Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
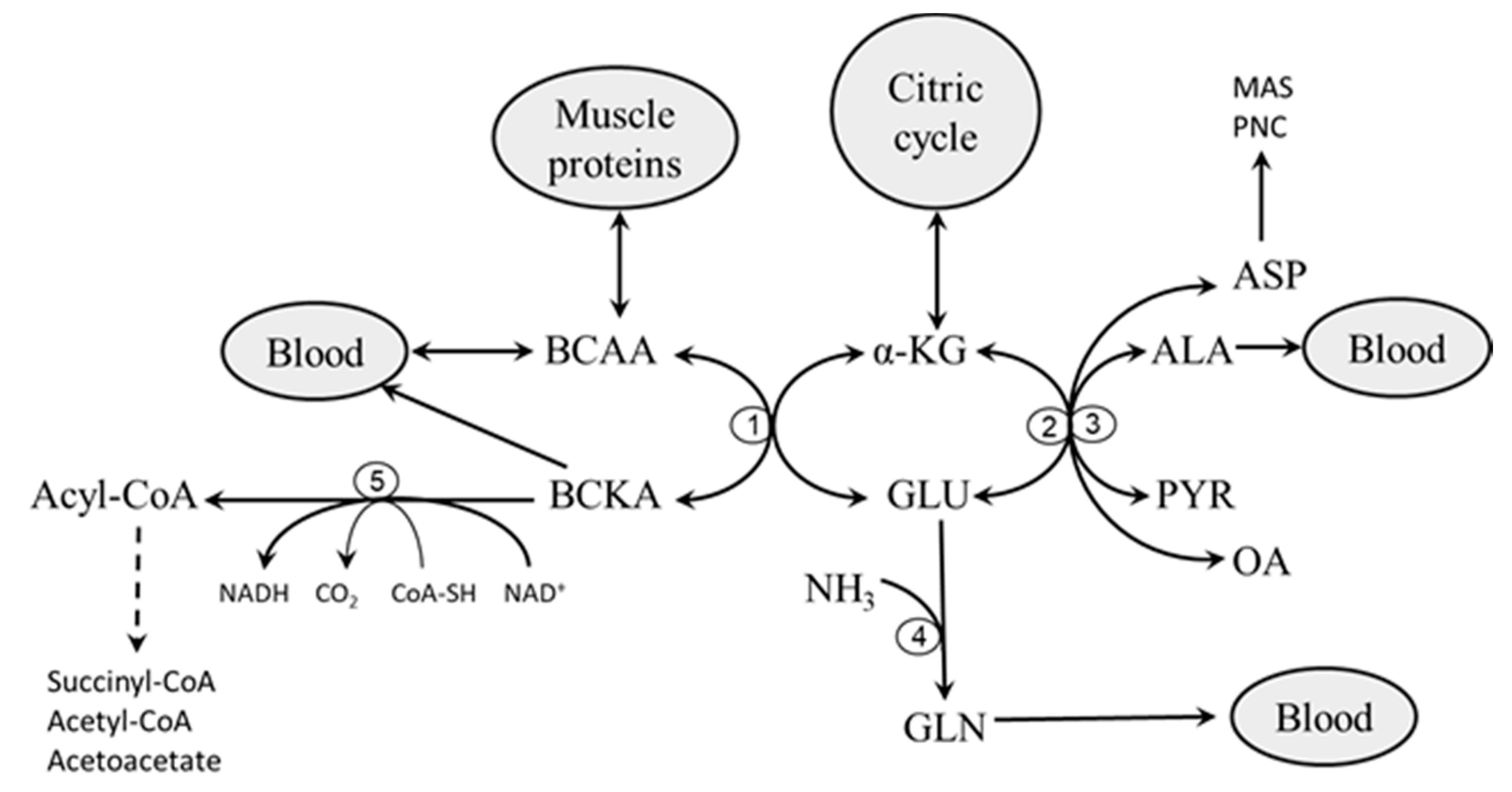
2. Catabolism of BCAAs
2.1. Branched-Chain Amino Acid Aminotransferase (BCAAT)
2.2. Branched-Chain α-Keto Acid Dehydrogenase (BCKAD)
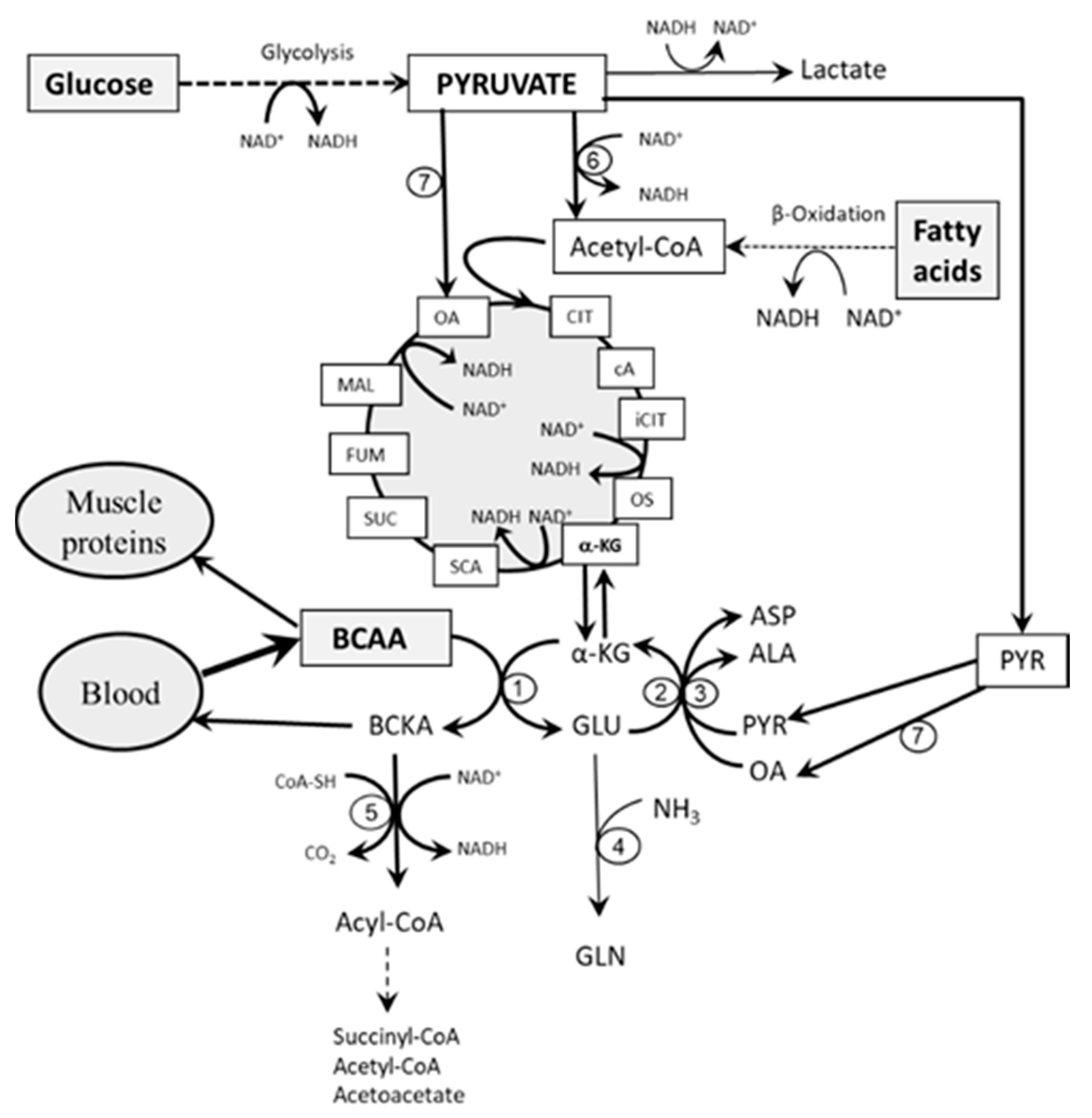
3. Role of Glycolysis and Fatty Acid Oxidation in BCAA Catabolism
3.1. The Role of Glycolysis
3.2. The Role of Fatty Acid Oxidation
4. Etiopathogenesis of Increased BCAA Levels in Starvation, T1DM, and T2DM—Common Features
- Impaired flux through the citric cycle due to decreased glycolysis and the inhibitory influence of NADH produced by fatty acid oxidation. The consequence should be a decreased supply of α-KG for the BCAAT reaction.
- Decreased supply of OA and PYR for conversion of GLU to α-KG due to decreased glycolysis. The consequence should be cataplerosis of α-KG and the shift of GLU metabolism towards GLN synthesis. The benefit for the body might be the use of GLN for ammonia synthesis by the kidneys and the subsequent increase in the buffering capacity of urine, which helps to compensate effects of increased production of ketone bodies on the acid-base balance during starvation and in T1DM. Increased GLN synthesis and expression of GLN synthetase in muscles have been reported in diabetic rats [52,53].
- Alterations in BCAA catabolism in the liver. Since BCAAT is absent in the liver, increased breakdown of hepatic proteins due to starvation or diabetes may result in increased release of BCAAs from the liver to the blood [9,56]. This suggestion is supported by increased contents of BCAAs in the liver