
The Circular Life of Human CD38: From Basic Science to Clinics and Back

 ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Premise
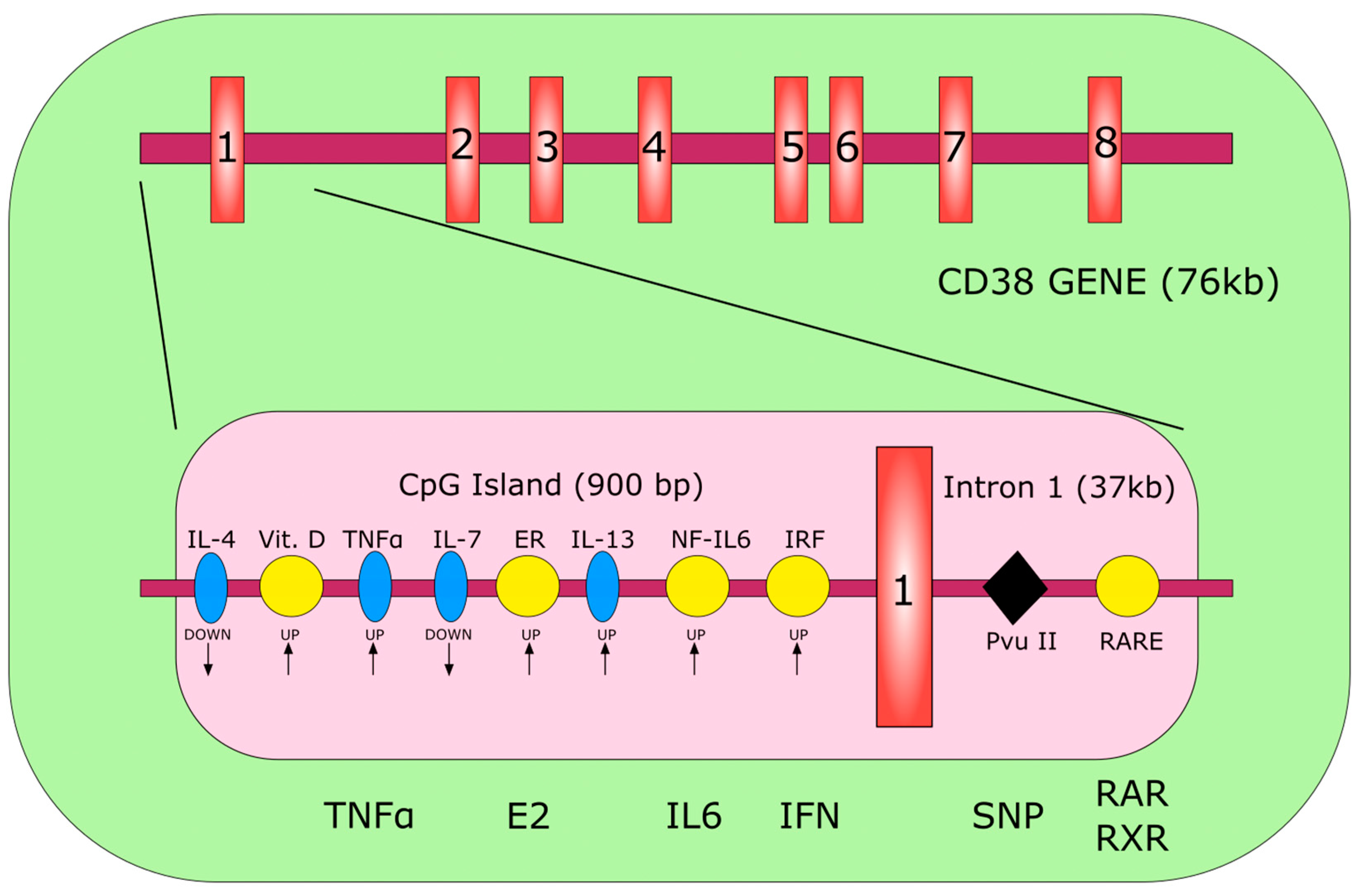
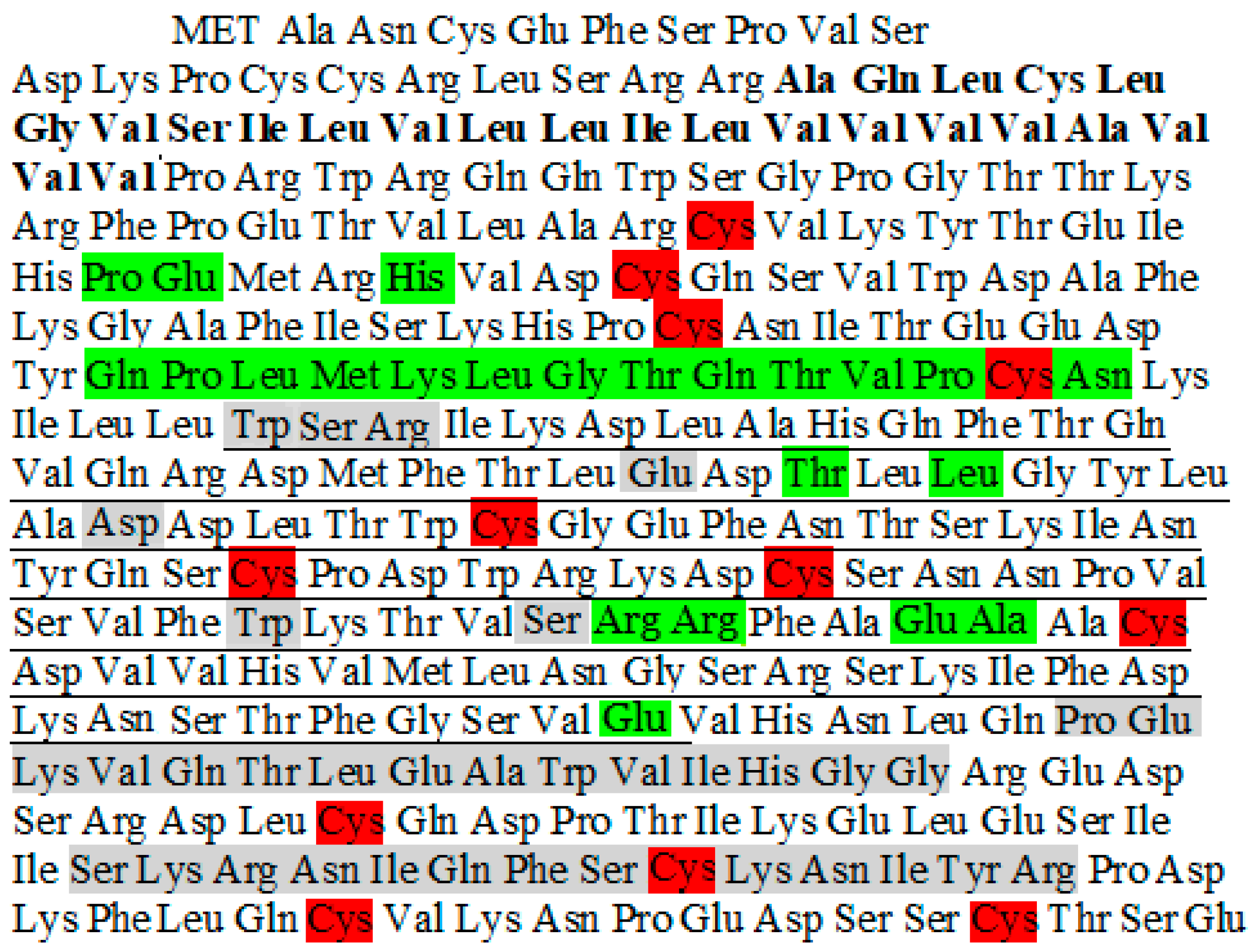
2. Gene and Structure
3. Gene Regulation
4. CD38 Functions
4.1. As a Receptor
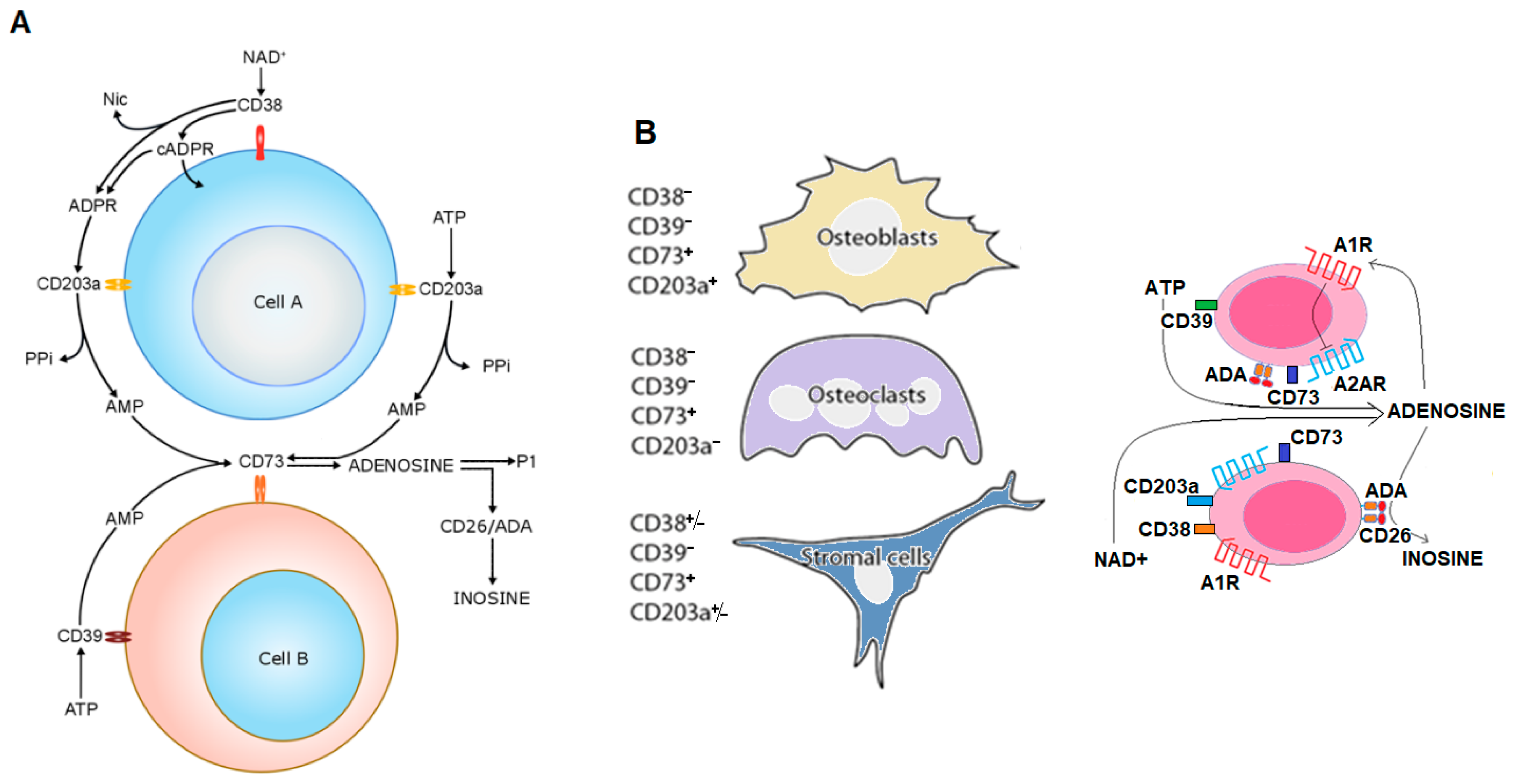
4.2. As an Ectoenzyme
4.3. CD38 Connections
4.4. Cytolytic and Catalytic CD38-Dependent Effects of Therapeutic mAbs in MM
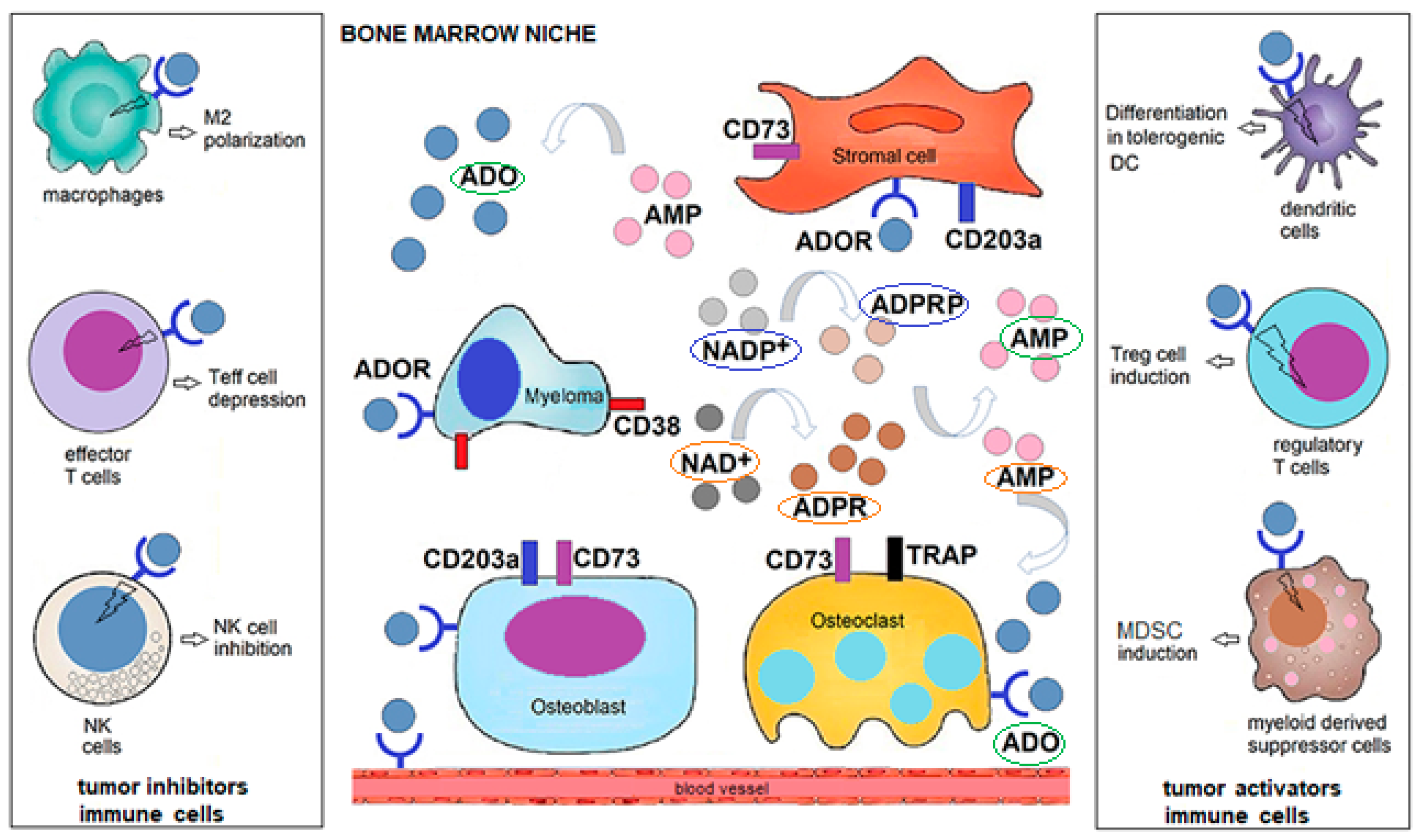
5. CD38-Controlled Activities and Metabolic Adaptation during Diseases
6. CD38 in the Polarization and Release of Extracellular Vesicles (EV)
7. Clinical Applications outside MM
8. Self-Vaccination
9. Conclusions
Funding
Conflicts of Interest
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nat. Cell Biol. 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Iii, R.O.W.; Tucker, H. Formulation strategies in immunotherapeutic pharmaceutical products. World J. Clin. Oncol. 2020, 11, 275–282. [Google Scholar] [CrossRef] [PubMed]
- De Winde, C.M.; Elfrink, S.; Van Spriel, A.B. Novel insights into membrane targeting of B cell lymphoma. Trends Cancer 2017, 3, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Lanza, F.; Maffini, E.; Rondoni, M.; Massari, E.; Faini, A.C.; Malavasi, F. CD22 Expression in B-Cell acute lymphoblastic leukemia: Biological significance and implications for inotuzumab therapy in adults. Cancers 2020, 12, 303. [Google Scholar] [CrossRef]
- Giuliani, N.; Malavasi, F. Editorial: Immunotherapy in multiple myeloma. Front. Immunol. 2019, 10, 1945. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Drach, J.; Zhao, S.; Malavasi, F.; Mehta, K. Rapid induction of CD38 antigen on myeloid leukemia cells by all trans-retinoic acid. Biochem. Biophys. Res. Commun. 1993, 195, 545–550. [Google Scholar] [CrossRef]
- Drach, J.; McQueen, T.; Engel, H.; Andreeff, M.; Robertson, K.A.; Collins, S.J.; Malavasi, F.; Mehta, K. Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res. 1994, 54, 1746–1752. [Google Scholar]
- Malavasi, F. Editorial: CD38 and retinoids: A step toward a cure. J. Leukoc. Biol. 2011, 90, 217–219. [Google Scholar] [CrossRef]
- Ferrero, E.; Faini, A.C.; Malavasi, F. A phylogenetic view of the leukocyte ectonucleotidases. Immunol. Lett. 2019, 205, 51–58. [Google Scholar] [CrossRef]
- Terhorst, C.; Van Agthoven, A.; LeClair, K.; Snow, P.; Reinherz, E.; Schlossman, S. Biochemical studies of the human thymocyte cell-surface antigens T6, T9 and T10. Cell 1981, 23, 771–780. [Google Scholar] [CrossRef]
