3.3. Characterization of Compounds
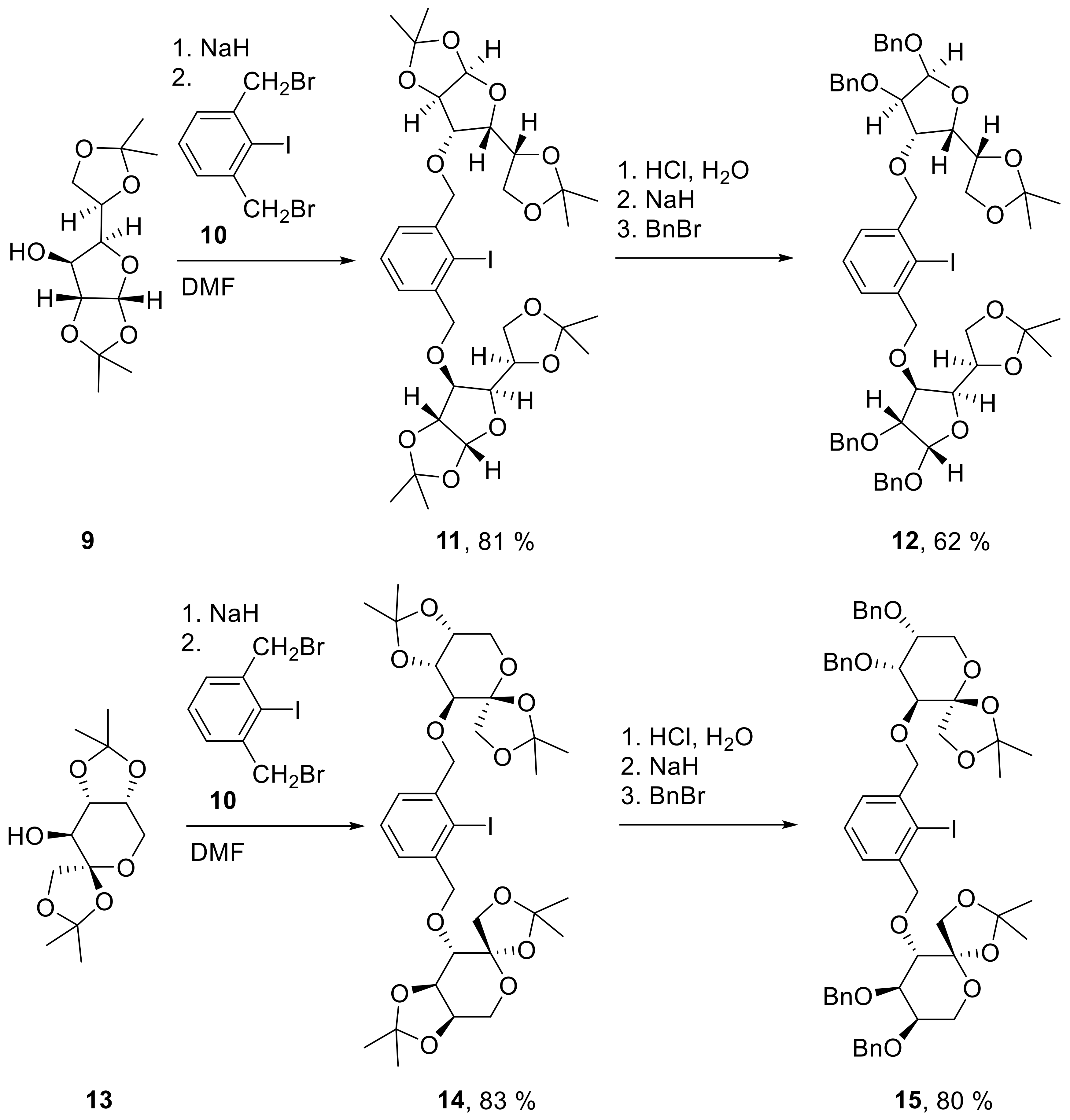
11: Prepared from 9 (599 mg, 2.30 mmol) and 10 (390 mg, 1.00 mmol) accordingly to general procedure A; colorless solid; 81% (604 mg) yield after column chromatography (PE/EtOAc, 5/1); Rf = 0.53 (PE/EtOAc, 5/1); mp = 56 °C (PE, EtOAc); [α]20D = −23.3° (c = 1.0, CHCl3); 1H-NMR (300 MHz, CDCl3) δ = 7.29–7.47 (m, 3H, H-Ar), 5.93 (d, J = 3.8 Hz, 2H, H-1), 4.63–4.81 (m, 6H, CH2Ar, H-2), 4.35–4.45 (m, 2H, H-5), 4.08–4.21 (m, 6H, H-3, H-4, H-6b), 3.98–4.04 (m, 2H, H-6a), 1.52 (s, 6H, CH3), 1.44 (s, 6H, CH3), 1.35 (s, 6H, CH3), 1.35 (s, 6H, CH3); 13C-NMR (75 MHz, CDCl3) δ = 140.4, 128.1, 128.0 (C-Ar), 111.9, 109.1 (C(CH3)2), 105.3 (C-1), 100.9 (CI), 82.5 (C-2), 82.2 (C-5), 81.3 (C-3), 76.6 (CH2Ar), 72.4 (C-4), 67.5 (C-6), 26.8, 26.8, 26.3, 25.5 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C32H45IO12Na: 771.18480, found: 771.18465; Anal calcd for C32H45IO12: C 51.34, H 6.06, found: C 51.37, H 6.20.
12: Prepared from 11 (408 mg, 0.545 mmol) accordingly to general procedure B; colorless syrup; 62% (348 mg) yield after column chromatography (PE/EtOAc, 5/1 → 3/1); Rf = 0.41 (PE/EtOAc, 3/1); [α]20D = −34.3° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.15–7.41 (m, 23H, H-Ar), 5.89 (d, J = 3.8 Hz, 2H, H-1), 4.64–4.79 (m, 6H, CH2Ar, H-2), 4.57 (s, 4H, CH2Ar), 4.44 (dd, J = 8.5, 11.9 Hz, 4H, CH2Ar), 4.30 (dd, J = 2.9, 9.2 Hz, 2H, H-4), 4.16 (d, J = 2.9 Hz, 2H, H-3), 3.97–4.04 (m, 2H, H-5), 3.91 (dd, J = 1.8, 10.6 Hz, 2H, H-6a), 3.67 (dd, J = 5.4, 10.6 Hz, 2H, H-6b), 1.48 (s, 6H, CH3), 1.31 (s, 6H, CH3); 13C-NMR (101 MHz, CD2Cl2) δ = 141.2, 139.4, 139.3, 128.8, 128.7, 128.4, 128.1, 128.0, 128.0, 127.9 (C-Ar), 112.3 (C(CH3)2), 105.8 (C-1), 101.1 (CI), 82.7 (C-3), 82.2 (C-2), 79.5 (C-4), 76.8 (CH2Ar), 76.3 (C-5), 73.9, 72.9 (CH2Ar), 71.5 (C-6), 27.1, 26.6 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C54H61IO12Na: 1051.31000, found: 1051.31032; Anal calcd for C54H61IO12: C 63.03, H 5.98, found: C 63.31, H 6.12.
14: Prepared from 13 (599 mg, 2.30 mmol) and 10 (390 mg, 1.00 mmol) accordingly to general procedure A; colorless solid; 83% (620 mg) yield after column chromatography (PE/EtOAc, 4/1); Rf = 0.59 (PE/EtOAc, 2/1); mp = 148 °C (PE, EtOAc); [α]20D = −79.3° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.40–7.48 (m, 2H, H-Ar), 7.26–7.34 (m, 1H, H-Ar), 4.99–5.08 (m, 2H, CH2Ar), 4.63–4.74 (m, 2H, CH2Ar), 4.39 – 4.44 (m, 2H, H-4), 4.25 (dd, J = 2.1, 5.6 Hz, 2H, H-5), 4.12–4.19 (m, 4H, H-1b, H6-b), 3.99–4.07 (m, 2H, H-6a), 3.93 (d, J = 8.6 Hz, 2H, H-1a), 3.59–3.65 (m, 2H, H-3), 1.61 (s, 6H, CH3), 1.51 (s, 6H, CH3), 1.44(s, 6H, CH3), 1.46 (s, 6H, CH3); 13C-NMR (101 MHz, CDCl3) δ = 141.1, 128.3, 127.9, (C-Ar), 112.2, 109.1 (C(CH3)2), 104.4 (C-2), 101.1 (CI), 77.6 (C-4), 76.9 (C-3), 73.9 (C-5), 72.9 (CH2Ar), 72.0 (C-1), 60.2 (C-6), 28.2, 26.9, 26.3, 26.1 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C32H45IO12Na: 771.18480, found: 771.18507; Anal calcd for C32H45IO12: C 51.34, H 6.06, found: C 51.45, H 6.12.