in both starvation and diabetes [13,15,56,57]. Alterations in hepatic BCKAD activity might also play a role. Both activation [58,59] and suppression [60,61,62] have been reported in rats with T1DM and T2DM. In my opinion, alterations in the liver are not sufficient to explain the pathogenesis of increased BCAA levels in starvation and diabetes. Just as occurs after food intake, enhanced amounts of the BCAA released from the liver to the blood should be efficiently removed by skeletal muscle if its metabolic functions are not impaired.
- Alterations in BCAA catabolism in adipose tissue. In adipose tissue, where leucine and isoleucine are substrates for fatty acid synthesis, insulin increases the activity of BCKAD [63,64]. Recent studies in obesity and IR have demonstrated downregulation of the expression of BCAA catabolizing enzymes in adipose tissue and suggest their role in the pathogenesis of increased BCAA levels [65,66,67,68]. However, considering that BCAAT activity in adipose tissue is much lower than in muscles and BCKAD activity is also very low [12,69], decreased BCAA oxidation in adipose tissue could have a minor role in the pathogenesis of increased BCAA levels in starvation and diabetes. The shortcomings of the hypothesis are also increased fat mass in obese people, which compensates for decreased activities of BCAA catabolic enzymes in adipocytes, and succinyl-CoA, the end-product of valine catabolism, which is not ketogenic. Therefore, adipose tissue may contribute to alterations in BCAA levels in insulin-deficient and IR states, but cannot play a major role.