- Alessio, M.; Roggero, S.; Funaro, A.; De Monte, L.B.; Peruzzi, L.; Geuna, M.; Malavasi, F. CD38 molecule: Structural and biochemical analysis on human T lymphocytes, thymocytes, and plasma cells. J. Immunol. 1990, 145, 878–884. [Google Scholar] [PubMed]
- Hara-Yokoyama, M.; Kukimoto-Niino, M.; Terasawa, K.; Harumiya, S.; Podyma-Inoue, K.A.; Hino, N.; Sakamoto, K.; Itoh, S.; Hashii, N.; Hiruta, Y.; et al. Tetrameric interaction of the ectoenzyme CD38 on the cell surface enables its catalytic and raft-association activities. Structure 2012, 20, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kriksunov, I.A.; Graeff, R.; Munshi, C.; Hao, Q.; Hao, Q. Crystal structure of human CD38 extracellular domain. Structure 2005, 13, 1331–1339. [Google Scholar] [CrossRef]
- Funaro, A.; Horenstein, A.L.; Calosso, L.; Morra, M.; Tarocco, R.P.; Franco, L.; De Flora, A.; Malavasi, F. Identification and characterization of an active soluble form of human CD38 in normal and pathological fluids. Int. Immunol. 1996, 8, 1643–1650. [Google Scholar] [CrossRef]
- Horenstein, L.A.; Stockinger, H.; Imhof, A.B.; Malavasi, F. CD38 binding to human myeloid cells is mediated by mouse and human CD31. Biochem. J. 1998, 330 Pt 3, 1129–1135. [Google Scholar] [CrossRef]
- Deaglio, S.; Dianzani, U.; Horenstein, A.L.; Fernández, J.E.; Van Kooten, C.; Bragardo, M.; Funaro, A.; Garbarino, G.; Di Virgilio, F.; Banchereau, J.; et al. Human CD38 ligand. A 120-KDA protein predominantly expressed on endothelial cells. J. Immunol. 1996, 156, 727–734. [Google Scholar]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar]
- Deaglio, S.; Mallone, R.; Baj, G.; Donati, D.; Giraudo, G.; Corno, F.; Bruzzone, S.; Geuna, M.; Ausiello, C.; Malavasi, F. Human CD38 and its ligand CD31 define a unique lamina propria T lymphocyte signaling pathway. FASEB J. 2001, 15, 580–582. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, H.; Fang, C.; Li, C.; Xhyliu, F.; Dysert, H.; Bodo, J.; Habermehl, G.; Russell, B.E.; Li, W.; et al. Targeting of CD38 by the tumor suppressor miR-26a serves as a novel potential therapeutic agent in multiple myeloma. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Zubiaur, M.; Izquierdo, M.; Terhorst, C.; Malavasi, F.; Sancho, J. CD38 ligation results in activation of the Raf-1/mitogen-activated protein kinase and the CD3-zeta/zeta-associated protein-70 signaling pathways in Jurkat T lymphocytes. J. Immunol. 1997, 159, 193–205. [Google Scholar] [PubMed]
