
Recognize Yourself—Innate Sensing of Non-LTR Retrotransposons

Abstract
1. Introduction
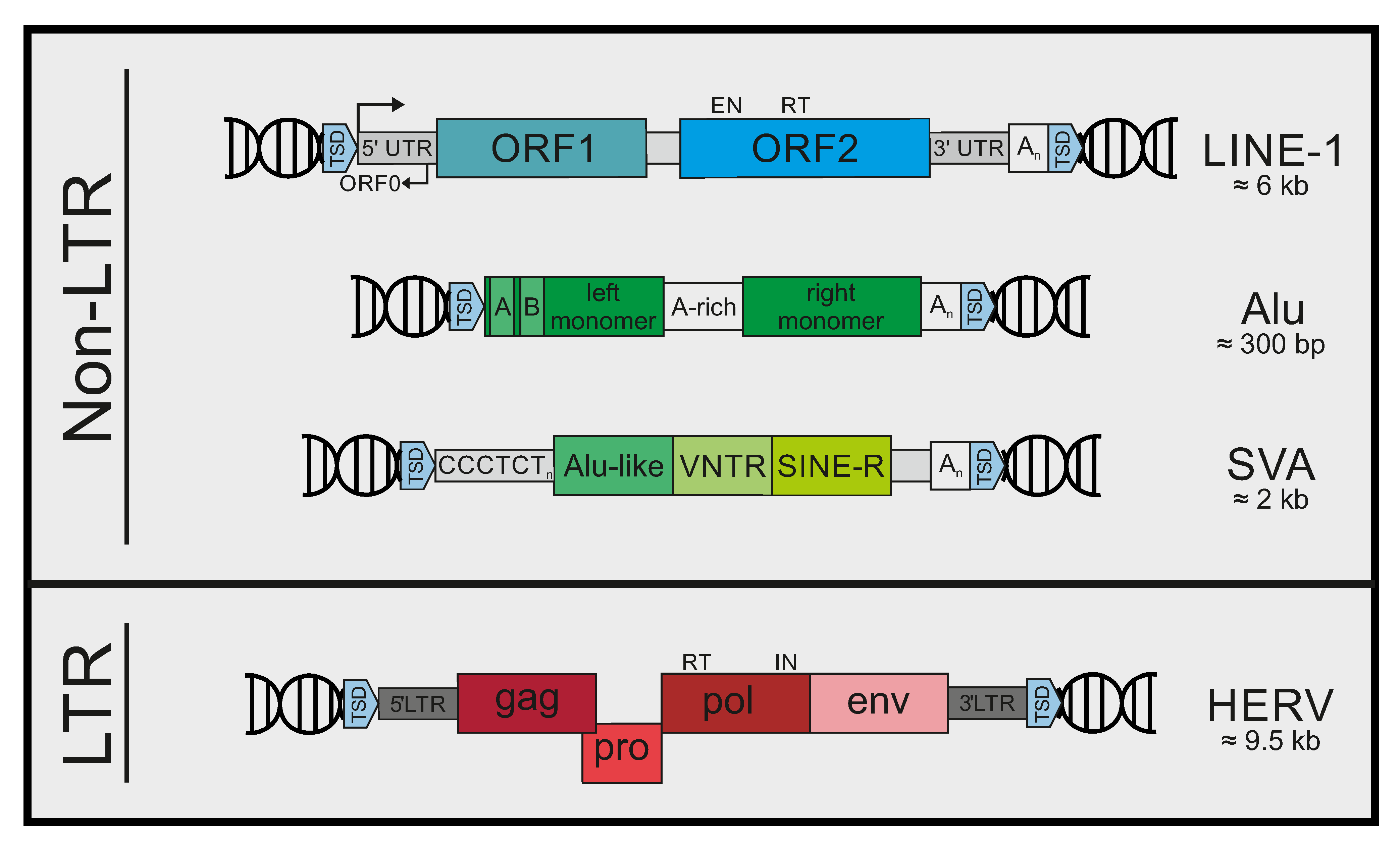
2. Mobile Genetic Elements in Humans
3. Controlling Mobile Elements
4. Non-LTR Retrotransposons in Inflammatory Diseases
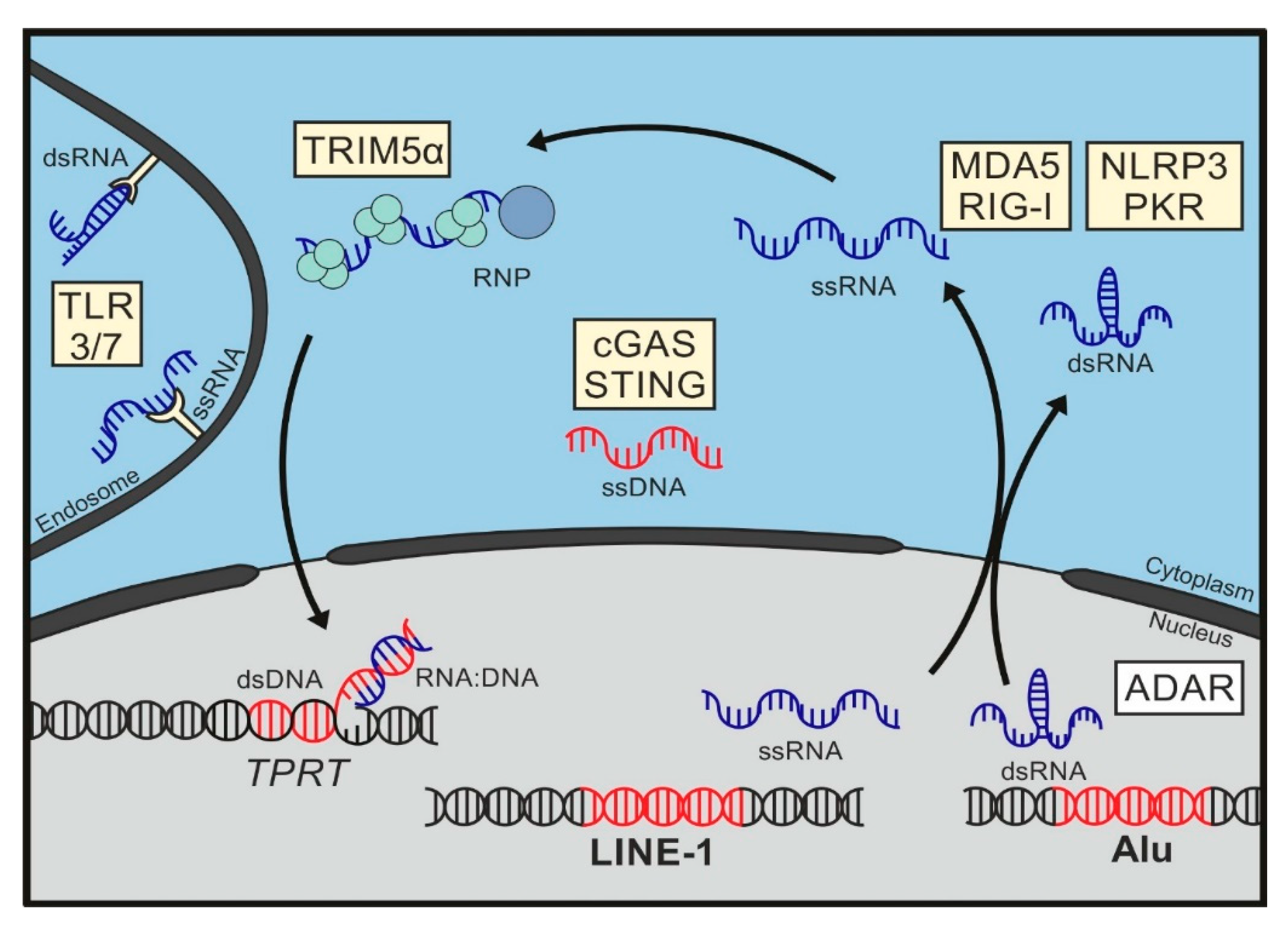
5. Sensing of Mobile Genetic Elements
6. Pattern Recognition Receptors
7. Immunogenic Patterns in LINE-1
7.1. LINE-1 DNA
7.2. LINE RNA
7.3. LINE-1 RNA/DNA Hybrids
7.4. LINE-1 RNPs
8. Immunogenic Patterns in Alu Elements
9. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.R.; Burns, K.H.; Boeke, J.D. Active transposition in genomes. Annu. Rev. Genet. 2012, 46, 651–675. [Google Scholar] [CrossRef] [PubMed]
- Enriquez-Gasca, R.; Gould, P.A.; Rowe, H.M. Host Gene Regulation by Transposable Elements: The New, the Old and the Ugly. Viruses 2020, 12, 1089. [Google Scholar] [CrossRef]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef]
- Payer, L.M.; Burns, K.H. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019, 20, 760–772. [Google Scholar] [CrossRef]
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genom. Hum. Genet. 2006, 7, 149–173. [Google Scholar] [CrossRef]
- Martin, M.A.; Bryan, T.; Rasheed, S.; Khan, A.S. Identification and cloning of endogenous retroviral sequences present in human DNA. Proc. Natl. Acad. Sci. USA 1981, 78, 4892–4896. [Google Scholar] [CrossRef]
- Smit, A.F. Identification of a new, abundant superfamily of mammalian LTR-transposons. Nucleic Acids Res. 1993, 21, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Feschotte, C. Co-option of endogenous viral sequences for host cell function. Curr. Opin. Virol. 2017, 25, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Curty, G.; Marston, J.L.; de Mulder Rougvie, M.; Leal, F.E.; Nixon, D.F.; Soares, M.A. Human Endogenous Retrovirus K in Cancer: A Potential Biomarker and Immunotherapeutic Target. Viruses 2020, 12, 726. [Google Scholar] [CrossRef] [PubMed]
- Lezhnyova, V.R.; Martynova, E.V.; Khaiboullin, T.I.; Urbanowicz, R.A.; Khaiboullina, S.F.; Rizvanov, A.A. The Relationship of the Mechanisms of the Pathogenesis of Multiple Sclerosis and the Expression of Endogenous Retroviruses. Biology 2020, 9, 464. [Google Scholar] [CrossRef] [PubMed]
- Dewannieux, M.; Harper, F.; Richaud, A.; Letzelter, C.; Ribet, D.; Pierron, G.; Heidmann, T. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006, 16, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, G.J.; Garcia-Perez, J.L. L1 Mosaicism in Mammals: Extent, Effects, and Evolution. Trends Genet. 2017, 33, 802–816. [Google Scholar] [CrossRef]
