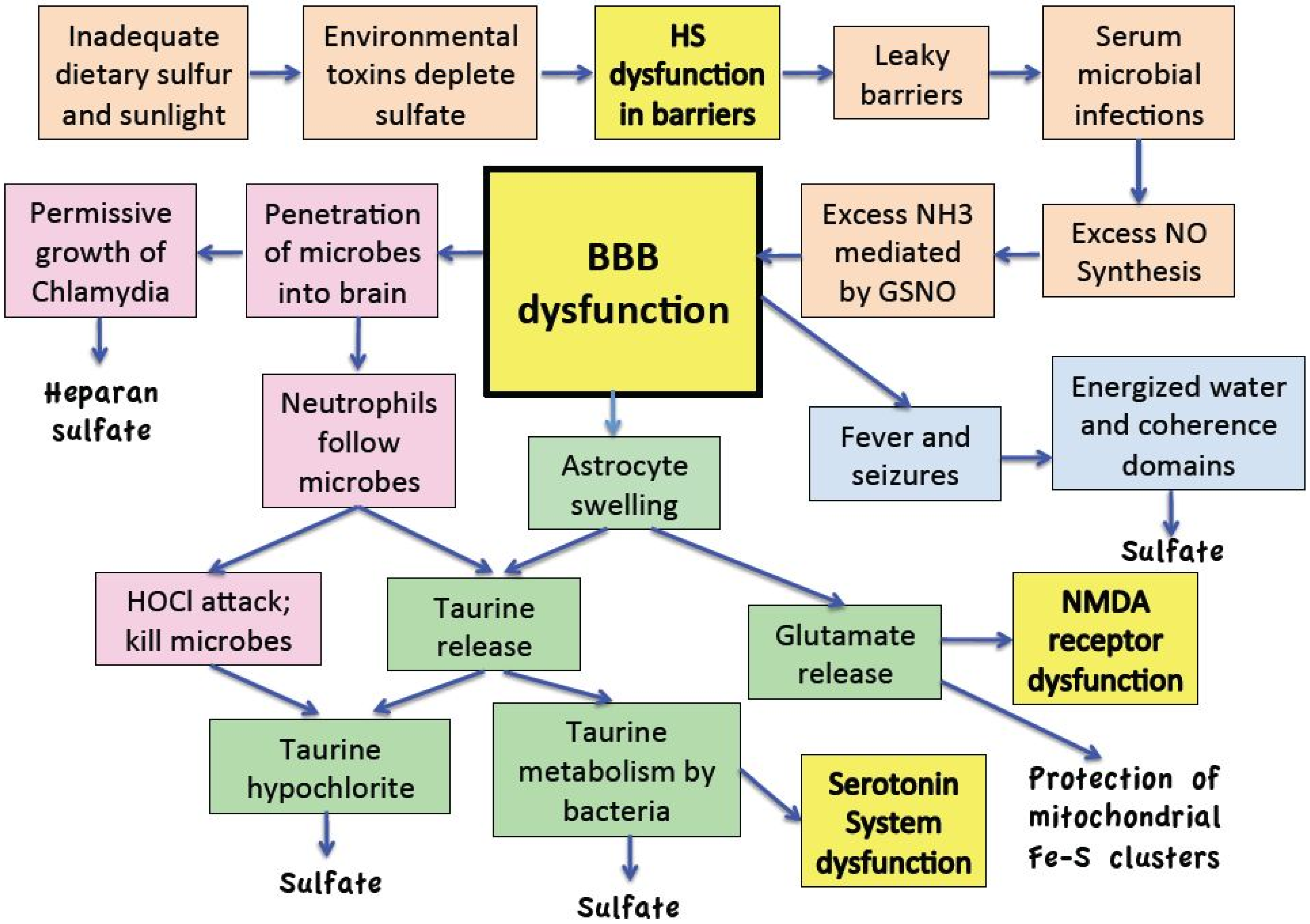
:
{kind=link}
{kind=link}
1. Introduction
2. ASD, Sulfur Metabolism, Glutathione and Ammonia
3. The Crucial Roles of Heparan Sulfate Proteoglycans
4. Insights from Hepatic Encephalopathy
5. Glutamate as a Neurotransmitter and an Energy Source
6. Taurine’s Dual Roles in Detoxification and Sulfate Renewal
7. Seizures, Electromagnetic Fields, and Sulfate Synthesis by RBCs
8. Environmental Factors
9. Anergy and Serotonin Impairment
10. The Reaction Cascade
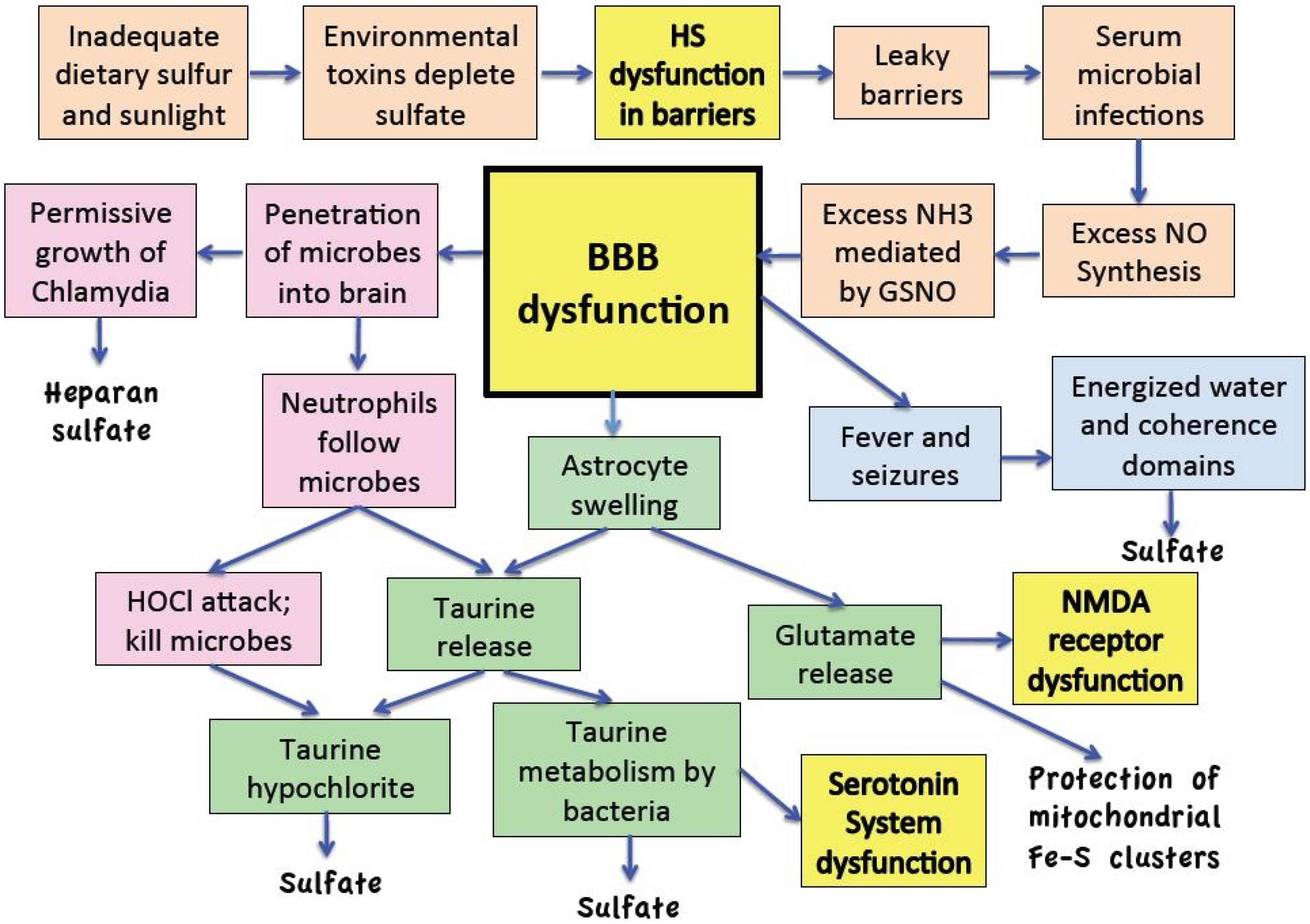
11. Discussion
12. Conclusions
Acknowledgements
References
- Caronna, E.B.; Milunsky, J.M.; Tager-Flusberg, H. Autism spectrum disorders: Clinical and research frontiers. Arch. Dis. Child. 2008, 93, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Horvath, K.; Papadimitriou, J.C.; Rabsztyn, A.; Drachenberg, C.; Tildon, J.T. Gastrointestinal abnormalities in children with autistic disorder. J. Pediatr. 1999, 135, 559–563. [Google Scholar] [CrossRef]
- Wakefield, A.J.; Puleston, J.M.; Montgomery, S.M.; Anthony, A.; O’Leary, J.J.; Murch, S.H. Review article: The concept of entero-colonic encephalopathy, autism and opioid receptor ligands. Aliment. Pharmacol. Ther. 2002, 16, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Doyle, R. Autism, gut-blood-brain barrier, and mast cells. J. Clin. Psychopharm. 2008, 2, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Zhang, B. Hypothesis: Neuro-inflammation, blood-brain barrier, seizures and autism. J. Neuroinflam. 2011, 8, 168. [Google Scholar] [CrossRef] [PubMed]
- Dettmer, K.; Hanna, D.; Whetstone, P.; Hansen, R.; Hammock, B.D. Autism and urinary exogenous neuropeptides: development of an on-line SPE-HPLC-tandem mass spectrometry method to test the opioid excess theory. Anal. Bioanal. Chem. 2007, 388, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Bennetto, L.; Scolding, N. Inflammatory/post-infectious encephalomyelitis. J. Neurol. Neurosurg. Psychiatry 2004, 75, i22–i28. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet. Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef]
