
Whole-Genome Analyses Identifies Multiple Reassortant Rotavirus Strains in Rwanda Post-Vaccine Introduction

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Preparation of Purified dsRNA and cDNA for Rotavirus Whole-Genome Sequencing
2.4. DNA Library Preparations and Whole-Genome Sequencing
2.5. Computational Analysis
3. Results
3.1. Genome Constellations
3.2. The VP4 and VP7 Antigenic Region Analyses
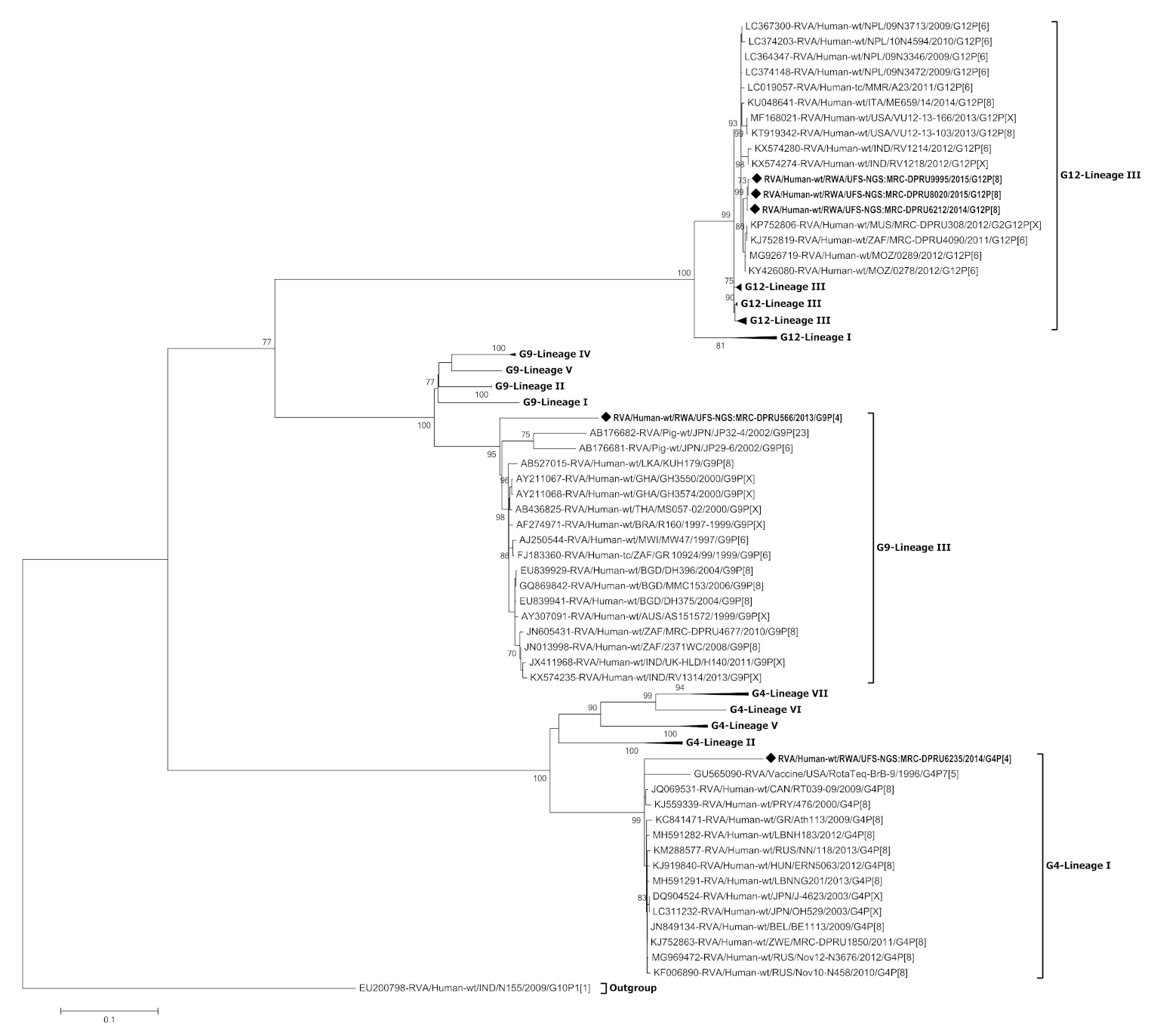
3.3. Phylogenetic Analysis of the VP7 Gene of G4, G9 and G12
3.4. Phylogenetic Analysis of the VP4 Gene of P[4] and P[8]
3.5. Phylogenetic Analyses of the VP1-VP3 and VP6 Genes
3.6. Phylogenetic Analyses of the NSP1-NSP5 Genes
4. Discussion
5. Conclusions
6. Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958. [Google Scholar] [CrossRef] [PubMed]
- Fischer Walker, C.L.; Perin, J.; Aryee, M.J.; Boschi-Pinto, C.; Black, R.E. Diarrhea incidence in low- and middle-income countries in 1990 and 2010: A systematic review. BMC Public Health 2012, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, K.N.C.; Babji, S.; Ganesan, S.K. Impact of rotavirus vaccines in low and middle-income countries. Curr. Opin. Infect. Dis. 2017, 30, 473–481. [Google Scholar] [CrossRef] [PubMed]
- GAVI. Rwanda Introduces New Vaccine Against a Leading Childhood Killer. Available online: https://www.gavi.org/rwanda-introduces-new-vaccine-against-a-leading-childhood-killer (accessed on 8 July 2020).