- Denli, A.M.; Narvaiza, I.; Kerman, B.E.; Pena, M.; Benner, C.; Marchetto, M.C.; Diedrich, J.K.; Aslanian, A.; Ma, J.; Moresco, J.J.; et al. Primate-specific ORF0 contributes to retrotransposon-mediated diversity. Cell 2015, 163, 583–593. [Google Scholar] [CrossRef]
- Wei, W.; Gilbert, N.; Ooi, S.L.; Lawler, J.F.; Ostertag, E.M.; Kazazian, H.H.; Boeke, J.D.; Moran, J.V. Human L1 retrotransposition: Cis preference versus trans complementation. Mol. Cell. Biol. 2001, 21, 1429–1439. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Moran, J.V. Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat. Struct. Mol. Biol. 2006, 13, 655–660. [Google Scholar] [CrossRef]
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 1993, 72, 595–605. [Google Scholar] [CrossRef]
- Sultana, T.; van Essen, D.; Siol, O.; Bailly-Bechet, M.; Philippe, C.; Zine El Aabidine, A.; Pioger, L.; Nigumann, P.; Saccani, S.; Andrau, J.C.; et al. The Landscape of L1 Retrotransposons in the Human Genome Is Shaped by Pre-insertion Sequence Biases and Post-insertion Selection. Mol. Cell 2019, 74, 555–570.e7. [Google Scholar] [CrossRef] [PubMed]
- Flasch, D.A.; Macia, A.; Sanchez, L.; Ljungman, M.; Heras, S.R.; Garcia-Perez, J.L.; Wilson, T.E.; Moran, J.V. Genome-wide de novo L1 Retrotransposition Connects Endonuclease Activity with Replication. Cell 2019, 177, 837–851.e28. [Google Scholar] [CrossRef] [PubMed]
- Weichenrieder, O.; Wild, K.; Strub, K.; Cusack, S. Structure and assembly of the Alu domain of the mammalian signal recognition particle. Nature 2000, 408, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Garcia-Perez, J.L.; Badge, R.M.; Moran, J.V. LINE-1 elements in structural variation and disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 187–215. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Tie, C.H.C.; Conde, L.; Tunbak, H.; Husovsky, C.; Tchasovnikarova, I.A.; Timms, R.T.; Herrero, J.; Lehner, P.J.; Rowe, H.M. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 2018, 28, 836–845. [Google Scholar] [CrossRef]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef]
- Jacobs, F.M.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef]
- Quenneville, S.; Turelli, P.; Bojkowska, K.; Raclot, C.; Offner, S.; Kapopoulou, A.; Trono, D. The KRAB-ZFP/KAP1 system contributes to the early embryonic establishment of site-specific DNA methylation patterns maintained during development. Cell Rep. 2012, 2, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Teissandier, A.; Pérez-Palacios, R.; Bourc’his, D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife 2016, 5, e11418. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Idica, A.; Sevrioukov, E.A.; Zisoulis, D.G.; Hamdorf, M.; Daugaard, I.; Kadandale, P.; Pedersen, I.M. MicroRNA miR-128 represses LINE-1 (L1) retrotransposition by down-regulating the nuclear import factor TNPO1. J. Biol. Chem. 2017, 292, 20494–20508. [Google Scholar] [CrossRef]
