
Genetically Determined Circulating Lactase/Phlorizin Hydrolase Concentrations and Risk of Colorectal Cancer: A Two-Sample Mendelian Randomization Study
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Genetic Instruments
2.3. Outcome Data Sources
2.4. Statistical Power Calculation
2.5. Statistical Analysis
3. Results
3.1. FinnGen Dataset
3.2. PLCO Dataset
3.3. Pan-UK Biobank Dataset
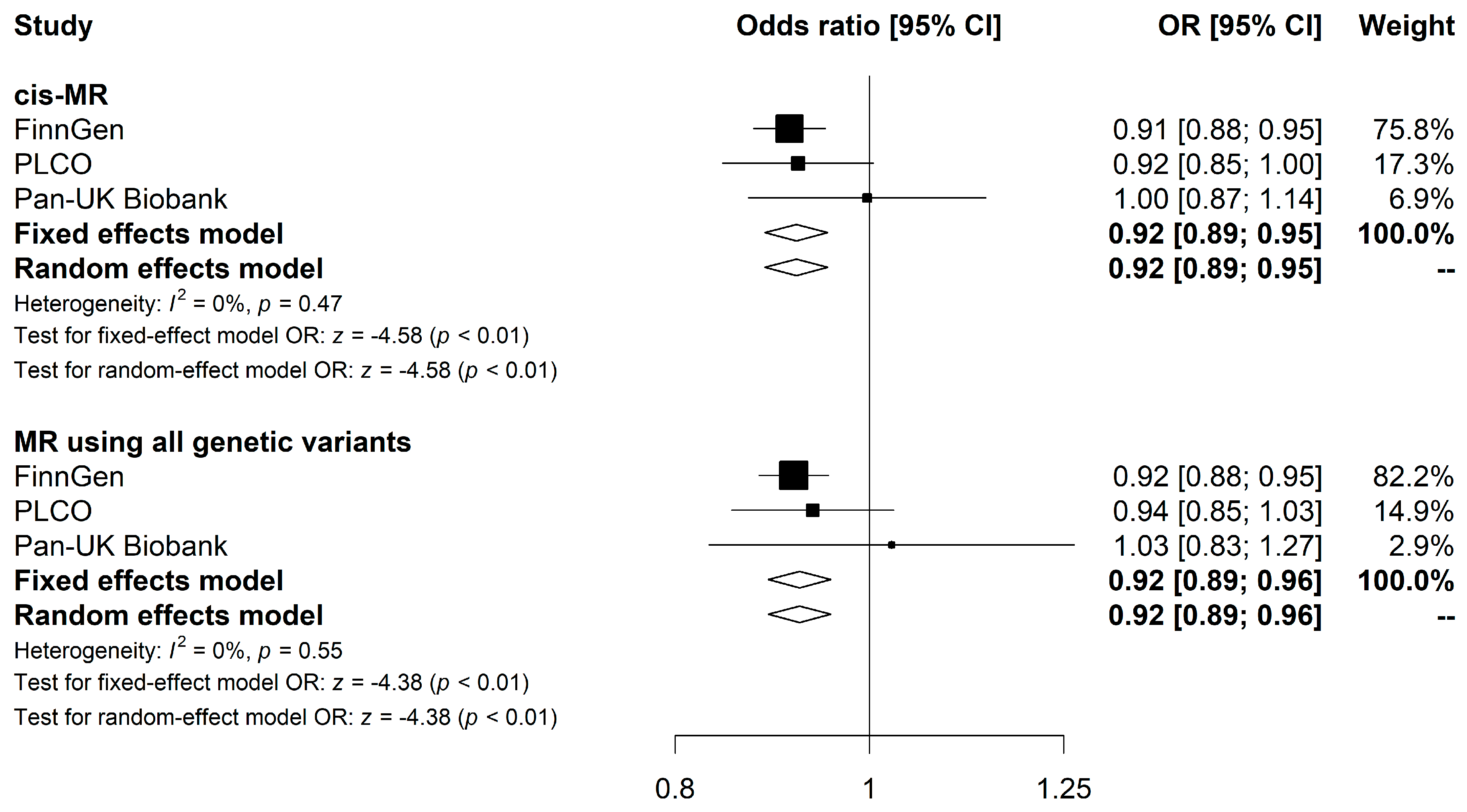
3.4. Meta-Analysis Combining FinnGen, PLCO, and Pan-UK Biobank Results
3.5. CRC Subtype-Specific MR Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC: | Colorectal cancer |
| LPH: | Lactase-phlorizin hydrolase |
| LNP: | Lactase non-persistence |
| SNP: | Single nucleotide polymorphism |
| MR: | Mendelian Randomization |
| IV: | Instrumental variable |
| GWAS: | Genome-wide association study |
| PLCO: | the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial |
| STROBE-MR: | Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization |
| EBI: | European Bioinformatics Institute |
| NHGRI: | Human Genome Research Institute |
| SD: | Standard deviation |
| LD: | Linkage disequilibrium |
| MAF: | Minor allele frequency |
| EAF: | Effect allele frequency |
| IVW: | Inverse-variance weighted |
| InSIDE: | Instrument Strength Independent of Direct Effect |
| OR: | Odds ratio |
| CI: | Confidence interval |
| CaSR: | Calcium-sensing receptor |
| 15-PDGH: | 15-hydroxyprostaglandin dehydrogenase |
| VDR: | Vitamin D receptor |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Zhang, X.; Giovannucci, E. Calcium, vitamin D and colorectal cancer chemoprevention. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 485–494. [Google Scholar]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef]
- de la Chapelle, A. Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 2004, 4, 769–780. [Google Scholar] [CrossRef]
- Eklund, E.A.; Bode, L.; Freeze, H.H. 4.19-Diseases Associated with Carbohydrates/Glycoconjugates*. In Comprehensive Glycoscience; Kamerling, H., Ed.; Elsevier: Oxford, UK, 2007; pp. 339–371. [Google Scholar]
- Catanzaro, R.; Sciuto, M.; Marotta, F. Lactose intolerance: An update on its pathogenesis, diagnosis, and treatment. Nutr. Res. 2021, 89, 23–34. [Google Scholar] [CrossRef]
- Kuokkanen, M.; Enattah, N.S.; Oksanen, A.; Savilahti, E.; Orpana, A.; Järvelä, I. Transcriptional regulation of the lactase-phlorizin hydrolase gene by polymorphisms associated with adult-type hypolactasia. Gut 2003, 52, 647–652. [Google Scholar] [CrossRef]
- Anguita-Ruiz, A.; Aguilera, C.M.; Gil, Á. Genetics of Lactose Intolerance: An Updated Review and Online Interactive World Maps of Phenotype and Genotype Frequencies. Nutrients 2020, 12, 2689. [Google Scholar] [CrossRef]
