Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach

 ,
,  ,
,  and
and 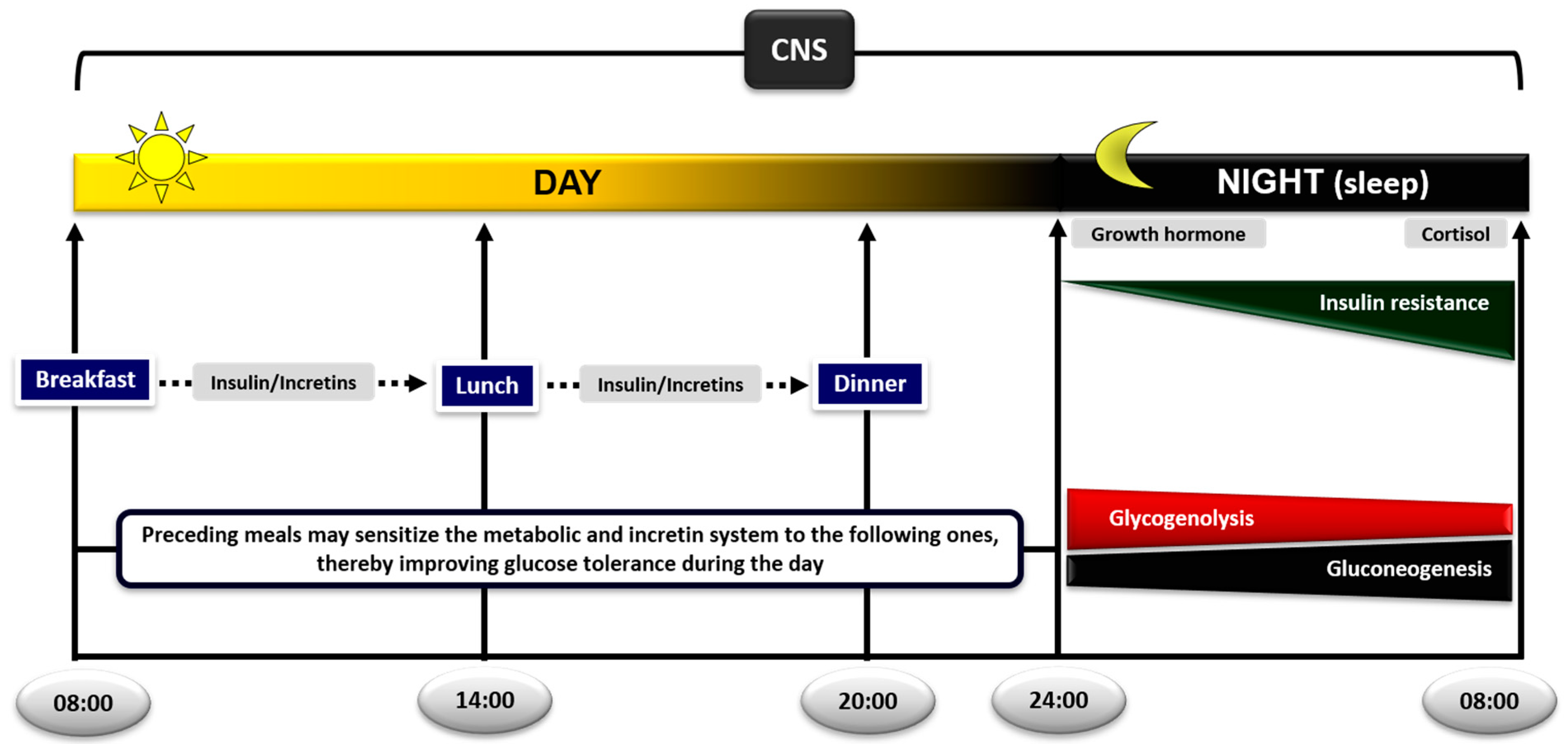{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Physiology of Insulin Effects on Target Tissues
2.1. Liver
2.2. Kidneys
2.3. Skeletal Muscle and Adipose Tissue
3. The Postabsorptive (Fasting) State
4. The Postprandial State
4.1. Oral Glucose Loads versus Mixed Meals
4.2. The Role of the Liver
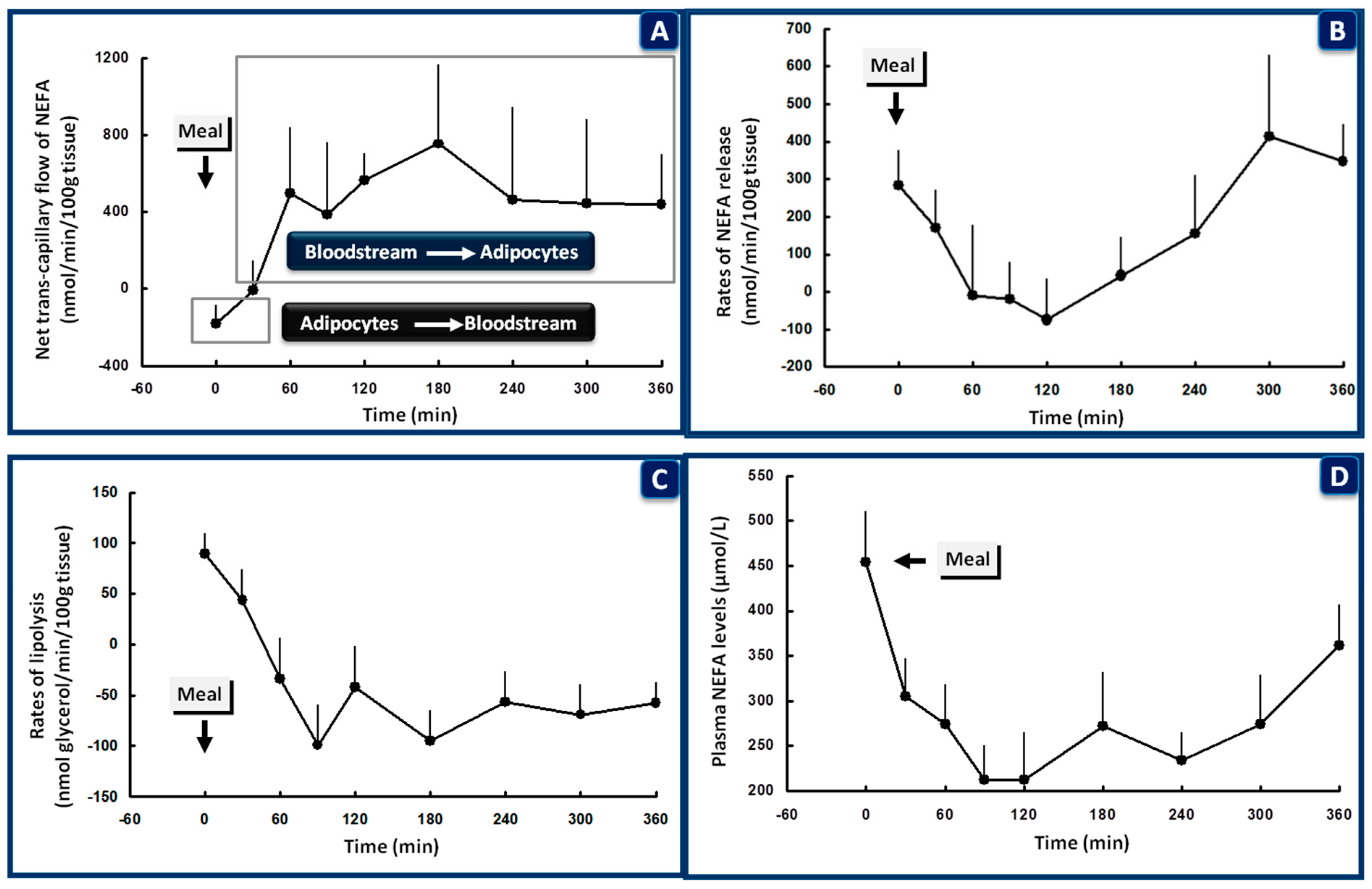
4.3. The Role of Skeletal Muscle and Adipose Tissue
4.4. The Role of de Novo Lipogenesis
4.5. The Role of the Kidneys
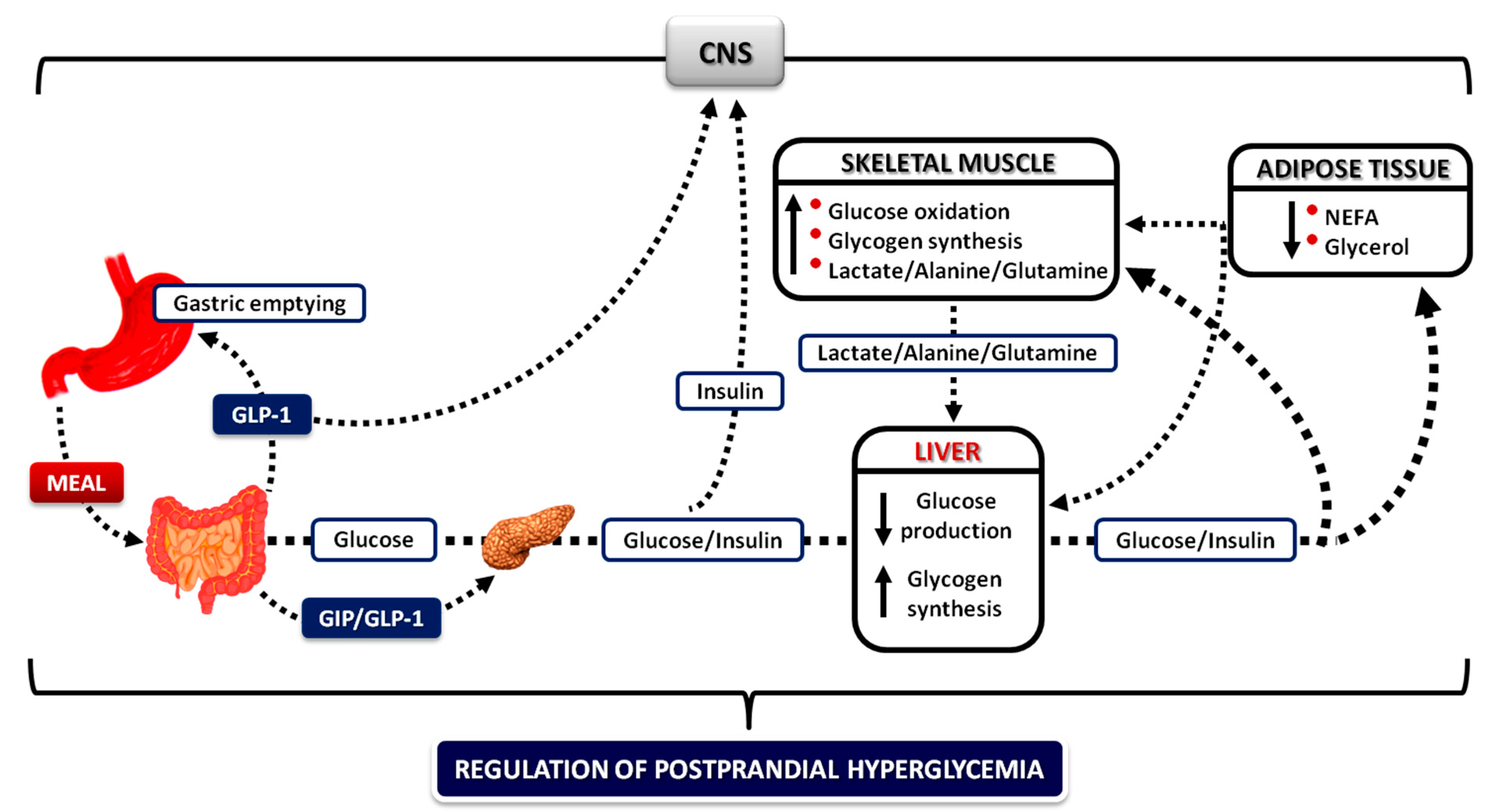
4.6. The Role of the Gastrointestinal Tract
4.6.1. Gastric Emptying
4.6.2. Gastrointestinal Hormones
Ghrelin
Intestinal Hormones
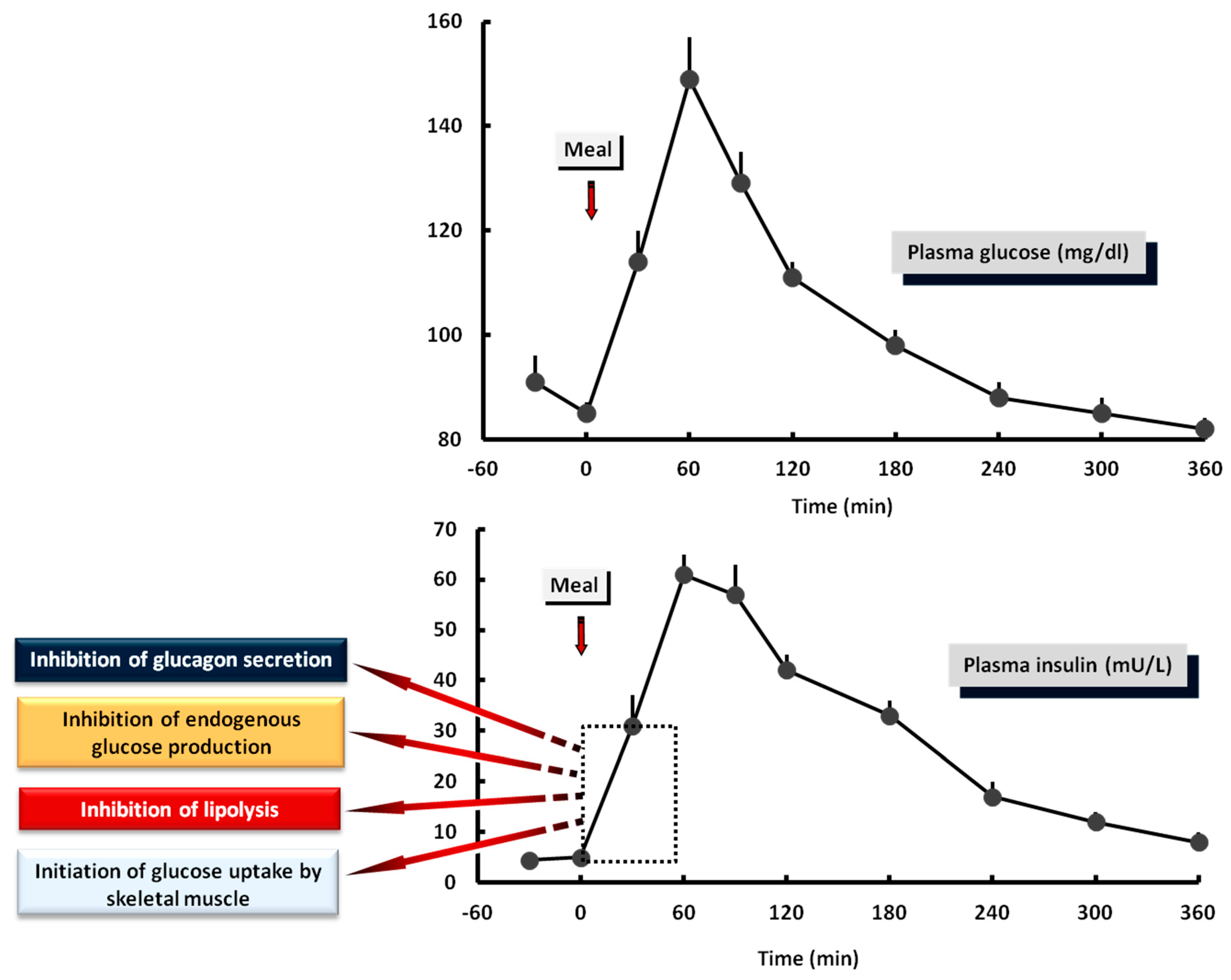
4.7. The Biphasic Manner of Insulin Secretion
4.8. The Role of Hyperglycemia
4.9. The Role of Meal Sequence and Composition
4.9.1. Meal Sequence within the Day
4.9.2. Nutrient Sequence within the Meal
4.9.3. Meal Composition
4.10. The Importance of Insulin Sensitivity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bender, D. Why eat? In Introduction to Nutrition and Metabolism, 2nd ed.; Taylor & Francis: London, UK; Bristol, PA, USA, 1997; pp. 1–10. [Google Scholar]
- Newsholme, E.A.; Dimitriadis, G. Integration of biochemical and physiologic effects of insulin on glucose metabolism. Exp. Clin. Endocrinol. Diabetes 2001, 109, S122–S134. [Google Scholar] [CrossRef] [PubMed]
- Menting, J.G.; Whittaker, J.; Margetts, M.B.; Whittaker, L.J.; Kong, G.K.; Smith, B.J.; Watson, C.J.; Zakova, L.; Kletvikova, E.; Jiracek, J.; et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013, 493, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Haring, H.U.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef]
- Ward, C.W.; Lawrence, M.C. Landmarks in insulin research. Front. Endocrinol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, A. Control of glucose uptake and release by the liver in vivo. Diabetes 1999, 48, 1198–1214. [Google Scholar] [CrossRef]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Agius, L. Hormonal and metabolite regulation of hepatic glucokinase. Annu. Rev. Nutr. 2016, 36, 389–415. [Google Scholar] [CrossRef] [PubMed]
- Rizza, R.; Mandarino, L.J.; Gerich, J.E. Dose-response characteristics for effects of insulin on glucose production and utilization of glucose in man. Am. J. Physiol. Endocrinol. Metab. 1981, 240, E630–E639. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Hendler, R.; Felig, P. Regulation of splachnic and peripheral glucose uptake by insulin and hyperglycemia in man. Diabetes 1983, 32, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bondy, P.K.; James, D.F.; Farrar, B.W. Studies on the role of the liver in human carbohydrate metabolism by the venous catheter technic. I. Normal subjects under fasting conditions and following the injection of glucose. J. Clin. Investig. 1949, 28, 238–244. [Google Scholar] [CrossRef]
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F. Liver and kidney metabolism during prolonged starvation. J. Clin. Investig. 1969, 48, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Exton, J. Gluconeogenesis. Metabolism 1972, 21, 945–990. [Google Scholar] [CrossRef]
- Dietze, G.; Wicklmayer, M.; Hepp, K.; Bogner, W.; Mehnert, H.; Czempiel, H.; Henftling, H. On gluconeogenesis of human liver: Accelerated hepatic glucose formation by increased precursor supply. Diabetologia 1976, 12, 555–561. [Google Scholar] [CrossRef]
- Jenssen, T.; Nurjahan, N.; Consoli, A.; Gerich, J. Failure of substrate-induced gluconeogenesis to increase overall glucose appearance in normal humans. J. Clin. Investig. 1990, 86, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Kalsbeek, A.; Bruinstroop, E.; Yi, C.X.; Klieverik, L.P.; la Fleur, S.E.; Fliers, E. Hypothalamic control of energy metabolism via the autonomic nervous system. Ann. N. Y. Acad. Sci. 2010, 1212, 114–129. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Kahn, R.C. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Edgerton, D.S.; Cherrington, A.D. Is brain insulin action relevant to the control of plasma glucose in humans? Diabetes 2015, 64, 696–699. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E. Regulation of hepatic glucose metabolism in humans. Diabetes Metab. Rev. 1987, 3, 415–459. [Google Scholar] [CrossRef]
- Ferrannini, E.; Barrett, E.J.; Bevilacqua, S.; DeFronzo, R.A. Effect of fatty acids on glucose production and utilization in man. J. Clin. Investig. 1983, 72, 1737–1747. [Google Scholar] [CrossRef]
- Groop, L.C.; Bonadonna, R.C.; DelPrato, S.; Ratheiser, K.; Zyck, K.; Ferrannini, E.; DeFronzo, R.A. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1989, 84, 205–213. [Google Scholar] [CrossRef]
- Williamson, J.R.; Kreisberg, R.A.; Felts, P.W. Mechanism for the stimulation of gluconeogenesis by fatty acids in perfused rat liver. Proc. Natl. Acad. Sci. USA 1966, 56, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.; Goodman, E.H.; Weinhouse, S. Effects of insulin and fatty acids on gluconeogenesis in the rat. J. Biol. Chem. 1967, 242, 3620–3627. [Google Scholar] [CrossRef]
- Wahren, J.; Hagenfeldt, L.; Felig, P. Splachnic and leg exchange of glucose, amino acids and free fatty acids during exercise in diabetes mellitus. J. Clin. Investig. 1975, 55, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Nurjahan, N.; Consoli, A.; Gerich, J.E. Increased lipolysis and its consequences on gluconeogenesis in non-insulin dependent diabetes mellitus. J. Clin. Investig. 1992, 89, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Rizza, R.A.; Mandarino, L.J.; Gerich, J.E. Effects of growth hormone on insulin action in man: Mechanisms of insulin resistance, impaired suppression of glucose production and impaired stimulation of glucose utilization. Diabetes 1982, 31, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Rizza, R.A.; Mandarino, L.J.; Gerich, J.E. Cortisol-induced insulin resistance in man: Impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor defect of insulin action. J. Clin. Endocrinol. Metab. 1982, 54, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Cryer, P.; Rizza, R.A. Hormonal mechanisms in acute glucose counterregulation: The relative roles of glucagon, epinephrine, norepinephrine, growth hormone and cortisol. Metabolism 1980, 29, 1164–1175. [Google Scholar] [CrossRef]
- Shamoon, H.; Hendler, R.; Sherwin, R.S. Synergistic interactions among anti-insulin hormones in the pathogenesis of stress hyperglycemia in humans. J. Clin. Endocrinol. Metab. 1981, 52, 1235–1241. [Google Scholar] [CrossRef]
- Rizza, R.A.; Cryer, P.E.; Gerich, J.E. Role of glucagon, catecholamines and growth hormone in human glucose counterregulation: Effects of somatostatin and combined α- and β-adrenergic blockade on plasma glucose recovery and glucose flux rates following insulin-induced hypoglycemia. J. Clin. Investig. 1979, 64, 62–71. [Google Scholar] [CrossRef]
- Gerich, J.; Woerle, H.; Meyer, C.; Stumvoll, M. Renal gluconeogenesis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef]
- Stumvoll, M.; Chintalapudi, U.; Perriello, G.; Welle, S.; Gutierrez, O.; Gerich, J.E. Uptake and release of glucose by the human kidney: Postabsorptive rates and responses to epinephrine. J. Clin. Investig. 1995, 96, 2528–2533. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Dostou, J.; Nadkarni, V.; Gerich, J.E. Effects of physiological hyperinsulinemia on systemic, renal and hepatic substrate metabolism. Am. J. Physiol. 1998, 275, F915–F921. [Google Scholar] [CrossRef]
- Meyer, C.; Stumvoll, M.; Welle, S.; Woerle, H.J.; Haymond, M.; Gerich, J.E. Relative importance of liver, kidney and substrates in epinephrine-induced increased gluconeogenesis in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E819–E826. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, P.; Kahn, B. Glucose transporters and insulin action. N. Engl. J. Med. 1999, 341, 248–257. [Google Scholar] [CrossRef] [PubMed]