- Fung, L.; Guzman, H.; Sevrioukov, E.; Idica, A.; Park, E.; Bochnakian, A.; Daugaard, I.; Jury, D.; Mortazavi, A.; Zisoulis, D.G.; et al. miR-128 Restriction of LINE-1 (L1) Retrotransposition Is Dependent on Targeting hnRNPA1 mRNA. Int. J. Mol. Sci. 2019, 20, 1955. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Orecchini, E.; Frassinelli, L.; Galardi, S.; Ciafre, S.A.; Michienzi, A. Post-transcriptional regulation of LINE-1 retrotransposition by AID/APOBEC and ADAR deaminases. Chromosome Res. 2018, 26, 45–59. [Google Scholar] [CrossRef]
- Schumann, G.G. APOBEC3 proteins: Major players in intracellular defence against LINE-1-mediated retrotransposition. Biochem. Soc. Trans. 2007, 35, 637–642. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Harris, R.S. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J. Biol. Chem. 2006, 281, 16837–16841. [Google Scholar] [CrossRef]
- Majer, C.; Schussler, J.M.; Konig, R. Intertwined: SAMHD1 cellular functions, restriction, and viral evasion strategies. Med. Microbiol. Immunol. 2019, 208, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Wittmann, S.; Thomas, D.; Shepard, C.N.; Kim, B.; Ferreiros, N.; Gramberg, T. The SAMHD1-mediated block of LINE-1 retroelements is regulated by phosphorylation. Mob. DNA 2018, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Li, J.; Xu, F.; Mei, S.; Le Duff, Y.; Yin, L.; Pang, X.; Cen, S.; Jin, Q.; Liang, C.; et al. SAMHD1 Inhibits LINE-1 Retrotransposition by Promoting Stress Granule Formation. PLoS Genet. 2015, 11, e1005367. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Du, J.; Han, X.; Goodier, J.L.; Li, P.; Zhou, X.; Wei, W.; Evans, S.L.; Li, L.; Zhang, W.; et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutieres syndrome-related SAMHD1. Cell Rep. 2013, 4, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Peng, Y.; Wang, S.; Hou, J.; Wang, Y.; Sun, T.; Zhao, K. Nucleocytoplasmic shuttling of SAMHD1 is important for LINE-1 suppression. Biochem. Biophys. Res. Commun. 2019, 510, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef]
- Goodier, J.L.; Cheung, L.E.; Kazazian, H.H., Jr. MOV10 RNA helicase is a potent inhibitor of retrotransposition in cells. PLoS Genet. 2012, 8, e1002941. [Google Scholar] [CrossRef]