- Gable, M.S.; Gavali, S.; Radner, A.; Tilley, D.H.; Lee, B.; Dyner, L.; Collins, A.; Dengel, A.; Dalmau, J.; Glaser, C.A. Anti-NMDA receptor encephalitis: Report of ten cases and comparison with viral encephalitis. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Oller, J.W., Jr.; Oller, S.D.; Oller, S.N. Autism: The Diagnosis, Treatment, & Etiology of the Undeniable Epidemic; Jones & Bartlett Learning: Sudbury, MA, USA, 2010. [Google Scholar]
- Vojdani, A.; Pangborn, J.B.; Vojdani, E.; Cooper, E.L. Infections, toxic chemicals and dietary peptides binding to lymphocyte receptors and tissue enzymes are major instigators of autoimmunity in autism. Int. J. Immunopath. Ph. 2003, 16, 189–199. [Google Scholar]
- Dufault, R.; Schnoll, R.; Lukiw, W.J.; LeBlanc, B.; Cornett, C.; Patrick, L.; Wallinga, D.; Gilbert, S.G.; Crider, R. Mercury exposure, nutritional deficiencies and metabolic disruptions may affect learning in children. Behav. Brain Funct. 2009, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Dufault, R.; Lukiw, W.J.; Crider, R.; Schnoll, R.; Wallinga, D.; Deth, R.A. Macroepigenetic approach to identify factors responsible for the autism epidemic in the United States. Clin. Epigenet. 2012, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, S.; Seneff, S. Impaired sulfate metabolism and epigenetics: Is there a link in autism? Entropy 2012, 14, 1953–1977. [Google Scholar] [CrossRef]
- Seneff, S.; Davidson, R.M.; Liu, J. Is cholesterol sulfate deficiency a common factor in preeclampsia, autism, and pernicious anemia? Entropy 2012, 14, 2265–2290. [Google Scholar] [CrossRef]
- Davidson, R.M.; Seneff, S. The initial common pathway of inflammation, disease, and sudden death. Entropy 2012, 14, 1399–1442. [Google Scholar] [CrossRef]
- Beckham, J.D.; Tyler, K.L. Encephalitis. In Principles and Practice of Infectious Diseases, 7th ed.; Mandell, G.L., Bennett, J.E., Dolin, R., Eds.; Elsevier Churchill Livingstone: Philadelphia, PA, USA, 2009; Chapter 87. [Google Scholar]
- Ghaziuddin, M.; Al-Khouri, I.; Ghaziuddin, N. Autistic symptoms following herpes encephalitis. Eur. Child Adolesc Psy. 2002, 11, 142–146. [Google Scholar] [CrossRef]
- Gilberg, I.C. Onset at age 14 of a typical autistic syndrome: A case report of a girl with herpes simplex encephalitis. J. Autism Dev. Disord. 1986, 16, 369–375. [Google Scholar] [CrossRef]
- Long-term cognitive and behavioral consequences of neonatal encephalopathy following perinatal asphyxia: A review. Eur J Pediatr. 2007, 166, 645–654.