15: Prepared from 14 (363 mg, 0.485 mmol) accordingly to general procedure B; colorless solid; 80% (400 mg) yield after column chromatography (PE/EtOAc, 4/1); Rf = 0.27 (PE/EtOAc, 3/1); mp = 54 °C (n-heptane, CH2Cl2); [α]20D = −61.7° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.22–7.51 (m, 23H, H-Ar), 5.07 (d, J = 13.0 Hz, 2H, H-1a), 4.60–4.75 (m, 8H, H-1b, CH2Ar), 4.55 (d, J = 11.5 Hz, 2H, CH2Ar), 4.08 (d, J = 8.4 Hz, 2H, CH2Ar), 3.76–4.02 (m, 12H, CH2Ar, H-3, H-4, H-5, H-6a, H-6b), 1.48 (s, 6H, CH3), 1.43 (m, 6H, CH3); 13C-NMR (101 MHz, CD2Cl2) δ = 141.9, 139.1, 139.0, 128.9, 128.8, 128.5, 128.4, 128.3, 128.2, 128.2, 128.0, 127.9 (C-Ar), 112.3 (C(CH3)2), 106.4 (C-2), 100.1 (CI), 80.5(C-5), 79.9 (C-1), 76.0 (C-3), 74.5 (C-4), 72.6, 72.3, 72.1 (CH2Ar), 61.8 (C-6), 27.4, 26.8 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C54H61IO12Na: 1051.31000, found: 1051.30988, Anal calcd for C54H61IO12: C 63.03, H 5.98; found: C 63.07, H 6.09.
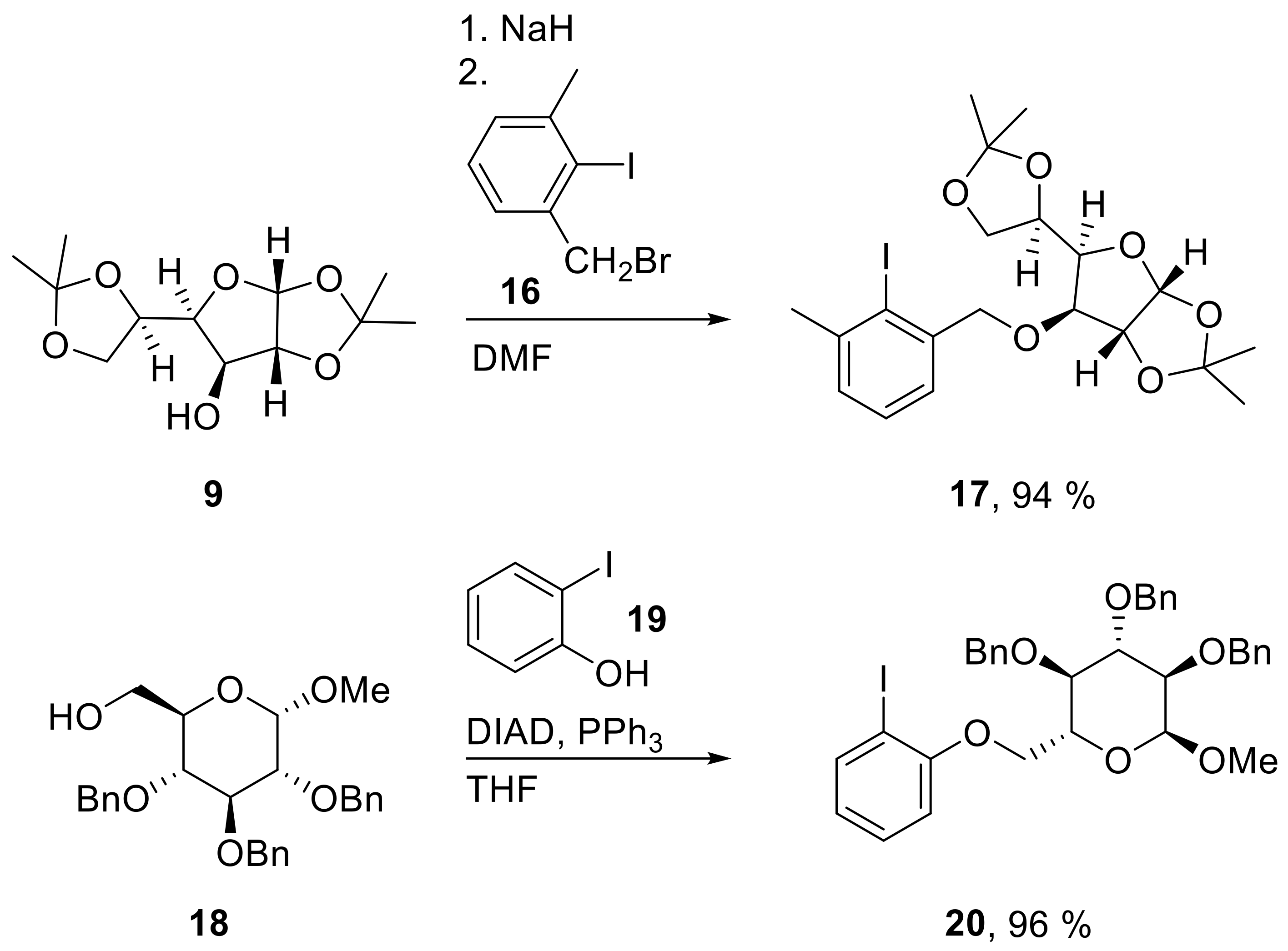
17: Prepared from 9 (312 mg, 1.20 mmol) and 16 (311 mg, 1.00 mmol) accordingly to general procedure A; slight yellow oil; 94% (460 mg) yield after column chromatography (PE/EtOAc, 5/1); Rf = 0.66 (PE/EtOAc, 2/1); [α]20D = −13.0° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.21–7.35 (m, 3H, H-Ar), 5.97 (d, J = 3.7 Hz, 1H, H-1), 4.67–4.81 (m, 3H, H-2, CH2), 4.42–4.48 (m, 1H, H-5), 4.13–4.24 (m, 3H, H-4, H-6b), 4.05 (dd, J = 6.0, 8.4 Hz, 1H, H-6a), 2.52 (s, 3H, CH3Ar), 1.56 (s, 3H, CH3), 1.48 (s, 3H, CH3), 1.40 (s, 3H, CH3), 1.38 (s, 3H, CH3); 13C-NMR (101 MHz, CDCl3) δ = 142.1, 140.5, 129.0, 129.0 (Ar-C), 127.9 (Ar-C),125.9, 125.9 (C-Ar), 111.9, 109.0 (C(CH3)2), 105.3 (C-1), 104.6 (CI), 82.5 (C-2), 82.1 (C-3), 81.3 (C-4), 77.0 (CH2Ar), 72.5 (C-5), 67.4 (C-6), 29.2 (CH3Ar), 26.9, 26.8, 26.3, 25.4 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C20H27IO6Na: 513.07446, found: 513.07419; Anal calcd for C20H27IO6: C 48.99, H 5.55, found: C 48.92, H 5.79.
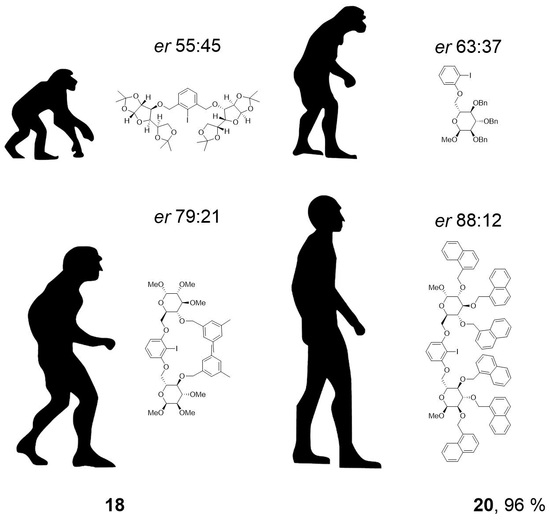
20: Prepared from 18 (550 mg, 1.18 mmol) and 19 (238 mg, 1.08 mmol) accordingly to general procedure C; colorless oil; 96% (688 mg) yield after column chromatography (PhMe/EtOAc, 40/1 → 10/1); Rf = 0.27 (PhMe/EtOAc, 30/1); [α]20D = +53.9° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.77 (dd, J = 1.5, 7.7 Hz, 1H, H-Ar), 7.11–7.43 (m, 15H, H-Ar), 6.74 (d, J = 7.7 Hz, 2H, H-Ar), 4.99 (d, J = 10.9 Hz, 1H, CH2Ar), 4.93 (d, J = 11.2 Hz, 1H, CH2Ar), 4.85 (d, J = 10.9 Hz, 1H, CH2Ar), 4.74–4.81 (m, 2H, H-1, CH2Ar), 4.61–4.72 (m, 2H, CH2Ar), 4.18 (dd, J = 1.7, 10.3 Hz, 1H, H-6a), 4.10 (dd, J = 4.5, 10.3 Hz, 1H, H-6b), 3.91–4.04 (m, 2H, H-4, H-5), 3.78–3.87 (m, 1H, H-3), 3.64 (dd, J = 3.5, 9.5 Hz, 1H, H-2), 3.45 (s, 3H, CH3); 13C-NMR (101 MHz, CD2Cl2) δ = 157.7, 139.9, 139.5, 139.0, 130.0, 129.5, 128.9, 128.8, 128.7, 128.6, 128.5, 128.4, 128.3, 128.1, 128.1, 123.1, 112.4 (C-Ar), 98.5 (C-1), 86.6 (CI), 82.5 (C-4), 81.0 (C-2), 78.3 (C-3), 76.1, 75.6, 73.6 (CH2Ar), 69.8 (C-5), 68.5 (C-6), 55.7 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C34H35IO6Na: 689.13705, found: 689.13711; Anal calcd for C34H35IO6: C 61.27, H 5.29, found: C 61.39, H 5.39.