- Carroccio, A.; Montalto, G.; Cavera, G.; Notarbatolo, A. Lactose intolerance and self-reported milk intolerance: Relationship with lactose maldigestion and nutrient intake. Lactase Deficiency Study Group. J. Am. Coll. Nutr. 1998, 17, 631–636. [Google Scholar] [CrossRef]
- Dewiasty, E.; Setiati, S.; Agustina, R.; Roosheroe, A.G.; Abdullah, M.; Istanti, R.; de Groot, L.C. Prevalence of lactose intolerance and nutrients intake in an older population regarded as lactase non-persistent. Clin. Nutr. ESPEN 2021, 43, 317–321. [Google Scholar] [CrossRef]
- Kato, K.; Ishida, S.; Tanaka, M.; Mitsuyama, E.; Xiao, J.-Z.; Odamaki, T. Association between functional lactase variants and a high abundance of Bifidobacterium in the gut of healthy Japanese people. PLoS ONE 2018, 13, e0206189. [Google Scholar]
- Heine-Bröring, R.C.; Winkels, R.M.; Renkema, J.M.; Kragt, L.; van Orten-Luiten, A.C.; Tigchelaar, E.F.; Chan, D.S.; Norat, T.; Kampman, E. Dietary supplement use and colorectal cancer risk: A systematic review and meta-analyses of prospective cohort studies. Int. J. Cancer 2015, 136, 2388–2401. [Google Scholar] [CrossRef]
- Chau, R.; Dashti, S.G.; Ait Ouakrim, D.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Winship, I.M.; Young, J.P.; Giles, G.G.; Macrae, F.A.; et al. Multivitamin, calcium and folic acid supplements and the risk of colorectal cancer in Lynch syndrome. Int. J. Epidemiol. 2016, 45, 940–953. [Google Scholar] [CrossRef]
- Cruz-Pierard, S.M.; Nestares, T.; Amaro-Gahete, F.J. Vitamin D and Calcium as Key Potential Factors Related to Colorectal Cancer Prevention and Treatment: A Systematic Review. Nutrients 2022, 14, 4934. [Google Scholar] [CrossRef]
- Arayici, M.E.; Basbinar, Y.; Ellidokuz, H. Vitamin D Intake, Serum 25-Hydroxyvitamin-D (25(OH)D) Levels, and Cancer Risk: A Comprehensive Meta-Meta-Analysis Including Meta-Analyses of Randomized Controlled Trials and Observational Epidemiological Studies. Nutrients 2023, 15, 2722. [Google Scholar] [CrossRef]
- Fratila, T.D.; Ismaiel, A.; Dumitrascu, D.L. Microbiome modulation in the prevention and management of colorectal cancer: A systematic review of clinical interventions. Med. Pharm. Rep. 2023, 96, 131–145. [Google Scholar] [CrossRef]
- Tsilidis, K.K.; Branchini, C.; Guallar, E.; Helzlsouer, K.J.; Erlinger, T.P.; Platz, E.A. C-reactive protein and colorectal cancer risk: A systematic review of prospective studies. Int. J. Cancer 2008, 123, 1133–1140. [Google Scholar]