- Seneff, S.; Davidson, R.; Mascitelli, L. Might cholesterol sulfate deficiency contribute to the development of autistic spectrum disorder? Med. Hypotheses 2012, 8, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Horan, F.E.; Hirsch, F.G.; Wood, L.A.; Wright, L.S. Surface effects on blood-clotting components as determined by Zeta-potentials. J. Clin. Invest. 1950, 29, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Lauritzen, A.; Davidson, R.; Lentz-Marino, L. Is endothelial nitric oxide synthase a moonlighting protein whose day job is cholesterol sulfate synthesis? Implications for cholesterol transport, diabetes and cardiovascular disease. Entropy 2012, 2492, 2492–2530. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J. Cerebr. Blood F. Met. 2012, 32, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; oude Egbrink, M.G.A. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. Eur. J. Physiol. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Chemical pathology of homocysteine V: Thioretinamide, thioretinaco, and cystathionine synthase function in degenerative diseases. Ann. Clin. Lab. Sci. 2011, 41, 300–313. [Google Scholar]
- Oja, S.S.; Saransaari, P. Taurine as osmoregulator and neuromodulator in the brain. Metab. Brain Dis. 1996, 11, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Sturman, J.A.; Hepner, G.W.; Hofmann, A.F.; Thomas, P.J. Metabolism of [35S] taurine in man. J. Nutr. 1975, 105, 1206–1214. [Google Scholar] [PubMed]
- Schoch, H.J.; Fischer, S.; Marti, H.H. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain 2002, 125, 2549–2557. [Google Scholar] [CrossRef] [PubMed]
- Skowrońska, M.; Albrecht, J. Alterations of blood brain barrier function in hyper-ammonemia: An overview. Neurotox. Res. 2012, 21, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Xaio, H.; Banks, W.A.; Niehoff, M.L.; Morley, J.E. Effect of LPS on the permeability of the blood brain barrier to insulin. Brain Res. 2001, 896, 36–42. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to open the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.D.; Egleton, R.D.; Davis, T.P. Molecular physiology and pathophysiology of tight junctions in the bloodbrain barrier. Trends Neurosci. 2001, 24, 719–725. [Google Scholar] [CrossRef]
- Misra, U.K.; Tan, C.T.; Kalita, J. Seizures in encephalitis. Neurol. Asia 2008, 13, 1–13. [Google Scholar]
- Chen, C.-A.; Wang, T.-Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.H.; Chen, Y.-R.; Druhan, J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Kern, J.K.; Garver, C.R.; Adams, J.B.; Audhya, T.; Nataf, R.; Geier, M.R. Biomarkers of environmental toxicity and susceptibility in autism. J. Neurolog. Sci. 2009, 280, 101–108. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [PubMed]
- Waring, R.H.; Klovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.-L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Downes, J.; Summanen, P.H. Microbiology of regressive autism. Anaerobe 2012, 18, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M. Desulfovibrio species are potentially important in regressive autism. Med. Hypotheses 2011, 77, 270–274. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2001, 6, 862–869. [Google Scholar] [CrossRef]
- Deth, R.; Muratore, C.; Benzecry, J.; Power-Charnitsky, V.-A.; Waly, M. How environmental and genetic factors combine to cause autism: A redox/ methylation hypothesis. Neurotoxicology 2008, 29, 190–201. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Hubanks, A.; Rose, S.; Gaylor, D.W. Abnormal transmethylation/transsulfuration metabolism and DNA hypomethylation among parents of children with autism. Autism Dev. Disord. 2008, 38, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Sweeten, T.L.; Posey, D.J.; Shankar, S.S.; McDougle, C.J. High nitric oxide production in autistic disorder: A possible role for interferon-γ. Biol. Psychiat. 2004, 55, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.-Z.; Lu, Q.; Deitch, E.A. Nitric oxide directly impairs intestinal barrier function. Shock 2002, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.; Pfeiffer, S.; Schrammel, A.; Koesling, D.; Schmidt, K.; Brunner, F. A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J. Biol. Chem. 1998, 273, 3264–3270. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and sulfated glycosaminoglycans. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 16. [Google Scholar]
- Vogl-Willis, C.A.; Edwards, I.J. High-glucose-induced structural changes in the heparan sulfate protegolycan, perlecan, of cultured human aortic endothelial cells. Biochim. Biophys. Acta 2004, 1672, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Stern, M. Insulin signaling and autism. Front. Endocrinol. 2011, 2, 54. [Google Scholar] [CrossRef] [PubMed]
- Fumitoshi Irie, F.; Hedieh Badie-Mahdavi, H.; Yamaguchi, Y. Autism-like sociocommunicative deficits and stereotypies in mice lacking heparan sulfate. Proc. Natl. Acad. Sci. USA 2012, 109, 5052–5056. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Gene. Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F. Lysosomal storage diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Bruggenwirth, H.T.; Olmer, R.; Wevers, R.A.; Verheijen, F.W.; Poorthuis, B.J.; Halley, D.J.; Wijburg, F.A. Mucopolysaccharidosis type IIIB may predominantly present with an attenuated clinical phenotype. Inherit. Metab. Dis. 2010, 33, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Mucopolysaccharidoses Fact Sheet. National Institute of Neurological Disorders and Stroke Website. Available online: http://www.ninds.nih.gov/disorders/mucopolysaccharidoses/detail/mucopolysacchari-doses.htm/ (accessed 2 April 2012).
- Gulbins, E.; Dreschers, S.; Wilker, B.; Grassme, H. Ceramide, membrane rafts and infections. J. Mol. Med. 2004, 82, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Bengheza, M.; Fauvarque, M.O.; Tournebize, R.; Froquet, R.; Marchetti, A.; Bergeret, E.; Lardy, B.; Klein, G.; Sansonetti, P.; Charette, S.J.; et al. Specific host genes required for the killing of Klebsiella bacteria by phagocytes. Cell Microbiol. 2006, 8, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Orellana, A.; Gil, G.; Hirschberg, C.B. Molecular cloning and expression of rat liver N-heparan sulfate sulfotransferase. J. Biol. Chem. 1992, 267, 15744–15750. [Google Scholar] [PubMed]
- Moseley, R.; Waddington, R.J.; Embery, G. Degradation of glycosaminoglycans by reactive oxygen species derived from stimulated polymorphonuclear leukocytes. BBA-Mol Basis. Dis. 1997, 1362, 221–231. [Google Scholar] [CrossRef]
- Ross, M.A.; Long, W.F.; Williamson, F.B. Inhibition by heparin of Fe(II)-catalysed free-radical peroxidation of linolenic acid. Biochem. J. 1992, 286, 717–720. [Google Scholar] [PubMed]
- Wakefield, A.J. The gut brain axis in childhood developmental disorders. J. Pediatr. Gastr. Nutr. 2002, 34, S14–S17. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Jayakumar, A.R.; Rama Rao, K.V.; Panickar, K.S. New concepts in the mechanism of ammonia-induced astrocyte swelling. Metab. Brain Dis. 2007, 22, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Master, S.; Gottstein, J.; Blei, A.T. Cerebral blood flow and the development of ammonia-induced brain edema in rats after portacaval anastomosis. Hepatology 1999, 30, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Panickar, K.S.; Murthy, C.R.K.; Norenberg, M.D. Oxidative stress and MAPK phosphorylation mediate ammonia-induced cell swelling and glutamate uptake inhibition in cultured astrocytes. J. Neurosci. 2006, 26, 4774–4784. [Google Scholar] [CrossRef] [PubMed]
- Oury, T.D.; Piantadosi, C.A.; Crapo, J.D. Cold-induced brain edema in mice: Involvement of extracellular superoxide dismutase and nitric oxide. J. Biol. Chem. 1993, 268, 15394–15398. [Google Scholar] [PubMed]
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. BBA-Bioenergetics 2004, 1658, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.N.; Peluffo, G.; Piacenza, L.; Radi, R. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: Consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J. Biol. Chem. 2011, 286, 6627–6640. [Google Scholar] [CrossRef] [PubMed]
- Poling, J.S.; Frye, R.E.; Shoffner, J.; Zimmerman, A.W. Developmental regression and mitochondrial dysfunction in a child with autism. J. Child Neurol. 2006, 21, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Good, P. Do salt cravings in children with autistic disorders reveal low blood sodium depleting brain taurine and glutamine? Med. Hypotheses 2011, 77, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Lia, X.; Chauhana, A.; Sheikha, A.M.; Patilb, S.; Chauhana, V.; Lib, X.-M.; Jia, L.; Browna, T.; Malika, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 201, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, J.T.; Man, K.-C.; Hall, D.E.; Colbourne, S.A.; Brosnan, M.E. Interorgan metabolism of amino acids in the streptozotocin-diabetic ketoacidotic rat. Am. J. Physiol. 1983, 244, E151–E158. [Google Scholar] [PubMed]
- Brosnan, J.T. Glutamate, at the interface between amino acid and carbohydrate metabolism. In Proc. International Symposium on Glutamate, Bergamo, Italy, 12–14 October 1998.