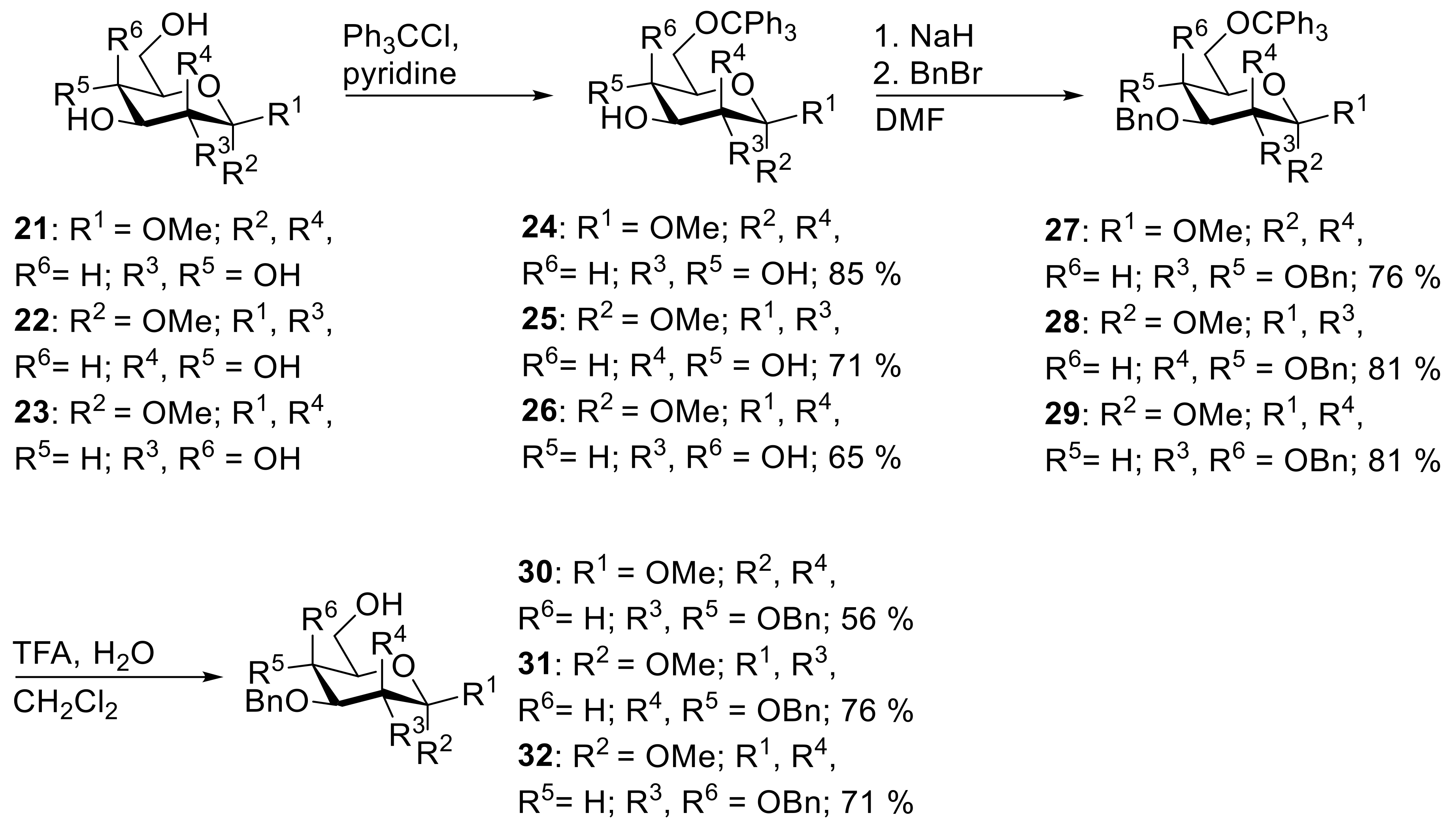
24: Prepared from 21 (5.08 g, 26.2 mmol) accordingly to general procedure E; colorless solid; 85% (9.70 g) yield after column chromatography (PhMe/EtOAc, 1/4); Rf = 0.30 (PhMe/EtOAc, 1/4); mp = 75 °C (n-heptane, EtOAc); [α]20D = −44.0° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.32–7.43 (m, 5H, H-Ar), 7.04–7.27 (m, 10H, H-Ar), 4.09 (d, J = 7.7 Hz, 1H, H-1), 4.03 (bs, 1H, OH), 3.56 (bs, 1H, OH), 3.44 (s, 3H, CH3), 3.19–3.41 (m, 7H, OH, H-2, H-3, H-4, H-5, H-6a, H-6b); 13C-NMR (101 MHz, CDCl3) δ = 143.7, 129.6, 129.0, 128.6, 128.2, 127.9, 127.1, 125.3 (C-Ar), 103.3 (C-1), 86.9 (CPh3), 76.3, 74.2, 73.4, 71.6 (C-2, C-3, C-4, C-5), 64.1 (C-6), 56.8 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C26H28O6Na: 459.17781, found: 459.17772; Anal calcd for C26H28O6: C 71.54, H 6.47, found: C 71.50, H 6.57.
25: Prepared from
22 (5.91 g, 30.0 mmol) accordingly to general procedure E; colorless solid; 71% (9.30 g) yield after column chromatography (PE/EtOAc, 1/2); analytical data were in good accordance with literature values [
29].
26: Prepared from 23 (5.91 g, 30.0 mmol) accordingly to general procedure E; colorless solid; 65% (8.52 g) yield after column chromatography (PhMe/EtOAc, 1/4 → EtOAc); Rf = 0.36 (EtOAc); mp = 127 °C (n-heptane, CH2Cl2); [α]20D = +53.0° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.30–7.44 (m, 5H, H-Ar), 7.08–7.29 (m, 10H, H-Ar), 4.71 (d, J = 3.8 Hz, 1H, H-1), 3.88–3.90 (m, 1H, H-3 or H-4), 3.69–3.76 (m, 2H, H-2, H-5), 3.59–3.66 (m, 1H, H-3 or H-4), 3.31–3.38 (m, 4H, CH3, H-6a), 3.28 (dd, J = 4.0, 9.6 Hz, 1H, H-6b); 13C-NMR (101 MHz, CDCl3) δ = 146.8, 143.7, 128.6, 127.9, 127.2, 127.1 (C-Ar), 99.4 (C-1), 87.0 (CPh3), 71.1, 69.7, 69.6, 69.1 (C-2, C-3, C-4, C-5), 63.2 (C-6), 55.3 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C26H28O6Na: 459.17781, found: 459.17816; Anal calcd for C26H28O6: C 71.54, H 6.47, found: C 71.55, H 6.51.
27: Prepared from 24 (3.64 g, 8.34 mmol) and BnBr (6.42 g, 37.5 mmol) accordingly to general procedure A; colorless syrup; 76% (4.50 g) yield after column chromatography (PE/EtOAc, 14/1 → PE/EtOAc, 3/1); Rf = 0.27 (PE/EtOAc, 14/1); [α]20D = +3.9° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.38–7.52 (m, 6H, H-Ar), 7.05–7.35 (m, 22H, H-Ar), 6.73–6.83 (m, 2H, H-Ar), 4.90 (d, J = 11.0 Hz, 1H, CH2Ph), 4.82 (d, J = 10.6 Hz, 1H, CH2Ph), 4.66–4.76 (m, 2H, CH2Ph), 4.62 (d, J = 10.3 Hz, 1H, CH2Ph), 4.24–4.36 (m, 2H, CH2Ph, H-1), 3.71–3.83 (m, 1H, H-4), 3.59 (s, 3H, CH3), 3.43–3.57 (m, 3H, H-2, H-3, H-6a), 3.30–3.36 (m, 1H, H-6), 3.17 (dd, J = 3.8, 10.1 Hz, 1H, H-6b); 13C-NMR (101 MHz, CDCl3) δ = 144.0, 138.7, 138.6, 137.9, 128.9, 128.5, 128.4, 128.3, 128.2, 128.1, 127.8, 127.8, 127.7 (C-Ar), 104.6 (C-1), 86.4 (CPh3), 84.7 (C-3), 82.7 (C-2), 77.9 (C-4), 76.0, 75.1, 74.9 (CH2Ph), 74.6 (C-5), 62.4 (C-6), 56.7 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C47H46O6Na: 729.31866, found: 729.31931; Anal calcd for C47H46O6: C 79.86, H 6.56, found: C 79.84, H 6.66.
