
A Fast and Reliable UHPLC–MS/MS-Based Method for Screening Selected Pharmacologically Significant Natural Plant Indole Alkaloids


Abstract
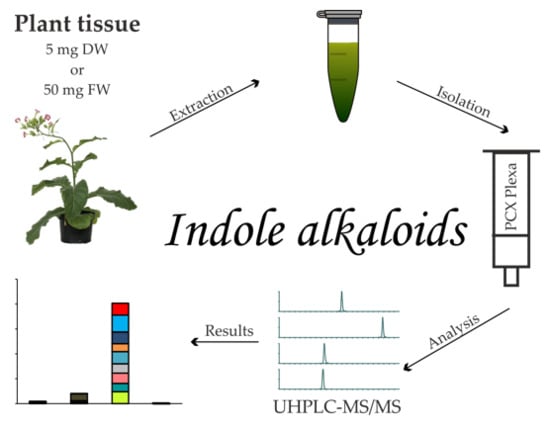
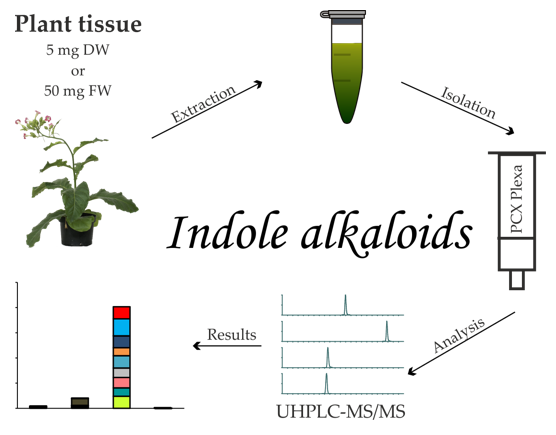
1. Introduction
2. Results
2.1. Extraction and Purification Procedure
2.2. MIA Analysis by Liquid Chromatography–Mass Spectrometry
2.3. Method Validation
2.3.1. Method Calibration and Linearity
2.3.2. Limit of Detection and Quantitation
2.3.3. Analytical Accuracy and Precision
2.3.4. Matrix Effect
2.4. MIA Determination in Tobbaco
2.5. Determination of MIAs in Tribulus terrestris
2.6. MIA Determination in Peganum harmala Seeds
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Chemicals
4.3. Extraction and Isolation Procedure
4.4. Indole Alkaloid Analysis by Ultra-High Performance Liquid Chromatography–Tandem Mass Spectrometry (UHPLC–MS/MS)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manske, R.H.F. The Alkaloids; Academic Press: New York, NY, USA, 1965; Volume I–XIII. [Google Scholar]
- Samuelsson, G.; Bohlin, L. Drugs of natural origin–A Treatise of Pharmacognosy, 7th ed.; Swedish Pharmaceutical Press: Stockholm, Sweden, 2015; pp. 625–738. ISBN 978-91-980942-5-1. [Google Scholar]
- O’Connor, S.E.; Maresh, J.J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 2006, 23, 532–547. [Google Scholar] [CrossRef]
- Shao, H.; Huang, X.; Zhang, Y.; Zhang, C. Main alkaloids of Peganum harmala L. and their different effects on dicot and monocot crops. Molecules 2013, 18, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, T.; Gonzáles, D.; Ancín-Azpilicueta, C.; Arán, V.J.; Guillén, H. ß-Carboline alkaloids in Peganum harmala and inhibition of human monoamine oxidase (MAO). Food Chem. Toxicol. 2010, 48, 839–845. [Google Scholar] [CrossRef]
- Itoh, A.; Kumashiro, T.; Yamaguchi, M. Indole alkaloids and other constituents of Rauwolfia. serpentine. J. Nat. Prod. 2005, 68, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Madhusudanan, K.P.; Banerjee, S.; Khanuja, S.P. Analysis of hairy root culture of Rauwolfia serpentina using direct analysis in real time mass spectrometric technique. Biomed. Chromatogr. 2008, 22, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Tripathi, A.K.; Pandey, R. Quantitative determination of reserpine, ajmaline, and ajmalicine in Rauwolfia serpentina by reversed-phase high-performance liquid chromatography. J. Chromatogr. Sci. 2006, 44, 557–560. [Google Scholar] [CrossRef]
- Wellens, H.J.; Dirk, D. Effect of procaine amide, quinidine, and ajmaline in the Wolff–Parkinson–White syndrome. Circulation 1974, 50, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Wellens, H.J.; Bär, F.W.; Gorgels, A.P.; Vanagt, E.J. Use of ajmaline in patients with the Wolff–Parkinson–White syndrome to disclose short refractory period of the accessory pathway. Am. J. Cardiol. 1980, 45, 130–133. [Google Scholar] [CrossRef]
- Stanford, J.L.; Martin, E.J.; Brinton, L.A.; Hoover, R.N. Rauwolfia use and breast cancer: A case-control study. J. Natl. Cancer Inst. 1986, 76, 817–822. [Google Scholar]
- Bemis, D.L.; Capodice, J.L.; Gorroochurn, P.; Katz, A.E.; Buttyan, R. Anti-prostate cancer activity of a beta-carboline alkaloid enriched extract from Rauwolfia. vomitoria. Int. J. Oncol. 2006, 29, 1065–1073. [Google Scholar] [CrossRef]