- Takata, M.A.; Goncalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Goodier, J.L.; Pereira, G.C.; Cheung, L.E.; Rose, R.J.; Kazazian, H.H., Jr. The Broad-Spectrum Antiviral Protein ZAP Restricts Human Retrotransposition. PLoS Genet. 2015, 11, e1005252. [Google Scholar] [CrossRef]
- Moldovan, J.B.; Moran, J.V. The Zinc-Finger Antiviral Protein ZAP Inhibits LINE and Alu Retrotransposition. PLoS Genet. 2015, 11, e1005121. [Google Scholar] [CrossRef] [PubMed]
- Kazazian, H.H., Jr.; Wong, C.; Youssoufian, H.; Scott, A.F.; Phillips, D.G.; Antonarakis, S.E. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 1988, 332, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Manel, N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat. Rev. Immunol. 2015, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Chase, D.S.; Lowenstein Schmidt, J.; Szynkiewicz, M.; Forte, G.M.; Gornall, H.L.; Oojageer, A.; Anderson, B.; Pizzino, A.; Helman, G.; et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 2015, 167A, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kirsch, M.A.; Gong, M.; Chowdhury, D.; Senenko, L.; Engel, K.; Lee, Y.A.; de Silva, U.; Bailey, S.L.; Witte, T.; Vyse, T.J.; et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007, 39, 1065–1067. [Google Scholar] [CrossRef]
- Rice, G.; Newman, W.G.; Dean, J.; Patrick, T.; Parmar, R.; Flintoff, K.; Robins, P.; Harvey, S.; Hollis, T.; O’Hara, A.; et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 2007, 80, 811–815. [Google Scholar] [CrossRef]
- Thomas, C.A.; Tejwani, L.; Trujillo, C.A.; Negraes, P.D.; Herai, R.H.; Mesci, P.; Macia, A.; Crow, Y.J.; Muotri, A.R. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017, 21, 319–331.e8. [Google Scholar] [CrossRef]
- Zhao, K.; Du, J.; Peng, Y.; Li, P.; Wang, S.; Wang, Y.; Hou, J.; Kang, J.; Zheng, W.; Hua, S.; et al. LINE1 contributes to autoimmunity through both RIG-I- and MDA5-mediated RNA sensing pathways. J. Autoimmun. 2018, 90, 105–115. [Google Scholar] [CrossRef]
- Lim, Y.W.; Sanz, L.A.; Xu, X.; Hartono, S.R.; Chédin, F. Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi-Goutières syndrome. eLife 2015, 4, e08007. [Google Scholar] [CrossRef]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824.e14. [Google Scholar] [CrossRef]