28: Prepared from 25 (3.86 g, 8.84 mmol) and BnBr (6.80 g, 39.8 mmol) accordingly to general procedure A; colorless solid; 81% (5.04 g) yield after column chromatography (PE/EtOAc, 9/1); Rf = 0.52 (PE/EtOAc, 4/1); mp = 112 °C (n-heptane, CH2Cl2); [α]20D = +17.8° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.40–7.49 (m, 6H, H-Ar), 7.36 (dd, J = 1.5, 7.8 Hz, 2H, H-Ar), 7.05–7.30 (m, 20H, H-Ar), 6.81 (dd, J = 1.5, 7.8 Hz, 2H, H-Ar), 4.7–4.80 (m, 2H, CH2Ph, H-1), 4.61–4.69 (m, 2H, CH2Ph), 4.56 (s, 2H, CH2Ph), 4.19 (d, J = 10.5 Hz, 1H, CH2Ph), 3.90–3.97 (m, 1H, H-4), 3.80 (dd, J = 3.2, 9.3 Hz, 1H, H-3), 3.74 (dd, J = 1.9, 3.2 Hz, 1H, H-2), 3.66–3.72 (m, 1H, H-5), 3.44 (dd, J = 1.7, 9.8 Hz, 1H, H-6a), 3.30 (s, 3H, CH3), 3.19 (dd, J = 5.3, 9.8 Hz, 1H, H-6b); 13C-NMR (101 MHz, CDCl3) δ = 144.2, 138.6, 138.6, 138.2, 128.8, 128.3, 128.3, 128.1, 127.7, 127.6, 127.5, 127.4, 127.4, 126.8 (C-Ar), 98.7 (C-1), 86.2 (CPh3), 80.2 (C-3), 75.4 (C-2), 75.0 (C-4), 72.7, 72.2, 71.7 (CH2Ph), 63.0 (C-6), 54.5 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C47H46O6Na: 729.31866, found: 729.31900; Anal calcd for C47H46O6: C 79.86, H 6.56, found: C 79.75, H 6.62.
29: Prepared from 26 (3.69 g, 8.45 mmol) and BnBr (6.50 g, 38.0 mmol) accordingly to general procedure A; colorless syrup; 81% (4.81 g) yield after column chromatography (PE/EtOAc, 9/1 → 3/1); Rf = 0.36 (PE/EtOAc, 6/1); [α]20D = +20.2° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.25–7.37 (m, 12H, H-Ar), 7.07–7.24 (m, 16H, H-Ar), 6.97–7.05 (m, 2H, H-Ar), 4.69–4.82 (m, 3H, CH2Ph), 4.52–4.67 (m, 3H, CH2Ph, H-1), 4.40 (d, J = 11.4 Hz, 1H, CH2Ph), 3.89 (dd, J = 3.7, 9.9, 1H, H-2), 3.77–3.84 (m, 2H, H-3, H-4), 3.63 (m, 1H, H-5), 3.33 (dd, J = 6.2, 9.7 Hz, 1H, H-6a), 3.29 (s, 3H, CH3), 3.05–3.09 (m, 1H, H-6b); 13C-NMR (101 MHz, CDCl3) δ = 143.9, 138.9, 138.5, 138.5, 128.6, 128.3, 128.3, 128.1, 128.1, 128.0, 127.8, 127.6, 127.5, 127.5, 127.4, 127.0 (C-Ar), 98.6 (C-1), 86.9 (CPh3), 79.0 (C-3), 76.4 (C-2), 75.6 (C-4), 74.6, 73.5, 73.3 (CH2Ph), 69.5 (C-5), 63.1 (C-6), 55.1 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C47H46O6Na: 729.31866, found: 729.31864; Anal calcd for C47H46O6: C 79.86, H 6.56, found: C 79.87, H 6.57.
30: Prepared from
27 (4.30 g, 6.08 mmol) accordingly to general procedure F; colorless solid; 56% (1.58 g) yield after column chromatography (PE/EtOAc, 3/1 → 2/1); analytical data were in good accordance with literature values [
30].
31: Prepared from
28 (3.14 g, 4.44 mmol) accordingly to general procedure F; colorless solid; 76% (1.56 g) yield after column chromatography (PE/EtOAc, 3/1 → 2/1); analytical data were in good accordance with literature values [
31].
32: Prepared from
29 (4.31 g, 6.10 mmol) accordingly to general procedure F; colorless syrup; 71% (2.00 g) yield after column chromatography (PE/EtOAc, 2/1 → 1/2); analytical data were in good accordance with literature values [
31].
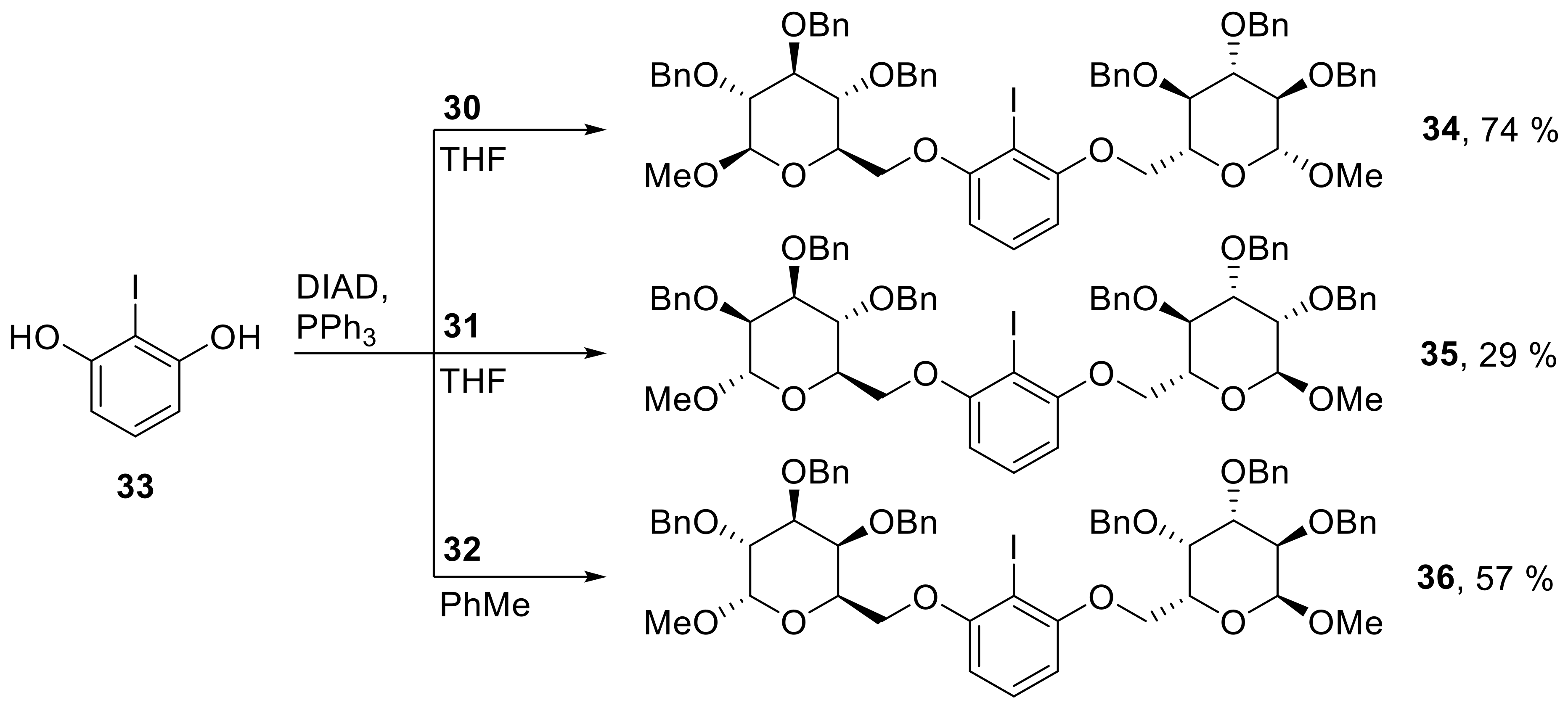
34: Prepared from 30 (585 mg, 1.26 mmol) and 33 (135 mg, 0.572 mmol) accordingly to general procedure C; colorless solid; 74% (480 mg) yield after column chromatography (PhMe/EtOAc, 10/1 → 6/1); Rf = 0.55 (PhMe/EtOAc, 6/1); mp = 145 °C (n-heptane, CH2Cl2); [α]20D = +49.4° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.15–7.43 (m, 31H, H-Ar), 6.49 (d, J = 8.3 Hz, 2H, H-Ar), 4.88–4.99 (m, 6H, CH2Ph), 4.83 (d, J = 11.1 Hz, 2H, CH2Ph), 4.74 (d, J = 11.1 Hz, 2H, CH2Ph), 4.65 (d, J = 11.1 Hz, 2H, CH2Ph), 4.39 (d, J = 7.8 Hz, 2H, H-1), 4.32 (dd, J = 1.6, 10.4 Hz, 2H, H-6a), 4.17 (dd, J = 4.3, 10.4 Hz, 2H, H-6b), 3.89–3.99 (m, 2H, H-4), 3.67–3.73 (m, 2H, H-3), 3.60–3.66 (m, 2H, H-5), 3.57 (s, 6H, CH3), 3.48 (dd, J = 7.8, 9.0 Hz, 2H, H-2); 13C-NMR (101 MHz, CH2Cl2) δ = 159.2, 139.4, 139.4, 139.0, 130.3, 128.8, 128.6, 128.5, 128.5, 128.1, 128.1, 105.9 (C-Ar), 105.4 (C-1), 85.1 (C-3), 82.8 (C-2), 79.0 (CI), 78.2 (C-4), 76.0, 75.6, 75.0 (CH2 Ph), 74.2 (C-5), 68.6 (C-6), 57.5 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C62H65IO12Na: 1151.34130, found: 1151.34152; Anal calcd for C62H65IO12: C 65.95, H 5.80, found: C 65.92, H 5.83.