- Gatera, M.; Bhatt, S.; Ngabo, F.; Utamuliza, M.; Sibomana, H.; Karema, C.; Mugeni, C.; Nutt, C.T.; Nsanzimana, S.; Wagner, C.M.; et al. Successive introduction of four new vaccines in Rwanda: High coverage and rapid scale up of Rwanda’s expanded immunization program from 2009 to 2013. Vaccine 2016, 34, 3420–3426. [Google Scholar] [CrossRef] [PubMed]
- WHO/UNICEF. Rwanda: WHO and UNICEF Estimates of Immunization Coverage: 2019 Revision. Available online: https://www.who.int/immunization/monitoring_surveillance/data/rwa.pdf (accessed on 15 September 2020).
- Sibomana, H.; Rugambwa, C.; Sayinzoga, F.; Iraguha, G.; Uwimana, J. Impact of routine rotavirus vaccination on all-cause and rotavirus hospitalizations during the first four years following vaccine introduction in Rwanda. Vaccine 2018, 36, 7135–7141. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Fields, D.M., Eds.; Wolters Kluwer Health/Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Desselberger, U. Rotaviruses. Virus Res. 2014, 190, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group a rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef]
- Nyaga, M.M.; Sabiu, S.; Ndze, V.N.; Dennis, F.E.; Jere, K.C. Report of the 1st African Enteric Viruses Genome Initiative (AEVGI) Data and Bioinformatics Workshop on whole-genome analysis of some African rotavirus strains held in Bloemfontein, South Africa. Vaccine 2020, 38, 5402–5407. [Google Scholar] [CrossRef]
- Ide, T.; Komoto, S.; Higo-Moriguchi, K.; Htun, K.W.; Myint, Y.Y.; Myat, T.W.; Thant, K.Z.; Thu, H.M.; Win, M.M.; Oo, H.N.; et al. Whole Genomic Analysis of Human G12P[6] and G12P[8] Rotavirus Strains that Have Emerged in Myanmar. PLoS ONE 2015, 10, e0124965. [Google Scholar] [CrossRef]
- Komoto, S.; Tacharoenmuang, R.; Guntapong, R.; Ide, T.; Tsuji, T.; Yoshikawa, T.; Tharmaphornpilas, P.; Sangkitporn, S.; Taniguchi, K. Reassortment of Human and Animal Rotavirus Gene Segments in Emerging DS-1-Like G1P[8] Rotavirus Strains. PLoS ONE 2016, 11, e0148416. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gomara, M.; Maes, P.; Patton, J.T.; et al. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- WHO. WHO Prequalifies New Rotavirus Vaccine. Available online: https://www.who.int/medicines/news/2018/prequalified_new-rotavirus_vaccine/en/ (accessed on 6 May 2020).
- Matthijnssens, J.; Joelsson, D.B.; Warakomski, D.J.; Zhou, T.; Mathis, P.K.; van Maanen, M.-H.; Ranheim, T.S.; Ciarlet, M. Molecular and biological characterization of the 5 human-bovine rotavirus (WC3)-based reassortant strains of the pentavalent rotavirus vaccine, RotaTeq®. Virology 2010, 403, 111–127. [Google Scholar] [CrossRef] [PubMed]
- PATH. Rotavirus Vaccine Country Introductions: Maps and List-PATH Vaccine Resource Library. Available online: https://vaccineresources.org/details.php?i=2235 (accessed on 6 December 2019).