- Ahmad, S.; Mu, X.; Yang, F.; Greenwald, E.; Park, J.W.; Jacob, E.; Zhang, C.Z.; Hur, S. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018, 172, 797–810.e13. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, C.P.; Sagalovskiy, I.; Guo, Q.; Nezos, A.; Kapsogeorgou, E.K.; Lu, P.; Liang Zhou, J.; Kirou, K.A.; Seshan, S.V.; Moutsopoulos, H.M.; et al. Expression of Long Interspersed Nuclear Element 1 Retroelements and Induction of Type I Interferon in Patients With Systemic Autoimmune Disease. Arthritis Rheumatol. 2016, 68, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Pratt, G.A.; Sundararaman, B.; Townsend, M.J.; Chaivorapol, C.; Bhangale, T.; Graham, R.R.; Ortmann, W.; Criswell, L.A.; Yeo, G.W.; et al. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 2015, 350, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Brégnard, C.; Guerra, J.; Déjardin, S.; Passalacqua, F.; Benkirane, M.; Laguette, N. Upregulated LINE-1 Activity in the Fanconi Anemia Cancer Susceptibility Syndrome Leads to Spontaneous Pro-inflammatory Cytokine Production. EBioMedicine 2016, 8, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Tossberg, J.T.; Heinrich, R.M.; Farley, V.M.; Crooke, P.S., 3rd; Aune, T.M. Adenosine-to-Inosine RNA Editing of Alu Double-Stranded (ds)RNAs Is Markedly Decreased in Multiple Sclerosis and Unedited Alu dsRNAs Are Potent Activators of Proinflammatory Transcriptional Responses. J. Immunol. 2020, 205, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885.e5. [Google Scholar] [CrossRef]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef]
- Kerur, N.; Fukuda, S.; Banerjee, D.; Kim, Y.; Fu, D.; Apicella, I.; Varshney, A.; Yasuma, R.; Fowler, B.J.; Baghdasaryan, E.; et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 2018, 24, 50–61. [Google Scholar] [CrossRef]
- Wang, A.T.; Smogorzewska, A. SnapShot: Fanconi anemia and associated proteins. Cell 2015, 160, 354–354.e1. [Google Scholar] [CrossRef]
- Bogliolo, M.; Surralles, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, R.; Brokstad, K.A.; Jonsson, M.V.; Delaleu, N.; Skarstein, K. Current concepts on Sjogren’s syndrome—Classification criteria and biomarkers. Eur. J. Oral. Sci. 2018, 126 (Suppl. 1), 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Bannert, N.; Hofmann, H.; Block, A.; Hohn, O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Alcazer, V.; Bonaventura, P.; Depil, S. Human Endogenous Retroviruses (HERVs): Shaping the Innate Immune Response in Cancers. Cancers 2020, 12, 610. [Google Scholar] [CrossRef]
- Greenig, M. HERVs, immunity, and autoimmunity: Understanding the connection. PeerJ 2019, 7, e6711. [Google Scholar] [CrossRef]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef]
- Lee-Kirsch, M.A. The Type I Interferonopathies. Annu. Rev. Med. 2017, 68, 297–315. [Google Scholar] [CrossRef]