- Murphy, N.; Carreras-Torres, R.; Song, M.; Chan, A.T.; Martin, R.M.; Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Bradbury, K.E. Circulating levels of insulin-like growth factor 1 and insulin-like growth factor binding protein 3 associate with risk of colorectal cancer based on serologic and mendelian randomization analyses. Gastroenterology 2020, 158, 1300–1312.e1320. [Google Scholar]
- Mehta, R.S.; Song, M.; Bezawada, N.; Wu, K.; Garcia-Albeniz, X.; Morikawa, T.; Fuchs, C.S.; Ogino, S.; Giovannucci, E.L.; Chan, A.T. A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J. Natl. Cancer Inst. 2014, 106, dju016. [Google Scholar]
- Kakourou, A.; Koutsioumpa, C.; Lopez, D.S.; Hoffman-Bolton, J.; Bradwin, G.; Rifai, N.; Helzlsouer, K.J.; Platz, E.A.; Tsilidis, K.K. Interleukin-6 and risk of colorectal cancer: Results from the CLUE II cohort and a meta-analysis of prospective studies. Cancer Causes Control. 2015, 26, 1449–1460. [Google Scholar]
- Piepoli, A.; Schirru, E.; Mastrorilli, A.; Gentile, A.; Cotugno, R.; Quitadamo, M.; Merla, A.; Congia, M.; Usai Satta, P.; Perri, F. Genotyping of the lactase-phlorizin hydrolase c/t-13910 polymorphism by means of a new rapid denaturing high-performance liquid chromatography-based assay in healthy subjects and colorectal cancer patients. J. Biomol. Screen 2007, 12, 733–739. [Google Scholar] [CrossRef]
- Rasinperä, H.; Forsblom, C.; Enattah, N.S.; Halonen, P.; Salo, K.; Victorzon, M.; Mecklin, J.P.; Järvinen, H.; Enholm, S.; Sellick, G.; et al. The C/C-13910 genotype of adult-type hypolactasia is associated with an increased risk of colorectal cancer in the Finnish population. Gut 2005, 54, 643–647. [Google Scholar] [CrossRef]
- Gençdal, G.; Salman, E.; Özütemiz, Ö.; Akarca, U.S. Association of LCT-13910 C/T Polymorphism and Colorectal Cancer. Ann. Coloproctol. 2017, 33, 169–172. [Google Scholar] [CrossRef]
- Bácsi, K.; Hitre, E.; Kósa, J.P.; Horváth, H.; Lazáry, A.; Lakatos, P.L.; Balla, B.; Budai, B.; Lakatos, P.; Speer, G. Effects of the lactase 13910 C/T and calcium-sensor receptor A986S G/T gene polymorphisms on the incidence and recurrence of colorectal cancer in Hungarian population. BMC Cancer 2008, 8, 317. [Google Scholar] [CrossRef]
- Tarabra, E.; Pazienza, P.; Borghesio, E.; Actis, G.C.; Tappero, G.; Framarin, L.; Ayoubi, M.; Castellino, F.; Leone, N.; Sansoè, G.; et al. LCT-13910C>T polymorphism-associated lactose malabsorption and risk for colorectal cancer in Italy. Dig. Liver Dis. 2010, 42, 741–743. [Google Scholar] [CrossRef]
- Larsson, S.C.; Mason, A.M.; Kar, S.; Vithayathil, M.; Carter, P.; Baron, J.A.; Michaëlsson, K.; Burgess, S. Genetically proxied milk consumption and risk of colorectal, bladder, breast, and prostate cancer: A two-sample Mendelian randomization study. BMC Med. 2020, 18, 370. [Google Scholar] [CrossRef]