- James, D.; Brown, R.; Navarro, J.; Pilch, P.F. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 1988, 333, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Beitner, R.; Kalant, N. Stimulation of glycolysis by insulin. J. Biol. Chem. 1971, 246, 500–503. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Parry-Billings, M.; Bevan, S.; Dunger, D.; Piva, T.; Krause, U.; Wegener, G.; Newsholme, E.A. Effects of IGF-1 on the rates of glucose transport and utilization in rat skeletal muscle in-vitro. Biochem. J. 1992, 285, 269–274. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Printz, R.L.; Cusi, K.A.; Kinchington, P.; O’Doherty, R.M.; Osawa, H.; Seweli, C.; Consoli, A.; Granner, D.K.; DeFronzo, R.A. Regulation of hexokinase II and glycogen synthase mRNA, protein and activity in human muscle. Am. J. Physiol. Endocrinol. Metab. 1995, 269, E701–E708. [Google Scholar] [CrossRef]
- Vogt, C.; Yki-Jarvinen, H.; Iozzo, P.; Pipek, R.; Pendergrass, M.; Koval, J.; Ardehali, H.; Printz, R.; Granner, D.; DeFronzo, R.; et al. Effects of insulin on subcellular localization of hexokinase II in human skeletal muscle in-vivo. J. Clin. Endocrinol. Metab. 1998, 83, 230–234. [Google Scholar] [CrossRef]
- Shulman, G.; Rossetti, L.; Rothman, D.; Blair, J.; Smith, D. Quantitative analysis of glycogen repletion by nuclear magnetic resonance spectroscopy in the conscious rat. J. Clin. Investig. 1987, 80, 387–393. [Google Scholar] [CrossRef]
- Rossetti, L.; Giaccari, A. Relative contribution of glycogen synthesis and glycolysis to insulin‑mediated glucose uptake. J. Clin. Investig. 1990, 85, 1785–1792. [Google Scholar] [CrossRef]
- Consoli, A.; Nurjahan, N.; Gerich, J.E.; Mandarino, L.J. Skeletal muscle is a major site of lactate uptake and release during hyperinsulinemia. Metabolism 1992, 41, 176–179. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Leech, T. Carbohydrate metabolism. In Functional Biochemistry in Health and Disease; Willey-Blackwell: Oxford, UK, 2009; Chapter 6; pp. 97–127. [Google Scholar]
- Frayn, K.; Coppack, S.; Humphreys, S.M.; Whyte, P.L. Metabolic characteristics of adipose tissue in vivo. Clin. Sci. 1989, 76, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Cori, C.F. The glucose-lactic acid cycle and gluconeogenesis. Curr. Top. Cell. Regul. 1981, 18, 377–387. [Google Scholar]
- Newsholme, E.A.; Crabtree, B. Substrate cycles in metabolic regulation and in heat generation. Biochem. Soc. Symp. 1976, 41, 61–109. [Google Scholar]
- Felig, P.; Pozefsk, T.; Marlis, E.; Cahill, G.F. Alanine: Key role in gluconeogenesis. Science 1970, 167, 1003–1004. [Google Scholar] [CrossRef]
- Garber, A.J.; Karl, I.E.; Kipnis, D.M. Alanine and glutamine synthesis and release from skeletal muscle. J. Biol. Chem. 1976, 251, 826–835. [Google Scholar] [CrossRef]
- Felig, P. The glucose-alanine cycle. Metabolism 1973, 22, 179–207. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Coppack, S.W.; Patel, J.N.; Lawrence, V.J. Nutritional regulation of lipid metabolism in human adipose tissue. Exp. Clin. Endocrinol. Diabetes 2001, 109, S202–S214. [Google Scholar] [CrossRef]
- Frayn, K.N.; Shadid, S.; Hamlani, R.; Humphreys, S.M.; Clark, M.L.; Fielding, B.A.; Boland, O.; Coppack, S.W. Regulation of fatty acid movement in human adipose tissue in the postabsorptive to postprandial transition. Am. J. Physiol. Endocrinol. Metab. 1994, 266, E308–E317. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N. Adipose tissue as a buffer for daily lipid flux. Diabetologia 2002, 45, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Coppack, S.W.; Jensen, M.D.; Miles, J.M. The in-vivo regulation of lipolysis in humans. J. Lipid Res. 1994, 35, 177–193. [Google Scholar] [PubMed]
- Eckel, R.H. Lipoprotein lipase: A multifactorial enzyme relevant to common metabolic diseases. N. Engl. J. Med. 1989, 320, 1060–1068. [Google Scholar] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty acid cycle: Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Frayn, K.N. The glucose-fatty acid cycle: A physiological perspective. Biochem. Soc. Trans. 2003, 31, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef]
- Shulman, G. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Janssen, I.; Heymsfield, S.B.; Wang, Z.; Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. 2000, 89, 81–88. [Google Scholar] [CrossRef]
- Frayn, K. Fat as a fuel: Emerging understanding of the adipose tissue-skeletal muscle axis. Acta Physiol. 2010, 199, 509–518. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93, 52–59. [Google Scholar] [CrossRef]
- Baron, A.; Steinberg, H.; Brechtel, G.; Johnson, A. Skeletal muscle blood flow independently modulates insulin-mediated glucose uptake. Am. J. Physiol. 1994, 266, E248–E253. [Google Scholar] [CrossRef] [PubMed]
- Coggins, M.; Lindner, J.; Rattigan, S.; Jahn, L.; Fasy, E.; Kaul, S.; Barrett, E. Physiological hyperinsulinemia enhances human skeletal muscle perfusion by capillary recruitment. Diabetes 2001, 50, 2682–2690. [Google Scholar] [CrossRef]
- Frayn, K.N.; Karpe, F. Regulation of human subcutaneous adipose tissue blood flow. Int. J. Obes. 2014, 38, 1019–1026. [Google Scholar] [CrossRef]
- Karpe, F.; Fielding, B.; Ilic, V.; Macdonald, I.A.; Summers, L.; Frayn, K.N. Adipose tissue blood flow response is related to aspects of insulin sensitivity. Diabetes 2002, 51, 2467–2473. [Google Scholar] [CrossRef]
- Wahren, J.; Felig, P.; Ahlborg, G.; Jorfeldt, L. Glucose metabolism during leg exercise in man. J. Clin. Investig. 1971, 50, 2715–2725. [Google Scholar] [CrossRef]
- Lambadiari, V.; Mitrou, P.; Maratou, E.; Raptis, A.; Raptis, S.A.; Dimitriadis, G. Increases in muscle blood flow after mixed meals are decreased at all stages of type 2 diabetes. Clin. Endocrinol. 2012, 76, 825–830. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Lambadiari, V.; Mitrou, P.; Maratou, E.; Boutati, E.; Panagiotakos, D.; Economopoulos, T.; Raptis, S.A. Impaired postprandial blood flow in adipose tissue may be an early marker of insulin resistance in type 2 diabetes. Diabetes Care 2007, 30, 3128–3130. [Google Scholar] [CrossRef]
- Polonsky, K.S.; Given, B.D.; Van Gauter, E. Twenty-four hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J. Clin. Investig. 1988, 81, 442–448. [Google Scholar] [CrossRef]
- Gerich, J.E. Control of glycemia. Belliere’s Clin. Endocrinol. Metab. 1993, 7, 551–586. [Google Scholar] [CrossRef]
- Felig, P.; Wahren, J. Influence of endogenous insulin secretion on splachnic glucose and amino acid metabolism in man. J. Clin. Investig. 1971, 50, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Dinneen, S.; Gerich, J.E.; Rizza, R.A. Carbohydrate metabolism in non-insulin dependent diabetes mellitus. N. Engl. J. Med. 1992, 327, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Physiology of glucose homeostasis. Diabetes Obes. Metab. 2000, 2, 345–350. [Google Scholar] [CrossRef]
- Andres, R.; Cader, G.; Zierler, K.L. The quantitatively minor role of carbohydrate in oxidative metabolism by skeletal muscle in intact man in the basal state: Measurements of oxygen and glucose uptake and carbon dioxide and lactate production in the forearm. J. Clin. Investig. 1956, 35, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Kreisberg, R.A.; Pennington, L.F.; Boshell, B.R. Lactate turnover and gluconeogenesis in normal and obese humans. Diabetes 1970, 19, 53–63. [Google Scholar] [CrossRef]
- Consoli, A.; Nurjhan, N.; Reilly, J.J.; Bier, D.M.; Gerich, J.E. Contribution of liver and skeletal muscle to alanine and lactate metabolism in humans. Am. J. Physiol. Endocrinol. Metab. 1990, 259, E677–E684. [Google Scholar] [CrossRef]
- Garber, A.; Karl, I.; Kipnis, D. Alanine and glutamine synthesis and release from skeletal muscle. II The precursor role of amino acids in alanine and glutamine synthesis. J. Biol. Chem. 1976, 251, 836–843. [Google Scholar] [CrossRef]
- Nurjhan, N.; Bucci, A.; Perriello, G.; Stumvoll, M.; Dailey, G.; Bier, D.M.; Toft, I.; Jenssen, M.; Gerich, J.E. Glutamine: A major gluconeogenic precursor and vehicle for interorgan carbon transport in man. J. Clin. Investig. 1995, 95, 272–277. [Google Scholar] [CrossRef]
- Perriello, G.; Jorde, R.; Nurjhan, N.; Stumvoll, M.; Dailey, G.; Jenssen, T.; Bier, M.; Gerich, J.E. Estimation of glucose-alanine-lactate-glutamine cycles in postabsorptive humans: Role of skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 1995, 269, E443–E450. [Google Scholar] [CrossRef]
- Cahill, G.F.; Herrera, M.G.; Morgan, A.P.; Soeldner, J.S.; Steinke, J.; Levy, P.L.; Reichard, G.A.; Kipnis, D.M. Hormone-fuel interrelationship during fasting. J. Clin. Investig. 1966, 45, 1751–1769. [Google Scholar] [CrossRef]