- Mwenda, J.M.; Ntoto, K.M.; Abebe, A.; Enweronu-Laryea, C.; Amina, I.; Mchomvu, J.; Kisakye, A.; Mpabalwani, E.M.; Pazvakavambwa, I.; Armah, G.E.; et al. Burden and Epidemiology of Rotavirus Diarrhea in Selected African Countries: Preliminary Results from the African Rotavirus Surveillance Network. J. Infect. Dis. 2010, 202, S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Seheri, M.; Nemarude, L.; Peenze, I.; Netshifhefhe, L.; Nyaga, M.M.; Ngobeni, H.G.; Maphalala, G.; Maake, L.L.; Steele, A.D.; Mwenda, J.M.; et al. Update of Rotavirus Strains Circulating in Africa From 2007 Through 2011. Pediatr. Infect. Dis. J. 2014, 33, S76–S84. [Google Scholar] [CrossRef] [PubMed]
- Todd, S.; Page, N.A.; Steele, A.D.; Peenze, I.; Cunliffe, N.A. Rotavirus Strain Types Circulating in Africa: Review of Studies Published during 1997–2006. J. Infect. Dis. 2010, 202, S34–S42. [Google Scholar] [CrossRef]
- Seheri, L.M.; Magagula, N.B.; Peenze, I.; Rakau, K.; Ndadza, A.; Mwenda, J.M.; Weldegebriel, G.; Steele, A.D.; Mphahlele, M.J. Rotavirus strain diversity in Eastern and Southern African countries before and after vaccine introduction. Vaccine 2018, 36, 7222–7230. [Google Scholar] [CrossRef]
- Agbemabiese, C.A.; Nakagomi, T.; Nguyen, M.Q.; Gauchan, P.; Nakagomi, O. Reassortant DS-1-like G1P[4] Rotavirus A strains generated from co-circulating strains in Vietnam, 2012/2013. Microbiol. Immunol. 2017, 61, 328–336. [Google Scholar] [CrossRef]
- Cowley, D.; Donato, C.M.; Roczo-Farkas, S.; Kirkwood, C.D. Emergence of a novel equine-like G3P[8] intergenogroup reassortant rotavirus strain associated with gastroenteritis in Australian children. J. Gen. Virol. 2016, 97, 403–410. [Google Scholar] [CrossRef]
- Heiman, E.M.; McDonald, S.M.; Barro, M.; Taraporewala, Z.F.; Bar-Magen, T.; Patton, J.T. Group A human rotavirus genomics: Evidence that gene constellations are influenced by viral protein interactions. J. Virol. 2008, 82, 11106–11116. [Google Scholar] [CrossRef] [PubMed]
- Hoa-Tran, T.N.; Nakagomi, T.; Vu, H.M.; Do, L.P.; Gauchan, P.; Agbemabiese, C.A.; Nguyen, T.T.T.; Nakagomi, O.; Thanh, N.T.H. Abrupt emergence and predominance in Vietnam of rotavirus A strains possessing a bovine-like G8 on a DS-1-like background. Arch. Virol. 2016, 161, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Tacharoenmuang, R.; Guntapong, R.; Ide, T.; Sinchai, P.; Upachai, S.; Fukuda, S.; Yoshikawa, T.; Tharmaphornpilas, P.; Sangkitporn, S.; et al. Identification and characterization of a human G9P[23] rotavirus strain from a child with diarrhoea in Thailand: Evidence for porcine-to-human interspecies transmission. J. Gen. Virol. 2017, 98, 532–538. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary Dynamics of Human Rotaviruses: Balancing Reassortment with Preferred Genome Constellations. PLoS Pathog. 2009, 5, e1000634. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, P.N.; Mogotsi, M.T.; Rasebotsa, S.P.; Seheri, M.L.; Mphahlele, M.J.; Ndze, V.N.; Dennis, F.E.; Jere, K.C.; Nyaga, M.M. Uncovering the first atypical ds-1-like g1p[8] rotavirus strains that circulated during pre-rotavirus vaccine introduction era in South Africa. Pathogens 2020, 9, 391. [Google Scholar] [CrossRef]