- Lumsden, A.L.; Mulugeta, A.; Hyppönen, E. Milk consumption and risk of twelve cancers: A large-scale observational and Mendelian randomisation study. Clin. Nutr. 2023, 42, 1–8. [Google Scholar] [CrossRef]
- Shepherd, S.J.; Lomer, M.C.; Gibson, P.R. Short-chain carbohydrates and functional gastrointestinal disorders. Am. J. Gastroenterol. 2013, 108, 707–717. [Google Scholar] [CrossRef]
- Misselwitz, B.; Pohl, D.; Frühauf, H.; Fried, M.; Vavricka, S.R.; Fox, M. Lactose malabsorption and intolerance: Pathogenesis, diagnosis and treatment. United Eur. Gastroenterol. J. 2013, 1, 151–159. [Google Scholar] [CrossRef]
- Holma, R.; Laatikainen, R.; Orell, H.; Joensuu, H.; Peuhkuri, K.; Poussa, T.; Korpela, R.; Österlund, P. Consumption of Lactose, Other FODMAPs and Diarrhoea during Adjuvant 5-Fluorouracil Chemotherapy for Colorectal Cancer. Nutrients 2020, 12, 407. [Google Scholar]
- Österlund, P.; Ruotsalainen, T.; Peuhkuri, K.; Korpela, R.; Ollus, A.; Ikonen, M.; Joensuu, H.; Elomaa, I. Lactose intolerance associated with adjuvant 5-fluorouracil-based chemotherapy for colorectal cancer. Clin. Gastroenterol. Hepatol. 2004, 2, 696–703. [Google Scholar] [CrossRef]
- Wright, E.M.; Martín, M.N.G.; Turk, E. Intestinal absorption in health and disease—Sugars. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 943–956. [Google Scholar]
- Grover, S.; Del Greco, M.F.; Stein, C.M.; Ziegler, A. Mendelian Randomization. Methods Mol. Biol. 2017, 1666, 581–628. [Google Scholar] [CrossRef]
- Smith, G.D.; Timpson, N.; Ebrahim, S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann. Med. 2008, 40, 524–541. [Google Scholar] [CrossRef]
- Hingorani, A.; Humphries, S. Nature’s randomised trials. Lancet 2005, 366, 1906–1908. [Google Scholar] [CrossRef]
- Gill, D.; Georgakis, M.K.; Laffan, M.; Sabater-Lleal, M.; Malik, R.; Tzoulaki, I.; Veltkamp, R.; Dehghan, A. Genetically determined FXI (factor XI) levels and risk of stroke. Stroke 2018, 49, 2761–2763. [Google Scholar]
- Gill, D.; Burgess, S. Use of a genetic variant related to circulating FXa (Activated Factor X) levels to proxy the effect of FXa inhibition on cardiovascular outcomes. Circ. Genom. Precis. Med. 2020, 13, 551–553. [Google Scholar]
- Gill, D.; Georgakis, M.K.; Walker, V.M.; Schmidt, A.F.; Gkatzionis, A.; Freitag, D.F.; Finan, C.; Hingorani, A.D.; Howson, J.M.; Burgess, S. Mendelian randomization for studying the effects of perturbing drug targets. Wellcome Open Res. 2021, 6, 16. [Google Scholar]