- Owen, O.E.; Reichard, G.A. Human forearm metabolism during progressive starvation. J. Clin. Investig. 1971, 50, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Bolli, G.B.; de Feo, P.; de Cosmo, S.; Perriello, G.; Ventura, M.M.; Calcinaro, F.; Lolli, C.; Campbell, P.; Brunetti, P.; Gerich, J.E. Demonstration of a dawn phenomenon in normal human volunteers. Diabetes 1984, 33, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.I.; Lin, Q.X.; Gwynne, J.T.; Jacobs, S. Fasting early morning rise in peripheral insulin: Evidence of the dawn phenomenon in nondiabetes. Diabetes Care 1984, 7, 32–35. [Google Scholar] [CrossRef]
- De Feo, P.; Perrriello, G.; Ventura, M.M.; Calcinaro, F.; Basta, G.; Lolli, C.; Cruciani, C.; Dell’Olio, A.; Santeusanio, F.; Brunetti, P.; et al. Studies on overnight insulin requirements and metabolic clearance rate of insulin in normal and diabetic man: Relevance to the pathogenesis of the dawn phenomenon. Diabetologia 1986, 29, 475–480. [Google Scholar] [CrossRef][Green Version]
- Frayn, K.N.; Humphreys, S.M. Metabolic characteristics of human subcutaneous abdominal adipose tissue after overnight fast. Am. J. Physiol. Endocrinol. Metab. 2011, 302, E468–E475. [Google Scholar] [CrossRef]
- Miles, J.M.; Wooldridge, D.; Grellner, W.J.; Windsor, S.; Isley, W.L.; Klein, S.; Harris, W.S. Nocturnal and postprandial free fatty acid kinetics in normal and type2 diabetic subjects. Diabetes 2003, 52, 675–681. [Google Scholar] [CrossRef]
- Mitrakou, A.; Ryan, C.; Veneman, T.; Mokan, M.; Jenssen, T.; Kiss, I.; Durrant, J.; Cryer, P.; Gerich, J.E. Hierarchy of glycemic thresholds for counterregulatory hormone secretiom, symptoms and cerebral dysfunction. Am. J. Physiol. Endocrinol. Metab. 1991, 260, E67–E74. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain metabolism during fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Rothman, D.L.; Magnusson, I.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 1991, 254, 573–576. [Google Scholar] [CrossRef]
- Landau, B.R.; Wahren, J.; Chandrainouli, V.; Schuman, W.C.; Ekberg, K.; Kalhan, S.C. Contribution of gluconeogenesis to glucose production in the fasting state. J. Clin. Investig. 1996, 98, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Cauter, E.V.; Blackman, J.D.; Roland, D.; Spire, J.P.; Refetoff, S.; Polonsky, K.S. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J. Clin. Investig. 1991, 88, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Bolli, G.B.; Cryer, P.; Gerich, J.E. Pathogenesis of the dawn phenomenon in patients with insulin-dependent diabetes mellitus. Accelerated glucose production and impaired glucose utilization due to nocturnal surges in growth hormone secretion. N. Engl. J. Med. 1985, 312, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Perriello, G.; De Feo, P.; Torlone, E.; Fanelli, C.; Santeusanio, F.; Brunetti, P.; Bolli, G.B. Nocturnal spikes of growth hormone secretion cause the dawn phenomenon in type 1 (insulin-dependent) diabetes mellitus by decreasing hepatic (and extrahepatic) seneitivity to insulin in the absence of insulin waning. Diabetologia 1990, 33, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Moller, N.; Butler, P.C.; Antsiferov, M.A.; Alberti, K.G.M.M. Effects of growth hormone on insulin sensitivity and forearm metabolism in normal man. Diabetologia 1989, 32, 105–110. [Google Scholar] [CrossRef]
- Oster, H.; Challet, E.; Ott, V.; Arvat, E.; de Kloet, E.R.; Dijk, D.; Lightman, S.; Vgontzas, A.; van Gauter, E. The functional and clinical significance of the 24-hour rhythm of circulating glucocorticoids. Endocr. Rev. 2017, 38, 3–45. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Parry-Billings, M.; Leighton, B.; Piva, T.; Dunger, D.; Calder, P.; Bond, J.; Newsholme, E.A. Studies on the effects of growth hormone administration in vivo on the rates of glucose transport and utilization in rat skeletal muscle. Eur. J. Clin. Investig. 1994, 24, 161–165. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Leighton, B.; Parry-Billings, M.; Sasson, S.; Young, M.; Krause, U.; Bevan, S.; Piva, T.; Wegener, G.; Newsholme, E.A. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochem. J. 1997, 321, 707–712. [Google Scholar] [CrossRef]
- Nielsen, S.; Moller, N.; Christiansen, J.S.; Jorgensen, J.O. Pharmacological antilipolysis restores insulin sensitivity during growth hormone exposure. Diabetes 2001, 50, 2301–2308. [Google Scholar] [CrossRef]
- Norrelund, H.; Nielsen, S.; Christiansen, J.S.; Jorgensen, J.O.; Moller, N. Modulation of basal glucose metabolism and insulin sensitivity by growth hormone and free fatty acids during short-term fasting. Eur. J. Endocrinol. 2004, 150, 779–787. [Google Scholar] [CrossRef][Green Version]
- Sakharova, A.A.; Horowitz, J.F.; Surya, S.; Goldenberg, N.; Harber, M.P.; Symons, K.; Barkan, A. Role of growth hormone in regulating lipolysis, proteolysis and hepatic glucose production during fasting. J. Clin. Endocrinol. Metab. 2008, 93, 2755–2759. [Google Scholar] [CrossRef] [PubMed]
- Djurhuus, C.B.; Gravholt, C.H.; Nielsen, S.; Mengel, A.; Christiansen, J.S.; Schmitz, O.E.; Moller, N. Effects of cortisol on lipolysis and regional interstitial glycerol levels in humans. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E172–E177. [Google Scholar] [CrossRef] [PubMed]
- Samra, J.S.; Clark, M.L.; Humphreys, S.M.; Macdolald, I.A.; Matthews, D.R.; Frayn, K.N. Effects of morning rise in cortisol concentration on regulation of lipolysis in subcutaneous adipose tissue. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E996–E1002. [Google Scholar] [CrossRef] [PubMed]
- Samra, J.S.; Clark, M.L.; Humphreys, S.M.; Macdonald, I.A.; Bannister, P.A.; Matthews, D.R.; Frayn, K.N. Suppression of the nocturnal rise in growth hormone reduces subsequent lipolysis in subcutaneous adipose tissue. Eur. J. Clin. Investig. 1999, 29, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.P.; Sandoval, D.A.; Seeley, R.J. Central nervous system integration of satiety signals. Curr. Biol. 2013, 23, R379–R388. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; DeFronzo, R. Insulin actions in vivo. In International Textbook of Diabetes Mellitus, 4th ed.; DeFronzo, R., Ferrannini, E., Zimmet, P., Alberti, G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; Chapter 14; pp. 211–233. [Google Scholar]
- McMahon, M.; Marsh, H.; Rizza, R. Comparison of the pattern of postprandial carbohydrate metabolism after ingestion of a glucose drink or a mixed meal. J. Clin. Endocrinol. Metab. 1989, 68, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.R.; Owens, D.R.; Luzio, S.; Williams, S.; Hayes, T.M. The glucose-dependent insulinotropic polypeptide response to oral glucose and mixed meals is increased in patients with type 2 (noninsulin dependent) diabetes mellitus. Diabetologia 1989, 32, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.C.; Zhao, X.T.; Wang, L. Fat absorption is not complete by midgut but is dependent on the load of fat. Am. J. Physiol. 1996, 271, G62–G67. [Google Scholar] [CrossRef]
- Woerle, H.J.; Meyer, C.; Dostou, J.M.; Gosmanov, N.R.; Islam, N.; Popa, E.; Wittlin, S.D.; Welle, S.L.; Gerich, J.E. Pathways for glucose disposal after meal ingestion in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E716–E725. [Google Scholar] [CrossRef]
- Najjar, S.; Perdomo, G. Hepatic insulin clearance: Mechanism and physiology. Physiology 2019, 34, 198–215. [Google Scholar] [CrossRef]
- Farmer, T.D.; Jenkins, E.C.; O’Brien, T.P.; McCoy, G.A.; Havlik, A.E.; Nass, E.R.; Nicholson, W.E.; Prinz, R.I.; Shiota, M. Comparison of the physiological relevance of systemic vs portal insulin delivery to evaluate whole body glucose flux during an insulin clamp. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E206–E222. [Google Scholar] [CrossRef]
- Felig, P.; Wahren, J.; Hendler, R. Influence of oral glucose ingestion on splachnic glucose and gluconeogenic substrate metabolism in man. Diabetes 1975, 24, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Tesfamariam, B.; Cohen, R.A. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am. J. Physiol. Endocrinol. Metab. 1992, 263, H321–H326. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Yamagishi, D.; Matsumura, T.; Kaneda, Y.; Yorek, M.; Beebe, D.; Oates, P.; Hammes, H.P.; Giardino, I.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycemia damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes and cardiovascular disease? The common soil hypothesis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 816–823. [Google Scholar] [CrossRef]