- Daikhin, Y.; Yudkoff, M. Compartmentation of brain glutamate metabolism in neurons and glia. In Proc. International Symposium on Glutamate, Bergamo, Italy, 12–14 October 1998.
- Schraufstitter, I.U.; Browne, K.; Harris, A.; Hyslop, P.A.; Jackson, J.H.; Quehenberger, O.; Cochrane, C.G. Mechanisms of hypochlorite injury of target cells. J. Clin. Invest. 1990, 85, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, G.R.; Tyson, R.L.; Auer, R.N. Truncation of the Krebs cycle during hypoglycemic coma. Med. Chem. 2008, 4, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Massieu, L.; Montiel, T.; Robles, G.; Quesada, O. Brain amino acids during hyponatremia in vivo: Clinical observations and experimental studies. Neurochem. Res. 2004, 29, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.H.; Andersen, M.L.; Cornett, C.; Gradinaru, R.; Grunnet, N. A role for taurine in mitochondrial function. J. Biomed. Sci. 2010, 17, S23–530. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.; Bender, A.S.; Norenberg, M.D. Ammonia stimulates the release of taurine from cultured astrocytes. Brain Res. 1994, 660, 228–232. [Google Scholar] [CrossRef]
- Albrecht, J. Roles of neuroactive amino acids in ammonia neurotoxicity. J. Neurosci. Res. 1993, 51, 133–138. [Google Scholar] [CrossRef]
- Foran, E.; Trotti, D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid. Redox Sign. 2009, 11, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate receptors and the induction of excitotoxic neuronal death. Prog. Brain Res. 1994, 100, 47–51. [Google Scholar] [PubMed]
- Blaylock, R.L.; Strunecka, A. Immune-glutamatergic dysfunction as a central mechanism of the autism spectrum disorders. Curr. Med. Chem. 2009, 16, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Monfort, P.; Muñoz, M.-D.; El Ayadi, A.; Kosenko, E.; Felipo, V. Effects of hyperammonemia and liver failure on glutamatergic neurotransmission. Metab. Brain Dis. 2002, 17, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.D.; Monfort, P.; Gaztelu, J.M.; Felipo, V. Hyperammonemia impairs NMDA receptor-dependent long-term potentiation in the CA1 of rat hippocampus in vitro. Neurochem. Res. 2000, 25, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.A.; Miñarro, J.; Felipo, V. Chronic moderate hyperammonemia impairs active and passive avoidance behavior and conditional discrimination learning in rats. Exp. Neurol. 2000, 161, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Romero-Vives, M.; Barrenechea, C.; Insausti, R.; Felipo, V.; Gaztelu, J.M. Sleep alterations in hepatic encephalopathy could be due to chronic hyperammonemia. J. Sleep Res. 1998, 7, 228. [Google Scholar]
- Creten, C.; van der Zwaan, S.; Blankenspoor, R.J.; Maatkamp, A.; van Os, J.; Schieveld, J.N.M. Late onset autism and anti-NMDA-receptor encephalitis. Lancet 2011, 378, 98–99. [Google Scholar] [CrossRef]
- Rasalam, A.D.; Hailey, H.; Williams, J.H.G.; Moore, S.J.; Turnpenny, P.D.; MooDeanre, J.C.S. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev. Med. Child Neurol. 2005, 47, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; King, J.; Cunningham, M.; Stephan, M.; Kerr, B.; Hersh, J.H. Fetal valproate syndrome and autism: additional evidence of an association. Dev. Med. Child Neurol. 2001, 43, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Wadzinski, J.; Franks, R.; Roane, D.; Bayard, M. Valproate-associated hyperammonemic encephalopathy. JABFM 2007, 20, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Yehya, N.; Saldarini, C.T.; Koski, M.E.; Davanzo, P. Valproate-induced hyperammonemic encephalopathy. Metab. Brain Dis. 2004, 43, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, T.; Kulangara, K.; Antoniello, K.; Markram, H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. USA 2007, 104, 13501–13506. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Kettle, A.J.; Winterbourn, C.C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase and bacterial killing. Blood 1998, 92, 3007–3017. [Google Scholar] [PubMed]
- Schuller-Levis, G.B.; Park, E. Taurine and Its Chloramine: Modulators of Immunity. Neurochem. Res. 2004, 29, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Olszowski, S.; Olszowska, E.; Kusior, D.; Szneler, E. Sulphoacetaldehyde as a product of taurine chloramine peroxidation at site of inflammation. Amino Acids 2002, 22, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Tipton, K.F.; Dixon, H.B.F. Conversion of taurine into N-chlorotaurine (taurine chloramine) and sulphoacetaldehyde in response to oxidative stress. Biochem. J. 1998, 330, 939–945. [Google Scholar] [PubMed]
- Ruff, J.; Denger, K.; Cook, A.M. Sulphoacetaldehyde acetyltransferase yields acid phosphate: Purification from Alcaligenes defragrans and gene clusters in taurine degradation. Biochem. J. 2003, 369, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S. The multifaceted and widespread pathology of magnesium deficiency. Med. Hypotheses 2001, 56, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Strambi, M.; Longini, M.; Hayek, J.; Berni, S.; Macucci, F.; Scalacci, E.; Vezzosi, P. Magnesium profile in autism. Biol. Trace Elem. Res. 2006, 109, 97–104. [Google Scholar] [CrossRef]
- Mousain-Bosc, M.; Roche, M.; Polge, A.; Pradal-Prat, D.; Rapin, J.; Bali, J.P. Improvement of neurobehavioural disorders in children supplemented with magnesium-B6. Magnesium Res. 2006, 19, 53–62. [Google Scholar]
- Hallak, M.; Berman, R.F.; Irtenkauf, S.M.; Evans, M.I.; Cotton, D.B. Peripheral magnesium sulfate enters the brain and increases the threshold for hippocampal seizures in rats. Am. J. Obstet. Gynecol. 1992, 167, 1605–1610. [Google Scholar] [CrossRef]
- Cotton, D.B.; Janusz, C.A.; Berman, R.F. Anticonvulsant effects of magnesium sulfate on hippocampal seizures: Therapeutic implications in preeclampsia-eclampsia. Am. J. Obstet. Gynecol. 1992, 166, 1127–1134. [Google Scholar] [CrossRef]