- Morra, M.; Zubiaur, M.; Terhorst, C.; Sancho, J.; Malavasi, F. CD38 is functionally dependent on the TCR/CD3 complex in human T cells. FASEB J. 1998, 12, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Capobianco, A.; Bergui, L.; Dürig, J.; Morabito, F.; Dührsen, U.; Malavasi, F. CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood 2003, 102, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Zubiaur, M.; Gregorini, A.; Bottarel, F.; Ausiello, C.M.; Dianzani, U.; Sancho, J.; Malavasi, F. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood 2002, 99, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Mallone, R.; Funaro, A.; Zubiaur, M.; Baj, G.; Ausiello, C.M.; Tacchetti, C.; Sancho, J.; Grossi, C.; Malavasi, F. Signaling through CD38 induces NK cell activation. Int. Immunol. 2001, 13, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Zilber, M.-T.; Gregory, S.; Mallone, R.; Deaglio, S.; Malavasi, F.; Charron, M.; Gelin, C. CD38 expressed on human monocytes: A coaccessory molecule in the superantigen-induced proliferation. Proc. Natl. Acad. Sci. USA 2000, 97, 2840–2845. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. ADP-ribosyl cyclase: An enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nat. Cell Biol. 2014, 509, 310–317. [Google Scholar] [CrossRef]
- Deng, Q.W.; Zhang, J.; Li, T.; He, W.M.; Fang, L.; Lee, H.C.; Zhao, Y.J. The transferrin receptor CD71 regulates type II CD38, revealing tight topological compartmentalization of intracellular cyclic ADP-ribose production. J. Biol. Chem. 2019, 294, 15293–15303. [Google Scholar] [CrossRef]
- Lee, H.C.; Zhao, Y.J. Resolving the topological enigma in Ca2+ signaling by cyclic ADP-ribose and NAADP. J. Biol. Chem. 2019, 294, 19831–19843. [Google Scholar] [CrossRef]
- Lund, F.E. Signaling properties of CD38 in the mouse immune system: Enzyme-dependent and -independent roles in immunity. Mol. Med. 2006, 12, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Liu, H.-X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nat. Cell Biol. 2007, 446, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Vaisitti, T.; Aydin, S.; Ferrero, E.; Malavasi, F. In-tandem insight from basic science combined with clinical research: CD38 as both marker and key component of the pathogenetic network underlying chronic lymphocytic leukemia. Blood 2006, 108, 1135–1144. [Google Scholar] [CrossRef]
- Martin, T.G.; Corzo, K.; Chiron, M.; Van De Velde, H.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells 2019, 8, 1522. [Google Scholar] [CrossRef] [PubMed]
- Van De Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Jiang, M.H.; Acharya, C.; Zhong, B.M.Y.; Cai, T.; Yang, G.; Song, Z.; Theilhaber, J.; Adrian, F.; Tai, Y.-T.; et al. SAR 650984, a therapeutic anti-CD38 monoclonal antibody, blocks CD38-CD31 interaction in multiple myeloma. Blood 2014, 124, 4729. [Google Scholar] [CrossRef]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The mechanism of action of the anti-CD38 monoclonal antibody Isatuximab in multiple myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef]