- Stegenga, M.E.; van der Crabben, S.N.; Levi, M.; de Vos, A.F.; Tank, M.W.; Sauerwein, H.P.; van der Poll, T. Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans. Diabetes 2006, 55, 1807–1812. [Google Scholar] [CrossRef]
- Lemkes, B.A.; Hermanides, J.; Devries, J.H.; Holleman, F.; Meijers, J.C.M.; Hoekstra, J.B.L. Hyperglycemia: A prothrombotic factor? J. Thromb. Haemost. 2010, 8, 1663–1669. [Google Scholar] [CrossRef]
- King, G.L.; Goodman, D.; Buznay, S.; Moses, A.; Kahn, R.C. Receptors and growth-promoting effects of insulin and insulin-like growth factors on cells from bovine retinal capillaries and aorta. J. Clin. Investig. 1985, 75, 1028–1036. [Google Scholar] [CrossRef]
- Monnier, L.; Colette, C. Glycemic variability. Diabetes Care 2008, 31, S150–S154. [Google Scholar] [CrossRef]
- Stout, R.W. Insulin-stimulated lipogenesis in arterial tissue in relation to diabetes and atheroma. Lancet 1968, 2, 283–290. [Google Scholar] [CrossRef]
- Dandona, P.; Chaudhuri, A.; Dhindsa, S. Proinflammatory and prothrombotic effects of hypoglycemia. Diabetes Care 2010, 33, 1686–1687. [Google Scholar] [CrossRef]
- Gample, J.M.; Chibrikov, E.; Twells, L.K.; Midodzi, W.K.; Young, S.W.; MacDonald, D.; Majumdar, S.R. Association of insulin dosage with mortality or major adverse cardiovascular events: A retrospective cohort study. Lancet Diabetes Endocrinol. 2016. [Google Scholar] [CrossRef]
- Ferrannini, E.; Galvan, A.Q.; Gastaldelli, A.; Camastra, S.; Sironi, A.M.; Toschi, E.; Baldi, S.; Frascerra, S.; Monzani, F.; Antonelli, A.; et al. Insulin: New roles for an ancient hormone. Eur. J. Clin. Investig. 1999, 29, 842–852. [Google Scholar] [CrossRef]
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinemia in man. Diabetologia 1985, 28, 70–75. [Google Scholar]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31, S262–S268. [Google Scholar] [CrossRef]
- Meyer, C.; Dostou, J.M.; Welle, S.L.; Gerich, J.E. Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E419–E427. [Google Scholar] [CrossRef]
- Ferrannini, E.; Bjorkman, O.; Reichard, G.A.; Pilo, A.; Olsson, M.; Wahren, J.; DeFronzo, R.A. The disposal of an oral glucose load in healthy subjects. Diabetes 1985, 34, 580–588. [Google Scholar] [CrossRef]
- Firth, R.G.; Bell, P.M.; Marsh, H.M.; Hansen, I.; Rizza, R.A. Postprandial hyperglycemia in patients with noninsulin-dependent diabetes mellitus: Role of hepatic and extrahepatic tissues. J. Clin. Investig. 1986, 77, 1525–1532. [Google Scholar] [CrossRef]
- Jackson, R.A.; Roshania, R.D.; Hawa, M.I.; Sim, B.M.; DiSilvio, L. Impact of glucose ingestion on hepatic and peripheral glucose metabolism in man: An analysis based on simultaneous use of the forearm and double isotope techniques. J. Clin. Endocrinol. Metab. 1986, 63, 541–549. [Google Scholar] [CrossRef]
- Frayn, K.N.; Coppack, S.W.; Humphreys, S.M.; Clark, M.L.; Evans, R.D. Periprandial regulation of lipid metabolism in insulin-treated diabetes mellitus. Metabolism 1993, 42, 504–510. [Google Scholar] [CrossRef]
- Magnusson, I.; Chandramouli, V.; Schumann, W.C.; Kumaran, K.; Wahren, J.; Landau, B. Quantitation of the pathways of hepatic glycogen formation on ingesting a glucose load. J. Clin. Investig. 1987, 80, 1748–1754. [Google Scholar] [CrossRef]
- Kelley, D.; Mitrakou, A.; Marsh, H.; Schwenk, F.; Benn, J.; Sonnenberg, G.; Arcangeli, M.; Aoki, T.; Sorensen, J.; Berger, M.; et al. Skeletal muscle glycolysis, oxidation and storage of an oral glucose load. J. Clin. Investig. 1988, 81, 1563–1571. [Google Scholar] [CrossRef]
- Magnusson, I.; Chandramouli, V.; Schuman, W.C.; Kumaran, K.; Wahren, J.; Landau, B.R. Pathways of hepatic glycogen formation in humans following ingestion of a glucose load in the fed state. Metabolism 1989, 38, 583–585. [Google Scholar] [CrossRef]
- Taylor, R.; Magnusson, I.; Rothman, D.L.; Cline, G.W.; Caumo, A.; Cobelli, C.; Shulman, G.I. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J. Clin. Investig. 1996, 97, 126–132. [Google Scholar] [CrossRef]
- Petersen, K.F.; Cline, G.W.; Gerard, D.P.; Magnusson, I.; Rothman, D.L.; Shulman, G.I. Contribution of net hepatic glycogen synthesis to disposal of an oral glucose load in humans. Metabolism 2001, 50, 598–601. [Google Scholar] [CrossRef]
- Shulman, G.I.; Cline, G.; Schumann, W.C.; Chandramouli, V.; Kumaran, K.; Landau, B.R. Quantitative comparison of pathways of hepatic glycogen repletion in fed and fasted humans. Am. J. Physiol. Endocrinol. Metab. 1990, 259, E335–E341. [Google Scholar] [CrossRef]
- Kruszynska, Y.T.; Mulford, M.I.; Yu, J.G.; Armstrong, D.A.; Olefsky, J.M. Effects of nonesterified fatty acids on glucose metabolism after glucose ingestion. Diabetes 1997, 46, 1586–1593. [Google Scholar] [CrossRef]
- Mitrakou, A.; Kelley, D.; Venema, T.; Jenssen, T.; Pangburn, T.; Reilly, J.; Gerich, J. Contribution of abnormal muscle and liver glucose metabolism to postprandial hyperglycemia in NIDDM. Diabetes 1990, 39, 1381–1390. [Google Scholar] [CrossRef]
- Sidossis, L.S.; Wolfe, R.R. Glucose and insulin-induced inhibition of fatty acid oxidation: The glucose-fatty acid cycle reversed. Am. J. Physiol. Endocrinol. Metab. 1996, 270, E733–E738. [Google Scholar] [CrossRef] [PubMed]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef]
- Kelley, D.E.; Mokan, M.; Simoneau, J.A.; Mandarino, L.J. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J. Clin. Investig. 1993, 92, 91–98. [Google Scholar] [CrossRef]
- Kelley, D.; Mandarino, L.J. Fuel selection in human skeletal muscle in insulin resistance. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef]
- Kelley, D. Skeletal muscle fat oxidation: Timing and flexibility are everything. J. Clin. Investig. 2005, 115, 1699–1702. [Google Scholar] [CrossRef]
- Taylor, R.; Price, T.B.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Direct measurement of change in muscle glycogen concentration after a mixed meal in normal subjects. Am. J. Physiol. Endocrinol. Metab. 1993, 265, E224–E229. [Google Scholar] [CrossRef] [PubMed]
- Thiebaud, D.; Jacot, E.; DeFronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.P. The effect of graded doses of insulin on total glucose uptake, glucose oxidation and glucose storage in man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Boutati, E.; Maratou, E.; Koukkou, E.; Tzanela, M.; Thalassinos, N.; Raptis, S.A. Glucose and lipid fluxes in the adipose tissue after meal ingestion in hyperthyroidism. J. Clin. Endocrinol. Metab. 2006, 91, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Wahren, J.; Felig, P.; DeFronzo, R.A. The role of fractional glucose extraction in the regulation of splachnic glucose metabolism in normal and diabetic man. Metabolism 1980, 29, 28–35. [Google Scholar] [CrossRef]
- Jenkins, D.J.A.; Wolever, T.M.S.; Collier, G.R.; Ocana, A.; Rao, A.V.; Buckley, G.; Lam, Y.; Mayer, A.; Thompson, L.U. Metabolic effects of a low-glycemic-index diet. Am. J. Clin. Nutr. 1987, 46, 968–975. [Google Scholar] [CrossRef]
- Brynes, A.E.; Adamson, J.; Dornhorst, A.; Frost, G.S. The beneficial effect of a diet with low glycaemic index on 24h glucose profiles in healthy young people as assessed by continuous glucose monitoring. Br. J. Nutr. 2005, 93, 179–182. [Google Scholar] [CrossRef]
- Agius, L. High-carbohydrate diets induce hepatic insulin resistance to protect the liver from substrate overload. Biochem. Pharmacol. 2013, 85, 306–312. [Google Scholar] [CrossRef]
- Daly, M.E.; Vale, C.; Walker, M.; Alberti, K.G.M.M.; Mathers, J.C. Dietary carbohydrates and insulin sensitivity: A review of the evidence and clinical implications. Am. J. Clin. Nutr. 1997, 66, 1072–1085. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, G.M.; Moore, S.M.; Hamley, S.; Selathurai, A.; Bruce, C.R. The effect of ingested glucose dose on the suppression of endogenous glucose production in humans. Diabetes 2017, 66, 2400–2406. [Google Scholar] [CrossRef] [PubMed]
- Nurjhan, N.; Campbell, P.J.; Kennedy, F.P.; Miles, J.M.; Gerich, J.E. Insulin dose-response characteristics for suppression of glycerol release and conversion to glucose in humans. Diabetes 1986, 35, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K. Integration of carbohydrate, fat, and protein metabolism in normal daily life. In Metabolic Regulation: A Human Perspective, 3rd ed.; Willey-Blackwell: Oxford, UK, 2010; Chapter 7; pp. 169–212. [Google Scholar]
