
Libertellenone H, a Natural Pimarane Diterpenoid, Inhibits Thioredoxin System and Induces ROS-Mediated Apoptosis in Human Pancreatic Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
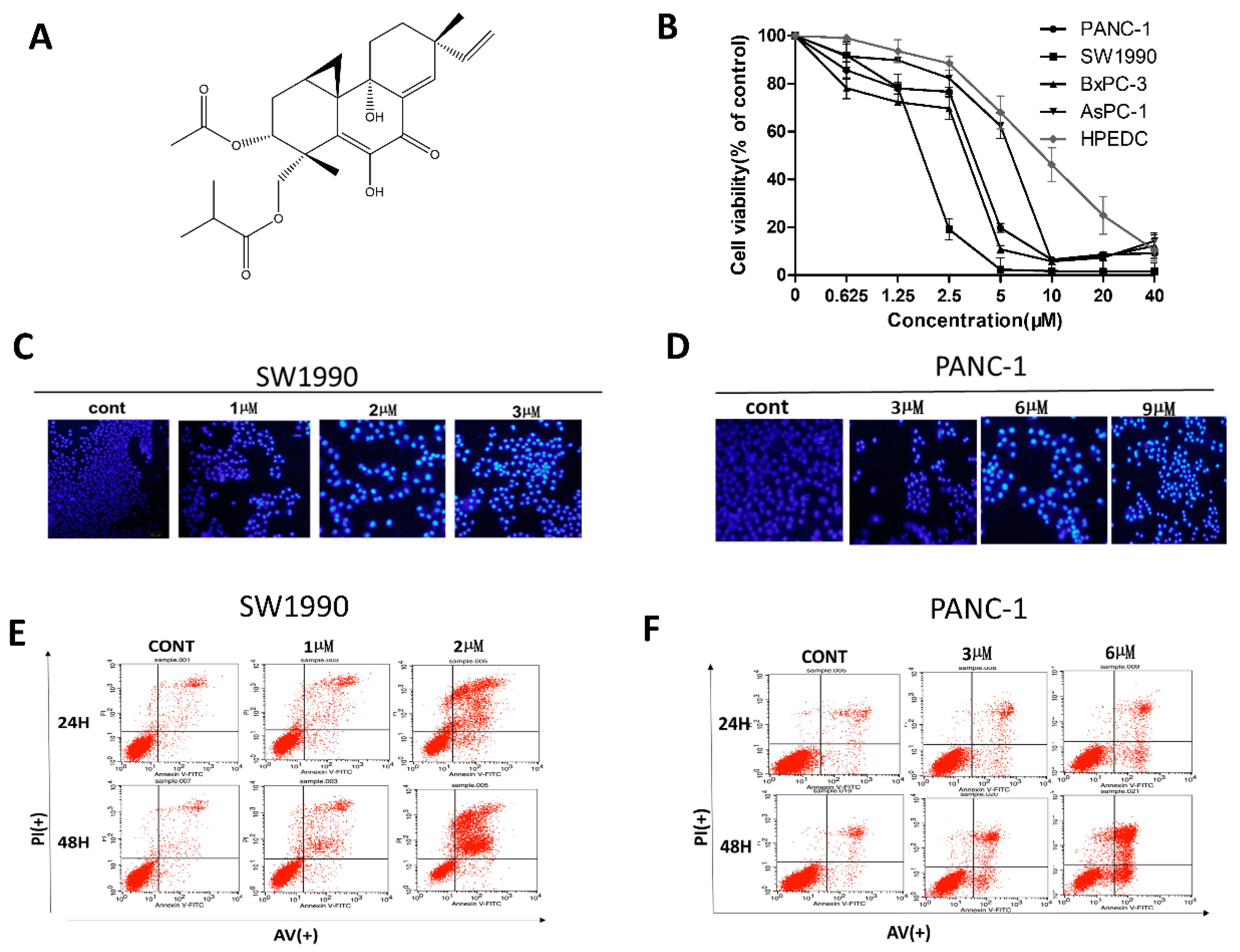
2.1. LH Inhibits Cell Growth in Human Pancreatic Cancer Cell Lines
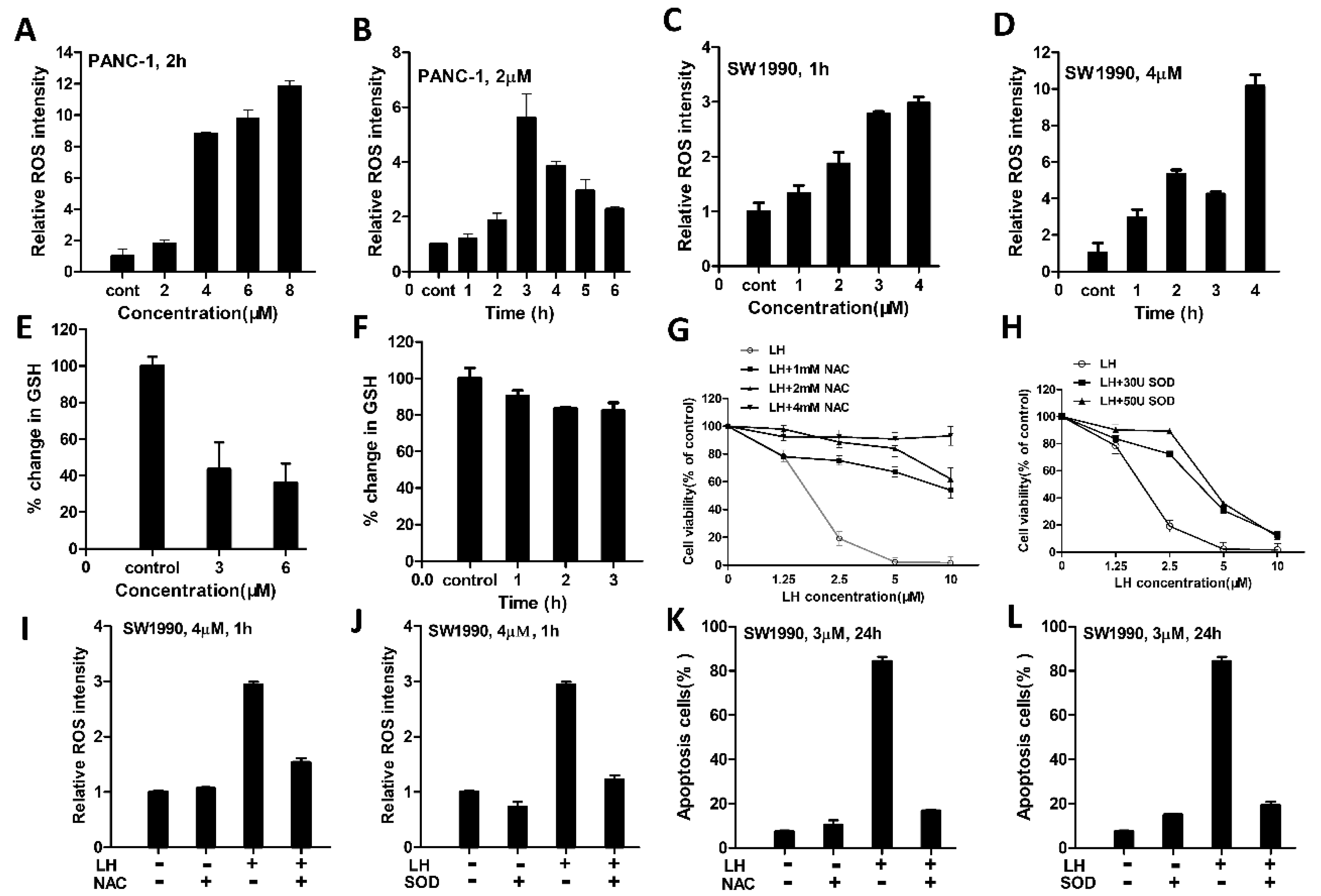
2.2. LH Induced-ROS Accumulation Involved in Its Antitumor Activity
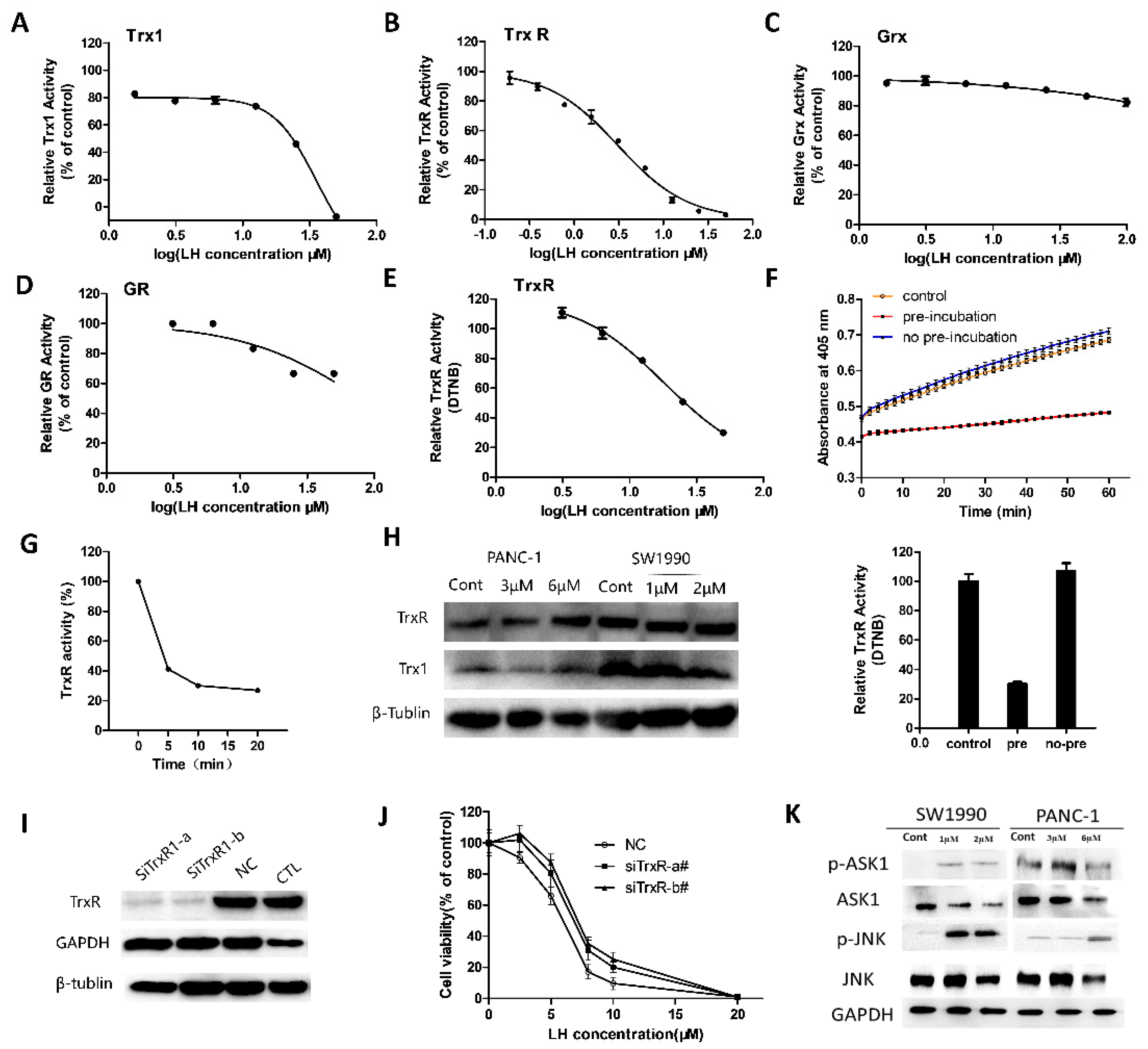
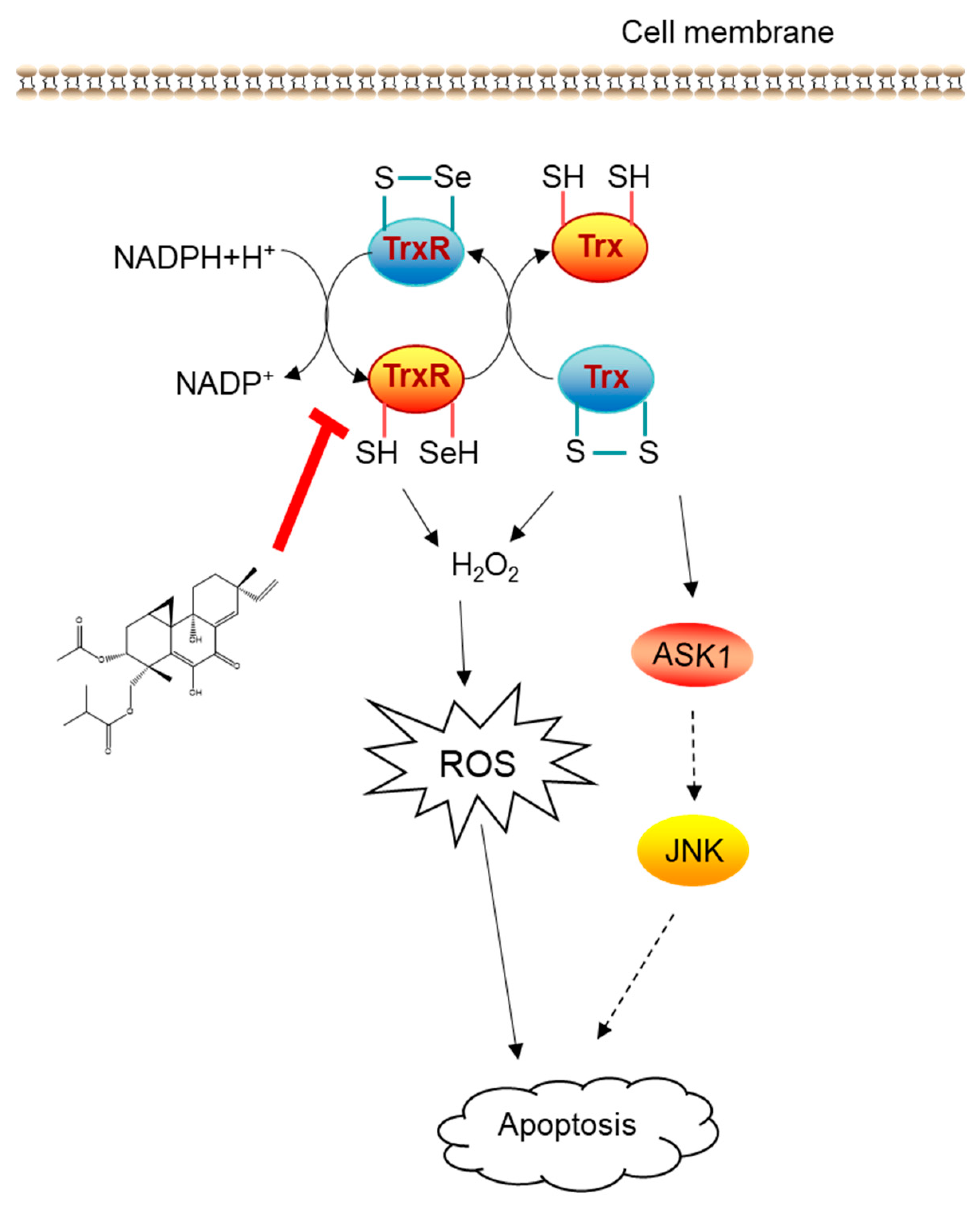
2.3. LH Inhibited Trx/TrxR System
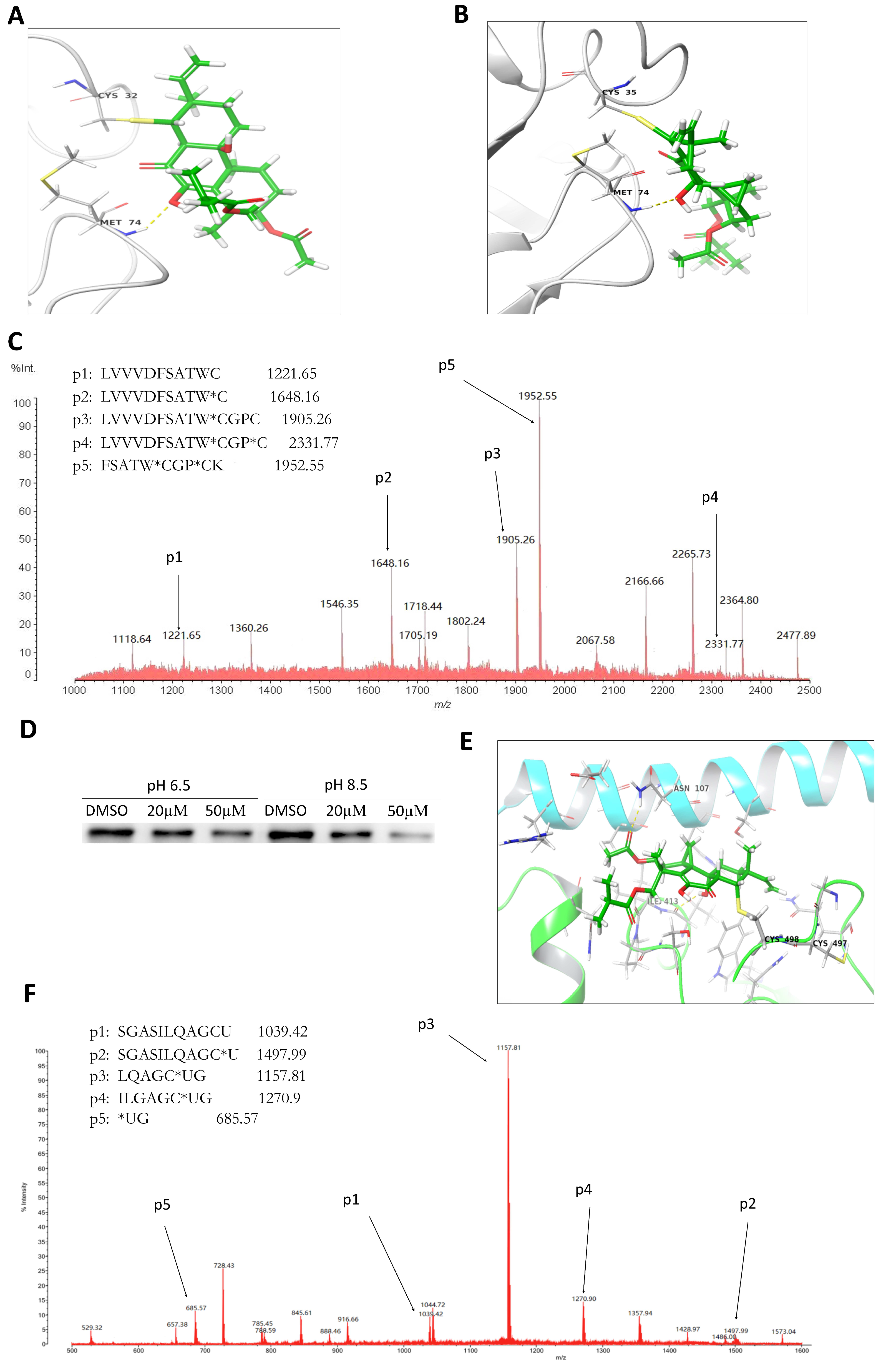
2.4. LH Covalently Bound to Cys32 and Cys35 Residues of Trx1
2.5. LH Covalently Bond to Sec498 Residues of TrxR
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Culture
4.3. Cell Proliferation
4.4. Cell Apoptosis Analysis
4.5. ROS Determination
4.6. Assessment of GSH Levels
4.7. Western Blot Analysis
4.8. Trx1 Activity Analysis
4.9. TrxR Enzyme Assay (Fluorescence Substrates Method)
4.10. TrxR Enzyme Assay (DTNB Method)
4.11. Grx1 Activity Analysis
4.12. GR Activity Analysis
4.13. RNA Interference
4.14. Molecular Docking
4.15. BIAM Labeling
4.16. MS/MS Analysis