- Malavasi, F.; Faini, A.C. Mechanism of action of a new anti-CD38 antibody: Enhancing myeloma immunotherapy. Clin. Cancer Res. 2019, 25, 2946–2948. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; Van De Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zito, A.; Mariani, V.; Faini, A.C.; Morandi, F.; Schiavoni, I.; Ausiello, C.M.; Malavasi, F. Antibody mimicry, receptors and clinical applications. Hum. Antibodies 2017, 25, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The multi-faceted ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in adenosinergic pathways and metabolic re-programming in human multiple myeloma cells: In-tandem insights from basic science to therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Van De Donk, N.W.C.J. Immunomodulatory effects of CD38-targeting antibodies. Immunol. Lett. 2018, 199, 16–22. [Google Scholar] [CrossRef]
- Yagui, K.; Shimada, F.; Mimura, M.; Hashimoto, N.; Suzuki, Y.; Tokuyama, Y.; Nata, K.; Tohgo, A.; Ikehata, F.; Takasawa, S.; et al. A missense mutation in the CD38 gene, a novel factor for insulin secretion: Association with Type II diabetes mellitus in Japanese subjects and evidence of abnormal function when expressed in vitro. Diabetologia 1998, 41, 1024–1028. [Google Scholar] [CrossRef][Green Version]
- Pham, A.; Mahindra, A. Solitary plasmacytoma: A review of diagnosis and management. Curr. Hematol. Malign. Rep. 2019, 14, 63–69. [Google Scholar] [CrossRef]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Bolzoni, M.; Toscani, D.; Costa, F.; Castella, B.; Faini, A.C.; Massaia, M.; Pistoia, V.; et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD+. OncoImmunology 2018, 7, e1458809. [Google Scholar] [CrossRef]
- Belli, S.I.; Goding, J.W. Biochemical characterization of human PC-1, an enzyme possessing alkaline phosphodiesterase I and nucleotide pyrophosphatase activities. JBIC J. Biol. Inorg. Chem. 1994, 226, 433–443. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. OncoImmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zito, A.; Roato, I.; Morandi, F.; Marimpietri, D.; Bolzoni, M.; Toscani, D.; Oldham, R.J.; et al. NAD+-metabolizing ectoenzymes in remodeling tumor–host interactions: The human myeloma model. Cells 2015, 4, 520–537. [Google Scholar] [CrossRef]