- Transamination of BCKAs to BCAAs. Since the BCAAT reaction is reversible and near equilibrium, increased supply of BCKA and GLN from muscles, as occurs in starvation and diabetes, may shift the BCAAT reaction towards BCAA synthesis. An interorgan cycle, in which muscles act as a source of BCKA and most other tissues aminate the BCKA into the corresponding BCAA has been postulated [70,71,72].
5. Etiopathogenesis of Increased BCAA Levels in Starvation, T1DM, and T2DM—Specific Features
5.1. Why Are BCAAs Increased in Starvation?
5.1.1. Early Starvation
- Higher increases in BCAA levels in muscles than in the plasma, liver, and heart of rats after 3 days of starvation [13].
- Increased BCAA and decreased GLN, GLU, and ALA concentrations in muscles of healthy volunteers after 72 h of fasting [73]. Results indicate impaired BCAA transamination.
- Increased release of BCAAs from forearm tissues to the blood in subjects fasted for 60 h [74].
- Incubation of muscles from 24 h-fasted chickens with acetoacetate and 3-hydroxybutyrate decreased glycolysis and PYR concentration, inhibited BCAA transamination and ALA formation, and increased GLU concentration and GLN release. The addition of PYR prevented the inhibitory effect of ketone bodies on BCAA transamination and ALA synthesis [78].
- Decreased ALA and GLN production by diaphragms obtained from 48 h-starved rats incubated with 3-hydroxybutyrate or acetate. Addition of PYR restored ALA and GLN production to control values [80].
- Parallel increments in BCAA and BCKA concentrations in blood plasma in the early stage of starvation [50]. This may reflect impaired BCAA transamination due to an insufficient rise in the flux of BCKA through BCKAD in muscles.
5.1.2. Prolonged Starvation
5.2. Why Are BCAAs Increased in T1DM?
- High BCAA levels in the muscles of patients with T1DM [4].
- Increased BCKA concentrations in blood plasma and muscles in rats with diabetes induced by alloxan [13]. Results indicate the role of impaired flux through BCKAD.
- Blunted activation of BCKAD by hyperleucinemia in muscles obtained from rats with streptozotocin-induced diabetes [16].
- Decreased RNA amounts of BCAAT and BCKAD in the liver and muscles of rabbits with T1DM induced by alloxan [92].
5.3. Why Are BCAAs Increased in T2DM?
- Decreased BCKA and α-KG and increased acylcarnitine concentrations in muscles of humans with IR [99]. Decreased BCKA and α-KG indicate altered activity of BCAAT; increased acylcarnitine concentrations indicate incomplete oxidation of fatty acids.
- Increased BCKA concentrations in plasma, muscle, and the liver [51]. Results indicate impaired flux of BCKA through BCKAD.
- Decreased BCAAT activity in muscles, but not in liver and adipose tissue, of rats with IR induced by high fructose diet [69].
- Decreased expression of genes encoding BCAAT and BCKAD in the muscles of patients with T2DM [100].
- Lower acetate oxidation (measure of citric cycle activity) by myotubes isolated from T2DM subjects when compared with controls [101].
6. Consequences of Increased BCAA Concentrations
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Adibi, S.A. Influence of dietary deprivations on plasma concentration of free amino acids of man. J. Appl. Physiol. 1968, 25, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Marliss, E.; Cahill, G.F. Plasma amino acid levels and insulin secretion in obesity. N. Engl. J. Med. 1969, 281, 811–816. [Google Scholar] [CrossRef]
- Carlsten, A.; Hallgren, B.; Jagenburg, R.; Svanborg, A.; Werkö, L. Amino acids and free fatty acids in plasma in diabetes. I. The effect of insulin on the arterial levels. Acta Med. Scand. 1966, 179, 361–370. [Google Scholar] [CrossRef]
- Borghi, L.; Lugari, R.; Montanari, A.; Dall’Argine, P.; Elia, G.F.; Nicolotti, V.; Simoni, I.; Parmeggiani, A.; Novarini, A.; Gnudi, A. Plasma and skeletal muscle free amino acids in type I, insulin-treated diabetic subjects. Diabetes 1985, 34, 812–815. [Google Scholar] [CrossRef]
- van den Berg, E.H.; Flores-Guerrero, J.L.; Gruppen, E.G.; de Borst, M.H.; Wolak-Dinsmore, J.; Connelly, M.A.; Bakker, S.J.L.; Dullaart, R.P.F. Non-alcoholic fatty liver disease and risk of incident type 2 diabetes: Role of circulating branched-chain amino acids. Nutrients 2019, 11, 705. [Google Scholar] [CrossRef]