- Schmidt, A.F.; Finan, C.; Gordillo-Marañón, M.; Asselbergs, F.W.; Freitag, D.F.; Patel, R.S.; Tyl, B.; Chopade, S.; Faraway, R.; Zwierzyna, M. Genetic drug target validation using Mendelian randomisation. Nat. Commun. 2020, 11, 3255. [Google Scholar]
- Gkatzionis, A.; Burgess, S.; Newcombe, P.J. Statistical methods for cis-Mendelian randomization with two-sample summary-level data. Genet. Epidemiol. 2023, 47, 3–25. [Google Scholar]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian randomization. JAMA 2017, 318, 1925–1926. [Google Scholar]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Timpson, N.J.; Higgins, J.P.T.; Dimou, N.; Langenberg, C.; et al. Strengthening the reporting of observational studies in epidemiology using mendelian randomisation (STROBE-MR): Explanation and elaboration. BMJ 2021, 375, n2233. [Google Scholar] [CrossRef]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Cortes, A.; Koprulu, M.; Wörheide, M.A.; Oerton, E.; Cook, J.; Stewart, I.D.; Kerrison, N.D.; et al. Mapping the proteo-genomic convergence of human diseases. Science 2021, 374, eabj1541. [Google Scholar] [CrossRef]
- Pietzner, M.; Wheeler, E.; Carrasco-Zanini, J.; Raffler, J.; Kerrison, N.D.; Oerton, E.; Auyeung, V.P.W.; Luan, J.; Finan, C.; Casas, J.P.; et al. Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun. 2020, 11, 6397. [Google Scholar] [CrossRef]
- Chen, Z.; Boehnke, M.; Wen, X.; Mukherjee, B. Revisiting the genome-wide significance threshold for common variant GWAS. G3 2021, 11, jkaa056. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Luo, J.; Thomassen, J.Q.; Bellenguez, C.; Grenier-Boley, B.; De Rojas, I.; Castillo, A.; Parveen, K.; Küçükali, F.; Nicolas, A.; Peters, O. Genetic Associations Between Modifiable Risk Factors and Alzheimer Disease. JAMA Netw. Open 2023, 6, e2313734. [Google Scholar]
- Papadimitriou, N.; Dimou, N.; Tsilidis, K.K.; Banbury, B.; Martin, R.M.; Lewis, S.J.; Kazmi, N.; Robinson, T.M.; Albanes, D.; Aleksandrova, K.; et al. Physical activity and risks of breast and colorectal cancer: A Mendelian randomisation analysis. Nat. Commun. 2020, 11, 597. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G. Use of allele scores as instrumental variables for Mendelian randomization. Int. J. Epidemiol. 2013, 42, 1134–1144. [Google Scholar] [CrossRef]
- Machiela, M.J.; Huang, W.-Y.; Wong, W.; Berndt, S.I.; Sampson, J.; De Almeida, J.; Abubakar, M.; Hislop, J.; Chen, K.-L.; Dagnall, C.; et al. GWAS Explorer: An open-source tool to explore, visualize, and access GWAS summary statistics in the PLCO Atlas. Sci. Data 2023, 10, 25. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Pan-UKB Team. Available online: https://pan.ukbb.broadinstitute.org (accessed on 2 May 2023).