- Bányai, K.; Mijatovic-Rustempasic, S.; Hull, J.J.; Esona, M.D.; Freeman, M.M.; Frace, A.M.; Bowen, M.D.; Gentsch, J.R. Sequencing and phylogenetic analysis of the coding region of six common rotavirus strains: Evidence for intragenogroup reassortment among co-circulating G1P[8] and G2P[4] strains from the United States. J. Med. Virol. 2011, 83, 532–539. [Google Scholar] [CrossRef]
- Dennis, F.E.; Fujii, Y.; Haga, K.; Damanka, S.; Lartey, B.; Agbemabiese, C.A.; Ohta, N.; Armah, G.E.; Katayama, K. Identification of Novel Ghanaian G8P[6] Human-Bovine Reassortant Rotavirus Strain by Next Generation Sequencing. PLoS ONE 2014, 9, e100699. [Google Scholar] [CrossRef]
- Heylen, E.; Likele, B.B.; Zeller, M.; Stevens, S.; De Coster, S.; Conceição-Neto, N.; Van Geet, C.; Jacobs, J.; Ngbonda, D.; Van Ranst, M.; et al. Rotavirus surveillance in Kisangani, the Democratic Republic of the Congo, reveals a high number of unusual genotypes and gene segments of animal origin in non-vaccinated symptomatic children. PLoS ONE 2014, 9, e100953. [Google Scholar] [CrossRef]
- Nakagomi, T.; Doan, Y.H.; Dove, W.; Ngwira, B.; Iturriza-Gómara, M.; Nakagomi, O.; Cunliffe, N.A. G8 rotaviruses with conserved genotype constellations detected in Malawi over 10 years (1997–2007) display frequent gene reassortment among strains co-circulating in humans. J. Gen. Virol. 2013, 94, 1273–1295. [Google Scholar] [CrossRef]
- Nyaga, M.M.; Tan, Y.; Seheri, M.L.; Halpin, R.A.; Akopov, A.; Stucker, K.M.; Fedorova, N.B.; Shrivastava, S.; Steele, A.D.; Mwenda, J.M.; et al. Whole-genome sequencing and analyses identify high genetic heterogeneity, diversity and endemicity of rotavirus genotype P[6] strains circulating in Africa. Infect. Genet. Evol. 2018, 63, 79–88. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.B.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-A self-parameterizing force field. Proteins Struct. Funct. Genet. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic Analyses Reveal Differences in the VP7 and VP4 Antigenic Epitopes between Human Rotaviruses Circulating in Belgium and Rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef]
- Aoki, S.T.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Donato, C.; Sequeira Trovão, N.; Cowley, D.; Heylen, E.; Donker, N.C.; McAllen, J.K.; Akopov, A.; Kirkness, E.F.; Lemey, P.; et al. Genome-Wide Evolutionary Analyses of G1P[8] Strains Isolated Before and After Rotavirus Vaccine Introduction. Genome Biol. Evol. 2015, 7, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Dormitzer, P.R.; Sun, Z.-Y.J.; Wagner, G.; Harrison, S.C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 2002, 21, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Das, B.K.; Bhan, M.K.; Glass, R.I.; Gentsch, J.R.; Bhambal, S.S.; Kerari, N.; Rawat, H.; Bahl, L.; Thakur, S. Great diversity of group A rotavirus strains and high prevalence of mixed rotavirus infections in India. J. Clin. Microbiol. 2001, 39, 3524–3529. [Google Scholar] [CrossRef]
- Kirkwood, C.D. Genetic and Antigenic Diversity of Human Rotaviruses: Potential Impact on Vaccination Programs. J. Infect. Dis. 2010, 202, S43–S48. [Google Scholar] [CrossRef]