35: Prepared from 31 (470 mg, 1.01 mmol) and 33 (109 mg, 0.460 mmol) accordingly to general procedure C; slight yellow syrup; 29% (153 mg) yield after column chromatography (PhMe/EtOAc, 15/1 → 6/1); Rf = 0.55 (PhMe/EtOAc, 6/1); [α]20D = +31.5° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.14–7.48 (m, 31H, H-Ar), 6.43 (d, J = 8.3 Hz, 2H, H-Ar), 4.96 (d, J = 11.1 Hz, 2H, CH2Ph), 4.83 (d, J = 1.6 Hz, 2H, H-1), 4.65–4.80 (m, 8H, CH2Ph), 4.63 (d, J = 11.2 Hz, 2H, CH2Ph), 4.24 (dd, J = 1.5, 10.2 Hz, 2H, H-6a), 4.09–4.21 (m, 4H, H-5, H-6b), 3.85–3.96 (m, 6H, H-2, H-3, H-4), 3.40 (s, 6H, CH3); 13C-NMR (101 MHz, CH2Cl2) δ = 159.3, 139.3, 139.1, 130.2, 128.9, 128.8, 128.8, 128.6, 128.4, 128.2, 128.1, 128.1, 128.0, 105.4 (C-Ar), 99.6 (C-1), 80.9 (C-3 or C-4), 78.7 (CI), 75.8 (C-2), 75.6 (CH2Ph), 75.4 (C-5), 73.4, 72.5 (CH2Ph), 71.2 (C-3 or C-4), 69.1 (C-6), 55.4 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C62H65IO12Na: 1151.34130, found: 1151.34185; Anal calcd for C62H65IO12: C 65.95, H 5.80, found: C 66.00, H 5.85.
36: Prepared from 32 (568 mg, 1.22 mmol) and 33 (116 mg, 0.490 mmol) accordingly to general procedure D; slight yellow syrup; 57% (315 mg) yield after column chromatography (PhMe/EtOAc, 15/1 → 10/1); Rf = 0.58 (PhMe/EtOAc, 6/1); [α]20D = +19.8° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.42–7.48 (m, 4H, H-Ar), 7.11–7.41 (m, 28H, H-Ar), 6.41 (d, J = 8.3 Hz, 2H, H-Ar), 4.99 (d, J = 11.0 Hz, 2H, CH2Ph), 4.89 (d, J = 12.0 Hz, 2H, CH2Ph), 4.78–4.84 (m, 6H, CH2Ph, H-1), 4.65 (d, J = 11.7 Hz, 2H, CH2Ph), 4.54 (d, J = 11.0 Hz, 2H, CH2Ph), 4.21–4.24 (m, 2H, H-3), 4.14–4.20 (m, 2H, H-5), 3.89–4.12 (m, 8H, H-2, H-4, H-6a, H-6b), 3.43 (s, 6H, CH3); 13C-NMR (101 MHz, CH2Cl2) δ = 158.9, 139.5, 139.4, 139.2, 130.5, 129.5, 128.9, 128.9, 128.8, 128.8, 128.7, 128.4, 128.2, 128.1, 128.1, 105.5 (C-Ar), 99.3 (C-1), 79.1 (C-2 or C-4), 78.2 (CI), 77.2 (C-2 or C-4), 76.0 (C-3), 75.6, 73.8, 73.6 (CH2Ph), 69.0 (C-5), 68.3 (C-6), 55.9 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C62H65IO12Na: 1151.34130, found: 1151.34058, Anal calcd for C62H65IO12: C 65.95, H 5.80; found: C 65.85, H 6.04.
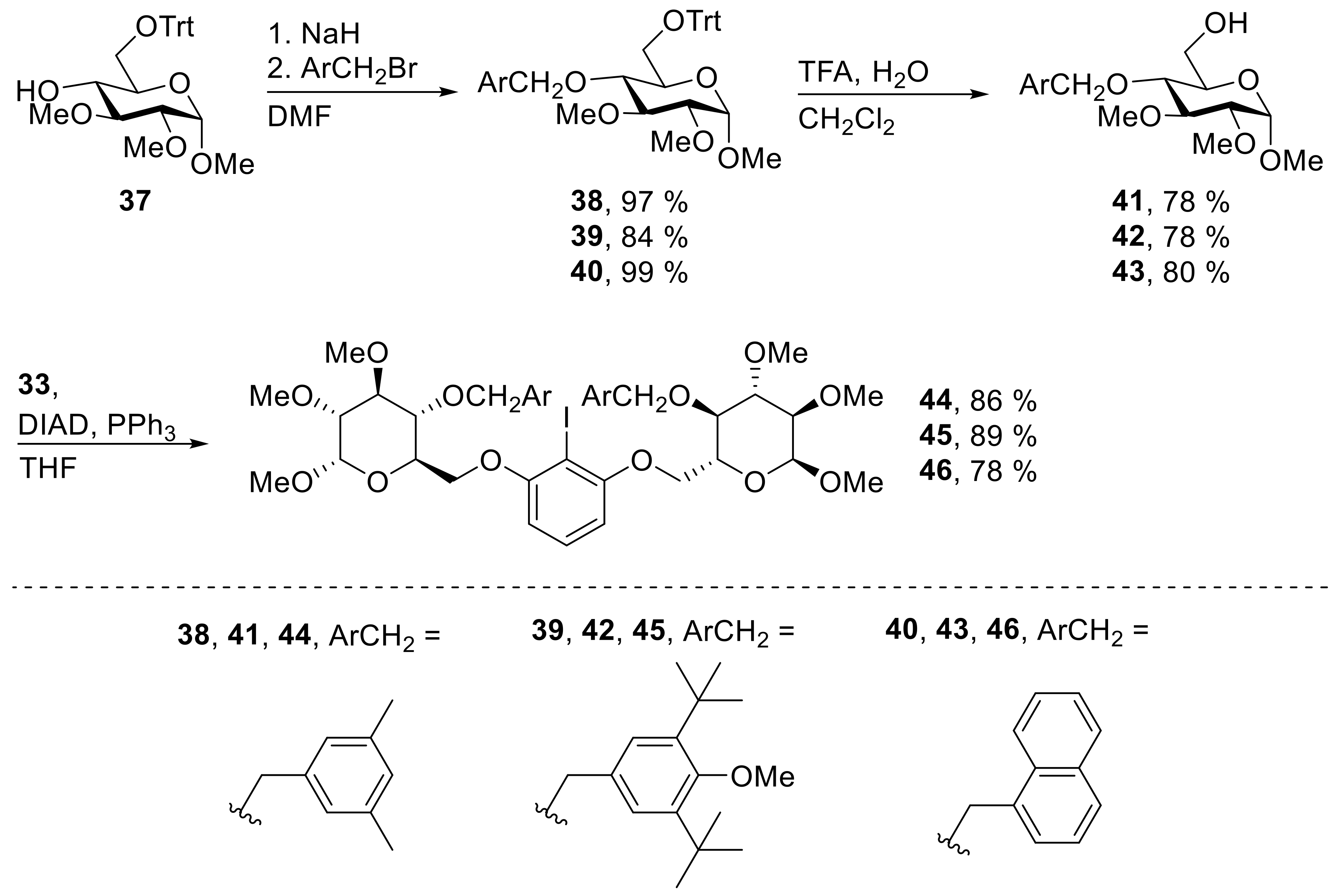
38: Prepared from 37 (2.02 g, 4.35 mmol) and 3,5-dimethyl-benzyl bromide (1.30 g, 6.53 mmol) accordingly to general procedure A; colorless solid; 97% (2.47 g) yield after column chromatography (PE/EtOAc, 5/1 → 4/1); Rf = 0.29 (PE/EtOAc, 5/1); mp = 53 °C (n-heptane, CH2Cl2); [α]20D = +81.7° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.37–7.48 (m, 6H, H-Ar), 7.09–7.27 (m, 9H, H-Ar), 6.79 (s, 1H, H-Ar), 6.50 (s, 2H, H-Ar), 4.87 (d, J = 3.6 Hz, 1H, H-1), 4.50 (d, J = 10.0 Hz, 1H, CH2Ar), 4.15 (d, J = 10.0 Hz, 1H, CH2Ar), 3.63 – 3.71 (m, 1H, H-5), 3.59 (s, 3H, CH3O), 3.48–3.56 (m, 5H, CH3O, H-3, H-4), 3.43 (dd, J = 1.8, 10.0 Hz, 1H, H-6a), 3.37 (s, 3H, CH3O), 3.27 (dd, J = 3.6, 9.3 Hz, 1H, H-2), 3.13 (dd, J = 4.3, 10.0 Hz, 1H, H-6b), 2.16 (s, 6H, CH3Ar); 13C-NMR (101 MHz, CHCl3) δ = 144.0, 137.7, 137.7, 129.3, 128.8, 127.8, 126.9, 126.0 (C-Ar), 97.3 (C-1), 86.2 (CPh3), 83.7 (C-3), 82.0 (C-2), 77.9 (C-4), 75.0 (CH2Ar), 70.1 (C-5), 62.4 (C-6), 61.2, 59.0, 54.8 (CH3O), 21.2 (CH3Ar); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C37H42O6Na: 605.28736, found: 605.28801; Anal calcd for C37H42O6: C 76.26, H 7.27, found: C 76.05, H 7.52.