- Kretschmer, S.; Lee-Kirsch, M.A. Type I interferon-mediated autoinflammation and autoimmunity. Curr. Opin. Immunol. 2017, 49, 96–102. [Google Scholar] [CrossRef]
- Moresco, E.M.; LaVine, D.; Beutler, B. Toll-like receptors. Curr. Biol. 2011, 21, R488–R493. [Google Scholar] [CrossRef] [PubMed]
- Streicher, F.; Jouvenet, N. Stimulation of Innate Immunity by Host and Viral RNAs. Trends Immunol. 2019, 40, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Bruns, A.M.; Horvath, C.M. Antiviral RNA recognition and assembly by RLR family innate immune sensors. Cytokine Growth Factor Rev. 2014, 25, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2020, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [PubMed]
- Hotter, D.; Bosso, M.; Jonsson, K.L.; Krapp, C.; Sturzel, C.M.; Das, A.; Littwitz-Salomon, E.; Berkhout, B.; Russ, A.; Wittmann, S.; et al. IFI16 Targets the Transcription Factor Sp1 to Suppress HIV-1 Transcription and Latency Reactivation. Cell Host Microbe 2019, 25, 858–872.e13. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Lahaye, X.; Gentili, M.; Silvin, A.; Conrad, C.; Picard, L.; Jouve, M.; Zueva, E.; Maurin, M.; Nadalin, F.; Knott, G.J.; et al. NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 2018, 175, 488–501.e22. [Google Scholar] [CrossRef]
- Michalski, S.; de Oliveira Mann, C.C.; Stafford, C.A.; Witte, G.; Bartho, J.; Lammens, K.; Hornung, V.; Hopfner, K.P. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 2020, 587, 678–682. [Google Scholar] [CrossRef]
- McKenna, S.A.; Lindhout, D.A.; Shimoike, T.; Puglisi, J.D. Biophysical and biochemical investigations of dsRNA-activated kinase PKR. Methods Enzym. 2007, 430, 373–396. [Google Scholar] [CrossRef]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J. Mol. Biol. 2008, 381, 351–360. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Stamp, G.; Robins, P.; Dulic, A.; Rosewell, I.; Hrivnak, G.; Daly, G.; Lindahl, T.; Barnes, D.E. Gene-targeted mice lacking the Trex1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell Biol. 2004, 24, 6719–6727. [Google Scholar] [CrossRef] [PubMed]
- Beck-Engeser, G.B.; Eilat, D.; Wabl, M. An autoimmune disease prevented by anti-retroviral drugs. Retrovirology 2011, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Achleitner, M.; Kleefisch, M.; Hennig, A.; Peschke, K.; Polikarpova, A.; Oertel, R.; Gabriel, B.; Schulze, L.; Lindeman, D.; Gerbaulet, A.; et al. Lack of Trex1 Causes Systemic Autoimmunity despite the Presence of Antiretroviral Drugs. J. Immunol. 2017, 199, 2261–2269. [Google Scholar] [CrossRef]
- Laguette, N.; Brégnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef]