- Medici, M.C.; Abelli, L.A.; Martella, V.; Martinelli, M.; Lorusso, E.; Buonavoglia, C.; Dettori, G.; Chezzi, C. Characterization of inter-genogroup reassortant rotavirus strains detected in hospitalized children in Italy. J. Med. Virol. 2007, 79, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Dóró, R.; László, B.; Martella, V.; Leshem, E.; Gentsch, J.; Parashar, U.; Bányai, K. Review of global rotavirus strain prevalence data from six years post vaccine licensure surveillance: Is there evidence of strain selection from vaccine pressure? Infect. Genet. Evol. 2014, 28, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Afrad, M.H.; Rahman, M.Z.; Matthijnssens, J.; Das, S.K.; Faruque, A.S.G.; Azim, T.; Rahman, M. High incidence of reassortant G9P[4] rotavirus strain in Bangladesh: Fully heterotypic from vaccine strains. J. Clin. Virol. 2013, 58, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Doan, Y.H.; Suzuki, Y.; Fujii, Y.; Haga, K.; Fujimoto, A.; Takai-Todaka, R.; Someya, Y.; Nayak, M.K.; Mukherjee, A.; Imamura, D.; et al. Complex reassortment events of unusual G9P[4] rotavirus strains in India between 2011 and 2013. Infect. Genet. Evol. 2017, 54, 417–428. [Google Scholar] [CrossRef]
- João, E.D.; Munlela, B.; Chissaque, A.; Chilaúle, J.; Langa, J.; Augusto, O.; Boene, S.S.; Anapakala, E.; Sambo, J.; Guimarães, E.; et al. Molecular Epidemiology of Rotavirus A Strains Pre- and Post-Vaccine (Rotarix®) Introduction in Mozambique, 2012–2019: Emergence of Genotypes G3P[4] and G3P[8]. Pathogens 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Lartey, B.L.; Damanka, S.; Dennis, F.E.; Enweronu-Laryea, C.C.; Addo-Yobo, E.; Ansong, D.; Kwarteng-Owusu, S.; Sagoe, K.W.; Mwenda, J.M.; Diamenu, S.K.; et al. Rotavirus strain distribution in Ghana pre- and post- rotavirus vaccine introduction. Vaccine 2018, 36, 7238–7242. [Google Scholar] [CrossRef]
- Pradhan, G.N.; Walimbe, A.M.; Chitambar, S.D. Molecular characterization of emerging G9P[4] rotavirus strains possessing a rare E6 NSP4 or T1 NSP3 genotype on a genogroup-2 backbone using a refined classification framework. J. Gen. Virol. 2016, 97, 3139–3153. [Google Scholar] [CrossRef]
- Quaye, O.; Mcdonald, S.; Esona, M.D.; Lyde, F.C.; Mijatovic-Rustempasic, S.; Roy, S.; Banegas, D.J.C.; Quiñonez, Y.M.; Chinchilla, B.L.; Santiago, F.G.; et al. Rotavirus G9P[4] in 3 countries in Latin America, 2009–2010. Emerg. Infect. Dis. 2013, 19, 1332. [Google Scholar] [CrossRef]
- Yamamoto, S.P.; Kaida, A.; Ono, A.; Kubo, H.; Iritani, N. Detection and characterization of a human G9P[4] rotavirus strain in Japan. J. Med. Virol. 2015, 87, 1311–1318. [Google Scholar] [CrossRef]
- Rose, T.L.; da Silva, M.F.M.; Goméz, M.M.; Resque, H.R.; Ichihara, M.Y.T.; de Mello Volotão, E.; Leite, J.P.G. Evidence of vaccine-related reassortment of rotavirus, Brazil, 2008–2010. Emerg. Infect. Dis. 2013, 19, 1843–1846. [Google Scholar] [CrossRef]
- Narang, A.; Bose, A.; Pandit, A.N.; Dutta, P.; Kang, G.; Bhattacharya, S.K.; Datta, S.K.; Suryakiran, P.V.; Delem, A.; Han, H.H.; et al. Immunogenicity, reactogenicity and safety of human rotavirus vaccine (RIX4414) in Indian infants. Hum. Vaccin. 2009, 5, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Taneja, D.; Malik, A. Burden of rotavirus in India-Is rotavirus vaccine an answer to it? Indian J. Public Health 2012, 56, 17. [Google Scholar] [CrossRef] [PubMed]
- Zaman, K.; Anh, D.D.; Victor, J.C.; Shin, S.; Yunus, M.; Dallas, M.J.; Podder, G.; Thiem, V.D.; Mai, L.T.P.; Luby, S.P.; et al. Efficacy of pentavalent rotavirus vaccine against severe rotavirus gastroenteritis in infants in developing countries in Asia: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 376, 615–623. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Nomenclature | VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RVA/Human-wt/RWA/UFS-NGS:MRC-DPRU6235/2014/G4P[4] (Non-Vaccinated)) | Genome constellations | G4 | P[4] | I1 | R2 | C2 | M2 | A2 | N2 | T1 | E1 | H2 |