- Angel, A.; Bray, G.A. Synthesis of fatty acids and cholesterol by liver, adipose tissue, and intestinal mucosa from obese and control patients. Eur. J. Clin. Investig. 1979, 9, 355–362. [Google Scholar] [CrossRef]
- Aas, V.; Kase, E.T.; Solberg, R.; Jensen, J.; Rustan, A.C. Chronic hyperglycaemia promotes lipogenesis and triacylglycerol accumulation in human skeletal muscle cells. Diabetologia 2004, 47, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Acheson, K.J.; Flatt, J.P.; Jequier, E. Glycogen synthesis versus lipogenesis after 500-gram carbohydrate meal in man. Metabolism 1982, 31, 1234–1240. [Google Scholar] [CrossRef]
- Acheson, K.J.; Schutz, Y.; Bessard, T.; Ravussin, E.; Jequier, E.; Flatt, J.P. Nutritional influences on lipogenesis and thermogenesis after a carbohydrate meal. Am. J. Physiol. Endocrinol. Metab. 1984, 246, E62–E70. [Google Scholar] [CrossRef]
- Aarsland, A.; Chinkes, D.; Wolfe, R.R. Hepatic and whole-body fat synthesis in humans during carbohydrate overfeeding. Am. J. Clin. Nutr. 1997, 65, 1774–1782. [Google Scholar] [CrossRef]
- Solinas, G.; Summermatter, S.; Mainieri, D.; Gubler, M.; Pirola, L.; Wymann, M.P.; Rusconi, S.; Montani, J.P.; Seydoux, J.; Dulloo, A.G. The direct effect of leptin on skeletal muscle thermogenesis is mediated by substrate cycling between de novo lipogenesis and lipid oxidation. FEBS Lett. 2004, 577, 539–544. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.H. Skeletal muscle triglyceride. An aspect of regional adiposity and insulin resistance. Diabetes Care 2001, 24, 933–941. [Google Scholar] [CrossRef]
- Dulloo, A.G.; Gubler, M.; Montani, J.P.; Seydoux, J.; Solinas, G. Substrate cycling between de novo lipogenesis and lipid oxidation: A thermogenic mechanism against skeletal muscle lipotoxicity and glucolipotoxicity. Int. J. Obes. 2004, 28, S29–S37. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Muller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef]
- Schoeller, D.A.; Cella, L.K.; Sinha, M.K.; Caro, J.F. Entrainment of the diurnal rhythm of plasma leptin to meal timing. J. Clin. Investig. 1997, 100, 1882–1887. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, E.A. A possible metabolic basis for the control of body weight. N. Engl. J. Med. 1980, 302, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Wirthensohn, G.; Gunder, W. Renal substrate metabolism. Physiol. Rev. 1986, 66, 469–497. [Google Scholar] [CrossRef]
- Meyer, C.; Nadkarni, V.; Stumvoll, M.; Gerich, J.E. Human kidney free fatty acid and glucose uptake: Evidence for a renal glucose-fatty acid cycle. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E650–E654. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, N.; Holdsworth, C.D.; Turner, D.S. New interpretation of oral glucose tolerance. Lancet 1964, 2, 20–21. [Google Scholar] [CrossRef]
- Elrick, H.; Stimmler, L.; Hlad, C.J.; Arai, Y. Plasma insulin responses to oral and intravenous glucose administration. J. Clin. Endocrinol. Metab. 1964, 24, 1076–1092. [Google Scholar] [CrossRef]
- Perley, M.J.; Kipnis, D.M. Plasma insulin responses to oral and intravenous glucose: Studies in normal and diabetic subjects. J. Clin. Investig. 1967, 46, 1954–1962. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Hendler, R.; Wahren, J.; Felig, P. Influence of hyperinsulinemia, hyperglycemia and the route of glucose administration on splachnic glucose. Proc. Natl Acad. Sci. USA 1978, 75, 5173–5177. [Google Scholar] [CrossRef]
- Ishida, T.; Chap, Z.; Chen, J. Differential effects of oral, peripheral intravenous and intraportal glucose on hepatic glucose uptake and insulin and glucagon extraction in conscious dogs. J. Clin. Investig. 1983, 72, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Marathe, C.S.; Rayner, C.K.; Jones, K.L.; Horowitz, M. Relationships between gastric emptying, postprandial glycemia and incretin hormones. Diabetes Care 2013, 36, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Lavin, J.H.; Wittert, G.A.; Sun, W.M.; Horowitz, M.; Morley, J.E.; Read, N.W. Appetite regulation by carbohydrate: Role of blood glucose and gastrointestinal hormones. Am. J. Physiol. Endocrinol. Metab. 1996, 271, E209–E214. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.G.; Andrews, J.M.; Jones, K.L.; Wittert, G.A.; Chapman, I.M.; Morley, J.E.; Horowitz, M. Effects of small intestinal nutrient infusion on appetite and pyloric motility are modified by age. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 273, R755–R761. [Google Scholar] [CrossRef] [PubMed]
- Brener, W.; Hendrix, T.R.; McHugh, P.R. Regulation of the gastric emptying of glucose. Gastroenterology 1983, 85, 76–82. [Google Scholar] [CrossRef]
- Schvarcz, E. Physiological hyperglycemia slows gastric emptying in normal subjects and patients with insulin-dependent diabetes mellitus. Gastroenterology 1997, 113, 60–66. [Google Scholar] [CrossRef]
- Corvilain, B.; Abramowicz, M.; Fery, F.; Schoutens, A.; Verlinden, M.; Balasse, E.; Horowitz, M. Effect of short-term starvation on gastric emptying in humans: Relationship to oral glucose tolerance. Am. J. Physiol. Gastrointest. Liver Physiol. 1995, 32, G512–G517. [Google Scholar] [CrossRef]
- Russo, A. Insulin-induced hypoglycemia accelerates gastric emptying of solids and liquids in long-standing type 1 diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 4489–4495. [Google Scholar] [CrossRef] [PubMed]
- Levin, F.; Edholm, T.; Schmidt, P.T.; Gryback, P.; Jacobsson, H.; Degerblad, M.; Hoybye, C.; Holst, J.J.; Rehfetd, J.F.; Hellstrom, P.M.; et al. Ghrelin stimulates gastric emptying and hunger in normal-weight humans. J. Clin. Endocrinol. Metab. 2006, 91, 3296–3302. [Google Scholar] [CrossRef] [PubMed]
- Chey, W.Y.; Hitanant, S.; Hendricks, J.; Lorber, S.H. Effect of secretin and cholecystokinin on gastric emptying and gastric secretion in man. Gastroenterology 1970, 58, 820–827. [Google Scholar] [CrossRef]
- Foxx-Orenstein, A.; Camilleri, M.; Stephens, D.; Burton, D. Effect of a somatostatin analogue on gastric motor and sensory functions in healthy humans. Gut 2002, 52, 1555–1561. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wettergren, A.; Schjoldager, B.; Mortensen, P.E.; Myhre, J.; Christiansen, J.; Holst, J.J. Truncated GLP-1 (proglucagon 78-107-amide) inhibits gastric and pancreatic functions in man. Dig. Dis. Sci. 1993, 38, 665–673. [Google Scholar] [PubMed]
- Savage, A.P.; Adrian, T.E.; Carolan, G.; Chaterjee, V.K.; Bloom, S.R. Effects of peptide YY (PYY) on mouth to caecum intestinal transit time and on the rate of gstric emptying in healthy volunteers. Gut 1987, 28, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.W.; Saslow, S.B.; Camilleri, M.; Thomforde, G.M.; Dinneen, S.; Rizza, R.A. Mechanism of accelerated gastric emptying of liquids and hyperglycemia in patients with type 2 diabetes mellitus. Gastroenterology 1995, 109, 755–765. [Google Scholar] [CrossRef]
- Woodyatt, R.T.; Sansum, W.D.; Wilder, R.M. Prolonged and accurately timed intravenous injections of sugar. JAMA 1915, 65, 2067–2070. [Google Scholar] [CrossRef]
- McDonald, G.W.; Fisher, G.F.; Burnham, C. Reproducibility of the oral glucose tolerance test. Diabetes 1965, 14, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.G.; Wingate, D.L.; Thomas, M.; Harrison, D. Gastric emptying as a determinant of the oral glucose tolerance test. Gastroenterology 1982, 82, 51–55. [Google Scholar] [CrossRef]
- Mourot, J.; Thouvenot, P.; Couet, C.; Antoine, J.M.; Krobicka, A.; Debry, G. Relationship between the rate of gastric emptying and glucose and insulin responses to starchy foods in young healthy adults. Am. J. Clin. Nutr. 1988, 48, 1035–1040. [Google Scholar] [CrossRef]
- Horowitz, M.; Edelbroek, M.A.L.; Wishart, J.M.; Straathof, J.W. Relationship between oral glucose tolerance and gastric emptying in normal healthy subjects. Diabetologia 1993, 36, 857–862. [Google Scholar] [CrossRef]
- O’Donovan, D.G.; Doran, S.; Feinle-Bisset, C.; Jones, K.L.; Meyer, J.H.; Wishart, J.M.; Morris, H.A.; Horowitz, M. Effect of variations in small intestinal glucose delivery on plasma glucose, insulin and incretin hormones in healthy subjects and type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3431–3435. [Google Scholar] [CrossRef]