- Iwasa, M.; Ishihara, T.; Mifuji-Moroka, R.; Fujita, N.; Kobayashi, Y.; Hasegawa, H.; Iwata, K.; Kaito, M.; Takei, Y. Elevation of branched-chain amino acid levels in diabetes and NAFL and changes with antidiabetic drug treatment. Obes. Res. Clin. Pract. 2015, 9, 293–297. [Google Scholar] [CrossRef]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: Novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Holeček, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. (Lond.) 2018, 15, 33. [Google Scholar]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Laferrère, B.; Reilly, D.; Arias, S.; Swerdlow, N.; Gorroochurn, P.; Bawa, B.; Bose, M.; Teixeira, J.; Stevens, R.D.; Wenner, B.R.; et al. Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci. Transl. Med. 2011, 3, 80re2. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.E.; Miller, R.H.; Block, K.P. Branched-chain amino acid metabolism. Annu Rev. Nutr. 1984, 4, 409–454. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Harper, A.E. Blood and tissue branched-chain amino and alpha-keto acid concentrations: Effect of diet, starvation, and disease. Am. J. Clin. Nutr. 1981, 34, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, T.; Alvarez, B.; Busquets, S.; Carbó, N.; López-Soriano, F.J.; Argilés, J.M. The increased skeletal muscle protein turnover of the streptozotocin diabetic rat is associated with high concentrations of branched-chain amino acids. Biochem. Mol. Med. 1997, 61, 87–94. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Man, K.C.; Hall, D.E.; Colbourne, S.A.; Brosnan, M.E. Interorgan metabolism of amino acids in streptozotocin-diabetic ketoacidotic rat. Am. J. Physiol. 1983, 244, E151–E158. [Google Scholar] [CrossRef]
- Aftring, R.P.; Miller, W.J.; Buse, M.G. Effects of diabetes and starvation on skeletal muscle branched-chain alpha-keto acid dehydrogenase activity. Am. J. Physiol. 1988, 254, E292–E300. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Odessey, R. Oxidation of amino acids by diaphragms from fed and fasted rats. Am. J. Physiol. 1972, 223, 1384–1391. [Google Scholar] [CrossRef]
- Buse, M.G.; Biggers, J.F.; Drier, C.; Buse, J.F. The effect of epinephrine, glucagon, and the nutritional state on the oxidation of branched chain amino acids and pyruvate by isolated hearts and diaphragms of the rat. J. Biol. Chem. 1973, 248, 697–706. [Google Scholar]
- Wagenmakers, A.J.; Schepens, J.T.; Veerkamp, J.H. Effect of starvation and exercise on actual and total activity of the branched-chain 2-oxo acid dehydrogenase complex in rat tissues. Biochem. J. 1984, 223, 815–821. [Google Scholar] [CrossRef]
- Fryburg, D.A.; Barrett, E.J.; Louard, R.J.; Gelfand, R.A. Effect of starvation on human muscle protein metabolism and its response to insulin. Am. J. Physiol. 1990, 259, E477–E482. [Google Scholar] [CrossRef]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.; Newgard, C.B.; et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J. Regulatory interactions between lipids and carbohydrates: The glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 1998, 14, 263–283. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. Branched-chain amino acids: Enzyme and substrate regulation. J. Nutr. 2006, 136, 207S–211S. [Google Scholar] [CrossRef]
- Kadowaki, H.; Knox, W.E. Cytosolic and mitochondrial isoenzymes of branched-chain amino acid aminotransferase during development of the rat. Biochem. J. 1982, 202, 777–783. [Google Scholar] [CrossRef]
- Shimomura, Y.; Obayashi, M.; Murakami, T.; Harris, R.A. Regulation of branched-chain amino acid catabolism: Nutritional and hormonal regulation of activity and expression of the branched-chain alpha-keto acid dehydrogenase kinase. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 419–423. [Google Scholar] [CrossRef]
- Smith, R.J.; Larson, S.; Stred, S.E.; Durschlag, R.P. Regulation of glutamine synthetase and glutaminase activities in cultured skeletal muscle cells. J. Cell Physiol. 1984, 120, 197–203. [Google Scholar] [CrossRef]
- Safer, B.; Williamson, J.R. Mitochondrial-cytosolic interactions in perfused rat heart. Role of coupled transamination in repletion of citric acid cycle intermediates. J. Biol. Chem. 1973, 248, 2570–2579. [Google Scholar]
- Paxton, R.; Harris, R.A. Regulation of branched-chain alpha-ketoacid dehydrogenase kinase. Arch. Biochem. Biophys. 1984, 231, 48–57. [Google Scholar] [CrossRef]
- Sketcher, R.D.; Fern, E.B.; James, W.P. The adaptation in muscle oxidation of leucine to dietary protein and energy intake. Br. J. Nutr. 1974, 31, 333–342. [Google Scholar] [CrossRef]
- Hutson, S.M.; Fenstermacher, D.; Mahar, C. Role of mitochondrial transamination in branched chain amino acid metabolism. J. Biol. Chem. 1988, 263, 3618–3625. [Google Scholar] [PubMed]