- Burgess, S. Sample size and power calculations in Mendelian randomization with a single instrumental variable and a binary outcome. Int. J. Epidemiol. 2014, 43, 922–929. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.; Timpson, N.; Davey Smith, G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar]
- Burgess, S.; Bowden, J.; Dudbridge, F.; Thompson, S.G. Robust instrumental variable methods using multiple candidate instruments with application to Mendelian randomization. arXiv 2016, arXiv:1606.03729. [Google Scholar]
- Slob, E.A.W.; Burgess, S. A comparison of robust Mendelian randomization methods using summary data. Genet. Epidemiol. 2020, 44, 313–329. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar]
- Burgess, S.; Thompson, S.G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef]
- Rees, J.M.B.; Wood, A.M.; Dudbridge, F.; Burgess, S. Robust methods in Mendelian randomization via penalization of heterogeneous causal estimates. PLoS ONE 2019, 14, e0222362. [Google Scholar] [CrossRef]
- Higgins, J.P.; Thompson, S.G.; Deeks, J.J.; Altman, D.G. Measuring inconsistency in meta-analyses. BMJ 2003, 327, 557–560. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 6 January 2023).
- Yavorska, O.O.; Burgess, S. MendelianRandomization: An R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 2017, 46, 1734–1739. [Google Scholar]
- Balduzzi, S.; Rücker, G.; Schwarzer, G. How to perform a meta-analysis with R: A practical tutorial. BMJ Ment. Health 2019, 22, 153–160. [Google Scholar]
- Vissers, L.E.T.; Sluijs, I.; van der Schouw, Y.T.; Forouhi, N.G.; Imamura, F.; Burgess, S.; Barricarte, A.; Boeing, H.; Bonet, C.; Chirlaque, M.D.; et al. Dairy Product Intake and Risk of Type 2 Diabetes in EPIC-InterAct: A Mendelian Randomization Study. Diabetes Care 2019, 42, 568–575. [Google Scholar] [CrossRef]
- Bergholdt, H.K.M.; Larsen, M.K.; Varbo, A.; Nordestgaard, B.G.; Ellervik, C. Lactase persistence, milk intake, hip fracture and bone mineral density: A study of 97 811 Danish individuals and a meta-analysis. J. Intern. Med. 2018, 284, 254–269. [Google Scholar] [CrossRef]
- Yuan, S.; Sun, J.; Lu, Y.; Xu, F.; Li, D.; Jiang, F.; Wan, Z.; Li, X.; Qin, L.Q.; Larsson, S.C. Health effects of milk consumption: Phenome-wide Mendelian randomization study. BMC Med. 2022, 20, 455. [Google Scholar] [CrossRef]
- Cho, E.; Smith-Warner, S.A.; Spiegelman, D.; Beeson, W.L.; van den Brandt, P.A.; Colditz, G.A.; Folsom, A.R.; Fraser, G.E.; Freudenheim, J.L.; Giovannucci, E.; et al. Dairy foods, calcium, and colorectal cancer: A pooled analysis of 10 cohort studies. J. Natl. Cancer Inst. 2004, 96, 1015–1022. [Google Scholar] [CrossRef]
- Aune, D.; Lau, R.; Chan, D.S.M.; Vieira, R.; Greenwood, D.C.; Kampman, E.; Norat, T. Dairy products and colorectal cancer risk: A systematic review and meta-analysis of cohort studies. Ann. Oncol. 2012, 23, 37–45. [Google Scholar] [CrossRef]
- Thorning, T.K.; Raben, A.; Tholstrup, T.; Soedamah-Muthu, S.S.; Givens, I.; Astrup, A. Milk and dairy products: Good or bad for human health? An assessment of the totality of scientific evidence. Food Nutr. Res. 2016, 60, 32527. [Google Scholar] [CrossRef]