39: Prepared from 37 (1.82 g, 3.91 mmol) and 3,5-ditbutyl-4-methoxy-benzyl bromide (1.47 g, 4.69 mmol) accordingly to general procedure A; colorless solid; 84% (2.29 g) yield after column chromatography (PE/EtOAc, 5/1 → 4/1); Rf = 0.37 (PE/EtOAc, 5/1); mp = 69 °C (n-heptane, CH2Cl2); [α]20D = +49.3° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.39–7.48 (m, 6H, H-Ar), 7.10–7.27 (m, 9H, H-Ar), 6.88 (s, 2H, H-Ar), 4.88 (d, J = 3.5 Hz, 1H, H-1), 4.45 (d, J = 9.7 Hz, 1H, CH2Ar), 4.05 (d, J = 9.7 Hz, 1H, CH2Ar), 3.63–3.70 (m, 1H, H-5), 3.62 (s, 3H, CH3O), 3.49–3.60 (m, 8H, 2 × CH3O, H-3, H-4), 3.46 (dd, J = 1.7, 10.1 Hz, 1H, H-6a), 3.36 (s, 3H, CH3O), 3.29 (dd, J = 3.5, 9.2 Hz, 1H, H-2), 3.14 (dd, J = 3.9, 10.1 Hz, 1H, H-6b), 1.29 (s, 18H, C(CH3)3); 13C-NMR (101 MHz, CHCl3) δ = 159.1, 144.0, 143.3, 132.0, 128.8, 127.7, 127.0, 126.8 (C-Ar), 97.3 (C-1), 86.2 (CPh3), 83.5 (C-3), 82.3 (C-2), 78.1 (C-4), 75.7 (CH2Ar), 70.1 (C-5), 64.2 (CH3O), 62.4 (C-6), 61.4, 59.0, 54.9 (CH3O), 35.6 (C(CH3)3), 32.0 (C(CH3)3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C44H56O7Na: 719.39182, found: 719.39276; Anal calcd for C44H56O7: C 75.83, H 8.10, found: C 75.83, H 8.36.
40: Prepared from 37 (2.02 g, 4.35 mmol) and 1-(bromomethyl)-naphthalene (1.44 g, 6.53 mmol) accordingly to general procedure A; colorless solid; 99% (2.60 g) yield after column chromatography (PE/EtOAc, 5/1 → 4/1); Rf = 0.19 (PE/EtOAc, 5/1); mp = 65 °C (n-heptane, CH2Cl2); [α]20D = +95.5° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.78 (d, J = 8.3 Hz, 1H, H-Ar), 7.74 (d, J = 8.1 Hz, 1H, H-Ar), 7.67 (d, J = 8.2 Hz, 1H, H-Ar), 7.30–7.43 (m, 7H, H-Ar), 7.07–7.28 (m, 11H, H-Ar), 6.95 (d, J = 6.5 Hz, 1H, H-Ar), 5.10 (d, J = 11.0 Hz, 1H, CH2Ar), 4.87 (d, J = 3.6 Hz, 1H, H-1), 4.63 (d, J = 11.1 Hz, 1H, CH2Ar), 3.67–3.75 (m, 1H, H-5), 3.58–3.63 (m, 1H, H-4), 3.48–3.57 (m, 7H, 2 × CH3O, H-3), 3.44 (dd, J = 1.8, 10.1 Hz, 1H, H-6a), 3.38 (s, 3H, CH3O), 3.30 (dd, J = 3.6, 9.4 Hz, 1H, H-2), 3.08 (dd, J = 4.7, 10.1 Hz, 1H, H-6b); 13C-NMR (101 MHz, CHCl3) δ = 143.9, 133.7, 133.5, 131.4, 128.7, 128.3, 128.3, 127.7, 126.9, 126.6, 126.1, 125.5, 125.1, 123.9 (C-Ar), 97.1 (C-1), 86.3 (CPh3), 83.5 (C-3), 82.5 (C-2), 77.8 (C-4), 72.6 (CH2Ar), 70.2 (C-5), 62.7 (C-6), 61.3, 58.9, 54.9 (CH3O); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C39H40O6Na: 627.27171, found: 627.27231; Anal calcd for C39H40O6: C 77.46, H 6.67, found: C 77.51, H 7.01.
41: Prepared from 38 (2.34 g, 4.02 mmol) accordingly to general procedure F; colorless solid; 78% (1.07 g) yield after column chromatography (PE/EtOAc, 1/1 → 1/2); Rf = 0.25 (PE/EtOAc, 1/1); mp = 109 °C (n-heptane, CH2Cl2); [α]20D = +115.6° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 6.89–7.02 (m, 3H, H-Ar), 4.78–4.86 (m, 2H, CH2Ar, H-1), 4.58 (d, J = 10.8 Hz, 1H, CH2Ar), 3.80 (dd, J = 2.8, 11.7 Hz, 1H, H-6a), 3.73 (dd, J = 3.9, 11.7 Hz 1H), 3.59–3.69 (m, 5H, CH3O, H-3, H-4), 3.54 (s, 3H, CH3O), 3.36–3.47 (m, 4H, CH3O, H-4), 3.22 (dd, J = 3.5, 9.5 Hz, 1H, H-2), 2.32 (s, 6H, CH3Ar); 13C-NMR (101 MHz, CHCl3) δ = 138.1, 137.9, 129.5, 126.0 (C-Ar), 97.5 (C-1), 83.6 (C-3), 82.0 (C-2), 77.2 (C-4), 75.0 CH2Ar, 70.6 (C-5), 61.9 (C-6), 61.0, 59.0, 55.1 (CH3O), 21.2 (CH3Ar); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C18H28O6Na: 363.17781, found: 363.17822; Anal calcd for C18H28O6: C 63.51, H 8.29, found: C 63.46, H 8.40.
42: Prepared from 39 (2.15 g, 3.09 mmol) accordingly to general procedure F; colorless solid; 78% (1.10 g) yield after column chromatography (PE/EtOAc, 1/1 → 1/2); Rf = 0.20 (PE/EtOAc, 1/1); mp = 139 °C (n-heptane, CH2Cl2); [α]20D = +92.7° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 7.24 (s, 2H, H-Ar), 4.77–4.85 (m, 2H, CH2Ar, H-1), 4.56 (d, J = 10.5 Hz, 1H, CH2Ar), 3.78 (dd, J = 2.8, 11.8 Hz, 1H, H-6a) 3.60–3.74 (m, 9H, 2 × CH3O, H-3, H-5, H-6b), 3.55 (s, 3H, CH3O), 3.40–3.45 (m, 4H, CH3O, H-4), 3.23 (dd, J = 3.6, 9.5 Hz, 1H, H-2), 1.43 (s, 18H, C(CH3)3); 13C-NMR (101 MHz, CHCl3) δ = 159.4, 143.8, 131.9, 126.9 (C-Ar), 97.5 (C-1), 83.7 (C-3), 82.1 (C-2), 77.2 (C-4), 75.5 (CH2Ar), 70.5 (C-5), 64.2 (CH3O), 61.8 (C-6), 61.1, 59.0, 55.1 (CH3O), 35.7 (C(CH3)3), 32.0 (C(CH3)3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C25H42O7Na: 477.28277, found: 477.28230; Anal calcd for C25H42O7: C 66.05, H 9.31, found: C 66.06, H 9.53.