- Kelley, D.; Mitrakou, A.; Marsh, H.; Schwenk, F.; Benn, J.; Sonnenberg, G.; Arcangeli, M.; Aoki, T.; Sorensen, J.; Berger, M. Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J. Clin. Investig. 1988, 81, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Wahren, J.; Felig, P.; Hagenfeldt, L. Effect of protein ingestion on splanchnic and leg metabolism in normal man and in patients with diabetes mellitus. J. Clin. Investig. 1976, 57, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Holecek, M.; Kovarik, M. Alterations in protein metabolism and amino acid concentrations in rats fed by a high-protein (casein-enriched) diet—Effect of starvation. Food Chem. Toxicol. 2011, 49, 3336–3342. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Berger, M. The formation of glutamine and alanine in skeletal muscle. J. Biol. Chem. 1974, 249, 5500–5506. [Google Scholar]
- Odessey, R.; Goldberg, A.L. Oxidation of leucine by rat skeletal muscle. Am. J. Physiol. 1972, 223, 1376–1383. [Google Scholar] [CrossRef]
- Durschlag, R.P.; Smith, R.J. Regulation of glutamine production by skeletal muscle cells in culture. Am. J. Physiol. 1985, 248, C442–C448. [Google Scholar] [CrossRef]
- Holecek, M.; Siman, P.; Vodenicarovova, M.; Kandar, R. Alterations in protein and amino acid metabolism in rats fed a branched-chain amino acid- or leucine-enriched diet during postprandial and postabsorptive states. Nutr. Metab. (Lond.) 2016, 13, 12. [Google Scholar] [CrossRef]
- Beatty, C.H.; West, E.S.; Bocek, R.M. Effect of succinate, fumarate, and oxalacetate on ketone body production by liver slices from non-diabetic and diabetic rats. J. Biol. Chem. 1958, 230, 725–733. [Google Scholar]
- Spydevold, S.; Davis, E.J.; Bremer, J. Replenishment and depletion of citric acid cycle intermediates in skeletal muscle. Indication of pyruvate carboxylation. Eur. J. Biochem. 1976, 71, 155–165. [Google Scholar] [CrossRef]
- Palmer, T.N.; Caldecourt, M.A.; Snell, K.; Sugden, M.C. Alanine and inter-organ relationships in branched-chain amino and 2-oxo acid metabolism. Rev. Biosci. Rep. 1985, 5, 1015–1033. [Google Scholar] [CrossRef] [PubMed]
- Zinneman, H.H.; Nuttall, F.Q.; Goetz, F.C. Effect of endogenous insulin on human amino acid metabolism. Diabetes 1966, 15, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.C.; Bessey, P.Q.; Black, P.R.; Aoki, T.T.; Wilmore, D.W. Insulin stimulates branched chain amino acid uptake and diminishes nitrogen flux from skeletal muscle of injured patients. J. Surg. Res. 1986, 40, 395–405. [Google Scholar] [CrossRef]
- Fukagawa, N.K.; Minaker, K.L.; Young, V.R.; Rowe, J.W. Insulin dose-dependent reductions in plasma amino acids in man. Am. J. Physiol. 1986, 250, E13–E17. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.S.; Björkman, O. Influence of insulin on peripheral uptake of branched chain amino acids in the 60-hour fasted state. Clin. Nutr. 1993, 12, 217–222. [Google Scholar] [CrossRef]
- Hijikata, Y.; Shiozaki, Y.; Sameshima, Y. Changes in plasma amino acids during the oral glucose tolerance test and the effect of these changes on hepatic encephalopathy. J. Clin. Chem. Clin. Biochem. 1985, 23, 259–264. [Google Scholar] [CrossRef]
- Rice, D.E.; Flakoll, P.J.; May, M.M.; Hill, J.O.; Abumrad, N.N. The opposing effects of insulin and hyperglycemia in modulating amino acid metabolism during a glucose tolerance test in lean and obese subjects. Metabolism 1994, 43, 211–216. [Google Scholar] [CrossRef]
- Marchesini, G.; Cassarani, S.; Checchia, G.A.; Bianchi, G.; Bua, V.; Zoli, M.; Pisi, E. Insulin resistance in aged man: Relationship between impaired glucose tolerance and decreased insulin activity on branched-chain amino acids. Metabolism 1987, 36, 1096–1100. [Google Scholar] [CrossRef]
- Vazquez, J.A.; Morse, E.L.; Adibi, S.A. Effect of dietary fat, carbohydrate, and protein on branched-chain amino acid catabolism during caloric restriction. J. Clin. Investig. 1985, 76, 737–743. [Google Scholar] [CrossRef]
- Schauder, P.; Herbertz, L.; Langenbeck, U. Serum branched chain amino and keto acid response to fasting in humans. Metabolism 1985, 34, 58–61. [Google Scholar] [CrossRef]
- She, P.; Olson, K.C.; Kadota, Y.; Inukai, A.; Shimomura, Y.; Hoppel, C.L.; Adams, S.H.; Kawamata, Y.; Matsumoto, H.; Sakai, R.; et al. Leucine and protein metabolism in obese Zucker rats. PLoS ONE 2013, 8, e59443. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.; Perlman, D.F.; McLaughlin, P.M.; King, P.A.; Cha, C.J. Muscle glutamine production in diabetic ketoacidotic rats. Biochem. J. 1983, 214, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Banner, C.; Max, S.R. Effect of diabetes on glutamine synthetase expression in rat skeletal muscles. Am. J. Physiol. 1990, 258, E762–E766. [Google Scholar] [CrossRef] [PubMed]
- Aseervatham, J.; Palanivelu, S.; Panchanadham, S. Semecarpus anacardium (bhallataka) alters the glucose metabolism and energy production in diabetic rats. Evid. Based Complement. Alternat. Med. 2011, 2011, 142978. [Google Scholar] [CrossRef]
- Chandramohan, G.; Al-Numair, K.S.; Veeramani, C.; Alsaif, M.A.; Almajwal, A.M. Protective effect of kaempferol, a flavonoid compound, on oxidative mitochondrial damage in streptozotocin-induced diabetic rats. Prog. Nutr. 2015, 17, 238–244. [Google Scholar]