4.17. Datal Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules–From Biology to Health and Disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Eklund, H.; Gleason, F.K.; Holmgren, A. Structural and functional relations among thioredoxins of different species. Proteins 1991, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Elias, S.J.A.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Gallegos, A.; Gasdaska, J.R.; Taylor, C.W.; Paine-Murrieta, G.D.; Goodman, D.; Gasdaska, P.Y.; Berggren, M.; Briehl, M.M.; Gowis, G. Transfection with Human Thioredoxin Increases Cell Proliferation and a Dominant-negative Mutant Thioredoxin Reverses the Transformed Phenotype of Human Breast Cancer Cells. Cancer Res. 1996, 56, 5765–5770. [Google Scholar]
- Yan, C.; Siegel, D.; Newsome, J.; Chilloux, A.; Moody, C.J.; Ross, D. Antitumor indolequinones induced apoptosis in human pancreatic cancer cells via inhibition of thioredoxin reductase and activation of redox signaling. Mol. Pharmacol. 2012, 81, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin Reductase 1 Deficiency Reverses Tumor Phenotype and Tumorigenicity of Lung Carcinoma Cells. J. Biol. Chem. 2006, 281, 13005–13008. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of Apoptosis by ASK1, a Mammalian MAPKKK That Activates SAPK/JNK and p38 Signaling Pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Bellamy, W.T.; Briehl, M.M.; Powis, G. The Redox Protein Thioredoxin-1 (Trx-1) Increases Hypoxia-inducible Factor 1α Protein Expression Trx-1 Overexpression Results in Increased Vascular Endothelial Growth Factor Production and Enhanced Tumor Angiogenesis. Cancer Res. 2002, 62, 5089. [Google Scholar] [PubMed]
- Kim, W.J.; Cho, H.; Lee, S.W.; Kim, Y.J.; Kim, K.W. Antisense-thioredoxin inhibits angiogenesis via pVHL-mediated hypoxia-inducible factor-1alpha degradation. Int. J. Oncol. 2005, 26, 1049–1052. [Google Scholar] [PubMed]
- Christina, K.T.; Fay, T.K. Thioredoxin and Cancer: A Role for Thioredoxin in all States of Tumor Oxygenation. Cancers 2010, 2, 209–232. [Google Scholar] [CrossRef] [Green Version]
- Kakolyris, S.; Giatromanolaki, A.; Koukourakis, M.; Powis, G.; Souglakos, J.; Sivridis, E.; Georgoulias, V.; Gatter, K.C.; Harris, A.L. Thioredoxin expression is associated with lymph node status and prognosis in early operable non-small cell lung cancer. Clin. Cancer Res. 2001, 7, 3087–3091. [Google Scholar]
- Raffel, J.; Bhattacharyya, A.K.; Gallegos, A.; Cui, H.; Einspahr, J.G.; Alberts, D.S.; Powis, G. Increased expression of thioredoxin-1 in human colorectal cancer is associated with decreased patient survival. J. Lab. Clin. Med. 2003, 142, 46–51. [Google Scholar] [CrossRef]