43: Prepared from 40 (2.46 g, 4.7 mmol) accordingly to general procedure F; colorless solid; 80% (1.18 g) yield after column chromatography (PE/EtOAc, 1/1 → 1/2); Rf = 0.19 (PE/EtOAc, 1/1); mp = 74 °C (n-heptane, CH2Cl2); [α]20D = +138.9° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 8.16 (d, J = 8.4 Hz, 1H, H-Ar), 7.79–7.92 (m, 2H, H-Ar), 7.41–7.60 (m, 4H, H-Ar), 5.41 (d, J = 11.4 Hz, 1H, CH2Ar), 5.08 (d, J = 11.4 Hz, 1H, CH2Ar), 4.83 (d, J = 3.6 Hz, 1H, H-1), 3.51–3.73 (m, 11H, 2 × CH3O, H-3, H-4, H-5, H-6a, H-6b), 3.39 (s, 3H, CH3O), 3.27 (dd, J = 3.6, 9.5 Hz, 1H, H-2); 13C-NMR (101 MHz, CHCl3) δ = 133.8, 133.7, 131.6, 128.8, 128.6, 126.9, 126.3, 125.8, 125.3, 123.9 (C-Ar), 97.4 (C-1), 83.6 (C-3), 82.3 (C-2), 76.8 (C-4), 72.7 (CH2Ar), 70.5 (C-5), 61.7 (C-6), 61.1, 58.9, 55.1 (CH3O); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C20H26O6Na: 385.16216, found: 385.16198; Anal calcd for C20H26O6: C 66.28, H 7.23, found: C 66.23, H 7.36.
44: Prepared from 41 (561 mg, 1.65 mmol) and 33 (194 mg, 0.824 mmol) accordingly to general procedure C; colorless solid; 86% (624 mg) yield after column chromatography (PE/EtOAc, 4/1 → 1/1); Rf = 0.26 (PE/EtOAc, 1/1); mp = 107 °C (n-heptane, CH2Cl2); [α]20D = +120.0° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.22 (t, J = 8.3 Hz, 1H, H-Ar), 6.94 (s, 2H, H-Ar), 6.78 (s, 4H, H-Ar), 6.41 (d, J = 8.3 Hz, 2H, H-Ar), 4.86 (d, J = 3.6 Hz, 2H, H-1), 4.80 (d, J = 11.0 Hz, 2H, CH2Ar), 4.51 (d, J = 11.0 Hz, 2H, CH2Ar), 4.17 (dd, J = 1.6, 10.3 Hz, 2H, H-6a), 4.07 (dd, J = 4.4, 10.3 Hz, 2H, H-6b), 3.81–3.88 (m, 2H, H-5), 3.68–3.77 (m, 2H, H-6), 3.63 (s, 6H, CH3O), 3.52–3.58 (m, 2H, H-3), 3.48 (s, 6H, CH3O), 3.43 (s, 6H, CH3O), 3.29 (dd, J = 3.6, 9.5 Hz, 2H, H-2), 2.21 (s, 12H, CH3Ar); 13C-NMR (101 MHz, CH2Cl2) δ = 159.2, 138.9, 138.4, 130.3, 129.7, 126.6, 105.4 (C-Ar), 98.1 (C-1), 84.3 (C-3), 82.5 (C-2), 78.5 (CI), 78.1 (C-4), 75.6 (CH2Ar), 69.8 (C-5), 68.7 (C-6), 61.2, 58.9, 55.6 (CH3O), 21.5 (CH3Ar); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C42H57IO12Na: 903.27870, found: 903.27835; Anal calcd for C42H57IO12: C 57.27, H 6.52, found: C 57.05, H 6.55.
45: Prepared from 42 (636 mg, 1.40 mmol) and 33 (157 mg, 0.661 mmol) accordingly to general procedure C; colorless solid; 89% (660 mg) yield after column chromatography (PE/EtOAc, 3/1 → 2/1); Rf = 0.55 (PE/EtOAc, 1/1); mp = 69 °C (n-heptane, CH2Cl2); [α]20D = +87.6° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 7.21 (t, J = 8.2 Hz, 1H, H-Ar), 7.11 (s, 4H, H-Ar), 6.47 (d, J = 8.2 Hz, 2H, H-Ar), 4.86 (d, J = 3.6 Hz, 2H, H-1), 4.78 (d, J = 10.4 Hz, 2H, CH2Ar), 4.48 (d, J = 10.4 Hz, 2H, CH2Ar), 4.22 (dd, J = 1.6, 10.3 Hz, 2H, H-6a), 4.16 (dd, J = 4.5, 10.3 Hz, 2H, H-6b), 3.82–3.89 (m, 2H, H-5), 3.70–3.79 (m, 2H, H-4), 3.65 (s, 6H, CH3O), 3.63 (s, 6H, CH3O), 3.54–3.59 (m, 2H, H-3), 3.48 (s, 6H, CH3O), 3.42 (s, 6H, CH3O), 3.30 (dd, J = 3.6, 9.5 Hz, 2H, H-2), 1.36 (s, 36H, C(CH3)3); 13C-NMR (101 MHz, CH2Cl2) δ = 159.7, 159.2, 144.1, 133.0, 130.5, 127.3, 105.6 (C-Ar), 98.1 (C-1), 84.3 (C-3), 82.5 (C-2), 78.6 (CI), 78.2 (C-4), 76.1 (CH2Ar), 69.8 (C-5), 68.8 (C-6), 64.8, 61.2, 58.9, 55.6 (CH3O), 36.1 (C(CH3)3), 32.4 (C(CH3)3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C56H85IO14Na: 1131.48763, found: 1131.48813; Anal calcd for C56H85IO14: C 60.64, H 7.72, found: C 60.59, H 8.02.
46: Prepared from 43 (600 mg, 1.66 mmol) and 33 (178 mg, 0.750 mmol) accordingly to general procedure C; colorless solid; 78% (541 mg) yield after column chromatography (PE/EtOAc, 2/1 → 1/1); Rf = 0.26 (PE/EtOAc, 1/1); mp = 78 °C (n-heptane, CH2Cl2); [α]20D = +120.3° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 8.02 (d, J = 8.4 Hz, 2H, H-Ar), 7.83 (d, J = 8.2 Hz, 2H, H-Ar), 7.75 (d, J = 7.9 Hz, 2H, H-Ar), 7.26–7.48 (m, 8H, H-Ar), 7.08 (t, J = 8.3 Hz, 1H, H-Ar), 6.08 (d, J = 8.3 Hz, 2H, H-Ar), 5.39 (d, J = 11.4 Hz, 2H, CH2Ar), 5.05 (d, J = 11.4 Hz, 2H, CH2Ar), 4.86 (d, J = 3.5 Hz, 2H, H-1), 4.04 (dd, J = 1.2, 10.0 Hz, 2H, H-6a), 3.85–3.94 (m, 4H, H-4, H-6a), 3.80–3.85 (m, 2H, H-5), 3.68 (s, 6H, CH3), 3.60–3.64 (m, 2H, H-3), 3.49 (s, 6H, CH3), 3.41 (s, 6H, CH3), 3.34 (dd, J = 3.5, 9.5 Hz, 2H, H-2); 13C-NMR (101 MHz, CH2Cl2) δ = 158.9, 134.6, 134.2, 132.3, 130.3, 129.1, 129.0, 127.8, 126.7, 126.2, 125.8, 124.7, 105.3 (C-Ar), 98.0 (C-1), 84.4 (C-3), 82.8 (C-2), 78.4 (CI), 77.4 (C-4), 73.3 (CH2Ar), 69.6 (C-5), 68.5 (C-6), 61.2, 58.8, 55.6 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C46H53IO12Na: 947.24740, found: 947.24650; Anal calcd for C46H53IO12: C 59.74, H 5.78, found: C 59.81, H 6.08.