- Findling, R.L.; Maxwell, K.; Scotese-Wojtila, L.; Huang, J.; Yamashita, T.; Wiznitzer, M. High-dose pyridoxine and magnesium administration in children with autistic disorder: An absence of salutary effects in a double-blind, placebo-controlled study. J. Autism Dev. Disord. 1997, 27, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Lelong, E.; Marchetti, A.; Gueho, A.; Lima, W.C.; Sattler, N.; Molmeret, M.; Hagedorn, M.; Soldati, T.; Cosson, P. Role of magnesium and a phagosomal P-type ATPase in intracellular bacterial killing. Cell. Microbiol. 2011, 13, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.T.; Komarov, A.M.; Wagner, T.L.; Stafford, R.E.; Dickens, B.F.; Weglicki, W.B. Enhanced NO production during Mg deficiency and its role in mediating red blood cell glutathione loss. Am. J. Physiol. 1996, 271, C385–C390. [Google Scholar] [PubMed]
- Chai, B.-H.; Zheng, J.-M.; Zhao, Q.; Pollack, G.H. Spectroscopic studies of solutes in aqueous solution. J. Phys. Chem. A 2008, 112, 2242–2247. [Google Scholar] [CrossRef] [PubMed]
- Pollack, G.H.; Figueroa, X.; Zhao, Q. Review: Molecules, water, and radiant energy: New clues for the origin of life. Int. J. Mol. Sci. 2009, 10, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Chai, B.; Yoo, H.; Pollack, G.H. Effect of Radiant Energy on Near-Surface Water. J. Phys. Chem. B 2009, 113, 13953–13958. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, E.; Spinetti, P.R.; Tedeschi, A. Water dynamics at the root of metamorphosis in living organisms. Water 2010, 2, 566–586. [Google Scholar] [CrossRef]
- Zheng, J.-M.; Chin, W.-C.; Khijniak, E.; Khijniak, E., Jr.; Pollack, G.H. Surfaces and interfacial water: Evidence that hydrophilic surfaces have long-range impact. Adv. Colloid Interface Sci. 2006, 127, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Guckenberger, R.; Heim, M.; Cevc, G.; Knapp, H.F.; Wiegrbe, W.; Hillebrand, A. Scanning tunneling microscopy of insulators and biological specimens based on lateral conductivity of ultrathin water films. Science 1994, 266, 1538–1540. [Google Scholar] [CrossRef] [PubMed]
- Markovitch, O.; Chen, H.; Izvekov, S.; Paesani, S.; Voth, G.A.; Agmon, N. Special Pair Dance and Partner Selection: Elementary Steps in Proton Transport in Liquid Water. J. Phys. Chem. B 2008, 112, 9456–9466. [Google Scholar] [CrossRef] [PubMed]
- Verdel, N.; Jerman, I.; Bukovec, P. The autothixotropic phenomenon of water and its role in proton transfer. Int. J. Mol. Sci. 2011, 12, 7481–7494. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Schulz, R.; Rassaf, T.; Lauer, T.; Dejam, A.; Jax, T.; Kumara, I.; Gharini, P.; Kabanova, S.; zuyaman, B.O.; et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood 2006, 107, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Raman, C.S.; Glaser, C.B.; Blasko, E.; Young, A.; Parkinson, J.F.; Whitlow, M.; Poulos, T.L. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J. Biol. Chem. 1999, 274, 21276–21284. [Google Scholar] [CrossRef] [PubMed]
- Faber, S.; Zinn, G.M.; Kern, J.C., II; Kingston, H.M.S. The plasma zinc/serum copper ratio as a biomarker in children with autism spectrum disorders. Biomarkers 2009, 14, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Rohwerder, T.; Sand, W. The sulfane sulfur of persulfides is the actual substrate of the sulfur-oxidizing enzymes from Acidithiobacillus and Acidiphilium spp. Microbiology 2003, 149, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Gratton, J.-P.; Fontana, J.; O’Connor, D.S.; García-Cardeña, G.; McCabe, T.J.; Sessa, W.C. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. JBC 2000, 275, 22268–22272. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Gratton, J.-P.; Sessa, W.C. Post-translational control of endothelial nitric oxide synthase: Why isn’t calcium/calmodulin enough? J. Pharmacol. Exp. Ther. 2001, 299, 818–824. [Google Scholar] [PubMed]
- Muñoz, S.; Sebastían, J.L.; Sancho, M.; Martínez, G. Analysis of radiofrequency energy stored in the altered shapes: Stomatocyteechinocyte of human erythrocytes. Bioelectrochemistry 2010, 77, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Helt, M.; Kelley, E.; Kinsbourne, M.; Pandey, J.; Boorstein, H.; Herbert, M.; Fein, D. Can children with autism recover? If so, how? Neuropsych. Rev. 2008, 18, 339–366. [Google Scholar] [CrossRef] [PubMed]
- Frosini, M. Changes in CSF composition during heat stress and fever in conscious rabbits. Prog. Brain Res. 2007, 162, 449–457. [Google Scholar] [PubMed]
- Fukushima, M.; Mohri, K.; Kataoka, T.; Matsumoto, M. Milli gauss pulsed magnetic field applied phosphate buffered saline solution elevates intracellular Ca2+ level and stimulates phagocytic activity of human neutrophils. Trans. Magn. Soc. Jpn. 2002, 2, 15–18. [Google Scholar] [CrossRef]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; White, H.S. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef]
- Fukushima, M.; Kataoka, T.; Sugiyama, N.; Mohri, K. Milligauss Magnetic Field Applied Pure Water Exert Firefly Luciferin-Luciferase Luminescence and Induce Intracellular Calcium Elevation of CHO Cells Without ATP. IEEE Trans. Magn. 2005, 41, 4188–4190. [Google Scholar] [CrossRef]
- Kovács, R.; Rabanus, A.; Otáhal; Patzak, A.; Kardos, J.; Albus, K.; Heinemann, U.; Kann, O. Endogenous nitric oxide is a key promoting factor for initiation of seizure-like events in hippocampal and entorhinal cortex slices. J. Neurosci. 2009, 29, 8565–8577. [Google Scholar] [CrossRef] [PubMed]
- Fukahori, M.; Itoh, M. Effects of dietary zinc status on seizure susceptibility and hippocampal zinc content in the El (epilepsy) mouse. Brain Res. 1990, 529, 16–22. [Google Scholar] [CrossRef]
- Noseworthy, M.D.; Bray, T.M. Zinc deficiency exacerbates loss in blood-brain barrier integrity induced by hyperoxia measured by dynamic MRI. Proc. Soc. Exp. Biol. Med. 2000, 223, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Rink, L.; Gabriel, P. Extracellular and immunological actions of zinc. BioMetals 2001, 14, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.H.; MacDonald, T.T.; Walker-Smith, J.A.; Levin, M.; Lionetti, P.; Klein, N.J. Disruption of sulphated glycosaminoglycans in intestinal inflammation. Lancet 1993, 341, 711–714. [Google Scholar] [CrossRef]