- Matos, J.; West, S.C. Holliday junction resolution: Regulation in space and time. DNA Repair 2014, 19, 176–181. [Google Scholar] [CrossRef]
- Rice, G.I.; Meyzer, C.; Bouazza, N.; Hully, M.; Boddaert, N.; Semeraro, M.; Zeef, L.A.H.; Rozenberg, F.; Bondet, V.; Duffy, D.; et al. Reverse-Transcriptase Inhibitors in the Aicardi–Goutières Syndrome. N. Engl. J. Med. 2018, 379, 2275–2277. [Google Scholar] [CrossRef]
- Benitez-Guijarro, M.; Lopez-Ruiz, C.; Tarnauskaite, Z.; Murina, O.; Mian Mohammad, M.; Williams, T.C.; Fluteau, A.; Sanchez, L.; Vilar-Astasio, R.; Garcia-Canadas, M.; et al. RNase H2, mutated in Aicardi-Goutieres syndrome, promotes LINE-1 retrotransposition. EMBO J. 2018, 37, e98506. [Google Scholar] [CrossRef]
- Volkmann, B.; Wittmann, S.; Lagisquet, J.; Deutschmann, J.; Eissmann, K.; Ross, J.J.; Biesinger, B.; Gramberg, T. Human TRIM5α senses and restricts LINE-1 elements. Proc. Natl. Acad. Sci. USA 2020, 117, 17965–17976. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Nishikura, K. Extensive adenosine-to-inosine editing detected in Alu repeats of antisense RNAs reveals scarcity of sense-antisense duplex formation. FEBS Lett. 2006, 580, 2301–2305. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; DeCerbo, J.N.; Carmichael, G.G. Alu element-mediated gene silencing. Embo J. 2008, 27, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, R.A.; Li, W.; Tian, B.; Maquat, L.E. STAU1 binding 3’ UTR IRAlus complements nuclear retention to protect cells from PKR-mediated translational shutdown. Genes Dev. 2013, 27, 1495–1510. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef]
- Wang, Q.; Khillan, J.; Gadue, P.; Nishikura, K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 2000, 290, 1765–1768. [Google Scholar] [CrossRef]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellåker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef]
- George, C.X.; Ramaswami, G.; Li, J.B.; Samuel, C.E. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J. Biol. Chem. 2016, 291, 6158–6168. [Google Scholar] [CrossRef]
- Van Eyck, L.; De Somer, L.; Pombal, D.; Bornschein, S.; Frans, G.; Humblet-Baron, S.; Moens, L.; de Zegher, F.; Bossuyt, X.; Wouters, C.; et al. Brief Report: IFIH1 Mutation Causes Systemic Lupus Erythematosus with Selective IgA Deficiency. Arthritis Rheumatol. 2015, 67, 1592–1597. [Google Scholar] [CrossRef]
- Rice, G.I.; Del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef]