| Contig length | 1065 | 2359 | 1353 | 3303 | 2729 | 3302 | 3302 | 1044 | 1073 | 750 | 673 | |
| Reads mapped to contigs | 4013 | 16,451 | 6652 | 17,536 | 13,647 | 19,920 | 7673 | 3907 | 5391 | 3371 | 1892 | |
| Genome constellations | G9 | P[4] | I1 | R2 | C2 | M2 | A1 | N1 | T1 | E1 | H1 | |
| RVA/Human-wt/RWA/UFS-NGS:MRC-DPRU566/2013/G9P[4] (Non-Vaccinated) | Contig length | 1061 | 2359 | 1352 | 3302 | 2726 | 2591 | 1567 | 1059 | 1074 | 748 | 664 |
| Reads mapped to contigs | 4324 | 7950 | 7548 | 19,078 | 11,493 | 11,132 | 4451 | 3604 | 4152 | 3879 | 722 | |
| Genome constellations | G12 | P[8] | I1 | R2 | C2 | M1 | A1 | N2 | T1 | E2 | H3 | |
| RVA/Human-wt/RWA/UFS-NGS:MRC-DPRU8020/2015/G12P[8] (Vaccinated) | Contig length | 1062 | 2359 | 1360 | 3304 | 2707 | 3302 | 3302 | 1059 | 1074 | 751 | 667 |
| Reads mapped to contigs | 3888 | 6742 | 5761 | 20,328 | 12,815 | 11,949 | 12,849 | 3172 | 3484 | 2600 | 1170 | |
| Genome constellations | G12 | P[8] | I1 | R2 | C2 | M1 | A1 | N2 | T1 | E2 | H3 | |
| RVA/Human-wt/RWA/UFS-NGS:MRC-DPRU9995/2015/G12P[8] (Vaccinated) | Contig length | 1062 | 2359 | 1356 | 3302 | 2687 | 3302 | 3302 | 1059 | 1074 | 751 | 668 |
| Reads mapped to contigs | 25,828 | 45,020 | 42,925 | 92,061 | 65,575 | 26,672 | 18,505 | 20,815 | 12,752 | 7205 | 11,688 | |
| Genome constellations | G12 | P[8] | I1 | R1 | C1 | M1 | A2 | N2 | T2 | E1 | H1 | |
| RVA/Human-wt/RWA/UFS-NGS:MRC-DPRU6212/2014/G12P[8] (Vaccinated) | Contig length | 1061 | 2359 | 1356 | 3301 | 2735 | 2591 | 3302 | 1059 | 1066 | 749 | 669 |
| Reads mapped to contigs | 22,656 | 60,092 | 26,846 | 62,970 | 55,548 | 59,218 | 26,988 | 15,855 | 20,750 | 24,216 | 4705 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasebotsa, S.; Uwimana, J.; Mogotsi, M.T.; Rakau, K.; Magagula, N.B.; Seheri, M.L.; Mwenda, J.M.; Mphahlele, M.J.; Sabiu, S.; Mihigo, R.; et al. Whole-Genome Analyses Identifies Multiple Reassortant Rotavirus Strains in Rwanda Post-Vaccine Introduction. Viruses 2021, 13, 95. https://doi.org/10.3390/v13010095
Rasebotsa S, Uwimana J, Mogotsi MT, Rakau K, Magagula NB, Seheri ML, Mwenda JM, Mphahlele MJ, Sabiu S, Mihigo R, et al. Whole-Genome Analyses Identifies Multiple Reassortant Rotavirus Strains in Rwanda Post-Vaccine Introduction. Viruses. 2021; 13(1):95. https://doi.org/10.3390/v13010095
Chicago/Turabian StyleRasebotsa, Sebotsana, Jeannine Uwimana, Milton T. Mogotsi, Kebareng Rakau, Nonkululeko B. Magagula, Mapaseka L. Seheri, Jason M. Mwenda, M. Jeffrey Mphahlele, Saheed Sabiu, Richard Mihigo, and et al. 2021. "Whole-Genome Analyses Identifies Multiple Reassortant Rotavirus Strains in Rwanda Post-Vaccine Introduction" Viruses 13, no. 1: 95. https://doi.org/10.3390/v13010095
APA StyleRasebotsa, S., Uwimana, J., Mogotsi, M. T., Rakau, K., Magagula, N. B., Seheri, M. L., Mwenda, J. M., Mphahlele, M. J., Sabiu, S., Mihigo, R., Mutesa, L., & Nyaga, M. M. (2021). Whole-Genome Analyses Identifies Multiple Reassortant Rotavirus Strains in Rwanda Post-Vaccine Introduction. Viruses, 13(1), 95. https://doi.org/10.3390/v13010095