- Clayton, T.A. Metabolic differences underlying two distinct rat urinary phenotypes, a suggested role for gut microbial metabolism of phenylalanine and a possible connection to autism. FEBS Lett. 2012, 586, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.M.; Weaver, L.M. The shikimate pathway. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 473–503. [Google Scholar] [CrossRef] [PubMed]
- Michalowicz, J.; Duda, W. Phenols sources and toxicity. Polish J. Environ. Stud. 2007, 16, 347–362. [Google Scholar]
- Shehata, A.A.; Schrödl, W.; Aldin, A.A.; Hafez, H.M.; Kruger, M. The effect of glyphosate on potential pathogens and beneficial members of poultry microbiota in vitro. Curr. Microbiol 2012, in press. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C. Effects of Clostridium difficile toxins on epithelial cell barrier. Ann. New York Acad. Sci. 2000, 915, 347–356. [Google Scholar] [CrossRef]
- Clayton, T.A.; Baker, D.; Lindon, J.C.; Everett, J.R.; Nicholson, J.K. Pharmacometabonomic identification of a significant hostmicrobiome metabolic interaction affecting human drug metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 14728–14733. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, R.E. Effects of glyphosate on metabolism of phenolic compounds: VI. Effects of glyphosine and glyphosate metabolites on phenylalanine ammonia-lyase activity, growth, and protein, chlorophyll, and anthocyanin levels in soybean (Glycine max) seedlings. Weed Sci. 1980, 28, 393–400. [Google Scholar]
- Duke, S.O.; Hoagland, R.E. Effects of glyphosate on metabolism of phenolic compounds I. Induction of phenylalanine ammonia-lyase activity in dark-grown maize roots. Plant Sci. Lett. 1978, 11, 185–190. [Google Scholar] [CrossRef]
- Hoyumpa, A.M., Jr. Mechanisms of thiamin deficiency in chronic alcoholism. Am. J. Clin. Nutr. 1980, 33, 2750–2761. [Google Scholar] [PubMed]
- Fattal-Valevski, A.; Bloch-Mimouni, A.; Kivity, S.; Heyman, E.; Brezner, A.; Strausberg, R.; Inbar, D.; Kramer, U.; Goldberg-Stern, H. Epilepsy in children with infantile thiamine deficiency. Neurology 2009, 73, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Yamakura, S.; Hirano, K.; Okumura, Y.; Aiba, H. Case of infantile autism with pediatric Wernicke’s encephalopathy due to severe eating disorder (in Japanese). No To Hattatsu. 2009, 41, 43–46. [Google Scholar] [PubMed]
- Mattioli, S.; Miglioli, M.; Montagna, P.; Lerro, M.F.; Pilotti, V.; Gozzetti, G. Wernicke’s encephalopathy during total parenteral nutrition: observation in one case. J. Parenter. Enteral. Nutr. 1988, 12, 626–627. [Google Scholar] [CrossRef]
- Moore, J.K.; Braymer, H.D.; Larson, A.D. Isolation of a Pseudomonas sp. which utilizes the phosphonate herbicide glyphosate. Appl. Environ. Microbiol. 1983, 46, 316–320. [Google Scholar] [PubMed]
- Siegel, N.; Haug, A. Aluminum interaction with calmodulin. Evidence for altered structure and function from optical and enzymatic studies. Biochim. Biophys. Acta 1983, 744, 36–45. [Google Scholar] [CrossRef]
- Tomljenovic, L.; Christopher A. Shaw, C.A. Do aluminum vaccine adjuvants contribute to the rising prevalence of autism? J. Inorg. Biochem. 2011, 105, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Blaylock, R.L. Aluminum induced immunoexcitotoxicity in neurodevelopmental and neurodegenerative disorders. Curr. Inorg. Chem. 2012, 2, 46–53. [Google Scholar] [CrossRef]
- Johnson, V.J.; Sharma, R.P. Aluminum disrupts the proinflammatory cytokine/neurotrophin balance in primary brain rotation-mediated aggregate cultures: Possible role in neurodegeneration. Neurotoxicology 2003, 24, 261–268. [Google Scholar] [CrossRef]
- Nayak, P.; Chatterjee, A.K. Effects of aluminum exposure on brain glutamate and GABA systems: An experimental study in rats. Food Chem. Toxicol. 2001, 39, 1285–1289. [Google Scholar] [CrossRef]
- Tsunoda, M.; Sharma, R.P. Modulation of tumor necrosis factor alpha expression in mouse brain after exposure to aluminum in drinking water. Arch. Toxicol. 1999, 73, 419–426. [Google Scholar] [CrossRef] [PubMed]
- El-Rahman, S.S. Neuropathology of aluminum toxicity in rats (glutamate and GABA impairment). Pharmacol. Res. 2003, 47, 189–194. [Google Scholar] [CrossRef]
- Bondy, S.C. The neurotoxicity of environmental aluminum is still an issue. Neurotoxicology 2010, 31, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.; Becaria, A.; Lahiri, D.K.; Sharman, K.; Bondy, S.C. Chronic exposure to aluminum in drinking water increases inflammatory parameters selectively in the brain. J. Neurosci. Res. 2004, 75, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Mundy, W.R.; Freudenrich, T.M.; Kodavanti, P.R.S. Aluminum potentiates glutamate-induced calcium accumulation and iron-induced oxygen free radical formation in primary neuronal cultures. Mol. Chem. Neuropathol. 1997, 32, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Oteiza, P.I. A mechanism for the stimulatory effect of aluminum on iron-induced lipid peroxidation. Arch. Biochem. Biophys. 1994, 308, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Davidson, R.M.; Liu, J. Empirical Data Confirm Autism Symptoms Related to Aluminum and Acetaminophen Exposure. Entropy 2012, 14, 2227–2253. [Google Scholar] [CrossRef]
- Schubert, J.; Riley, E.J.; Tyler, S.A. Combined effects in toxicology--a rapid systematic testing procedure: cadmium, mercury, and lead. J. Toxicol. Environ. Health 1978, 4, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Heeney, G.M.; Woolf, M.M. Encephalopathy from lead poisoning masquerading as a flu-like syndrome in an autistic child. Pediatr. Emerg. Care 2010, 26, 370–373. [Google Scholar]
- Mushak, P.; Davis, J.M.; Crocetti, A.F.; Grant, L.D. Prenatal and postnatal effects of low-level lead exposure: Integrated summary of a report to the U.S. congress on childhood lead poisoning. Environ. Res. 1989, 50, 11–36. [Google Scholar] [CrossRef]
- Sharifi, A.M.; Baniasadi, S.; Jorjani, M.; Rahimi, F.; Bakhshayesh, M. Investigation of acute lead poisoning on apoptosis in rat hippocampus. Neurosci. Lett. 2002, 329, 45–48. [Google Scholar] [CrossRef]