- Quarona, V.; Ferri, V.; Chillemi, A.; Bolzoni, M.; Mancini, C.; Zaccarello, G.; Roato, I.; Morandi, F.; Marimpietri, D.; Faccani, G.; et al. Unraveling the contribution of ectoenzymes to myeloma life and survival in the bone marrow niche: Ectoenzymes and the myeloma niche. Ann. N. Y. Acad. Sci. 2015, 1335, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Morandi, F.; Bracci, C.; Pistoia, V.; Malavasi, F. Functional insights into nucleotide-metabolizing ectoenzymes expressed by bone marrow-resident cells in patients with multiple myeloma. Immunol. Lett. 2019, 205, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, E.; Horenstein, A.L.; Canzonetta, C.; Costa, F.; Morandi, F. Canonical and non-canonical adenosinergic pathways. Immunol. Lett. 2018, 205, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zini, R.; Quarona, V.; Bianchi, N.; Manfredini, R.; Gambari, R.; Malavasi, F.; Ferrari, D. Cytokine-induced killer cells express CD39, CD38, CD203a, CD73 Ectoenzymes and P1 adenosinergic receptors. Front. Pharmacol. 2018, 9, 196. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16−NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+T cell proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef]
- Morandi, F.; Morandi, B.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zaccarello, G.; Carrega, P.; Ferlazzo, G.; Mingari, M.C.; Moretta, L.; et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015, 6, 25602–25618. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine generated in the bone marrow niche through a CD38-mediated pathway correlates with progression of human myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef]
- Yang, R.; Elsaadi, S.; Misund, K.; Abdollahi, P.; Vandsemb, E.N.; Moen, S.H.; Kusnierczyk, A.; Slupphaug, G.; Standal, T.; Waage, A.; et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J. Immunother. Cancer 2020, 8, e000610. [Google Scholar] [CrossRef]
- Chillemi, A.; Quarona, V.; Zito, A.; Morandi, F.; Marimpietri, D.; Cuccioloni, M.; Robert, O.J.; Mark, C.S.; Bolzoni, M.; Toscani, D.; et al. Generation and characterization of microvesicles after daratumumab interaction with myeloma cells. Blood 2015, 126, 1849. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; Van Egmond, M.; Van Bueren, J.J.L.; Mutis, T.; Groen, R.W.J.; Breij, E.; Martens, A.C.M.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–320. [Google Scholar] [CrossRef]
- Matas-Céspedes, A.; Vidal-Crespo, A.; Rodriguez, V.; Villamor, N.; Delgado, J.; Giné, E.; Roca-Ho, H.; Menéndez, P.; Campo, E.; López-Guillermo, A.; et al. The human CD38 monoclonal antibody daratumumab shows antitumor activity and hampers leukemia–microenvironment interactions in chronic lymphocytic leukemia. Clin. Cancer Res. 2016, 23, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Manna, A.; Aulakh, S.; Jani, P.; Ahmed, S.; Akhtar, S.; Coignet, M.; Heckman, M.G.; Meghji, Z.; Bhatia, K.; Sharma, A.; et