- Farina, A.R.; Tacconelli, A.; Cappabianca, L.; Masciulli, M.P.; Holmgren, A.; Beckett, G.J.; Gulino, A.; Mackay, A.R. Thioredoxin alters the matrix metalloproteinase/tissue inhibitors of metalloproteinase balance and stimulates human SK-N-SH neuroblastoma cell invasion. Eur J. Biochem. 2001, 268, 405–413. [Google Scholar] [CrossRef]
- Farina, A.R.; Cappabianca, L.; Desantis, G.; Ianni, N.D.; Ruggeri, P.; Ragone, M.; Merolle, S.; Tonissen, K.F.; Gulino, A.; Mackay, A.R. Thioredoxin stimulates MMP-9 expression, de-regulates the MMP-9/TIMP-1 equilibrium and promotes MMP-9 dependent invasion in human MDA-MB-231 breast cancer cells. FEBS Lett. 2011, 585, 3328–3336. [Google Scholar] [CrossRef]
- Ghareeb, H.; Metanis, N. The Thioredoxin System: A Promising Target for Cancer Drug Development. Chemistry 2020, 26, 10175–10184. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.; Peng, S.; Liu, R.; Li, X.; Hou, Y.; Han, X.; Fang, J. Thioredoxin reductase inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 547–556. [Google Scholar] [CrossRef]