- Jenkins, D.; Wolever, T.; Ocana, A. Metabolic effects of reducing rate of glucose ingestion by single bolus versus continuous sipping. Diabetes 1990, 39, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Argyrakopoulou, G.; Simati, S.; Dimitriadis, G.; Kokkinos, A. How important is eating rate in the physiological response to food intake, control of body weight and glycemia? Nutrients 2020, 12, 1734. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, R.; Tamakoshi, K.; Yatsuya, H.; Wada, K.; Matsuhita, K.; OuYang, P.; Hotta, Y.; Takefuji, S.; Mitsuhashi, H.; Sugiura, K.; et al. Eating fast leads to insulin resistance: Findings in middle-aged Japanese women. Prev. Med. 2008, 19, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.M.; Greene, G.W.; Melanson, K.J. Eating slowly led to decreases in energy intake within meals in healthy women. J. Am. Diet. Assoc. 2008, 108, 1186–1191. [Google Scholar] [CrossRef]
- Kokkinos, A.; Le Roux, C.W.; Alexiadou, K.; Tentolouris, N.; Vincent, R.P.; Kyriaki, D.; Perrea, D.; Ghatei, M.A.; Bloom, S.R.; Katsilambros, N. Eating slowly increases postprandial response of the anorexigenic gut hormones, peptide YY and glucagon-like peptide-1. J. Clin. Endocrinol. Metab. 2010, 95, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Gribble, F.; Horowitz, M.; Rayner, C.K. Roles of the gut in glucose homeostasis. Diabetes Care 2016, 39, 884–892. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20, 5–21. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, M.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Ariyasu, H.; Takaya, K.; Tagami, T.; Ogawa, Y.; Hosoda, K.; Akamizu, T.; Suda, M.; Koh, T.; Natsui, K.; Toyooka, S.; et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity. J. Clin. Endocrinol. Metab. 2001, 86, 4753–4758. [Google Scholar] [CrossRef]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef]
- Tschop, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef]
- Wren, A.M.; Seal, L.J.; Cohen, M.A.; Brynes, A.E.; Frost, G.S.; Murphy, K.G.; Dhillo, W.S.; Ghatei, M.A.; Bloom, S.R. Ghrelin enhances appetite and increases food intake in humans. J. Clin. Endocrinol. Metab. 2001, 86, 5992–5995. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Purnell, J.Q.; Frayo, R.S.; Schmidova, K.; Wisse, B.E.; Weigle, D.S. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 2001, 50, 1714–1719. [Google Scholar] [PubMed]
- Tschop, M.; Wawarta, R.; Riepl, R.L.; Friedrich, S.; Bildlingmeier, M.; Landgraf, R.; Folwaczny, C. Postprandial decrease of circulating human ghrelin levels. J. Endocrinol. Investig. 2001, 24, RC19–RC21. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Wang, G.; Englander, E.W.; Kojima, M.; Greeley, G.H. Ghrelin, a new gastrointestinal endocrine peptide that stimulates insulin secretion: Enteric distribution, ontogeny, influence of endocrine and dietary manipulations. Endocrinology 2002, 143, 185–190. [Google Scholar] [CrossRef]
- Murdolo, G.; Lucidi, P.; di Loreto, C.; Parlanti, N.; de Cicco, A.; Fatone, C.; Fanelli, C.G.; Bolli, G.B.; Santeusanio, F.; de Feo, P. Insulin is required for prandial ghrelin suppression in humans. Diabetes 2003, 52, 2923–2927. [Google Scholar] [CrossRef]
- Watterson, K.R.; Bestow, D.; Gallagher, J.; Lee Hamilton, D.; Ashford, F.B.; Meakin, P.J.; Ashford, M.L.J. Anorexigenic and orexigenic hormone modulation of mammalian target of rapamycin complex 1 activity and the regulation of hypothalamic agouti-related protein mRNA expression. Neurosignals 2013, 21, 28–41. [Google Scholar] [CrossRef]
- Kelesidis, T.; Kelesidis, I.; Chou, S.; Mantzoros, C.S. The role of leptin in human physiology: Emerging clinical applications. Ann. Intern. Med. 2010, 152, 93–100. [Google Scholar] [CrossRef]
- Pliquett, R.U.; Fuhrer, D.; Falk, S.; Zysset, S.; von Cramon, D.Y.; Stumvoll, M. The effects of insulin on the central nervous system —Focus of appetite regulation. Horm. Metab. Res. 2006, 38, 442–446. [Google Scholar]
- Giu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Ronnekleiv, O.K.; Kelly, M. Insulin excites anorexigenic propiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab. 2014, 19, 682–693. [Google Scholar]
- Klok, M.D.; Jokobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.T.; Kalra, P.S.; Crowley, W.R.; Kalra, S.P. Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 1984, 115, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Shintani, M.; Ogawa, Y.; Ebihara, K.; Aizawa-Abe, M.; Miyanaga, F.; Takaya, K.; Hayashi, T.; Inoue, G.; Hosoda, K.; Kojima, M.; et al. Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes 2001, 50, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Morton, G.J.; Steams, W.H.; Rhodes, C.J.; Myers, M.G.; Schwartz, M.W. Intracellular signaling. Key enzyme in leptin-induced anorexia. Nature 2001, 413, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Niswender, K.D.; Morrison, C.D.; Clegg, D.J.; Olson, R.; Baskin, D.G.; Myers, M.G.; Seeley, R.J.; Schwartz, M.W. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: A key mediator of insulin-induced anorexia. Diabetes 2003, 52, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Sarraf, P.; Wright, M.; Yao, K.M.; Mueller, E.; Solanes, G.; Lowell, B.B.; Spiegelman, B.M. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Investig. 1998, 101, 1–9. [Google Scholar] [CrossRef]
- Harvey, J.; McKenna, F.; Herson, P.S.; Spanswick, D.; Ashford, L. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line CRI-G1. J. Physiol. 1997, 504, 527–535. [Google Scholar] [CrossRef]
- Hosoda, H.; Kojima, M.; Matsuo, H.; Kangawa, K. Ghrelin and des-acyl ghrelin: Two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem. Biophys. Res. Commun. 2000, 279, 909–913. [Google Scholar] [CrossRef]
- Hosoda, H.; Kojima, M.; Mizushima, T.; Shimizu, S.; Kangawa, K. Structural divergence of human ghrelin. Identification of multiple ghrelin-derived molecules produced by post-translational processing. J. Biol. Chem. 2003, 278, 64–70. [Google Scholar] [CrossRef]
- De Vriese, C.; Gregoire, F.; Lema-Kisoka, R.; Waelbroeck, M.; Robberecht, P.; Delporte, C. Ghrelin degradation by serum and tissue homogenates: Identification of the cleavage sites. Endocrinology 2004, 145, 4997–5005. [Google Scholar]
- Lucidi, P.; Murdolo, G.; di Loreto, C.; Parlanti, N.; de Cicco, A.; Ranchelli, A.; Fatone, C.; Taglioni, C.; Fanelli, C.; Santeusanio, F.; et al. Meal intake similarly reduces concentrations of octanoyl and total ghrelin in humans. J. Endocrinol. Investig. 2004, 27, RC12–RC15. [Google Scholar] [CrossRef]
- Broglio, F.; Gottero, C.; Prodam, F.; Gauna, C.; Muccioli, G.; Papoti, M.; Abribat, T.; van der Lely, A.J.; Ghigo, E. Non-acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J. Clin. Endocrinol. Metab. 2004, 89, 3062–3065. [Google Scholar] [CrossRef]
- Liu, J.; Prudom, C.E.; Nass, R.; Pezzoli, S.S.; Oliveri, M.C.; Johnson, M.L.; Veldhuis, P.; Gordon, D.A.; Howard, A.D.; Witcher, D.R.; et al. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J. Clin. Endocrinol. Metab. 2008, 93, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Inui, A.; Fujimiya, M.; Sakamaki, R.; Shinfuku, N.; Ueta, Y.; Meguid, M.M.; Kasuga, M. Stomach regulates energy balance via acylated ghrelin and desacylated ghrelin. Gut 2005, 54, 18–24. [Google Scholar] [CrossRef]
- Inhoff, T.; Monnikes, H.; Noetzel, S.; Stengel, A.; Goebel, M.; Dinh, Q.T.; Riedl, A.; Bannert, N.; Wisser, A.S.; Wiedenmann, B.; et al. Desacyl ghrelin inhibits the orexigenic effect of peripherally injected ghrelin in rats. Peptides 2008, 29, 2159–2168. [Google Scholar] [PubMed]
- Gauna, C.; Meyler, F.M.; Janssen, J.A.; Delhanty, P.J.D.; Abribat, T.; van Koetsveld, P.; Hofland, L.J.; Broglio, F.; Ghigo, E.; van der Lely, A.J. Administration of acylated ghrelin reduces insulin sensitivity, whereas the combination of acylated plus unacylated ghrelin strongly improves insulin sensitivity. J. Clin. Endocrinol. Metab. 2004, 89, 5035–5042. [Google Scholar] [CrossRef]
- Vestergaard, E.T.; Gormsen, L.C.; Jessen, N.; Lund, S.; Hansen, T.K.; Moller, N.; Jorgensen, J.O.L. Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes 2008, 57, 3205–3210. [Google Scholar]
- Gauna, C.; Delhanty, P.J.; Hofland, L.J.; Janssen, J.A.; Broglio, F.; Ross, R.J.; Ghigo, E.; van der Lely, A.J. Ghrelin stimulates, whereas des-octanoyl ghrelin inhibits, glucose output by primary hepatocytes. J. Clin. Endocrinol. Metab. 2005, 90, 1055–1060. [Google Scholar] [CrossRef]
- Barazzoni, R.; Zanetti, M.; Ferreira, C.; Vinci, P.; Pirulli, A.; Mucci, M.; Dore, F.; Fonda, M.; Ciocci, B.; Cattin, L.; et al. Relationships between desacylated and acylated ghrelin and insulin sensitivity in the metabolic syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 3935–3940. [Google Scholar] [CrossRef]