al. Targeting CD38 enhances the antileukemic activity of ibrutinib in chronic lymphocytic leukemia. Clin. Cancer Res. 2019, 25, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Paulus, A.; Manna, A.; Akhtar, S.; Paulus, S.M.; Sharma, M.; Coignet, M.V.; Jiang, L.; Roy, V.; Witzig, T.E.; Ansell, S.M.; et al. Targeting CD38 with daratumumab is lethal to Waldenström macroglobulinaemia cells. Br. J. Haematol. 2018, 183, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Manna, A.; Kellett, T.; Aulakh, S.; Lewis-Tuffin, L.J.; Dutta, N.; Knutson, K.; Chini, E.; Pinilla-Ibarz, J.; Lamanna, N.; Manochakian, R.; et al. Targeting CD38 is lethal to Breg-like chronic lymphocytic leukemia cells and Tregs, but restores CD8+ T-cell responses. Blood Adv. 2020, 4, 2143–2157. [Google Scholar] [CrossRef]
- Tolbert, V.P.; Goldsby, R.; Huang, J.; Shimano, K.; Melton, A.; Willert, J.; Horn, B.N.; Dvorak, C.C.; Wahlstrom, J.T. Daratumumab is effective in the treatment of refractory post-transplant autoimmune hemolytic anemia: A pediatric case report. Blood 2016, 128, 4819. [Google Scholar] [CrossRef]
- Cooling, L.; Hugan, S. Daratumumab in combination with standard treatment for autoimmune hemolytic anemia in a pediatric patient. Transfusion 2019, 59, 3801–3802. [Google Scholar] [CrossRef]
- Even-Or, E.; Eddin, A.N.; Shadur, B.; Schejter, Y.D.; Najajreh, M.; Zelig, O.; Zaidman, I.; Stepensky, P. Successful treatment with daratumumab for post-HSCT refractory hemolytic anemia. Pediatr. Blood Cancer 2019, 67, e28010. [Google Scholar] [CrossRef]
- Schuetz, C.; Hoenig, M.; Moshous, D.; Weinstock, C.; Castelle, M.; Bendavid, M.; Shimano, K.; Tolbert, V.; Schulz, A.S.; Dvorak, C.C. Daratumumab in life-threatening autoimmune hemolytic anemia following hematopoietic stem cell transplantation. Blood Adv. 2018, 2, 2550–2553. [Google Scholar] [CrossRef]
- Blennerhassett, R.; Sudini, L.; Gottlieb, D.; Bhattacharyya, A. Post-allogeneic transplant Evans syndrome successfully treated with daratumumab. Br. J. Haematol. 2019, 187, e48–e51. [Google Scholar] [CrossRef]
- Buteyn, N.J.; Fatehchand, K.; Santhanam, R.; Fang, H.; Dettorre, G.M.; Gautam, S.; Harrington, B.; Henderson, S.E.; Merchand-Reyes, G.; Mo, X.; et al. Anti-leukemic effects of all-trans retinoic acid in combination with Daratumumab in acute myeloid leukemia. Int. Immunol. 2018, 30, 375–383. [Google Scholar] [CrossRef]