- Tonissen, K.F.; Trapani, G.D. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Du, Z.Y.; Huang, Z.S.; Lee, K.S.; Gu, L.Q. Inhibition of thioredoxin reductase by curcumin analogs. Biosci. Biotechnol. Biochem. 2008, 72, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, X.X.; Jiang, Z.B.; Loo, W.T.; Yao, X.J.; Leung, E.L.; Chow, L.W.; Liu, L. Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol. Res. 2017, 115, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Papp, L.V.; Fang, J.; Rodriguez-Nieto, S.; Zhivotovsky, B.; Holmgren, A. Inhibition of Mammalian Thioredoxin Reductase by Some Flavonoids: Implications for Myricetin and Quercetin Anticancer Activity. Cancer Res. 2006, 66, 4410–4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Tuladhar, A.; Rolle, S.; Lai, Y.; Rey, F.R.D.; Zavala, C.E.; Liu, Y.; Rein, K.S. Brevetoxin-2, is a unique inhibitor of the C-terminal redox center of mammalian thioredoxin reductase-1. Toxicol. Appl. Pharmacol. 2017, 329, 58–66. [Google Scholar] [CrossRef]
- Sun, X.; Wang, W.; Chen, J.; Cai, X.; Yang, J.; Yang, Y.; Yan, H.; Cheng, X.; Ye, J.; Lu, W.; et al. The Natural Diterpenoid Isoforretin A Inhibits Thioredoxin-1 and Triggers Potent ROS-Mediated Antitumor Effects. Cancer Res. 2017, 77, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, R.K.; Abbruzzese, J.; Dragovich, T.; Kirkpatrick, L.; Guillen, J.M.; Baker, A.F.; Pestano, L.A.; Green, S.; Von