- Bloxam, D.L. Nutritional aspects of amino acid metabolism. 2. The effects of starvation on hepatic portal-venous differences in plasma amino acid concentration and on liver amino acid concentrations in the rat. Br. J. Nutr. 1972, 27, 233–247. [Google Scholar] [CrossRef]
- Wijekoon, E.P.; Skinner, C.; Brosnan, M.E.; Brosnan, J.T. Amino acid metabolism in the Zucker diabetic fatty rat: Effects of insulin resistance and of type 2 diabetes. Can. J. Physiol. Pharmacol. 2004, 82, 506–514. [Google Scholar] [CrossRef]
- Harris, R.A.; Goodwin, G.W.; Paxton, R.; Dexter, P.; Powell, S.M.; Zhang, B.; Han, A.; Shimomura, Y.; Gibson, R. Nutritional and hormonal regulation of the activity state of hepatic branched-chain alpha-keto acid dehydrogenase complex. Ann. N. Y. Acad. Sci. 1989, 573, 306–313. [Google Scholar] [CrossRef]
- May, M.E.; Mancusi, V.J.; Aftring, R.P.; Buse, M.G. Effects of diabetes on oxidative decarboxylation of branched-chain keto acids. Am. J. Physiol. 1980, 239, E215–E222. [Google Scholar] [CrossRef]
- Gibson, R.; Zhao, Y.; Jaskiewicz, J.; Fineberg, S.E.; Harris, R.A. Effects of diabetes on the activity and content of the branched-chain alpha-ketoacid dehydrogenase complex in liver. Arch. Biochem. Biophys. 1993, 306, 22–28. [Google Scholar] [CrossRef]
- Kuzuya, T.; Katano, Y.; Nakano, I.; Hirooka, Y.; Itoh, A.; Ishigami, M.; Hayashi, K.; Honda, T.; Goto, H.; Fujita, Y.; et al. Regulation of branched-chain amino acid catabolism in rat models for spontaneous type 2 diabetes mellitus. Biochem. Biophys. Res. Commun. 2008, 373, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Bajotto, G.; Murakami, T.; Nagasaki, M.; Sato, Y.; Shimomura, Y. Decreased enzyme activity and contents of hepatic branched-chain alpha-keto acid dehydrogenase complex subunits in a rat model for type 2 diabetes mellitus. Metabolism 2009, 58, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Tischler, M.E.; Goldberg, A.L. Leucine degradation and release of glutamine and alanine by adipose tissue. J. Biol. Chem. 1980, 255, 8074–8081. [Google Scholar] [PubMed]
- Frick, G.P.; Goodman, H.M. Insulin regulation of the activity and phosphorylation of branched-chain 2-oxo acid dehydrogenase in adipose tissue. Biochem. J. 1989, 258, 229–235. [Google Scholar] [CrossRef]
- Pietiläinen, K.H.; Naukkarinen, J.; Rissanen, A.; Saharinen, J.; Ellonen, P.; Keränen, H.; Suomalainen, A.; Götz, A.; Suortti, T.; Yki-Järvinen, H.; et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: Pathways behind acquired obesity. PLoS Med. 2008, 5, e51. [Google Scholar] [CrossRef]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. 2007, 293, E1552–E1563. [Google Scholar] [CrossRef]
- Wiklund, P.; Zhang, X.; Pekkala, S.; Autio, R.; Kong, L.; Yang, Y.; Keinänen-Kiukaanniemi, S.; Alen, M.; Cheng, S. Insulin resistance is associated with altered amino acid metabolism and adipose tissue dysfunction in normoglycemic women. Sci. Rep. 2016, 6, 24540. [Google Scholar] [CrossRef]
- Herman, M.A.; She, P.; Peroni, O.D.; Lynch, C.J.; Kahn, B.B. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J. Biol. Chem. 2010, 285, 11348–11356. [Google Scholar] [CrossRef]
- David, J.; Dardevet, D.; Mosoni, L.; Savary-Auzeloux, I.; Polakof, S. Impaired skeletal muscle branched-chain amino acids catabolism contributes to their increased circulating levels in a non-obese insulin-resistant fructose-fed rat model. Nutrients 2019, 11, 355. [Google Scholar] [CrossRef]
- Holecek, M. The BCAA-BCKA cycle: Its relation to alanine and glutamine synthesis and protein balance. Nutrition 2001, 17, 70. [Google Scholar] [CrossRef]
- Holecek, M. Relation between glutamine, branched-chain amino acids, and protein metabolism. Nutrition 2002, 18, 130–133. [Google Scholar] [CrossRef]
- Holeček, M. Branched-chain amino acids and branched-chain keto acids in hyperammonemic states: Metabolism and as supplements. Metabolites 2020, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Giesecke, K.; Magnusson, I.; Ahlberg, M.; Hagenfeldt, L.; Wahren, J. Protein and amino acid metabolism during early starvation as reflected by excretion of urea and methylhistidines. Metabolism 1989, 38, 1196–1200. [Google Scholar] [CrossRef]
- Pozefsky, T.; Tancredi, R.G.; Moxley, R.T.; Dupre, J.; Tobin, J.D. Effects of brief starvation on muscle amino acid metabolism in nonobese man. J. Clin. Investig. 1976, 57, 444–449. [Google Scholar] [CrossRef] [PubMed]
- De Blaauw, I.; Deutz, N.E.; Von Meyenfeldt, M.F. In vivo amino acid metabolism of gut and liver during short and prolonged starvation. Am. J. Physiol. 1996, 270, G298–G306. [Google Scholar] [CrossRef]