- Mistry, J.J.; Moore, J.A.; Kumar, P.; Marlein, C.R.; Hellmich, C.; Pillinger, G.; Jibril, A.; Di Palma, F.; Collins, A.; Bowles, K.M.; et al. Daratumumab inhibits acute myeloid leukaemia metabolic capacity by blocking mitochondrial transfer from mesenchymal stromal cells. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, S.; Liu, F.; Wang, J. Daratumumab for quick and sustained remission in post-transplant relapsed/refractory acute lymphoblastic leukemia. Leuk. Res. 2020, 91, 106332. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.A.; McPhail, L.C.; Snyderman, R. Redistribution of protein kinase C activity in human monocytes: Correlation with activation of the respiratory burst. J. Immunol. 1985, 135, 3411–3416. [Google Scholar]
- Ofran, Y.; Ringelstein-Harlev, S.; Slouzkey, I.; Zuckerman, T.; Yehudai-Ofir, D.; Henig, I.; Beyar-Katz, O.; Hayun, M.; Frisch, A. Daratumumab for eradication of minimal residual disease in high-risk advanced relapse of T-cell/CD19/CD22-negative acute lymphoblastic leukemia. Leukemia 2020, 34, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Hari, P.; Raj, R.V.; Olteanu, H. Targeting CD38 in refractory extranodal natural killer cell–T-cell lymphoma. N. Engl. J. Med. 2016, 375, 1501–1502. [Google Scholar] [CrossRef] [PubMed]
- Paulus, A.; Akhtar, S.; Bashir, Y.; Paulus, S.M.; Yousaf, H.; Roy, V.; Ailawadhi, S.; Ansell, S.; Witzig, T.E.; Chanan-Khan, A.A. Drug resistance alters CD38 expression and in vitro response to daratumumab in waldenstrom macroglobulinemia cells. Blood 2016, 128, 3018. [Google Scholar] [CrossRef]
- Lecumberri, R.; Krsnik, I.; Askari, E.; Sirvent, M.; González-Pérez, M.S.; Escalante, F.; Pradillo, V.; Tamariz, L.E.; Cánovas, V.; Alegre, A.; et al. Treatment with daratumumab in patients with relapsed/refractory AL amyloidosis: A multicentric retrospective study and review of the literature. Amyloid 2020, 1–5, 1–5. [Google Scholar] [CrossRef]
- Canichella, M.; Serrao, A.; Annechini, G.; D’Elia, G.M.; De Luca, M.L.; Pulsoni, A. Long-term response to daratumumab in a patient with advanced immunoglobulin light-chain (AL) amyloidosis with organ damage. Ann. Hematol. 2018, 98, 1047–1048. [Google Scholar] [CrossRef]
- Deshpande, S.; Gertz, M.A.; Dispenzieri, A.; Kumar, S.S.; Parikh, S.A.; Muchtar, E. daratumumab as successful initial therapy for AL amyloidosis with nerve involvement. Leuk. Lymphoma 2020, 1–4, 1–4. [Google Scholar] [CrossRef]
- Roccatello, D.; Fenoglio, R.; Sciascia, S.; Naretto, C.; Rossi, D.; Ferro, M.; Barreca, A.; Malavasi, F.; Baldovino, S. CD38 and anti-CD38 monoclonal antibodies in AL amyloidosis: Targeting plasma cells and beyond. Int. J. Mol. Sci. 2020, 21, 4129. [Google Scholar] [CrossRef]
- Li, S.; England, C.G.; Ehlerding, E.B.; Kutyreff, C.J.; Engle, J.W.; Jiang, D.; Cai, W. ImmunoPET imaging of CD38 expression in hepatocellular carcinoma using 64Cu-labeled daratumumab. Am. J. Transl. Res. 2019, 11, 6007–6015. [Google Scholar] [PubMed]