- Barazzoni, R.; Gortan Cappellari, G.; Semolic, A.; Ius, M.; Mamolo, L.; Dore, F.; Giacca, M.; Zanetti, M.; Vinci, P.; Guarnieri, G. Plasma total and unacylated ghrelin predict 5-year changes in insulin resistance. Clin. Nutr. 2016, 35, 1168–1173. [Google Scholar] [CrossRef]
- Tong, J.; Prigeon, R.L.; Davis, H.W.; Bidlingmaier, M.; Tschop, M.H.; D’Alessio, D. Physiologic concentrations of exogenously infused ghrelin reduces insulin secretion without affecting insulin sensitivity in healthy humans. J. Clin. Endocrinol. Metab. 2013, 98, 2536–2543. [Google Scholar]
- Gauna, C.; Kiewiet, R.M.; Janssen, J.A.; van de Zande, B.; Delhanty, P.J.; Ghigo, E.; Hofland, L.J.; Themmen, A.P.; van der Lely, A.J. Unacylated ghrelin acts as a potent insulin secretagogue in glucose-stimulated conditions. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E697–E704. [Google Scholar] [CrossRef] [PubMed]
- Page, L.C.; Gastaldelli, A.; Gray, S.M.; D’Alessio, D.A.; Tong, J. Interaction of GLP-1 and Ghrelin on glucose tolerance in healthy humans. Diabetes 2018, 67, 1976–1985. [Google Scholar] [CrossRef]
- Spiegel, K.; Tasali, E.; Leproult, R.; Scherberg, N.; van Gauter, E. Twenty-four-hour profiles of acylated and total ghrelin: Relationship with glucose levels and impact of time of day and sleep. J. Clin. Endocrinol. Metab. 2011, 96, 486–493. [Google Scholar] [CrossRef]
- Togliatto, G.; Trombetta, A.; Dentelli, P.; Gallo, S.; Rosso, A.; Cotogni, P.; Granata, R.; Falcioni, R.; Delale, T.; Ghigo, E.; et al. Unacylated ghrelin induces oxidative stress resistance in a glucose intolerance and peripheral artery disease mouse model by restoring endothelial cell miR-126 expression. Diabetes 2015, 64, 1370–1382. [Google Scholar]
- Kreymann, B.; Ghatei, M.A.; Williams, G.; Bloom, S.R. Glucagon-like peptide-1 (7-36): A physiological incretin in man. Lancet 1987, 2, 1300–1305. [Google Scholar]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Eissele, R.; Göke, R.; Willemer, S.; Harthus, H.P.; Vermeer, H.; Arnold, R.; Göke, B. Glucagon-like peptide-1 cells in the gastrointestinal tract and pancreas of rat, pig and man. Eur. J. Clin. Investig. 1992, 22, 283–291. [Google Scholar]
- Fehmann, H.C.; Göke, R.; Göke, B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocrine. Rev. 1995, 16, 390–410. [Google Scholar]
- Thorens, B. Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8641–8645. [Google Scholar]
- Heller, R.S.; Kieffer, T.J.; Habener, J.F. Insulinotropic glucagon like peptide-1 receptor expression in glucagon producing alpha cells of the rat endocrine pancreas. Diabetes 1997, 46, 785–791. [Google Scholar] [CrossRef]
- Gremlich, S.; Porret, A.; Hani, E.H.; Cherif, D.; Vionnet, N.; Froguel, P.; Thorens, B. Cloning, functional expression and chromosomal localization of the human pancreatic islet glucose-dependent insulinotropic polypeptide receptor. Diabetes 1995, 44, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Moens, K.; Heimberg, H.; Flamez, D.; Huypens, P.; Quartier, E.; Ling, Z.; Pipeleers, D.; Gremlich, S.; Thorens, B.; Schuit, F. Expression and functional activity of glucagon, glucagon-like peptide-1, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 1996, 45, 257–261. [Google Scholar] [CrossRef]
- Gutniak, M.; Orskov, C.; Holst, J.J.; Ahren, B.; Efendic, S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36) in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic effects of GLP-1 and analogs on cell signaling, metabolism and function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef]
- Hayes, M.R.; Bradley, L.; Grill, H.J. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology 2009, 150, 2654–2659. [Google Scholar]
- Nauck, M.A.; Niedereichholz, U.; Ettler, R.; Holst, J.J.; Orskov, C.; Ritzel, R.; Schmiegel, W.H. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E981–E988. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Behle, K.; Holst, J.J.; Nauck, M.S.; Ritzel, R.; Hufner, M.; Schmiegel, W.H. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J. Clin. Endocrinol. Metab. 2002, 87, 1239–1246. [Google Scholar]
- Quddusi, S.; Vahl, T.P.; Hanson, K.; Prigeon, R.L.; D’Alessio, D.A. Differential effects of acute and extended infusions of glucagon-like peptide-1 on first and second-phase insulin secretion in diabetic and nondiabetic humans. Diabetes Care 2003, 26, 791–798. [Google Scholar]
- Larsson, H.; Holst, J.J.; Ahren, B. Glucagon-like peptide-1 reduces hepatic glucose production indirectly through insulin and glucagon in humans. Acta Physiol. Scand. 1997, 160, 413–422. [Google Scholar] [PubMed]
- Seghieri, M.; Rebelos, E.; Gastaldelli, A.; Astiarraga, B.D.; Casolaro, A.; Barsotti, E.; Pocai, A.; Nauck, M.; Muscelli, E.; Ferrannini, E. Direct effect of GLP-1 infusion on endogenous glucose production in humans. Diabetologia 2013, 56, 156–161. [Google Scholar] [CrossRef] [PubMed]
- D’ Alessio, D.A.; Kahn, S.E.; Leusner, C.R.; Ensinck, J.W. Glucagon-like peptide-1 enhances glucose tolerance both by stimulation of insulin release and by increasing insulin-independent glucose disposal. J. Clin. Investig. 1994, 93, 2263–2266. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010. [Google Scholar] [CrossRef] [PubMed]
- Vilsboll, T.; Krarup, T.; Madsbad, S.; Holst, J.J. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul. Pept. 2003, 114, 115–121. [Google Scholar] [CrossRef]
- Meier, J.J.; Goetze, O.; Anstipp, L.; Hagemann, D.; Holst, J.J.; Schmidt, W.E.; Gallwitz, B.; Nauck, M.A. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E621–E625. [Google Scholar] [CrossRef]
- Edholm, T.; Degerblad, M.; Gryback, P.; Hilsted, L.; Holst, J.J.; Jacobsson, H.; Efendic, S.; Schmidt, P.T.; Hellstrom, P.M. Differential incretin effects of GIP and GLP-1 on gastric emptying, appetite, and insulin-glucose homeostasis. Neurogastroenterol. Motil. 2010, 22, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J. Incretin hormones and the satiation signal. Int. J. Obes. 2013, 37, 1161–1168. [Google Scholar] [CrossRef]
- Meier, J.J.; Gallwitz, B.; Siepmann, N.; Holst, J.J.; Deacon, C.F.; Schmidt, W.E.; Nauck, M.A. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003, 46, 798–801. [Google Scholar] [CrossRef]
- Christensen, M.; Vedtofte, L.; Holst, J.J.; Visboll, T.; Knop, F.K. Glucose-dependent insulinotropic polypeptide. A bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes 2011, 60, 3103–3109. [Google Scholar] [CrossRef]
- Schirra, J.; Katschinski, M.; Weidmann, C.; Schäfer, T.; Wank, U.; Arnold, R.; Göke, B. Gastric emptying and release of incretin hormones after glucose ingestion in humans. J. Clin. Investig. 1996, 97, 92–103. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.; Arfuso, F.; Keane, K. Nutrient regulation of insulin secretion and action. J. Endocrinol. 2014, 221, R105–R120. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.C.; Boitard, C.; Efendic, S.; Ferrannini, E.; Steiner, D.F.; Cerasi, E. Insulin secretion: Movement at all levels. Diabetes 2002, 51, S1–S2. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, F.M.; Rorsman, P. Electrophysiology of the pancreatic β-cell. Prog. Biophys. Mol. Biol. 1989, 54, 87–143. [Google Scholar] [CrossRef]
- Curry, D.L.; Bennett, L.L.; Grodsky, G.M. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 1968, 83, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Investig. 2011, 121, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Steiner, K.E.; Mouton, S.M.; Bowles, C.R.; Williams, P.E.; Cherrington, A.D. The relative importance of first- and second-phase insulin secretion in countering the action of glucagon on glucose turnover in the conscious dog. Diabetes 1982, 31, 964–972. [Google Scholar] [CrossRef]
- Dimitriadis, G.; Boutati, E.; Lambadiari, V.; Mitrou, P.; Maratou, E.; Brunel, P.; Raptis, S.A. Restoration of early insulin secretion after a meal in type 2 diabetes: Effects on lipid and glucose metabolism. Eur. J. Clin. Investig. 2004, 34, 490–497. [Google Scholar]
- Picard, F.; Naimi, N.; Richard, D.; Deshaies, Y. Response of adipose tissue lipoprotein lipase to the cephalic phase of insulin secretion. Diabetes 1999, 48, 452–459. [Google Scholar] [CrossRef]