- Pulpati, H.; Biradar, Y.S.; Raiani, M. High performance thin layer chromatography densitometric method for the quantification of harmine, harmaline, vasicine, and vasicinone in Peganum. harmala. J. AOAC Int. 2008, 91, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Monsef-Esfahani, H.R.; Faramarzi, M.A.; Montezaee, V.; Amini, M.; Rouini, M.R. Determination of harmine and harmaline in Peganum harmala seeds by high performance liquid chromatography. J. Appl. Sci. 2008, 8, 1761–1765. [Google Scholar]
- Gerasimenko, I.; Sheludko, Y.; Unger, M.; Stöckigt, J. Development of an efficient system for the separation of indole alkaloids by high performance liquid chromatography and its applications. Phytochem. Anal. 2001, 12, 96–103. [Google Scholar] [CrossRef]
- Perrin, D.D. Dissociation constants of organic bases in aqueous solution. Butterworths: London, UK, 1965. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC–MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Baker, A.; Chen, P. Profiling the indole alkaloids in yohimbe bark with ultra performance liquid chromatography coupled with ion mobility quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Mitchelson, K.R. Improved separation of six harmane alkaloids by high-performance capillary electrophoresis. J. Chromatogr. A 1997, 761, 297–305. [Google Scholar] [CrossRef]
- Chhatre, S.; Nesari, T.; Somani, G.; Kanchan, D.; Sathaye, S. Phytopharmacological overview of Tribulus. terrestris. Pharmacogn. Rev. 2014, 8, 45–51. [Google Scholar] [CrossRef]
- Wagner, H.; Bladt, S. A thin layer chromatography Atlas. In Plant Drug Analysis, 2nd ed.; Springer: Verlag, Heidelberg, 1996. [Google Scholar]
- Grella, B.; Dukat, M.; Young, R.; Teitler, M.; Herrick-Davis, K.; Gauthier, C.B.; Glennon, R.A. Investigation of hallucinogenic and related β-carboline. Drug Alcohol. Depend. 1998, 50, 99–107. [Google Scholar] [CrossRef]
- Lamchouri, F.; Settaf, A.; Cherrah, Y.; Zemzani, M.; Lyoussi, B.; Zaid, A.; Atif, N.; Hassar, M. Antitumour principles from Peganum harmala seeds. Therapie 1999, 54, 753–758. [Google Scholar]
- Khan, F.A.; Maalik, A.; Iqbal, Z.; Malik, I. Recent pharmacological developments in beta-carboline alkaloid “harmaline”. Eur. J. Pharmacol. 2013, 721, 391–394. [Google Scholar] [CrossRef]
- Harrison, S.T.L. Cell disruption. In Comprehensive biotechnology, 2nd ed.; Moo-Young, M., Ed.; Elsevier: Oxford, UK, 2011; pp. 619–640. [Google Scholar]
- Kumar, S.; Singh, A.; Bajpai, V.; Srivastava, M.; Singh, B.P.; Kumar, B. Structural characterization of monoterpene indole alkaloids in ethanolic extracts of Rauwolfia species by liquid chromatography with quadrupole time-of-flight mass spectrometry. J. Pharmaceut. Anal. 2016, 6, 363–373. [Google Scholar] [CrossRef]
- Tarkowská, D.; Novák, O.; Oklestkova, J.; Strnad, M. The determination of 22 natural brassinosteroids in a minute sample of plant tissue by UHPLC-ESI-MS/MS. Anal. Bioanal. Chem. 2016, 408, 6799–6812. [Google Scholar] [CrossRef]