- Björck, I.; Granfeldt, Y.; Liljeberg, H.; Tovar, J.; Asp, N.-G. Food properties affecting the of carbohydrates, digestion and absorption. Am. J. Clin. Nutr. 1994, 59, 699S–705S. [Google Scholar] [PubMed]
- Kaya, M.; Küçük, M.; Bulut Kalayci, R.; Cimen, V.; Gürses, C.; Elmas, I.; Arican, N. Magnesium sulfate attenuates increased blood-brain barrier permeability during insulin-induced hypoglycemia in rats. Can. J. Physiol. Pharm. 2001, 79, 793–798. [Google Scholar] [CrossRef]
- Bindra, G.S.; Gibson, R.S.; Thompson, L.U. [Phytate][calcium]/[zinc] ratios in Asian immigrant lacto-ovo vegetarian diets and their relationship to zinc nutriture. Nutr. Res. 1986, 6, 475–483. [Google Scholar] [CrossRef]
- Famularo, G.; De Simone, C.; Pandey, V.; Sahu, A.R.; Minisola, G. Probiotic lactobacilli: an innovative tool to correct the malabsorption syndrome of vegetarians? Med. Hypotheses 2005, 65, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Saes Zobiole, L.H.; de Oliveira, R.S., Jr.; Huber, D.M.; Constantin, J.; de Castro, C.; de Oliveira, F.A.; de Oliveira, A., Jr. Glyphosate reduces shoot concentrations of mineral nutrients in glyphosate-resistant soybeans. Plant Soil 2010, 328, 57–69. [Google Scholar] [CrossRef]
- Le Bougu ́enec, C.; Schouler, C. Sugar metabolism, an additional virulence factor in enterobacteria. Int. J. Med. Microbiol. 2011, 301, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Palková, Z.; Vachova, L. Ammonia signaling in yeast colony formation. Int. Rev. Cytol. 2003, 225, 229–272. [Google Scholar] [PubMed]
- Nlu, M.; Kupletskaia, M.B.; Bab'eva, I.P.; Egorov, N.S. Phenylalanine ammonia-lyase of pigmented yeasts (in Russian). Mikrobiologiia 1980, 49, 269–273. [Google Scholar]
- Burrus, C.J. A biochemical rationale for the interaction between gastrointestinal yeast and autism. Med. Hypotheses 2012, 79, 784–785. [Google Scholar] [CrossRef] [PubMed]
- Jyonouchi, H.; Geng, L.; Ruby, A.; Reddy, C.; Zimmerman-Bier, B. Evaluation of an association between gastrointestinal symptoms and cytokine production against common dietary proteins in children with autism spectrum disorders. J. Pediatr. 2005, 146, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Wills, S.; Van de Water, J. The immune response in autism: A new frontier for autism research. J. Leukoc. Biol. 2006, 80, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; van de Water, J.; Paul Ashwood, P. Immune dysfunction in autism: A pathway to treatment. Neurotherapeutics 2010, 7, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Macfarlane, G.T. Role of intestinal bacteria in nutrient metabolism. Clin. Nutr. 1997, 16, 3–11. [Google Scholar] [CrossRef]
- Nicolson, G.L.; Gan, R.; Nicolson, N.L.; Haier, J. Evidence for Mycoplasma ssp., Chlamydia pneunomiae, and human Herpes Virus-6 coinfections in the blood of patients with autistic spectrum disorders. J. Neurosci. Res. 2007, 85, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen-Lathrop, S.J.; Koshiyama, K.; Phillips, N.; Stephens, R.S. Chlamydia-dependent biosynthesis of a heparan sulphate-like compound in eukaryotic cells. Cell. Microbiol. 2000, 2, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.; Bour, J.B.; Lidholt, K.; Gauthray, C.; Pothier, P. Heparin-like structures on respiratory syncytial virus are involved in its infectivity in vitro. J. Virol. 1998, 72, 7221–7227. [Google Scholar] [PubMed]
- Eichhorn, E.; van der Ploeg, J.R.; Kertesz, M.A.; Leisinger, T. Characterization of α-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J. Biol. Chem. 1997, 272, 23031–23036. [Google Scholar] [CrossRef] [PubMed]
- Van der Ploeg, J.R.; Eichhorn, E.; Leisinger, T. Sulfonate-sulfur metabolism and its regulation in Escherichia coli. Arch Microbiol. 2001, 176, 1–8. [Google Scholar] [CrossRef] [PubMed]
- McCusker, K.P.; Klinman, J.P. Facile synthesis of 1,1-[2h2]-2-methylaminoethane-1-sulfonic acid as a substrate for taurine α-ketoglutarate dioxygenase (TauD). Tetrahedron Lett. 2009, 50, 611–613. [Google Scholar] [CrossRef]
- Westergaard, N.; Sonnewald, U.; Schousboea, A. Release of -ketoglutarate, malate and succinate from cultured astrocytes: Possible role in amino acid neurotransmitter homeostasis. Neurosci. Lett. 1994, 176, e105–e109. [Google Scholar] [CrossRef]
- Cook, E.H., Jr.; Leventhal, B.L. The serotonin system in autism. Curr. Opin. Pediatr. 1996, 8, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Gutknecht, L.; Carlier, M.; Spitz, E.; Antoine, C.; Slama, F.; Carsalade, V.; Cohen, D.J.; Ferrari, P.; Roubertoux, P.L.; et al. Role of the serotonin transporter gene in the behavioral expression of autism. Mol. Psychiatr. 2001, 6, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Richerson, G.B. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat. Rev. Neurosci. 2004, 5, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Montagnier, L.; Jamal Aïssa, J.; Ferris, S.; Montagnier, J.-L.; Lavallée, C. Electromagnetic Signals Are Produced by Aqueous Nanostructures Derived from Bacterial DNA Sequences. Interdiscip. Sci. Comput. Life Sci. 2009, 1, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Janssen, B.; Buys, E.; Sips, P.; Brouckaert, P. Anaphylactic shock depends on PI3K and eNOS-derived NO. J. Clin. Invest. 2006, 116, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. Stress and disease. The Laryngoscope 1955, 65, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K.; Goderie, S.K.; Higman, S.; Pang, S.; Waniewski, R.A. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J. Neurosci. 1990, 10, 1583–1591. [Google Scholar] [PubMed]
- Viorritto, I.C.B.; Nikolov, N.P.; Siegel, R.M. Autoimmunity versus tolerance: Can dying cells tip the balance? Clin. Immunol. 2007, 122, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Westall, F.C. Molecular mimicry revisited: Gut bacteria and multiple sclerosis. J. Clin. Microbiol. 2006, 44, 2099–2104. [Google Scholar] [CrossRef] [PubMed]