- Holecek, M. Effect of starvation on branched-chain alpha-keto acid dehydrogenase activity in rat heart and skeletal muscle. Physiol. Res. 2001, 50, 19–24. [Google Scholar]
- Umpleby, A.M.; Scobie, I.N.; Boroujerdi, M.A.; Sönksen, P.H. The effect of starvation on leucine, alanine and glucose metabolism in obese subjects. Eur. J. Clin. Investig. 1995, 25, 619–626. [Google Scholar] [CrossRef]
- Wu, G.Y.; Thompson, J.R. The effect of ketone bodies on alanine and glutamine metabolism in isolated skeletal muscle from the fasted chick. Biochem. J. 1988, 255, 139–144. [Google Scholar] [CrossRef]
- Holecek, M.; Sprongl, L.; Tilser, I. Metabolism of branched-chain amino acids in starved rats: The role of hepatic tissue. Physiol. Res. 2001, 50, 25–33. [Google Scholar]
- Palaiologos, G.; Felig, P. Effects of ketone bodies on amino acid metabolism in isolated rat diaphragm. Biochem. J. 1976, 154, 709–716. [Google Scholar] [CrossRef]
- Karakelides, H.; Asmann, Y.W.; Bigelow, M.L.; Short, K.R.; Dhatariya, K.; Coenen-Schimke, J.; Kahl, J.; Mukhopadhyay, D.; Nair, K.S. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 2007, 56, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Felig, P.; Wahren, J.; Sherwin, R.; Palaiologos, G. Amino acid and protein metabolism in diabetes mellitus. Arch. Intern. Med. 1977, 137, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Nair, K.S. Protein metabolism in insulin-dependent diabetes mellitus. J. Nutr. 1998, 128, 323S–327S. [Google Scholar] [CrossRef]
- Lombardo, Y.B.; Thamotharan, M.; Bawani, S.Z.; Paul, H.S.; Adibi, S.A. Posttranscriptional alterations in protein masses of hepatic branched-chain keto acid dehydrogenase and its associated kinase in diabetes. Proc. Assoc. Am. Phys. 1998, 110, 40–49. [Google Scholar] [PubMed]
- Lombardo, Y.B.; Serdikoff, C.; Thamotharan, M.; Paul, H.S.; Adibi, S.A. Inverse alterations of BCKA dehydrogenase activity in cardiac and skeletal muscles of diabetic rats. Am. J. Physiol. 1999, 277, E685–E692. [Google Scholar] [CrossRef] [PubMed]
- Nair, K.S.; Garrow, J.S.; Ford, C.; Mahler, R.F.; Halliday, D. Effect of poor diabetic control and obesity on whole body protein metabolism in man. Diabetologia 1983, 25, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Zimmermann-Telschow, H.; Berchtold, P.; Drost, H.; Müller, W.A.; Gries, F.A.; Zimmermann, H. Blood amine acid levels in patients with insulin excess (functioning insulinoma) and insulin deficiency (diabetic ketosis). Metabolism 1978, 27, 793–799. [Google Scholar] [CrossRef]
- Jensen-Waern, M.; Andersson, M.; Kruse, R.; Nilsson, B.; Larsson, R.; Korsgren, O.; Essén-Gustavsson, B. Effects of streptozotocin-induced diabetes in domestic pigs with focus on the amino acid metabolism. Lab. Anim. 2009, 43, 249–254. [Google Scholar] [CrossRef]
- Buse, M.G.; Weigand, D.A.; Peeler, D.; Hedden, M.P. The effect of diabetes and the redox potential on amino acid content and release by isolated rat hemidiaphragms. Metabolism 1980, 29, 605–616. [Google Scholar] [CrossRef]
- Buse, M.G.; Herlong, H.F.; Weigand, D.A. The effect of diabetes, insulin, and the redox potential on leucine metabolism by isolated rat hemidiaphragm. Endocrinology 1976, 98, 1166–1175. [Google Scholar] [CrossRef]
- Aftring, R.P.; Manos, P.N.; Buse, M.G. Catabolism of branched-chain amino acids by diaphragm muscles of fasted and diabetic rats. Metabolism 1985, 34, 702–711. [Google Scholar] [CrossRef]
- Gürke, J.; Hirche, F.; Thieme, R.; Haucke, E.; Schindler, M.; Stangl, G.I.; Fischer, B.; Santos, A.N. Maternal diabetes leads to adaptation in embryonic amino acid metabolism during early pregnancy. PLoS ONE 2015, 10, e0127465. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.C.; Hsu, J.W.; Khoo, C.M.; Tai, E.S.; Yu, S.; Chacko, S.; Lai, O.F.; Jahoor, F. Alterations in branched-chain amino acid kinetics in nonobese but insulin-resistant Asian men. Am. J. Clin. Nutr. 2018, 108, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Seibert, R.; Abbasi, F.; Hantash, F.M.; Caulfield, M.P.; Reaven, G.; Kim, S.H. Relationship between insulin resistance and amino acids in women and men. Physiol. Rep. 2015, 3, e12392. [Google Scholar] [CrossRef]
- Würtz, P.; Soininen, P.; Kangas, A.J.; Rönnemaa, T.; Lehtimäki, T.; Kähönen, M.; Viikari, J.S.; Raitakari, O.T.; Ala-Korpela, M. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 2013, 36, 648–655. [Google Scholar] [CrossRef]
- Shaham, O.; Wei, R.; Wang, T.J.; Ricciardi, C.; Lewis, G.D.; Vasan, R.S.; Carr, S.A.; Thadhani, R.; Gerszten, R.E.; Mootha, V.K. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol. Syst. Biol. 2008, 4, 214. [Google Scholar] [CrossRef]