- Atanackovic, D.; Yousef, S.; Shorter, C.; Tantravahi, S.K.; Steinbach, M.; Iglesias, F.; Sborov, D.; Radhakrishnan, S.V.; Chiron, M.; Miles, R.; et al. In vivo vaccination effect in multiple myeloma patients treated with the monoclonal antibody isatuximab. Leukemia 2019, 34, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, E.; Suarez-Fueyo, A.; Bradley, S.J.; Mizui, M.; Marin, A.V.; Mulki, L.; Krishfield, S.; Malavasi, F.; Yoon, J.; Sui, S.J.H.; et al. The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T Cell function and identifies patients with SLE prone to infections. Cell Rep. 2020, 30, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Mehrotra, S. CD38: Modulating histone methyltransferase EZH2 activity in SLE. Trends Immunol. 2020, 41, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.U.; Kumar, D.T.; Siva, R.; Doss, C.G.P.; Younes, S.; Younes, N.; Sidenna, M.; Zayed, H. Dysregulation of signaling pathways due to differentially expressed genes from the B-Cell transcriptomes of systemic lupus erythematosus patients—A bioinformatics approach. Front. Bioeng. Biotechnol. 2020, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Korver, W.; Carsillo, M.; Yuan, J.; Idamakanti, N.; Wagoner, M.; Shi, P.; Xia, C.Q.; Smithson, G.; McLean, L.; Zalevsky, J.; et al. A Reduction in B, T, and natural killer cells expressing CD38 by TAK-079 inhibits the induction and progression of collagen-induced arthritis in cynomolgus monkeys. J. Pharmacol. Exp. Ther. 2019, 370, 182–196. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horenstein, A.L.; Faini, A.C.; Morandi, F.; Bracci, C.; Lanza, F.; Giuliani, N.; Paulus, A.; Malavasi, F. The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules 2020, 25, 4844. https://doi.org/10.3390/molecules25204844
Horenstein AL, Faini AC, Morandi F, Bracci C, Lanza F, Giuliani N, Paulus A, Malavasi F. The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules. 2020; 25(20):4844. https://doi.org/10.3390/molecules25204844
Chicago/Turabian StyleHorenstein, Alberto L., Angelo C. Faini, Fabio Morandi, Cristiano Bracci, Francesco Lanza, Nicola Giuliani, Aneel Paulus, and Fabio Malavasi. 2020. "The Circular Life of Human CD38: From Basic Science to Clinics and Back" Molecules 25, no. 20: 4844. https://doi.org/10.3390/molecules25204844
APA StyleHorenstein, A. L., Faini, A. C., Morandi, F., Bracci, C., Lanza, F., Giuliani, N., Paulus, A., & Malavasi, F. (2020). The Circular Life of Human CD38: From Basic Science to Clinics and Back. Molecules, 25(20), 4844. https://doi.org/10.3390/molecules25204844