- Urbanová, T.; Tarkowská, D.; Novák, O.; Hedden, P.; Strnad, M. Analysis of gibberellins as free acids by ultra performance liquid chromatography–tandem mass spectrometry. Talanta 2013, 112, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Zanolari, B.; Ndjoko, K.; Ioset, J.-R.; Marston, A.; Hostettmann, K. Qualitative and quantitative determination of yohimbine in authentic yohimbe bark and in commercial aphrodisiacs by HPLC–UV–API/MS methods. Phytochem Anal. 2003, 14, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Li, W.J.; Song, A.H.; Zhao, C.J. Determination of indole alkaloids and highly volatile compounds in Rauvolfia verticillata by HPLC–UV and GC–MS. J. Chromatogr. Sci. 2013, 51, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Klyushnichenko, V.E.; Yakimov, S.A.; Tuzova, T.P.; Syagailo, Y.V.; Kuzovkina, I.N.; Wulfson, A.N.; Miroshnikov, A.I. Determination of indole alkaloids from R. serpentina and R. vomitoria by high-performance liquid chromatography and high-performance thin-layer chromatography. J. Chromatogr. A 1995, 704, 357–362. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time (min) | Calibration Equation | Correlation Coefficient R2 | LOD (pg) | LOQ (pg) | Dynamic Range (pmol) |
|---|---|---|---|---|---|---|
| Harmaline | 2.13 ± 0.01 | log y = 0.9707 (log x) − 0.9819 | 0.9958 | 0.31 | 1.04 | 5–100 |
| Harmine | 2.16 ± 0.01 | log y = 0.7374 (log x) − 0.2451 | 0.9935 | 0.18 | 0.60 | 5–100 |
| Yohimbine | 2.98 ± 0.01 | log y = 1.0147 (log x) − 0.3091 | 0.9910 | 0.01 | 0.05 | 5–100 |
| Ajmalicine | 4.94 ± 0.01 | log y = 0.8678 (log x) − 0.2006 | 0.9985 | 0.11 | 0.36 | 5–100 |
| Compound | Determined Spiked Content (A) (pmol) | Analytical Precision (A) (%) | Analytical Accuracy (A) (%) | Determinedspiked Content (B) (pmol) | Analytical Precision (B) (%) | Analytical Accuracy (B) (%) |
|---|---|---|---|---|---|---|
| Harmaline | 125.5 ± 0.2 | 3.5 | 100.4 | 252.5 ± 1.4 | 2.5 | 101.0 |
| Harmine | 123.8 ± 0.9 | 1.5 | 99.0 | 245.5 ± 0.2 | 2.2 | 98.2 |
| Yohimbine | 125.0 ± 0.3 | 2.1 | 100.3 | 255.1 ± 0.9 | 2.9 | 102.0 |
| Ajmalicine | 122.5 ± 1.9 | 2.4 | 98.0 | 244.2 ± 1.2 | 2.3 | 97.7 |
| Sample Weight (mg) | Harmaline (pg·mg–1) | Harmine (pg·mg–1) | Yohimbine (μg·mg–1) | Ajmalicine (pg·mg–1) |
|---|---|---|---|---|
| 5 | ND | 32.34 ± 3.69 | 5.29 ± 0.63 | 3.46 ± 0.92 |
| 10 | ND | 25.93 ± 2.79 | 4.01 ± 0.34 | 7.46 ± 1.24 |
| 15 | ND | 13.59 ± 3.11 | 3.31 ± 0.37 | 10.51 ± 1.28 |
| Sample Weight (mg) | Matrix Factor (%) | |||
|---|---|---|---|---|
| Harmaline | Harmine | Yohimbine | Ajmalicine | |
| 5 | 76.57 ± 0.50 | 85.55 ± 0.16 | 94.05 ± 1.34 | 96.27 ± 1.12 |
| 10 | 69.20 ± 0.33 | 82.70 ± 0.30 | 74.74 ± 0.37 | 69.27 ± 0.50 |
| 15 | 58.68 ± 0.46 | 70.51 ± 0.21 | 65.48 ± 1.06 | 49.75 ± 0.48 |
| Tissue | Alkaloid Content (pg·mg–1) | |||
|---|---|---|---|---|
| Harmaline | Harmine | Yohimbine | Ajmalicine | |
| Flowers | 0.45 ± 0.04 | 0.53 ± 0.11 | 14.40 ± 1.68 | 0.0062 ± 0.0011 |
| 1st–3rd leaf | 0.38 ± 0.05 | 0.44 ± 0.15 | 7.76 ± 1.11 | 0.0111 ± 0.0010 |
| 4th leaf | 0.25 ± 0.09 | 0.59 ± 0.02 | 10.02 ± 1.36 | 0.0047 ± 0.0008 |
| 5th leaf | 0.41 ± 0.11 | 0.76 ± 0.09 | 12.38 ± 1.33 | 0.0135 ± 0.0002 |
| 6th leaf | 0.36 ± 0.05 | 0.70 ± 0.10 | 11.30 ± 1.61 | 0.0065 ± 0.0015 |
| 7th leaf | 0.19 ± 0.02 | 0.89 ± 0.04 | 14.65 ± 1.09 | 0.0080 ± 0.0016 |