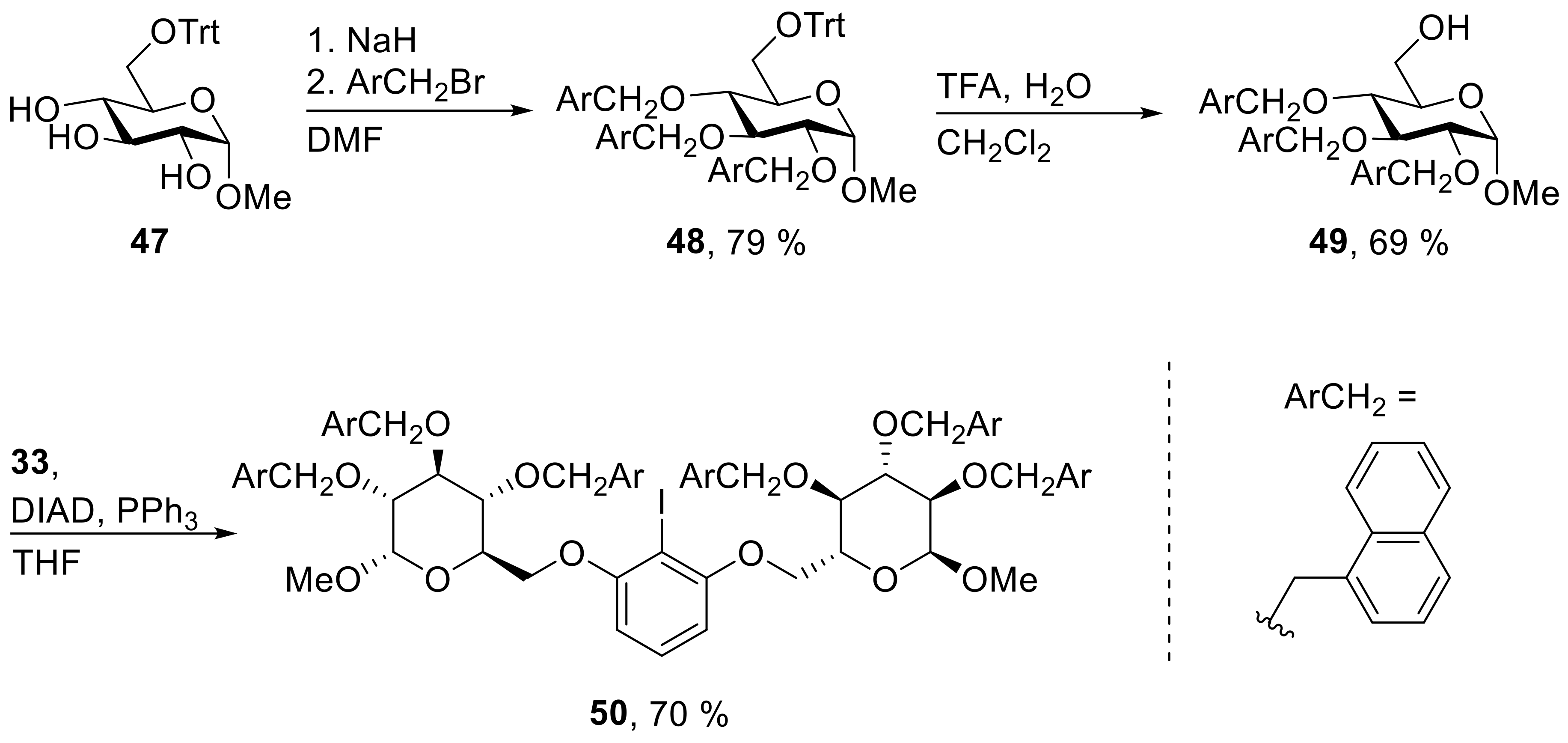
48: Prepared from 47 (3.00 g, 6.87 mmol) and 1-(bromomethyl)-naphthalene (5.32 g, 24.1 mmol) accordingly to general procedure A; colorless solid; 79% (4.65 g) yield after column chromatography (PhMe → PhMe/EtOAc, 20/1); Rf = 0.26 (PhMe); mp = 71 °C (PhMe, EtOH); [α]20D = +58.2° (c = 1.0, PhMe); 1H-NMR (400 MHz, PhMe-d8) δ = 7.87 (d, J = 8.3 Hz, 1H, H-Ar), 7.46–7.62 (m, 13H, H-Ar), 7.39 (dd, J = 6.7, 17.2 Hz, 2H, H-Ar), 6.87–7.28 (m, 28H, H-Ar), 5.42 (d, J = 11.6 Hz, 1H, CH2Ar), 5.25 (d, J = 12.1 Hz, 1H, CH2Ar), 5.05 (d, J = 11.8 Hz, 1H, CH2Ar), 4.98 (d, J = 11.8 Hz, 1H, CH2Ar), 4.87 (d, J = 11.8 Hz, 1H, CH2Ar), 4.79 (d, J = 12.1 Hz, 1H, CH2Ar), 4.73 (d, J = 3.5 Hz, 1H, H-1), 4.31 (t, J = 9.2 Hz, 1H, H-3), 4.01–4.06 (m, 1H, H-5), 3.83–3.91 (m, 1H, H-4), 3.80 (dd, J = 3.5, 9.5 Hz, 1H, H-2), 3.61 (dd, J = 1.7, 9.9 Hz, 1H, H-6a), 3.31 (dd, J = 5.0, 9.9 Hz, 1H, H-6b), 3.23 (s, 3H, CH3); 13C-NMR (101 MHz, PhMe-d8) δ = 144.7, 135.5, 134.8, 134.7, 134.2, 134.0, 133.8, 132.3, 132.0, 131.5, 129.3, 129.2, 129.1, 128.6, 128.5, 128.2, 128.0, 127.7, 127.1, 126.5, 126.2, 125.9, 125.9, 125.8, 125.8, 125.6, 125.5, 125.5, 125.4, 125.3, 124.9, 124.5, 123.9 (C-Ar), 98.3 (C-1), 86.9 (CPh3), 82.3 (C-3), 81.6 (C-2), 78.8 (C-4), 73.7, 72.6, 71.5 (CH2Ar), 71.0 (C-5), 63.6 (C-6), 54.8 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C59H52O6Na: 879.36561, found: 879.36644; Anal calcd for C59H52O6: C 82.68, H 6.12, found: C 82.45, H 6.34.
49: Prepared from 48 (4.45 g, 5.19 mmol) accordingly to general procedure F; colorless solid; 69% (2.19 g) yield after column chromatography (PhMe/EtOAc, 5/1); Rf = 0.31 (PhMe/EtOAc, 5/1); mp = 67 °C (CH2Cl2, EtOH); [α]20D = +23.0° (c = 1.0, CHCl3); 1H-NMR (400 MHz, CDCl3) δ = 8.12 (dd, J = 1.0 8.0 Hz, 1H, H-Ar), 7.96 (d, J = 8.4 Hz, 1H, H-Ar), 7.80 (d, J = 8.3 Hz, 1H, H-Ar), 7.62–7.76 (m, 6H, H-Ar), 7.11–7.43 (m, 13H, H-Ar), 5.44 (d, J = 11.5 Hz, 1H, CH2Ar), 5.12–5.29 (m, 3H, CH2Ar), 4.90–4.99 (m, 2H, CH2Ar), 4.23 (d, J = 3.5 Hz, 1H, H-1), 4.09–4.15 (m, 1H, H-3), 3.55–3.66 (m, 3H, H-2, H-4, H-5), 3.46 – 3.53 (m, 1H, H-6a), 3.41 (dd, J = 3.4, 10.9 Hz, 1H, H-6b), 3.07 (s, 3H, CH3); 13C-NMR (101 MHz, CHCl3) δ = 134.4, 133.8, 133.7, 133.5, 133.5, 133.4, 131.7, 131.3, 131.2, 129.1, 128.5, 128.2, 127.2, 126.2, 126.2, 126.1, 126.0, 125.8, 125.7, 125.7, 125.5, 125.3, 125.2, 125.1, 124.3, 123.8, 123.6 (C-Ar), 98.1 (C-1), 82.0 (C-2), 79.7 (C-3), 77.1 (C-4), 73.5, 72.6, 72.0 (CH2Ar), 70.6 (C-5), 61.7 (C-6), 55.1 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C40H38O6Na: 637.2560677, found: 637.25562; Anal calcd for C40H38O6: C 78.15, H 6.23, found: C 78.13, H 6.28.
50: Prepared from 49 (1062 mg, 1.73 mmol) and 33 (186 mg, 0.786 mmol) accordingly to general procedure C; colorless solid; 70% (784 mg) yield after column chromatography (PhMe/EtOAc, 30/1 → 20/1); Rf = 0.48 (PhMe/EtOAc, 10/1); mp = 98 °C (n-heptane, CH2Cl2); [α]20D = +54.5° (c = 1.0, CH2Cl2); 1H-NMR (400 MHz, CD2Cl2) δ = 8.19 (d, J = 8.3 Hz, 2H, H-Ar), 8.08 (d, J = 8.4 Hz, 2H, H-Ar), 7.75–7.97 (m, 12H, H-Ar), 7.64–7.74 (m, 2H, H-Ar), 7.54 (d, J = 6.7 Hz, 4H, H-Ar), 7.33–7.49 (m, 12H, H-Ar), 7.22–7.31 (m, 6H, H-Ar), 7.17–7.19 (m, 3H, H-Ar), 7.00 (t, J = 8.3 Hz, 1H, H-Ar), 5.96 (d, J = 8.3 Hz, 2H, H-Ar), 5.52 (d, J = 11.5 Hz, 2H, CH2Ar), 5.34 (d, J = 11.9 Hz, 2H, CH2Ar), 5.18–5.29 (m, 4H, CH2Ar), 5.13 (d, J = 11.7 Hz, 2H, CH2Ar), 5.02 (d, J = 11.9 Hz, 2H, CH2Ar), 4.71 (d, J = 3.4 Hz, 2H, H-1), 4.15–4.27 (m, 2H, H-3), 3.89–4.07 (m, 6H, H-4, H-5, H-6a), 3.76–3.88 (m, 4H, H-2, H-6b), 3.31 (s, 6H, CH3); 13C-NMR (101 MHz, CH2Cl2) δ = 158.8, 135.3, 134.5, 134.5, 134.3, 134.1, 132.3, 132.0, 132.0, 130.2, 129.5, 129.3, 128.9, 128.9, 128.9, 128.7, 128.6, 127.3, 127.2, 126.7, 126.6, 126.5, 126.5, 126.3, 126.1, 125.9, 125.8, 125.8, 124.8, 124.6, 124.4, 105.2 (C-Ar), 98.4 (C-1), 82.7 (C-3), 81.2 (C-2), 78.4 (CI), 77.9 (C-4), 73.9, 73.3, 71.9 (CH2Ar), 69.7 (C-5), 68.5 (C-6), 55.7 (CH3); HRMS (ESI-TOF) m/z [M + Na]+: calcd for C86H77IO12Na: 1451.43520, found: 1451.43439; Anal calcd for C86H77IO12: C 72.26, H 5.43, found: C 72.55, H 5.86.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