Hoff, D.D. A randomized phase II study of PX-12, an inhibitor of thioredoxin in patients with advanced cancer of the pancreas following progression after a gemcitabine-containing combination. Cancer Chemother. Pharmacol. 2011, 67, 503–509. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine Natural Products: A Source of Novel Anticancer Drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [Green Version]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, G.; Liu, W.; Yang, X.; Zhu, N.; Shen, J.; Wang, Z.; Liu, Z.; Liu, Y.; Cheng, S.; et al. Recent progress in research and development of marine drugs. Chin. Mar. Drugs 2019, 38, 35–69. [Google Scholar]
- Lu, X.L.; Liu, J.T.; Liu, X.Y.; Gao, Y.; Zhang, J.P.; Jiao, B.H.; Zheng, H. Pimarane diterpenes from the Arctic fungus Eutypella sp. D-1. J. Antibiot. 2014, 67, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Dörthe, B.; Siedler, F.; Diercks, T.; Kessler, H.; Moroder, L. The Redox Potential of Selenocystine in Unconstrained Cyclic Peptides. Angew. Chem. Int. Ed. Engl. 1997, 36, 883–885. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Nat. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef] [Green Version]
- Mavridou, D.A.I.; Saridakis, E.; Kritsiligkou, P.; Mozley, E.C.; Ferguson, S.J.; Redfield, C. An Extended Active-site Motif Controls the Reactivity of the Thioredoxin Fold. J. Biol. Chem. 2014, 289, 8681–8696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, G.; Foloppe, N.; Messens, J. Understanding the pKa of Redox Cysteines: The Key Role of Hydrogen Bonding. Antioxid. Redox Signal. 2013, 18, 94–127. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Zhu, Y.; Yu, H.; Liu, X.; Jiao, B.; Lu, X. Libertellenone H, a Natural Pimarane Diterpenoid, Inhibits Thioredoxin System and Induces ROS-Mediated Apoptosis in Human Pancreatic Cancer Cells. Molecules 2021, 26, 315. https://doi.org/10.3390/molecules26020315
Zhang W, Zhu Y, Yu H, Liu X, Jiao B, Lu X. Libertellenone H, a Natural Pimarane Diterpenoid, Inhibits Thioredoxin System and Induces ROS-Mediated Apoptosis in Human Pancreatic Cancer Cells. Molecules. 2021; 26(2):315. https://doi.org/10.3390/molecules26020315
Chicago/Turabian StyleZhang, Weirui, Yuping Zhu, Haobing Yu, Xiaoyu Liu, Binghua Jiao, and Xiaoling Lu. 2021. "Libertellenone H, a Natural Pimarane Diterpenoid, Inhibits Thioredoxin System and Induces ROS-Mediated Apoptosis in Human Pancreatic Cancer Cells" Molecules 26, no. 2: 315. https://doi.org/10.3390/molecules26020315