| 8th leaf | 0.23 ± 0.06 | 0.07 ± 0.02 | 9.53 ± 0.72 | 0.0067 ± 0.0007 |
| 9th leaf | 0.23 ± 0.07 | 0.09 ± 0.01 | 14.12 ± 1.71 | 0.0048 ± 0.0011 |
| Stem | 0.23 ± 0.07 | 0.07 ± 0.01 | 20.28 ± 1.22 | 0.0090 ± 0.0022 |
| Roots | ND | 0.07 ± 0.02 | 8.04 ± 1.07 | 0.0056 ± 0.0021 |
| Tissue | Alkaloid Content (pg·mg−1) | |||
|---|---|---|---|---|
| Harmaline | Harmine | Yohimbine | Ajmalicine | |
| Burrs + stems (Bulgaria) | 0.012 ± 0.005 | ND | 36.14 ± 2.35 | ND |
| Whole plant (China) | 0.004 ± 0.002 | 1.76 ± 0.18 | 62.74 ± 5.02 | ND |
| Burrs only (China) | 0.004 ± 0.001 | ND | 43.21 ± 0.77 | ND |
| Flower, burrs (India) | 0.005 ± 0.001 | 0.21 ± 0.05 | 57.59 ± 3.25 | ND |
| Alkaloid content (pg·mg–1) | |||
|---|---|---|---|
| Harmaline | Harmine | Yohimbine | Ajmalicine |
| 19.76 ± 0.44 | 1096.67 ± 14.77 | 7.25 ± 0.83 | 0.24 ± 0.03 |
| Instrumental Method Used | Amount of Tissue | Type of Tissue | Limit of Detection | Reference | Capacity of the Method Used |
|---|---|---|---|---|---|
| UHPLC–(+)ESI–MS/MS | 5 mg | whole plant | 0.01–0.31 pg | method presented | 4 IAs |
| HPLC–UV | n.a. | whole plant | 0.77–56 mg·g−1 | [5] | 2 IAs |
| HPLC–UV | 0.1 g | roots | 6–8 µg·mL−1 | [8] | 3 IAs |
| HPLTLC | n.a. | seeds | n.a. | [13] | 4 IAs |
| HPLC–UV | 1 g | seeds | 0.01–0.05 µg·mL−1 | [14] | 2 IAs |
| HPLC–UV | n.a. | cell culture | n.a. | [15] | 22 IAs |
| UPLC/IM–QTOF–MS * | 0.1 g | yohimbe bark | n.a. | [18] | 55 IAs |
| MEKC–UV/LIF | n.a. | none | n.a. | [19] | 6 IAs |
| HPLC–(+)ESI–QTOF–MS/MS | 50 g | roots | n.a. | [26] | 47 IAs |
| HPLC–UV–MS | 0.5 g | aphrodisiac products | 3–60 ng·mL−1 | [29] | yohimbine |
| HPLC–UV | 2 g | roots | 0.05–0.39 µg·mL−1 | [30] | 5 IAs |
| HPLC–UV | 5 g | roots | n.a. | [31] | 6 IAs |
| Compound | RT (min) | MRM (Q) | Cone Voltage (V) | Collision Energy (V) | MRM (C) | Cone Voltage (V) | Collision Energy (V) |
|---|---|---|---|---|---|---|---|
| Harmaline | 2.13 | 214.89 > 199.89 | 40 | 25 | 214.89 > 172.11 | 40 | 30 |
| Harmine | 2.16 | 212.94 > 198.00 | 40 | 25 | 212.94 > 169.84 | 40 | 30 |
| Yohimbine | 2.98 | 355.02 > 143.82 | 40 | 30 | 355.02 > 211.93 | 40 | 25 |
| Methyl-13C, D3-Yohimbine | 2.96 | 359.05 > 143.94 | 40 | 30 | 359.05 > 216.02 | 40 | 25 |
| Ajmalicine | 4.94 | 352. 94 > 144.07 | 40 | 25 | 352.94 > 177.91 | 40 | 25 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarkowská, D. A Fast and Reliable UHPLC–MS/MS-Based Method for Screening Selected Pharmacologically Significant Natural Plant Indole Alkaloids. Molecules 2020, 25, 3274. https://doi.org/10.3390/molecules25143274
Tarkowská D. A Fast and Reliable UHPLC–MS/MS-Based Method for Screening Selected Pharmacologically Significant Natural Plant Indole Alkaloids. Molecules. 2020; 25(14):3274. https://doi.org/10.3390/molecules25143274
Chicago/Turabian StyleTarkowská, Danuše. 2020. "A Fast and Reliable UHPLC–MS/MS-Based Method for Screening Selected Pharmacologically Significant Natural Plant Indole Alkaloids" Molecules 25, no. 14: 3274. https://doi.org/10.3390/molecules25143274
APA StyleTarkowská, D. (2020). A Fast and Reliable UHPLC–MS/MS-Based Method for Screening Selected Pharmacologically Significant Natural Plant Indole Alkaloids. Molecules, 25(14), 3274. https://doi.org/10.3390/molecules25143274