- Oda, H.; Nakagawa, K.; Abe, J.; Awaya, T.; Funabiki, M.; Hijikata, A.; Nishikomori, R.; Funatsuka, M.; Ohshima, Y.; Sugawara, Y.; et al. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 2014, 95, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, W.; Liu, Y.; Tan, X.; Li, X.; Zou, Q.; Xiao, Z.; Xu, H.; Wang, Y.; Yang, X. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 2018, 37, e99017. [Google Scholar] [CrossRef] [PubMed]
- Dema, B.; Charles, N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies 2016, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.; Reichlin, M.; Tomasi, T.B., Jr. Characterization of a soluble cytoplasmic antigen reactive with sera from patients with systemic lupus erythmatosus. J. Immunol. 1969, 102, 117–122. [Google Scholar] [PubMed]
- Alspaugh, M.; Maddison, P. Resolution of the identity of certain antigen-antibody systems in systemic lupus erythematosus and Sjögren’s syndrome: An interlaboratory collaboration. Arthritis Rheumatol. 1979, 22, 796–798. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease | Element | Nucleic Acid | Sensor | Reference |
|---|---|---|---|---|
| Aicardi-Goutières syndrome (AGS) | ||||
| Type 1 (TREX1) | LINE-1 | ssDNA | cGAS/STING | [57] |
| RNA | MDA5/RIG-I | [58] | ||
| RNA:DNA | unknown | [59] | ||
| Type 2-4 (RNaseH2) | LINE-1 | RNA | MDA5/RIG-I | [58] |
| RNA:DNA | unknown | [59] | ||
| Type 5 (SAMHD1) | LINE-1 | RNA | MDA5/RIG-I | [58] |
| RNA:DNA | unknown | [59] | ||
| Type 6 (ADAR) | LINE-1 | RNA | MDA5/RIG-I | [58] |
| Alu | IR-Alu | MDA5/PKR | [60] | |
| Type 7 (MDA5) | Alu | IR-Alu | MDA5 | [61] |
| Systemic Lupus Erythematosus (SLE) | LINE-1 | RNA | unknown | [62] |
| Alu | RNA | TLR7 | [63] | |
| Sjögren’s syndrome | LINE-1 | RNA | unknown | [62] |
| Fanconi Anemia | LINE-1 | ssDNA | cGAS/STING | [64] |
| Multiple sclerosis | Alu | dsRNA | RIG-I/TLR3 | [65] |
| Aging | LINE-1 | ssDNA | cGAS/STING | [66,67] |
| Age Macular Degeneration-Geographic Atrophy (GA) | Alu | RNA | NLRP3/cGAS | [68,69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagisquet, J.; Zuber, K.; Gramberg, T. Recognize Yourself—Innate Sensing of Non-LTR Retrotransposons. Viruses 2021, 13, 94. https://doi.org/10.3390/v13010094
Lagisquet J, Zuber K, Gramberg T. Recognize Yourself—Innate Sensing of Non-LTR Retrotransposons. Viruses. 2021; 13(1):94. https://doi.org/10.3390/v13010094
Chicago/Turabian StyleLagisquet, Justine, Kilian Zuber, and Thomas Gramberg. 2021. "Recognize Yourself—Innate Sensing of Non-LTR Retrotransposons" Viruses 13, no. 1: 94. https://doi.org/10.3390/v13010094
APA StyleLagisquet, J., Zuber, K., & Gramberg, T. (2021). Recognize Yourself—Innate Sensing of Non-LTR Retrotransposons. Viruses, 13(1), 94. https://doi.org/10.3390/v13010094