- Verdonk, C.A.; Rizza, R.A.; Gerich, J.E. Effects of plasma glucose concentration on glucose utilization and glucose clearance in normal man. Diabetes 1981, 30, 535–537. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Acute and chronic effects of hyperglycemia on glucose metabolism. Diabetologia 1990, 33, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rizza, R.A. Glucose effectiveness: Measurement in diabetic and non-diabetic humans. Exp. Clin. Endocrinol. Diabetes 2001, 109, S157–S165. [Google Scholar] [CrossRef] [PubMed]
- Ader, M.; Pacini, G.; Yang, Y.J.; Bergman, R.N. Importance of glucose per se to intravenous glucose tolerance: Comparison of the minimal model prediction with direct measurements. Diabetes 1985, 34, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Brechtel, G.; Wallace, P.; Edelman, S.V. Rates and tissue sites of non-insulin and insulin-mediated glucose uptake in humans. Am. J. Physiol. Endocrinol. Metab. 1988, 255, E769–E774. [Google Scholar] [CrossRef]
- Mathoo, J.M.R.; Shi, Z.Q.; Klip, A.; Vranic, M. Opposite effects of acute hypoglycemia and acute hyperglycemia on glucose transport and glucose transporters in perfused rat skeletal muscle. Diabetes 1999, 48, 1281–1288. [Google Scholar] [CrossRef]
- Rossetti, L.; Giaccari, A.; Barzilai, N.; Howard, K.; Sebel, G.; Hu, M. Mechanism by which hyperglycemia inhibits hepatic glucose production in conscious rats. J. Clin. Investig. 1993, 92, 1126–1134. [Google Scholar] [CrossRef]
- Petersen, K.F.; Laurent, D.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism by which glucose and insulin inhibit net hepatic glycogenolysis in humans. J. Clin. Investig. 1998, 101, 1203–1209. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Consoli, A.; Jain, A.; Kelley, D.E. Differential regulation of intracellular glucose metabolism by glucose and insulin in human muscle. Am. J. Physiol. Endocrinol. Metab. 1993, 265, E898–EE905. [Google Scholar] [CrossRef]
- Bonuccelli, S.; Muscelli, E.; Gastaldelli, A.; Barsotti, E.; Astiarraga, B.; Holst, J.J.; Mari, A.; Ferrannini, E. Improved tolerance to sequential glucose loading (Staub-Traugott effect): Size and mechanisms. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E532–E537. [Google Scholar] [CrossRef][Green Version]
- Jakubowicz, D.; Wainstein, J.; Ahren, B.; Landau, Z.; Bar-Dayan, Y.; Froy, O. Fasting until noon triggers increased postprandial hyperglycemia and impaired insulin response after lunch and dinner in individuals with type 2 diabetes: A randomized clinical trial. Diabetes Care 2015, 38, 1820–1826. [Google Scholar] [CrossRef]
- Moore, M.C.; Smith, M.S.; Farmer, B.; Coate, K.C.; Kraft, G.; Shiota, M.; Williams, P.E.; Cherrington, A.D. Morning hyperinsulinemia primes the liver for glucose uptake and glycogen storage later in the day. Diabetes 2018, 67, 1237–1245. [Google Scholar] [PubMed]
- Trico, D.; Baldi, S.; Tulipani, A.; Frascerra, S.; Macedo, M.P.; Mari, A.; Ferrannini, E.; Natali, A. Mechanisms through which a small protein and lipid preload improves glucose tolerance. Diabetologia 2015, 58, 2503–2512. [Google Scholar] [CrossRef] [PubMed]
- Gentilcore, D.; Chaikomin, R.; Jones, K.L.; Russo, A.; Feinle-Bisset, C.; Wishart, J.M.; Rayner, C.K.; Horowitz, M. Effects of fat on gastric emptying of and the glycemic, insulin, and incretin responses to a carbohydrate meal in type 2 diabetes. J. Clin. Endocrinol. Metab. 2006, 91, 2062–2067. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Stevens, J.E.; Cukier, K.; Maddox, A.F.; Wishart, J.M.; Jones, K.L.; Clifton, P.M.; Horowitz, M.; Rayner, C.K. Effects of a protein preload on gastric emptying, glycemia and gut hormones after a carbohydrate meal in diet-controlled type 2 diabetes. Diabetes Care 2009, 32, 1600–1602. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.P.; Iliescu, R.G.; Thomas, C.E.; Aronne, L.J. Food order has a significant impact on postprandial glucose and insulin levels. Diabetes Care 2015, 38, e98–e99. [Google Scholar] [PubMed]
- Sun, L.; Goh, H.J.; Govindharajulu, P.; Leow, M.K.; Henry, C.J. Postprandial glucose, insulin and incretin responses differ by test meal macronutrient ingestion sequence. Clin. Nutr. 2020, 39, 950–957. [Google Scholar]
- Weickert, M.O.; Pfeiffer, A.F.H. Metabolic effects of dietary fiber consumption and prevention of diabetes. J. Nutr. 2008, 138, 439–442. [Google Scholar] [CrossRef]
- Nesti, L.; Mengozzi, A.; Trico, D. Impact of nutrient type and sequence on glucose tolerance: Physiologic insights and therapeutic implications. Front. Endocrinol. 2019, 10, 144. [Google Scholar] [CrossRef]
- Vlachos, D.; Malisova, S.; Lindberg, F.A.; Karaniki, G. Glycemic Index (GI) or glycemic load (GL) and dietary interventions for optimizing postprandial hyperglycemia in patients with T2 diabetes: A review. Nutrients 2020, 12, 1561. [Google Scholar] [CrossRef]
- Korakas, E.; Dimitriadis, G.; Raptis, A.; Lambadiari, V. Dietary composition and cardiovascular risk: A mediator or a bystander? Nutrients 2018, 10, 1912–1942. [Google Scholar]
- Hyde, P.N.; Sapper, T.N.; Crabtree, C.D.; LaFountain, R.A.; Bowling, M.L.; Buga, A.; Fell, B.; McSwiney, F.T.; Dickerson, R.M.; Miller, V.J.; et al. Dietary carbohydrate restriction improves metabolic syndrome independent of weight loss. J. Clin. Investig. Insight 2019, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.; Morrison, D.J.; Betik, A.C.; Roberts-Thomson, K.; Kaur, G.; Wadley, G.D.; Shaw, C.S.; Keske, M.A. High glucose mixed nutrient meal ingestion impairs skeletal muscle microvascular blood flow in healthy young males. Am. J. Physiol. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Meng, H.; Matthan, N.R.; Ausman, L.M.; Lichtenstein, A.H. Effect of macronutrients and fiber on postprandial glycemic responses and meal glycemic index and glycemic load value determinations. Am. J. Clin. Nutr. 2017, 105, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Trico, D.; Natali, A. Modulation of postprandial glycemic responses by non-carbohydrate nutrients provides novel approaches to the prevention and treatment of type 2 diabetes. Am. J. Clin. Nutr. 2017, 106, 701–702. [Google Scholar]
- Bergman, R.N.; Phillips, L.S.; Cobelli, C. Physiologic evaluation of factors controlling glucose tolerance in man: Measurement of insulin sensitivity and β-cell glucose sensitivity from the response to intravenous glucose. J. Clin. Investig. 1981, 68, 1456–1467. [Google Scholar] [PubMed]
- Kahn, S.; Prigeon, R.L.; McCulloch, D.K.; Boyko, E.J.; Bergman, R.N.; Schwartz, M.W.; Neifing, J.L.; Ward, W.K.; Beard, J.C.; Palmer, J.P.; et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects. Diabetes 1993, 42, 1663–1672. [Google Scholar] [PubMed]
- Perseghin, G.; Price, T.B.; Petersen, K.F.; Roden, M.; Cline, G.W.; Gerow, K.; Rothman, D.L.; Shulman, G.I. Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N. Engl. J. Med. 1996, 335, 1357–1362. [Google Scholar] [CrossRef]
- Hayashi, T.; Wojtaszewski, J.F.; Goodyear, L.J. Exercise regulation of glucose transport in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 1997, 273, E1039–E1051. [Google Scholar] [CrossRef]
- Borghouts, L.B.; Keizer, H.A. Exercise and insulin sensitivity: A review. Int. J. Sports Med. 1999, 20, 1–12. [Google Scholar] [CrossRef]
- Holloszy, J. Exercise-induced increase in muscle insulin sensitivity. J. Appl. Physiol. 2005, 99, 338–343. [Google Scholar]
- Philippou, A.; Chryssanthopoulos, C.; Maridaki, M.; Dimitriadis, G.; Koutsilieris, M. Exercise metabolism in health and disease. In Cardiorespiratory Fitness in Cardiometabolic Diseases; Kokkinos, P., Narayan, P., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 57–96. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitriadis, G.D.; Maratou, E.; Kountouri, A.; Board, M.; Lambadiari, V. Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients 2021, 13, 159. https://doi.org/10.3390/nu13010159
Dimitriadis GD, Maratou E, Kountouri A, Board M, Lambadiari V. Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients. 2021; 13(1):159. https://doi.org/10.3390/nu13010159
Chicago/Turabian StyleDimitriadis, George D., Eirini Maratou, Aikaterini Kountouri, Mary Board, and Vaia Lambadiari. 2021. "Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach" Nutrients 13, no. 1: 159. https://doi.org/10.3390/nu13010159
APA StyleDimitriadis, G. D., Maratou, E., Kountouri, A., Board, M., & Lambadiari, V. (2021). Regulation of Postabsorptive and Postprandial Glucose Metabolism by Insulin-Dependent and Insulin-Independent Mechanisms: An Integrative Approach. Nutrients, 13(1), 159. https://doi.org/10.3390/nu13010159