- Wills, S.; Cabanlit, M.; Bennett, J.; Ashwood, P.; Amaral, D.G.; van de Water, J. Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain Behav. Immun. 2009, 23, 64–74. [Google Scholar] [CrossRef] [PubMed]
- McEntee, W.J.; Crook, T.H. Glutamate: Its role in learning, memory, and the aging brain. Psychopharmacol 1993, 111, 391–401. [Google Scholar] [CrossRef]
- Meló, T.M.; Sonnewald, U.; Touret, M.; Nehlig, A. Cortical glutamate metabolism is enhanced in a genetic model of absence epilepsy. J. Cererbr. Blood F. Met. 2006, 26, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Farrell, R.; Ciaran, P.K. Celiac Sprue. N. Engl. J. Med. 2002, 346, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.; Waring, R.H. The plasma cysteine/sulphate ratio: A possible clinical biomarker. J. Nutr. Environ. Med. 2003, 13, 215–229. [Google Scholar] [CrossRef]
- Heafield, M.T.; Fearn, S.; Steventon, G.B.; Waring, R.H.; Williams, A.C.; Sturman, S.G. Plasma cysteine and sulphate levels in patients with motor neurone, Parkinson’s and Alzheimer’s disease. Neurosci. Lett. 1990, 110, 216–220. [Google Scholar] [CrossRef]
- Ming, X.; Stein, P.; Barnes, V.; Rhodes, N.; Guo, L. Metabolic perturbance in autism spectrum disorders: A metabolomics study. J. Proteome Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.; Emerton, J.; Bhadravathi, V.; Weisblatt, E.; Pasco, G.; Willatt, L.R.; McMahon, R.; Yates, J.R. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J. Med. Genet. 2008, 45, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, Y.P.; Sheng, M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J. Neurosci. 1999, 19, 7415–7425. [Google Scholar] [PubMed]
- Ethell, I.M.; Yamaguchi, Y. Cell surface heparan sulfate proteoglycan syndecan-2 induces the maturation of dendritic spines in rat hippocampal neurons. J. Cell. Biol. 1999, 144, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Lauri, S.E.; Kaukinen, S.; Kinnunen, T.; Ylinen, A.; Imai, S.; Kaila, K.; Taira, T.; Rauvala, H. Regulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J. Neurosci. 1999, 19, 1226–1235. [Google Scholar] [PubMed]
- DeLong, G.R. Autism, amnesia, hippocampus, and learning. Neurosci. Biobehav. R. 1992, 16, 63–70. [Google Scholar] [CrossRef]
- Bolton, P.J.; Carcani-Rathwell, I.; Hutton, J.; Sue Goode, S.; Howlin, P.; Rutter, M. Epilepsy in autism: Features and correlates. BJP 2011, 198, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Gabisa, L.; Pomeroyb, J.; Andriolac, M.R. Autism and epilepsy: Cause, consequence, comorbidity, or coincidence? Epilepsy Behav. Dec. 2005, 7, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blmcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Shinohea, A.; Hashimotob, K.; Nakamuraa, K.; Tsujiic, M.; Iwataa, Y.; Tsuchiyaa, K.J.; Sekinea, Y.; Sudaa, S.; Suzukia, K.; Sugiharaa, G.I.; et al. Increased serum levels of glutamate in adult patients with autism. Prog. Neuro. Psychoph. 2006, 30, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pellegrino, C.; Rama, S.; Dumalska, I.; Salyha, Y.; Ben-Ari, Y.; Medina, I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol. 2006, 572, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, EÉ.; Millet, B.; S2D team; et al. Rare mutations in N-methyl-D-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.H.; Shankar, M.; Shuster, J.; Theriaque, D.; Burns, S.; Sherrill, L. The gluten-free, casein-free diet in autism: Results of a preliminary double blind clinical trial. J. Autism Dev. Disord. 2006, 36, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, D.; Shamberger, R.J.; Audhya, T. Treatment of autism spectrum children with thiamine tetrahydrofurfuryl disulfide: a pilot study. Neuro. Endocrinol. Lett. 2002, 23, 303–308. [Google Scholar] [PubMed]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; Barnhouse, S.; Lee, W. Effect of a vitamin/mineral supplement on children and adults with autism. BMC Pediatr. 2011, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Aneja, A.; Tierney, E. Autism: The role of cholesterol in treatment. Int. Rev. Psychiatr. 2008, 20, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Bukelis, I.; Porter, F.D.; Zimmerman, A.W.; Tierney, E. Smith-Lemli-Opitz syndrome and autism spectrum disorder. Am. J. Psychiatry 2007, 164, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Sikora, D.M.; Pettit-Kekel, K.; Penfield, J.; Merkens, L.S.; Steiner, R.D. The near universal presence of autism spectrum disorders in children with Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. Part A 2006, 140A, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Davis, W. Wheat Belly: Lose the Wheat, Lose the Weight, and Find Your Path Back to Health; Rodale Books: Emmaus, PA, USA, 2011. [Google Scholar]
- Grant, W.B.; Cannell, J.J. Autism prevalence in the United States with respect to solar UV-B doses: An ecological study. Dermatoendocrinology 2012, 4. in press. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Huang, E.S.; Ananthakrishnan, A.N.; Higuchi, L.; Richter, J.M.; Fuchs, C.S.; Chan, A.T. Geographical variation and incidence of inflammatory bowel disease among us women. Gut 2012, 61, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Tomljenovic, L; Shaw, C.A. Aluminum vaccine adjuvants: Are they safe? Curr. Med. Chem. 2011, 18, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Seneff, S.; Lauritzen, A.; Davidson, R.M.; Lentz-Marino, L. Is Encephalopathy a Mechanism to Renew Sulfate in Autism? Entropy 2013, 15, 372-406. https://doi.org/10.3390/e15010372
Seneff S, Lauritzen A, Davidson RM, Lentz-Marino L. Is Encephalopathy a Mechanism to Renew Sulfate in Autism? Entropy. 2013; 15(1):372-406. https://doi.org/10.3390/e15010372
Chicago/Turabian StyleSeneff, Stephanie, Ann Lauritzen, Robert M. Davidson, and Laurie Lentz-Marino. 2013. "Is Encephalopathy a Mechanism to Renew Sulfate in Autism?" Entropy 15, no. 1: 372-406. https://doi.org/10.3390/e15010372
APA StyleSeneff, S., Lauritzen, A., Davidson, R. M., & Lentz-Marino, L. (2013). Is Encephalopathy a Mechanism to Renew Sulfate in Autism? Entropy, 15(1), 372-406. https://doi.org/10.3390/e15010372