- Vieira, A.R.; Abar, L.; Chan, D.S.M.; Vingeliene, S.; Polemiti, E.; Stevens, C.; Greenwood, D.; Norat, T. Foods and beverages and colorectal cancer risk: A systematic review and meta-analysis of cohort studies, an update of the evidence of the WCRF-AICR Continuous Update Project. Ann. Oncol. 2017, 28, 1788–1802. [Google Scholar] [CrossRef]
- Barrubés, L.; Babio, N.; Becerra-Tomás, N.; Rosique-Esteban, N.; Salas-Salvadó, J. Association Between Dairy Product Consumption and Colorectal Cancer Risk in Adults: A Systematic Review and Meta-Analysis of Epidemiologic Studies. Adv. Nutr. 2019, 10, S190–S211. [Google Scholar] [CrossRef]
- Kim, J.W. Lactose Intolerance and Colorectal Cancer. Ann. Coloproctol. 2017, 33, 157–158. [Google Scholar] [CrossRef]
- Campbell, B.J.; Finnie, I.A.; Hounsell, E.F.; Rhodes, J.M. Direct demonstration of increased expression of Thomsen-Friedenreich (TF) antigen in colonic adenocarcinoma and ulcerative colitis mucin and its concealment in normal mucin. J. Clin. Investig. 1995, 95, 571–576. [Google Scholar] [CrossRef]
- Evans, R.C.; Fear, S.; Ashby, D.; Hackett, A.; Williams, E.; Van Der Vliet, M.; Dunstan, F.D.; Rhodes, J.M. Diet and colorectal cancer: An investigation of the lectin/galactose hypothesis. Gastroenterology 2002, 122, 1784–1792. [Google Scholar]
- Yang, W.; Liu, L.; Masugi, Y.; Qian, Z.R.; Nishihara, R.; Keum, N.; Wu, K.; Smith-Warner, S.; Ma, Y.; Nowak, J.A.; et al. Calcium intake and risk of colorectal cancer according to expression status of calcium-sensing receptor (CASR). Gut 2018, 67, 1475–1483. [Google Scholar] [CrossRef]
- Fleet, J.C.; Desmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar]
- Norat, T.; Riboli, E. Dairy products and colorectal cancer. A review of possible mechanisms and epidemiological evidence. Eur. J. Clin. Nutr. 2003, 57, 1–17. [Google Scholar] [CrossRef]
- Kozu, T.; Iinuma, G.; Ohashi, Y.; Saito, Y.; Akasu, T.; Saito, D.; Alexander, D.B.; Iigo, M.; Kakizoe, T.; Tsuda, H. Effect of Orally Administered Bovine Lactoferrin on the Growth of Adenomatous Colorectal Polyps in a Randomized, Placebo-Controlled Clinical TrialEffect of bLF on Colorectal Polyps. Cancer Prev. Res. 2009, 2, 975–983. [Google Scholar]
- Pufulete, M. Intake of dairy products and risk of colorectal neoplasia. Nutr. Res. Rev. 2008, 21, 56–67. [Google Scholar]
- Abdelali, H.; Cassand, P.; Soussotte, V.; Daubeze, M.; Bouley, C.; Narbonne, J.F. Effect of dairy products on initiation of precursor lesions of colon cancer in rats. Nutr. Cancer 1995, 24, 121–132. [Google Scholar]
- Parodi, P. Conjugated linoleic acid and other anticarcinogenic agents of bovine milk fat. J. Dairy Sci. 1999, 82, 1339–1349. [Google Scholar]
- Velázquez, O.C.; Zhou, D.; Seto, R.W.; Jabbar, A.; Choi, J.; Lederer, H.M.; Rombeau, J.L. In vivo crypt surface hyperproliferation is decreased by butyrate and increased by deoxycholate in normal rat colon: Associated in vivo effects on c-fos and c-jun expression. J. Parenter. Enter. Nutr. 1996, 20, 243–250. [Google Scholar]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Man Lei, Y.; Jabri, B.; Alegre, M.-L. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar]
- Burgess, S.; Davies, N.M.; Thompson, S.G. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016, 40, 597–608. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| LPH GWAS | CRC GWAS | Colon Cancer GWAS | Rectal Cancer GWAS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study | First Author (Year) | Sample Size | Population | Sex | Study | N Cases | N Controls | Population | Sex | CRC Ascertainment | N Cases | N Controls | N Cases | N Controls |