- Yamakado, M.; Nagao, K.; Imaizumi, A.; Tani, M.; Toda, A.; Tanaka, T.; Jinzu, H.; Miyano, H.; Yamamoto, H.; Daimon, T.; et al. Plasma free amino acid profiles predict four-year risk of developing diabetes, metabolic syndrome, dyslipidemia, and hypertension in Japanese population. Sci. Rep. 2015, 5, 11918. [Google Scholar] [CrossRef]
- Biolo, G.; Tessari, P.; Inchiostro, S.; Bruttomesso, D.; Sabadin, L.; Fongher, C.; Panebianco, G.; Fratton, M.G.; Tiengo, A. Fasting and postmeal phenylalanine metabolism in mild type 2 diabetes. Am. J. Physiol. 1992, 263, E877–E883. [Google Scholar] [CrossRef]
- Lerin, C.; Goldfine, A.B.; Boes, T.; Liu, M.; Kasif, S.; Dreyfuss, J.M.; De Sousa-Coelho, A.L.; Daher, G.; Manoli, I.; Sysol, J.R.; et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol. Metab. 2016, 5, 926–936. [Google Scholar] [CrossRef]
- Hernández-Alvarez, M.I.; Díaz-Ramos, A.; Berdasco, M.; Cobb, J.; Planet, E.; Cooper, D.; Pazderska, A.; Wanic, K.; O’Hanlon, D.; Gomez, A.; et al. Early-onset and classical forms of type 2 diabetes show impaired expression of genes involved in muscle branched-chain amino acids metabolism. Sci. Rep. 2017, 7, 13850. [Google Scholar] [CrossRef]
- Gaster, M. Reduced TCA flux in diabetic myotubes: A governing influence on the diabetic phenotype? Biochem. Biophys. Res. Commun. 2009, 387, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.; Krssak, M.; Bernroider, E.; Anderwald, C.; Brehm, A.; Meyerspeer, M.; Nowotny, P.; Roth, E.; Waldhäusl, W.; Roden, M. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes 2002, 51, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Yu, J.; Guo, Y.; Deng, J.; Li, K.; Du, Y.; Chen, S.; Zhu, J.; Sheng, H.; Guo, F. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism 2014, 63, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, F.; Marette, A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J. Biol. Chem. 2001, 276, 38052–38060. [Google Scholar] [PubMed]
- Lynch, C.J.; Adams, S.H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 2014, 10, 723–736. [Google Scholar] [CrossRef]
- Siddik, M.A.B.; Shin, A.C. Recent progress on branched-chain amino acids in obesity, diabetes, and beyond. Endocrinol. Metab. (Seoul) 2019, 34, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xie, G.; Jia, W.; Jia, W. Insulin resistance and the metabolism of branched-chain amino acids. Front. Med. 2013, 7, 53–59. [Google Scholar] [CrossRef]
- Giesbertz, P.; Daniel, H. Branched-chain amino acids as biomarkers in diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 48–54. [Google Scholar] [CrossRef]
- Aguer, C.; McCoin, C.S.; Knotts, T.A.; Thrush, A.B.; Ono-Moore, K.; McPherson, R.; Dent, R.; Hwang, D.H.; Adams, S.H.; Harper, M.E. Acylcarnitines: Potential implications for skeletal muscle insulin resistance. FASEB J. 2015, 29, 336–345. [Google Scholar] [CrossRef]
- White, P.J.; Lapworth, A.L.; An, J.; Wang, L.; McGarrah, R.W.; Stevens, R.D.; Ilkayeva, O.; George, T.; Muehlbauer, M.J.; Bain, J.R.; et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 2016, 5, 538–551. [Google Scholar] [CrossRef]
- Yoon, M.S. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients 2016, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Neinast, M. Branched Chain Amino Acids in Metabolic Disease. Curr. Diab. Rep. 2018, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- De Bandt, J.P.; Cynober, L. Therapeutic use of branched-chain amino acids in burn, trauma, and sepsis. J. Nutr. 2006, 136, 308S–313S. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.C.; Fajans, S.S.; Conn, J.W.; Knopf, R.F.; Rull, J. Stimulation of insulin secretion by amino acids. J. Clin. Investig. 1966, 45, 1487–1502. [Google Scholar] [CrossRef]
- Anthony, J.C.; Lang, C.H.; Crozier, S.J.; Anthony, T.G.; MacLean, D.A.; Kimball, S.R.; Jefferson, L.S. Contribution of insulin to the translational control of protein synthesis in skeletal muscle by leucine. Am. J. Physiol. 2002, 282, E1092–E1101. [Google Scholar] [CrossRef]
- Desikan, V.; Mileva, I.; Garlick, J.; Lane, A.H.; Wilson, T.A.; McNurlan, M.A. The effect of oral leucine on protein metabolism in adolescents with type 1 diabetes mellitus. Int. J. Pediatr. Endocrinol. 2010, 2010, 493258. [Google Scholar] [CrossRef]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holeček, M. Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes? Nutrients 2020, 12, 3087. https://doi.org/10.3390/nu12103087
Holeček M. Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes? Nutrients. 2020; 12(10):3087. https://doi.org/10.3390/nu12103087
Chicago/Turabian StyleHoleček, Milan. 2020. "Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes?" Nutrients 12, no. 10: 3087. https://doi.org/10.3390/nu12103087
APA StyleHoleček, M. (2020). Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes? Nutrients, 12(10), 3087. https://doi.org/10.3390/nu12103087