| Fenland Study | Pietzner (2021) [44] | 10,708 | 100% European | 53% Female | FinnGen Study | 6509 | 287,137 | 100% European | 42% Female | ICD10: C18–C20 | 3935 | 287,137 | 2361 | 287,137 |
| PLCO Atlas | 2065 | 67,500 | 100% European | 45% Female | ICD-O-2 Site: 180/182–189/199/209/212/218 | 1611 | 65,142 | 320 | 65,142 | |||||
| Pan-UK Biobank | 592 | 419,881 | 100% European | 44% Female | Self-reported diagnosis of large bowel cancer/colorectal caner | 1384 | 419,089 | 301 | 420,172 | |||||
| RSID | Position (GRCh38) | Effect Allele | Other Allele | EAF | R2 | F | Beta | SE | p-Value | Associated Gene | cis/trans Variant for LPH Levels |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs4988235 | chr2:135851076 | A | G | 0.31 | 33.28% | 5340.06 | 0.882 | 0.011 | 3 × 10−1451 | MCM6 | cis |
| rs516246 | chr19:48702915 | C | T | 0.49 | 0.81% | 87.01 | 0.127 | 0.013 | 2 × 10−22 | FUT2 | trans |
| rs532436 | chr9:133274414 | G | A | 0.20 | 1.27% | 137.41 | 0.199 | 0.016 | 3 × 10−35 | ABO | trans |
| rs641476 | chr18:32225445 | C | T | 0.61 | 1.07% | 115.85 | 0.150 | 0.013 | 5 × 10−30 | GAREM1 | trans |
| SNP Selected | Effect Allele | Beta | SE | p-Value |
|---|---|---|---|---|
| FinnGen | ||||
| rs4988235 | A | −0.081 | 0.020 | 0.001 |
| rs516246 | C | −0.024 | 0.020 | 0.291 |
| rs635634 (proxy) a | G | 0.006 | 0.025 | 0.386 |
| rs641476 | C | 0.004 | 0.020 | 0.967 |
| PLCO | ||||
| rs4988235 | A | −0.072 | 0.039 | 0.063 |
| rs516246 | C | −0.034 | 0.032 | 0.292 |
| rs532436 | G | 0.047 | 0.040 | 0.237 |
| rs641476 | C | 0.020 | 0.033 | 0.537 |
| Pan-UK Biobank | ||||
| rs4988235 | A | −0.002 | 0.061 | 0.971 |
| rs516246 | C | −0.072 | 0.051 | 0.162 |
| rs532436 | G | 0.144 | 0.066 | 0.030 |
| rs641476 | C | 0.061 | 0.053 | 0.245 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Yao, J.; Yamazaki, H.; Streicher, S.A.; Rao, J.; Nianogo, R.A.; Zhang, Z.; Huang, B.Z. Genetically Determined Circulating Lactase/Phlorizin Hydrolase Concentrations and Risk of Colorectal Cancer: A Two-Sample Mendelian Randomization Study. Nutrients 2024, 16, 808. https://doi.org/10.3390/nu16060808
Han S, Yao J, Yamazaki H, Streicher SA, Rao J, Nianogo RA, Zhang Z, Huang BZ. Genetically Determined Circulating Lactase/Phlorizin Hydrolase Concentrations and Risk of Colorectal Cancer: A Two-Sample Mendelian Randomization Study. Nutrients. 2024; 16(6):808. https://doi.org/10.3390/nu16060808
Chicago/Turabian StyleHan, Sihao, Jiemin Yao, Hajime Yamazaki, Samantha A. Streicher, Jianyu Rao, Roch A. Nianogo, Zuofeng Zhang, and Brian Z. Huang. 2024. "Genetically Determined Circulating Lactase/Phlorizin Hydrolase Concentrations and Risk of Colorectal Cancer: A Two-Sample Mendelian Randomization Study" Nutrients 16, no. 6: 808. https://doi.org/10.3390/nu16060808
APA StyleHan, S., Yao, J., Yamazaki, H., Streicher, S. A., Rao, J., Nianogo, R. A., Zhang, Z., & Huang, B. Z. (2024). Genetically Determined Circulating Lactase/Phlorizin Hydrolase Concentrations and Risk of Colorectal Cancer: A Two-Sample Mendelian Randomization Study. Nutrients, 16(6), 808. https://doi.org/10.3390/nu16060808